REVIEW: Functions of Noncoding Sequences in Mammalian Genomes
L. I. Patrushev* and T. F. Kovalenko
Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia; E-mail: patrush@mx.ibch.ru* To whom correspondence should be addressed.
Received October 1, 2014
Most of the mammalian genome consists of nucleotide sequences not coding for proteins. Exons of genes make up only 3% of the human genome, while the significance of most other sequences remains unknown. Recent genome studies with high-throughput methods demonstrate that the so-called noncoding part of the genome may perform important functions. This hypothesis is supported by three groups of experimental data: 1) approximately 10% of the sequences, most of which are located in noncoding parts of the genome, is evolutionarily conserved and thus can be of functional importance; 2) up to 99% of the mammalian genome is being transcribed forming short and long noncoding RNAs in addition to common mRNA; and 3) mutations in noncoding parts of the genome can be accompanied by progression of pathological states of the organism. In the light of these data, in the review we consider the functional role of numerous known sequences of noncoding parts of the genome including introns, DNA methylation regions, enhancers and locus control regions, insulators, S/MAR sequences, pseudogenes, and genes of noncoding RNAs, as well as transposons and simple repeats of centromeric and telomeric regions of chromosomes. The assumption is made that the intergenic noncoding sequences without definite/clear functions can be involved in spatial organization of genetic loci in interphase nuclei.
KEY WORDS: noncoding sequences, pervasive transcription, transcription regulation, promoter, 5′-UTR, 3′-UTR, DNA methylation, CpG islands, intron, splicing, transcription enhancer, LCR, insulator, S/MAR, noncoding RNA, siRNA, miRNA, lncRNA, pseudogene, repeated sequences, MGEDOI: 10.1134/S0006297914130021
Abbreviations: CAGE, cap analysis gene expression; ceRNA, competing endogenous RNA; CGI, CpG islands; ChIP, chromatin immunoprecipitation; CNV, copy-number variation; DHS, DNase hypersensitivity site; eRNA, enhancer RNA; GWAS, genome-wide association studies; hnRNP, heterogeneous nuclear RNP; HOR, higher order repeat; ICE, imprint control element; ICR, imprinting control region; IGS, intergap segments; IME, intron-mediated enhancement; intRNA, intron RNA; kb, thousand base pair; LCR, locus control region; lincRNA, long intergenic noncoding RNA; LINE, long interspersed nuclear elements; lncRNA, long noncoding RNA; Mb, million base pair; 5mC, 5-methylcytosine; MGE, mobile genetic elements; miRNA, microRNA; mtDNA, mitochondrial DNA; ncRNA, noncoding RNA; NGS, new generation sequencing; NIM, neutral indel model; NUMT, nuclear mitochondrial; satDNA, α-satellite DNA; shRNA, short RNA; SINE, short interspersed nuclear elements; siRNA, small interfering RNA; S/MAR, scaffold/matrix attachment regions; SNP, single-nucleotide polymorphism; snRNA, small nuclear RNA; SNV, single-nucleotide variant; TAD, topologically associating domain; tDNA, telomere DNA; TERRA, telomeric repeat-containing RNA; TERT, telomerase catalytic subunit; TR, telomerase RNA; TSS, transcription start site; uORF, upstream open reading frame; UTR, untranslated region (of RNA); vlincRNA, very long intergenic noncoding RNA.
The genome of a living organism stores and implements most of the
genetic (inherited) information in the course of ontogenesis.
Functioning of the genome – above all manifested through
efficient expression of the genes enclosed – forms the basis for
life of Earth. In eukaryotes, genes are sequences of nucleotides of
genomic DNA affecting a phenotypic trait [1, 2]; to work correctly, genes are to respond without
fail to the multiple endogenous and exogenous regulatory signals. This
function of the genome is fulfilled by the regulatory sequences
recognized by relevant regulatory molecules of the organism. In this
connection, a genome-wide search for regulatory elements is necessary
to understand genome organization and is one of the key problems of
modern functional genomics [3].
The human genome sequencing data obtained by now has unleashed several specific features of genome organization. Surprisingly, sequences of DNA nucleotides encoding exon regions of genes were found to make up only a small part (2.94%) of the genome, while exons, encoding only amino acid sequences (without the untranslated exons and their fragments) make up even less (1.2%) [4]. In this review, we will call the rest of the sequences, making up ~98% of the genome, noncoding sequences. The total number of genes turned out to be less than was expected (~25,000); however, the mechanisms regulating their expression and the regulatory sequences demonstrate extraordinary diversity [5]. Besides, sequences of individual human genomes turned out to be highly polymorphic. Comprehensive analysis of individual genomes, the total number of which reaches 1.5 thousands, revealed dozens of millions of single-nucleotide polymorphisms (SNP and SNV), millions of short insertions and deletions (indels), and multiple extended sequences present in a varying number of copies (CNV) [6, 7]. These results indicate that the primary structure of an individual’s genome is unique. Indeed, sequencing of each new genome detects up to 3,500,000 SNPs and ~1000 large (>500 bp) SNVs absent in the reference sequence. Up to 500,000 SNPs in each genome have not been recognized previously. On average, any sequenced genome of an apparently healthy human contains 25,000 SNPs in its coding parts. Up to 10,000 of them are nonsynonymous nucleotide substitutions, among which up to 100 SNPs have been referred to deleterious mutations resulting in pathological states in heterozygotes. Most polymorphic nucleotides are located in noncoding fragments of the genome, the function of which is yet unknown. It is believed that collectively individual features of genome organization, including the noncoding sequences, can determine many specific features of the activity of a human organism, although not unambiguously [8]. Today, the concept is developing in frames of a large unsolved genetic problem of interaction between the genotype and the phenotype [9].
The idea of the importance of genetic factors in etiology of complex multifactorial diseases has been supported by many modern studies on association of SNPs and the development of pathological processes. Lately, the system of genome-wide association studies (GWAS) to search for such associations has been actively developing and has already yielded fruits [10]. Within the approach, biochip technologies are used to study simultaneously the association of millions of SNPs, the clinical value of which is unknown, with a certain disease in control and clinical case groups of hundreds of thousands of participants. As a result, over 12,000 new genetic loci associated with studied pathological states have been revealed, which, in a number of cases, led to a paradigm shift in understanding of molecular mechanisms of the disease (www.genome.gov/gwastudies/). However, interpretation of the results obtained by GWAS encounters considerable difficulties. Correlation between an SNP allele frequencies and a disease status is weak (odds ratio typically does not exceed 1.2); besides, most SNPs are located in noncoding sequences of the genome [10]. This suggests that changes in the genome caused by these SNPs are actually associated with functional sequences involved in regulation of gene expression and can interfere with interactions between genes.
The recently discovered phenomenon of pervasive transcription [11] also evidences that the functional role of noncoding sequences in eukaryotic genomes has been underestimated. Analysis of the human transcriptome with modern techniques demonstrated that most sequences in studied eukaryotic genomes are being transcribed. Besides, high phylogenetic conservation of many of the noncoding genome regions between evolutionarily distant species also supports the idea of their functional significance [12].
In this review, we analyze the problem of functionality of noncoding sequences in mammalian genomes. We provide data supporting functional significance of most of the noncoding sequences and briefly consider the specific features of how the main regulatory elements, including 5′- and 3′-UTR, introns, transcription enhancers, insulators, DNA methylation regions, S/MAR sequences, noncoding RNA genes, and repeated sequences function. Finally, we review the mutations in noncoding parts of the genome associated with human diseases, which supports the functional importance of noncoding sequences.
FUNCTIONS OF NONCODING SEQUENCES IN HUMAN AND ANIMAL
GENOMES
The problem of human genome redundancy in terms of sequences of DNA nucleotides not coding for proteins remains unsolved. Till now, evolutionary conservation of the gigantic human genome (the size of a haploid genome is 3.3·109 bp), as well as those of other eukaryotes, all rich with noncoding sequences, appears to be a mystery. Attempts to explain the phenomenon have conceived multiple theories, which can be divided in two large groups. Supporters of the first group of theories believe that most of noncoding sequences are not functional and reflect evolutionary accumulation of shatters of genes no longer needed or represent selfishly multiplying DNA. Another group of researchers, to which the authors of this review belong, on the contrary, supposes that most of the noncoding sequences should be functional [13, 14].
With the elaboration of a reference human genome sequence, elucidation of all functionally important genome parts became a realistic task. To solve the task, an international consortium on the development of an ENCyclopedia Of DNA Elements (ENCODE, http://genome.ucsc.edu/ENCODE/) was created in 2003; the main goal of the consortium is to search for functional elements of the human genome controlling gene expression and to compile a comprehensive catalog of such elements. To detect functionally important sequences, the consortium members, as well as independent researchers, use three groups of methods: methods of evolutionary biology, biochemistry, and genetics [15]. In the frame of the evolutionary approach, methods of comparative structural genomics reveal the evolutionarily conserved parts of the mammalian genome under natural selection; biochemists produce evidence of molecular activity, while geneticists register phenotypical changes in the organism in response to mutational rearrangements of the primary structure of genomic DNA. All three approaches are highly informative and complement each other, although the contributions of each to human genome annotation are different. Analysis of DNA mutations leading to disease progression also contributes to our knowledge on the functional role of noncoding sequences.
Evolutionary Conservation of Noncoding Sequences in the Genome
The evolutionary approach to determination of functionally important DNA sequences in the human genome utilizes the αsel parameter, which denotes the share of the genome nucleotides under purifying (negative) selection among all genome nucleotides [16]. To determine the parameter, two groups of methods are used today. First, in comparative studies conserved genomic sequences in distant divergent species are revealed under the assumption that sequences that perform important functions remain unchanged for a long evolutionary period. The technique is poorly applicable to the analysis of rapidly evolving regulatory regions of the genome. In the latter case, the neutral indel model (NIM) [17] is used instead. In this model, the size of DNA fragments located between adjacent indel sites termed intergap segments (IGS) is evaluated genome-wide. The IGS length distribution is strongly shifted towards long sequences upon analysis of the whole genome, which is in sharp contrast with the IGS length distribution profile of neutrally evolving genome parts. This is interpreted as evidence for selection aimed at preservation of these long sequences in the genome and, consequently, their probable functional significance.
When human and murine genomes were first aligned and compared, the percentage of sequences under negative selection among short (50-100 bp) genome segments was found to be ~5% [18].
In a recent study on evolutionary conservation of genomic DNA sequence, the human genome was compared to 28 genome sequences of maximally diverged placental mammals [19]. Most of the genomes, the primary structure of which was unknown prior to the study, were sequenced in the course of the work. Alignment of the genome sequences showed that approximately 5.5% of the human genome has undergone purifying (negative) selection, and 4.2% are represented by conserved, presumably functional, sequences. These conserved DNA elements, totaling ~3.6 million, were mostly represented by exons comprising mRNA (~30%), gene introns (~30%), and intergenic segments (~40%). Synonymous nucleotide substitution constraints were detected in exons of over a quarter of all known human genes. In the latter case, sequences are highly conserved probably due to the necessity to maintain certain mRNA structure during splicing, interactions between regulatory microRNA (miRNA), or its editing at the posttranscriptional level. In the study, approximately 4000 new exon candidates were discovered among gene transcripts encoding proteins, their introns, and untranslated mRNA sequences (UTR). Over 1000 conserved sequences presumably associated with a new class of noncoding RNA as well as 2.7 million short presumably regulatory sequences were found. Besides, up to 40% of the nucleotides of the revealed conserved sequences remain uncharacterized in terms of their possible functionality.
With an advanced version of the above-mentioned NIM approach, which reveals the rapidly evolving genome parts, it was found that up to 8-9% to the total number of human genome nucleotides is under negative selection and thus may be functional [20]. Only 2% of sequences turned out to be conserved upon comparison of human and mouse genomes. These data indicated that there are many short-living species-specific sequences under negative selection in the studied genomes. Most of these sequences were found to be located in noncoding fragments of the genome and have been referred to known functional elements, including the DNase I hypersensitivity sites, transcription factor binding sites, promoters, enhancers, and long noncoding RNA (lncRNA) genes. The latter turned out to be the most evolutionarily unstable.
Therefore, modern methods of DNA sequencing and bioinformatics has allowed a new take on the problem of noncoding sequences, which are now viewed as rich with probably functional elements of the genome. Further development of molecular biology methods confirmed and broadened this new concept.
Pervasive Transcription
The first indication that most of the human genome is being transcribed was obtained upon study of transcription of small chromosomes 21 and 22 and was confirmed by the ENCODE consortium in an attempt to comprehensively analyze the functional activity of 1% of the human genome [5, 21-24]. At least 93% of the genome was found to be transcribed with varying efficiency jointly in cells of different human tissues. Non-annotated transcripts with unknown functions were termed “dark matter” [25]. Mining these data was made possible mainly by two modern methods of transcriptome investigation: cDNA hybridization with ordered oligonucleotide probes organized into high-density biochips (tiling arrays) and RNA-seq modification of DNA next generation sequencing (NGS) [26-28].
Modern biochips can contain millions of oligonucleotide probes, sequences of which cover the entire human genome. In the course of transcriptome analysis, total RNA isolated from a small number of cells of tissue under study is freed from the products of actively transcribed genes (especially rRNA) and used for cDNA synthesis, which is further amplified with PCR, fragmented, labeled with fluorescent dyes, and hybridized with the biochip. Fluorescence signals are registered and analyzed in an automated mode. Biochip technology allows for up to 109 individual hybridizations per chip, producing unique information on specific features of transcription of large genomes, including mapping of the transcripts with resolution of several base pairs [23, 24]. Despite the outstanding capabilities of biochip technology, it is not free from limitations. The high cost of individual experiments, the need to know the sequence of the genome under study, high background level of nonspecific cross-hybridizations, and limited range of quantitative measurements of gene transcription rate due to high background and fast saturation of the hybridization signal are to be mentioned among such limitations [29, 30].
The RNA sequencing (RNA-Seq) methodology competes successfully with the biochip technology in transcriptome studies. The main advantage of this approach is the direct determination of nucleotide sequences of cDNA under study with high-performance NGS and direct calculation of individual cDNA molecules in analyzed samples. RNA-Seq allows both mapping of the transcriptome and quantitative evaluation of transcription levels of individual genome fragments. In the course of the analysis, total or fractionated RNA is used to prepare a library of cDNA fragments, to which oligonucleotide adaptors are ligated at one or two ends, allowing for simultaneous solid-phase amplification and/or sequencing of millions of immobilized cDNA fragments at one or two ends using only one or two oligonucleotide primers [31, 32]. In general, application of RNA-Seq methods in functional transcriptomics does not require the knowledge of the primary structure of the genome under study and allows elucidation of structural variants (for example, SNPs) of its transcripts [33]. Besides, as opposed to biochip technology, RNA-seq methods are characterized by very low level of background signals. The main disadvantage of the methods are the artifacts associated with cDNA library preparation and the so far low efficiency of bioinformatics methods elaborated for the analysis of big data, leaving much to be desired [34]. For other approaches to transcriptome analysis, see works [35, 36].
Today the phenomenon of pervasive transcription has been reliably confirmed experimentally [11, 37]. In all studied human cells and tissues, both healthy and tumorous, RNA molecules with unknown function make up the majority of total RNA preparations after separation of rRNA and mitochondrial RNA [38]. Hundreds of genome regions, in which very long intergenic noncoding RNA (vlincRNA) exceeding 100 kb are transcribed, have been discovered; such regions spread over 4% of intergenic sequences of the genome. The sets of vlincRNA turn out to be tissues-specific, and most of the genome sequences are represented in total RNA isolated from many tissues. Surprisingly, sequencing of individual RNA molecules revealed that the fraction of intron (int) RNA makes up 30-50% of the total human RNA after separation from rRNA and mtRNA. Different genes and even separate fragments of introns differ by their ability to generate intRNA [38]. The comprehensive functional significance of intRNA is yet to be understood, however already it is clear that almost half of sequences of known miRNA, which we will speak of further on in a relevant section of this review, belongs to intRNA.
Mobile genetic elements (MGE) of the human genome, which make up ~45% of all its sequences, contribute significantly to the pervasive transcription. For a long period, investigation of the global transcription of repeated sequences has been hampered by difficulties in discriminating between different transcripts due to their cross-hybridization on biochips. Elaboration of the cap analysis gene expression (CAGE) system together with NGS techniques solved this methodological problem [39, 40]. It turned out that up to 30% of capped transcripts stem from the repeated sequences of the human genome. Overall, ~250,000 transcription start sites (TSS) have been detected in genomic retrotransposons; their distribution over the genome is tissues-specific; they cluster in gene-rich parts of the genome.
In general, despite the lack of knowledge on the functional significance of the newly discovered RNA of the mammalian transcriptome, it is clear that many noncoding RNAs perform important functions in regulation of gene expression and maintenance of genome stability. Structural and functional features of the main classes of noncoding RNAs will be considered below in relevant sections of this review.
Cis-Acting Regulatory Elements of Transcription and Translation
Protein-encoding gene expression is regulated at many levels, including transcription, co- and posttranscriptional mRNA processing, translation, and posttranslational processing of synthesized polypeptides [41]. Noncoding sequences termed regulatory genome elements play an important role in control of transcription and translation. Promoters and sequences encoding regulatory signals of 5′- and 3′-UTR, together with splicing sites and splicing enhancers/silencers, are some of these regulatory elements. Besides, regulatory elements performing global regulation of transcription, such as enhancers, locus control regions, insulators, S/MAR sequences, and specific regions of DNA methylation are referred to this group. Most of the regulatory elements are located in close proximity to genes they control, within the same chromosome domain, and consequently perform regulatory effect in cis, that is within the controlled region of the genome. Below we review the main structural and functional features of noncoding cis-acting regulatory elements, which make up a significant and indispensable part of the genome.
5′-Terminal regulatory elements of genes. Promoters. Gene promoters are specialized sequences of genomic DNA that enclose TSS, i.e. the first nucleotide (position +1) from which RNA synthesis starts [42]. The functional role of promoters is the provision for initiation complex assembly and further initiation of transcription, the key player in which is the DNA-dependent RNA polymerase. In humans, protein-coding genes and many noncoding RNAs are transcribed by RNA polymerase II.
The region around TSS is divided into upstream prolonged proximal promoter and minimal (or core) promoter surrounding the TSS. Proximal promoter locates binding sites for some proximal transcription factors, which in turn can group into clusters of cis-regulatory modules.
Core promoter is the minimal DNA sequence providing for precise initiation of transcription by RNA polymerase II [43]. Purified RNA polymerase II is not able to recognize core promoters and requires basic transcription factors to implement its function [44]. The factors include TFIIA (RNA polymerase II transcription factor A), TFIIB, TFIID, TFIIE, TFIIF, and TFIIH. The core sequence is recognized by TFIID, which comprises the TATA box-binding protein (TBP) and TBP-associated factor (TAF). In the presence of these factors, RNA polymerase II interacts with the core promoter and is enrolled in the large preinitiation complex (PIC).
Functions of the core promoter determine the specific sequences it comprises. The sequences have been termed elements (or motifs) of the core promoter. By now, no elements universal for all promoters have been elucidated. The most commonly occurring element of core promoters is the initiator (Inr) comprising the TSS (with a consensus sequence in humans of YYANWYY, where the A is at position +1), and it appears that basal transcription requires the interaction of TFIID with Inr. The consensus sequence of the TATA box is TATAWAAR, with the 5′-terminal T located at position –30 or –31 from the A of Inr. The TBP subunit of factor TFIID interacts with the sequence. Two TFIIB recognition elements (BRE) can be located both upstream (BREu) and downstream (BREd) from the TATA box, with which they are functionally linked, and produce both positive and negative effects on the basal level of transcription. Located close to each other and sometimes even overlapping, downstream core promoter element (DPE) and motif ten element (MTE) contact with subunits TAF6 and TAF9 of TFIID. These sequences contain four regions at positions 18-22 (CGANC) and 27-29 (CGG) of MTE and 27-29 and 30-33 of DPE (with respect to position +1 in the Inr) particularly important for their functionality. The presence of the first and the third regions is sufficient for a functional core promoter, although such a combination occurs rarely in natural promoters.
In addition to the indicated basic elements, promoters of some genes may contain specific sequences necessary for binding of certain activating factors. For example, E-box with a canonical sequence of CACGTG belongs to this group of elements. E-box is a binding site for proteins belonging to the helix-loop-helix family; it is present in many genes expressed in muscle and nervous tissues and in pancreas [45]. Recent studies demonstrated that E-box or similar elements occur in promoters of the so-called clock genes, expression of which obey circadian rhythms (that is, evolves cyclically during the day) [46]. Another example is the heat shock elements (HSEs) contained in promoters of heat shock protein genes. These elements are binding sites for transcription factors known as the heat shock factors. HSEs are represented by several copies of a pentanucleotide sequence NGAAN-3′ differently oriented with respect to each other. The number of pentanucleotide monomers can differ in promoters of different genes; however, three units are the least sufficient number for successful transcription activation [47].
In mammals, three types of promoters are distinguished by their nucleotide composition and functions. Promoters encompassing the TATA box with low GC content are referred to as type I promoters. In most promoters of this type, transcription initiation is limited by one or several closely located nucleotides (focused transcription), and the TATA box is located at a certain distance from the TSS, which, together with the initiator consensus sequence, determines the choice of TSS on the DNA. Type I promoters govern the tissue-specific transcription in adult organisms and are methylated in the active state.
Type II and III promoters are characterized by high GC content and the presence of multiple TSSs located within a 50-100-nt-long region (dispersed transcription). Type II promoters have short CpG islands (CGI), lack the TATA box, and direct transcription of most housekeeping genes. Type III promoters contain prolonged CGIs covering gene sequences, are marked by trimethylated histones H3K27me3, and are regulated by the Polycomb repression system. Promoters of this type control genes involved in regulation of development and cell differentiation.
An individual group is formed by the so-called TCT promoters typical for highly expressed genes of ribosomal proteins. Their name derives from the typical structure of the initiator, which contains a polypyrimidine tract YYC+1TYTTYYY (TSS corresponds to +1T) [48].
The 5′-terminal sequences of some genes can contain more than a single promoter. Alternative promoters of a single gene can function in different tissues or at different developmental stages of the organism [49]. Despite the fact that RNA transcripts obtained from alternative promoters may be of different size, the encoded proteins might be identical if the same translation initiation site is utilized [49]. Another type of alternative promoters are the promoters the activity of which results in various protein isoforms with different properties, for example, the methyltransferase gene DNMT1 promoter. The 5′-terminal region of the gene includes three promoters. In somatic cells, the 1s promoter is active, while in oocytes it is the 1o promoter. In the latter case translation starts from the ATG codon located in exon 4. The resulting protein is shorter than the enzyme synthesized in somatic cells by 118 N-terminal amino acids. In spermatocytes, promoter 1p is active. However, mRNA formed from this promoter is not translated. As a result, spermatocytes contain no DNMT1 enzyme [50].
The genome of eukaryotes and humans in particular contains many genes located in close proximity to each other and transcribed from different strands of the same DNA molecule in opposite directions. Promoter regions of such genes partially overlap, and the distance between their TSS does not exceed 1000 bp. The DNA fragment localized between the TSS of two genes transcribed from different strands was termed a bidirectional promoter [51, 52]. In particular, arrangement of many of DNA reparation system genes and chaperon genes implements bidirectional promoters. In most cases, bidirectional promoters provide for simultaneous expression of two genes; however, sometimes initiation of transcription of one gene of the gene pair suppresses the transcription of the other [51]. The primary structure of bidirectional promoters has some specific features. For example, only 9% of such promoters contain TATA boxes. The average GC content in bidirectional promoters is somewhat higher than in the usual ones (66 and 55%, respectively).
Mutations easily destroy functional integrity of the promoters due to their overload with regulatory elements interacting with transcription factors. For example, the TERT gene encodes a catalytic reverse transcription subunit of telomerase maintaining the telomere length (see below the section on telomeres). Elevated telomerase activity is among the specific features of tumorous tissues. The promoter of the TERT gene contains many binding sites for various activators and repressors. The A57C, C124T, and C146T mutations create a new overlapping site for binding transcription factors Ets and TCF, thus increasing the level of expression of the TERT gene. The indicated somatic mutations are incompatible and occur at different rates in various oncological diseases. The C124T mutation is the most frequent one in bladder cancer (occurs in 53.5% patients). In general, the three mutations occur in 65.4% of bladder cancer patients and in 80% of glioma patients. These genetic changes are associated with a more aggressive progression of the disease and negative prognosis [53].
Another example is the Leiden hemophilia B characterized by low content of blood clotting factor IX (60% of the normal concentration at most), in which the G26C and T20A SNPs in the promoter of the F9 gene impair the overlapping site of TATA/HNF-4 binding. HNF-4 is the hepatocyte nuclear receptor protein controlling the expression of the factor IX gene. Inability of HNF-4 to bind the modified site leads to hemophilia [54].
The 5′-terminal untranslated regions (5′-UTR), typically encoded by the first exon in eukaryotic genes, enclose sequences providing for regulated mRNA translation. The average length of human 5′-UTR is 210 nt, and the minimal one is 18 nt. The maximal length of a 5′-UTR sequence in humans (2858 nt) has been noted for the Tre oncogene mRNA [55]. The length of the 5′-UTR influences the efficiency of mRNA translation, since the ribosome has to hurdle the often highly structured region of the template between the initiatory AUG codon and the m7G cap group, where it is assembled. High GC content is typical of 5′-UTRs. Ten to fifteen percent of mammalian genes utilize alternative 5′-UTR owing to transcription initiation at different promoters. In another 13% of the genes, the same effect is reached via alternative splicing [56]. Utilization of an alternative 5′-UTR has important consequences for translation and expression of genes.
The secondary structure of 5′-UTRs plays a big role in regulation of expression of genes, in particular, transcription factor and growth factor genes, as well as protooncogenes and their receptors. Over 90% of mRNA molecules have constitutive stable secondary structures in their 5′-UTR sequences, typically located not far from the cap group. Regulatory proteins interact with these structures, which can be accompanied by inhibition of translation from the mRNA. RNA-binding proteins (RBP) (over 1000 have been discovered in humans) are divided into two large groups: (1) RBP essential for translation from all mRNAs and (2) specific RBP regulating translation of certain mRNAs. RBPs interacting with the same sequences can act as antagonists inhibiting or activating translation; CUGBP1 and calreticulin regulating the expression of p21 involved in cell cycle regulation provide an example [57].
The upstream open reading frames (uORFs), which are short ORFs located before the main reading frame carrying their own initiation codon but lacking a translation termination codon, also belong to the major regulatory elements of 5′-UTR. Up to 50% of RNAs of human and mouse transcriptomes contain uORFs, which are rather heterogeneous in terms of their size and position [58]. Functional activity of a uORF is determined by the distance from the cap group, its primary structure, the context in which it is located, its size, efficiency of the translation initiation site, and the number of AUG codons it carries. The uORFs modulate scanning of the mRNA 5′-terminus with ribosomes, in which uAUG codons act as traps. Besides, ribosomes are held back at the end of uORF upon termination of translation resulting in interactions between the attenuating peptides and the ribosomes [59]. Many human pathologies are associated with mutations affecting uORF, including the predisposition to melanoma and breast cancer, inherited thrombocytemia, Alzheimer’s disease, and hypotrichose (see reviews [58, 60] for references).
3′-Terminal regulatory elements of genes. The 3′-terminal noncoding elements of genes comprising the 3′-terminal untranslated regions (3′-UTR) of mRNA perform important regulatory functions at the posttranscriptional and translational levels of eukaryotic gene expression [61]. With their start being at the translation termination codon, these sequences are involved in mRNA processing, they control mRNA stability and localization of the transcripts in cells, and they influence the rate of translation. Analysis of the 3′-UTR of human genes revealed considerable size heterogeneity among them. Their average length is 1.3 kb or ~36% of the length of a mature mRNA (without the poly(A) tract), although it can exceed 5 kb (with the highest registered length of 8.5 kb) [55, 62]. This is almost twice the average size of 3′-UTR in other mammals, which is probably due to the presence of a larger number of regulatory sequences in human 3′-UTR and finer regulation of translation in humans.
When pre-mRNA synthesis is complete, its polyadenylation signals (poly(A) sites) with the consensus AAUAAA sequence direct the assembly of a ribonucleoprotein complex consisting of ~85 proteins, which shortens pre-mRNA at its 3′-terminus and ensures binding of a poly(A) sequence ~250-nt-long. Approximately 1/3 of mRNA molecules contain two or more poly(A) sites that can be used in the course of pre-mRNA processing as alternatives. The poly(A) sequence is a signal for binding of mRNA with regulatory poly(A)-binding proteins (PABP), which is necessary for mRNA export from the nucleus, regulation of its stability, and transcription surveillance, as well as positive and negative regulation of translation by ribosomes, including translation control by miRNA (reviewed in [63]). During initiation of the translation, the 5′-terminal cap group and 3′-UTR act synergistically, which is made possible by their interaction upon mRNA circularization. In the case of mRNA of the human p53 gene, there is a sequence in its 3′-UTR complementary to the 5′-UTR of the same mRNA that interacts with the RPL26 translation factor, which stimulates translation of the mRNA in response to DNA damage in a cell [64].
The 3′-UTRs are among the major targets of regulatory miRNAs and not only inhibit, but also stimulate translation of relevant mRNAs by facilitating their circularization (see relevant section below).
The necessity for 5′- and 3′-terminal interactions in mRNA, as well as the interactions with miRNA in the course of translation of mRNA, sets the requirements for the length and secondary structure of 3′-UTRs. In general, longer 3′-UTRs associate with lower level of translation of their mRNA, which is particularly due to the presence of a larger number of miRNA-binding sites in them. For example, some mRNA isoforms, such as transcripts of the Hip2 gene, use alternative 3′-UTRs of varying length that differ in the number of miRNA-binding sites [65].
The AU-rich element (ARE) located in 3′-UTRs containing a single or several (up to five) AUUUA sequences controls mRNA stability [66]. The UUAUUUAUU nonamer or its UUAUUUAWW analog are the minimal sequences leading to destabilization of mRNA, while the WWWUAUUUAUWWW 13-nucleotide sequence is considered a minimal ARE in humans. Increase in the number of copies of the pentamer motif in The ARE decreases mRNA stability. Multifunctional proteins interacting with ARE accelerate poly(A) sequence shortening to 30-60 nt, which is critical for mRNA stability; then, the process enters the second phase, when rapid degradation of the central part of the mRNA molecule occurs. Functionality of ARE-binding proteins, in turn, is regulated via posttranslational modifications (phosphorylation) or results from directed intracellular localization. The miRNA also regulates ARE.
In addition to the above-mentioned motifs, CU-rich element (CURE) and differentiation control element (DICE), similar in their modes of action, are present in 3′-UTRs of a number of genes. They interact with heterogeneous nuclear (hn) RNP, which is accompanied by inhibition of translation initiation due to specific suppression of 80S ribosome assembly. Phosphorylation of hnRNP in signal transduction activates translation.
GU-rich elements (GRE) of 3′-UTR consisting of 2-5 overlapping GUUUG pentamers are present in 5% of human mRNAs [67]. When protein-bound, GRE participates in deadenylation, degradation, and splicing of mRNA.
CA-rich elements (CARE) are among the most abundant dinucleotide repeats in the human genome in both coding and noncoding sequences. They produce a stabilizing effect on mRNA upon interaction with an hnRNP L, a global pre-mRNA processing regulator.
Iron responsive elements (IRE) controlling iron metabolism were found in 5′- and 3′-UTRs of 10 human genes, in particular, the genes of light and heavy ferritin polypeptides (FTL and AEP1, respectively) and transferrin receptor 1. Stem-loop secondary structure is typical for 26-30-nt-long IRE sequences; the structure interacts with two proteins regulating iron metabolism, iron regulatory proteins (IRP) 1 and 2. IRP1 binds 5′-UTR and suppresses translation, preventing the binding of minor and major ribosome subunits. IRP2 stabilizes mRNA through binding the 3′-UTR [68].
The 3′-UTR sequence containing the selenocysteine insertion sequence element (SECIS) forms the stem-loop structure recognized by proteins that ensure decoding of the UGA stop codon as a codon for the essential amino acid selenocysteine [69].
To conclude, it should be noted that many human 3′-UTRs contain Alu retroelements interacting with lncRNA that contain similar complementary sequence. As a result, a fragment of double-stranded RNA is created that binds the Staufen-1 (Stau1) protein, which directs mRNA target towards the degradation route. Besides, a site forming the intramolecular double-stranded structure in an RNA, which interacts with Stau1, can be present in the 3′-UTR as such [70].
Saturation of 3′-UTRs with functional elements makes them, similarly to promoters, rather sensitive to mutational substitutions of the nucleotides accompanying the development of many pathological states of the human organism, including immune system dysfunctions and innate cardiovascular diseases [60, 66]. For example, prothrombin (blood coagulation factor II) is a thrombin precursor. The latter plays the major role in fibrinogen transformation to fibrin upon clot formation and in other reactions ensuring hemostasis. The G20210A mutation in the prothrombin gene affects its 3′-UTR, which is accompanied by a 1.5-2-fold increase in prothrombin content in plasma. This, in turn, results in a 3-fold increase in thrombosis risk [71].
Polymorphism of C/T in the 3′-UTR of the estrogen receptor 1 (ESR-1) gene affects the miR-453 miRNA-binding site. Replacement of C with T decreases the efficiency of miR-453 binding with the site. As a result, the level of ESR-1 gene expression increases, which is associated with elevated risk of breast cancer [72].
DNA methylation regions. In animal DNAs, cytosine can be methylated at position 5 of the pyrimidine ring with high specificity, forming 5-methylcytosine (5mC), mainly in CpG sequences spread over the genome nonrandomly (see [73] for review). Methyl groups of 5mC are located in the DNA major groove, which makes them available for recognition by specific proteins possessing methyl-CpG binding domains (MBD). This, in turn, creates the conditions for interaction between these proteins and other protein complexes suppressing transcription via various mechanisms, including the heterochromatization of relevant DNA regions. Besides, the presence of 5mC can prevent binding of transcription factors to regulatory regions of DNA, although this is not a common mechanism of gene suppression [74]. In turn, the interaction of transcription factors with regulatory regions of DNA in a number of cases is necessary and sufficient to maintain CpG dinucleotides in these sequences nonmethylated, creating low-methylated regions (LMR) in the genome [75].
DNA methylation has other functions as well. Repression of transposon gene transcription through DNA methylation prevents their mobilization, which is an important mechanism of genome stability maintenance [76]. Besides, methylation of sequences inside the active genes can apparently produce regulatory effects on splicing and the choice of alternative promoters [77]. In somatic tissues, up to 80% of CpG dinucleotides is methylated. Mapping of DNA methylation regions in the genome by high-throughput methods demonstrated that, in accordance with the above-mentioned functions, satellite and other repeating sequences, including transposons and their remnants, unique intergenic sequences, and other noncoding sequences, as well as gene exons, are methylated in somatic cells. The level and patterns of methylation in these sequences in compliance with CpG dinucleotide localization in them can differ considerably between individuals of the same biological species, representing genomic polymorphisms and epimutations [78, 79].
Maintenance DNA methyltransferases, such as DNA methyltransferase 1 (Dnmt1), maintain characteristic patterns of DNA methylation (cell methylome) for many cell generations. Inactivation of the enzyme by gene knockout in mouse embryonic stem cells is accompanied by global demethylation of DNA and is lethal for the developing embryo. Being an important part of the cellular epigenome, the methylome is highly dynamic in the course of the life cycle of a living organism, tissue specific, and can change upon enzymatic methylation and de novo demethylation of DNA [73, 80].
The so-called CpG islands (CGI), 0.2-2 kb CG-rich sequences located close to promoters and 5′-terminal sequences of genes, as well as in the intergenic regions of the genome, are an exclusion from the globally methylated sequences of the genome (see [81] for review). Presumably, functionally important are the sequences located at a distance of up to 2 kb (the so-called shores) and 2-4 kb (shelves) from the CGI that are differentially methylated in cells of different tissues [82, 83]. Approximately 60% of mammalian genes have CGI promoters, and their CGI remain nonmethylated in most somatic tissues, in germ line cells, and during early embryogenesis. Maintenance of the nonmethylated state of CGI supposedly involves a CpG-binding protein CFP1 (CxxC finger protein 1) and a histone demethylase KDM2A, both possessing the characteristic zinc-finger CXXC domains. These proteins bind specifically CpG dinucleotides, change the pattern of histone methylation in a given genome region, and activate relevant promoters [84]. Trimethylated histones H3K4me3 are dominant in nucleosomes associated with CGI, with H3K36me2 histones being demethylated, which is typical for actively transcribed genes. In mammalian genomes, 20,000 CGI were found, making ~5% of the genomic CpG dinucleotides or ~1% of the total genome [85].
In contrast to CGI of active genes, global methylation of CGI accompanies inactivation of one of the X chromosomes necessary for dose compensation of sex chromosome gene expression in female mammals [86]. Comparison of DNA from cells with a single active X chromosome (Xa) (45,X karyotype – Turner’s syndrome) and normal cells with a single active (Xa) and a single inactivated X chromosome (Xi) revealed that only 7% CGI demonstrated decreased methylation in Xi while methylation of the rest of CGI was much higher than in control Xa from Turner’s syndrome cells. Both intergenic and intragenic CGI demonstrated high level of methylation that was maximal in promoters of inactivated genes. In addition to methylation, noncoding RNA (ncRNA) play an important role in X chromosome inactivation.
One of the bright examples of functional importance of methylation is parental imprinting, a phenomenon of inheriting part of the parental genes in inactivated state [87]. Realization of this mechanism is necessary for correct embryonic and neonatal development in mammals. Today, about 150 imprinted genes located in clusters on 17 chromosomes have been discovered in mice. Expression of each of the cluster (either entire cluster or its part) is regulated by special sequences termed imprinting control regions (ICR), as well as lncRNA, genes of which are located close to ICRs and are expressed only on homologous chromosomes not subjected to imprinting. Accordingly, in most cases, ICRs regulate lncRNA expression positively. In general, according to modern perceptions, ICRs function as insulators in imprinting by preventing interaction between enhancers and promoters of imprinted genes of a cluster, while lncRNA provides the heterochromatization of genes not being expressed in the course of imprinting. (For more details on insulators and lncRNA, see sections below.)
ICRs are divided into two classes – germ line ICRs (gICRs) and somatic ICRs (sICRs) [87a]. Allele-specific methylation of the former occurs during gametogenesis and is maintained in the course of embryonic development. On the contrary, methylation status of sICR is established in ontogenesis and often is tissues-specific. In the mouse genome, 55 ICRs varying between 1 and 5 kb in length have been identified [87].
Methylation of DNA in CGIs of promoters of types II and III (see above) is accompanied by suppression of transcription of adjacent genes. At the same time, CpG methylation in type I promoters does not affect considerably the synthesis of relevant RNAs.
DNA methylation in mammals does not always lead to suppression of transcription. Recent studies demonstrated that specific methylation of DNA in intergenic regions of the genome, as well as in introns, can be accompanied by activation of genes. For example, hypermethylation of the first intron of the EGR2 gene produces a stimulating effect on its expression, while hypomethylation of the indicated sequence associates with suppression of the transcription. The mechanism of the effect has not been studied yet [88]. The inactive state of the BCL6 oncogene is maintained by the CTCF transcription factor, which blocks the enhancer from interacting with CGI of intron 1 when the latter is nonmethylated. Methylation of the sequence in tumors results in activation of the oncogene transcription due to the blocking of CTCF binding [88].
Noncoding sequences of the genome subjected to specific methylation perform important regulatory functions. At the cellular level, methylation ensures the cell differentiation program operates correctly and maintains the genome stability through suppression of MGE activity. At the molecular level, 5mC functions depend on many parameters, the main of which is the genomic context in which the methylated sequences reside, in particular, in promoters, introns, exons, or intergenic regions of the genome. These regions of the genome are labeled by specific sequences, particularly, with CGIs in the vicinity of genes and ICEs controlling imprinting, which altogether make up a considerable fraction of noncoding sequences of the mammalian genome. Indeed, although the size of the human epigenome has not been estimated reliably yet, it is enormous. A diploid set contains >108 potentially methylated C residues, 107 of which comprise the CpG dinucleotides; a considerable part of methylated C is localize in noncoding regions of the genome. Besides, there are >108 potentially modified terminal amino acid sequences of histones [89].
Epigenetic modifications of DNA and histones are inherited in a number of cell generations and can be destroyed upon epimutations, which is accompanied by progression of pathologic conditions. At the same time, the diseases are often accompanied by changes in the epigenome; thus, it is often difficult to distinguish which are the cause and the effect. Variations in methylation of CpG sites termed methylation variable positions (MVP) are epigenetic analogs of SNP. Changes in methylation status of several CpG dinucleotides in a row are called differentially methylated regions (DMR). Such changes are particularly typical for oncological diseases, when aberrant methylation of CGIs, loss of imprinting, and epigenetic rearrangement of repeats, especially in satellite DNA, are often observed [89a]. Technologies allowing for genome-wide search for correlation between changes in the epigenome and human diseases (epigenome-wide association studies, EWAS) have been developed [89]. Application of this group of methods promises a deeper knowledge on the effect of MVPs and DMRs on human health in the nearest future and already promotes generation of new data [90].
Introns. Introns are transcribed intragenic sequences that, as a rule, are not included in mature mRNAs and are removed from their precursors during splicing [91]. Since most introns carry no information about amino acid sequences, they can be referred to the noncoding part of eukaryotic genome. On average, each human gene contains 8-9 introns; human genes are among the most intron-rich genes if compared to other biological species. In particular, the gene of titin (connectin), a gigantic muscular protein built from 244 protein domains, contains 362 introns [92]; the length of the longest human introns reaches 1 Mb. Only ~300 annotated human genes are intron-free; half of them are related to signal transduction pathways and one fifth encodes histones [93]. The total fraction of introns in the human genome is ~24% [94, 95].
To answer comprehensively the question on functional significance of introns, one should elucidate the reasons for their evolutionary genesis and widespread occurrence among eukaryotic organisms. Twenty year-long debates between the supporters of the early and late origin of introns can end with reconciliation of the parties [96]. Although the self-excision introns (introns that are removed upon self-splicing) might have originated early in the developing world of ancient RNAs, modern introns probably appeared late in the process of eukaryogenesis, and their global functional significance for eukaryotes is yet to be understood [97].
One of the popular explanations for the evolutionary securing of introns in the genome is the hypothesis of facilitation of recombinant exchange between individual exons, combination of which into new sequences could speed up the emergence of the new genes [98]. In particular, this hypothesis is supported by the phenomenon of alternative splicing widely spread in eukaryotes, which results in joining of exons and their parts in various combinations at the level of processed RNA and further formation of isoforms of the polypeptide product with different activities [99]. The emergence of these mechanisms considerably increases the information capacity of the eukaryotic genome: in humans, 95% of multi-exon genes support alternative splicing with formation of 10-11 isoforms of mRNA/gene on average [100]. One or two mRNA isoforms dominate in a certain cell line; therefore, their expression is tissue-specific. Overall, 20,687 human genes direct the formation of ~100,000 proteins, which evidences the 5-fold increase in information capacity of the genome due to alternative splicing [101]. Besides, alternative splicing can be involved in negative regulation of gene expression through formation of nonfunctional or rapidly degrading RNAs [102], as well as provide for intracellular localization of the transcripts through introduction of sequences necessary for correct transport of mature mRNAs [103]. The advantages that alternative splicing affords in eukaryotes do not explain the high size heterogeneity of introns. Besides, many alternative transcripts remain functionally uncharacterized, and the question on the level of information noise resulting from aberrant alternative splicing remains open [104, 105].
Trans-splicing can be defined as a type of alternative splicing that results in joining of exons of pre-mRNAs of two different genes into a single chimeric molecule [106]. In humans, chimeric mRNAs and proteins combining sequences of adjacent genes occur most often. This results from RNA polymerase skipping the transcription terminator without transcription termination, which leads to formation of a long chimeric transcript. Further intergenic splicing forms chimeric mRNAs combining exons of different genes, and their translation is accompanied by synthesis of chimeric proteins [107]. In humans, hundreds of incidents of intergenic splicing have been found in different tissues, and the process of formation of chimeric RNAs is a regulated and tissue-specific one. It results in formation of new bifunctional proteins and new RNAs, translation of which is regulated the same way one of the genes is regulated depending on the sequence of 5′- or 3′-UTR included in the transcript, as well as in the suppression of an upstream gene via the nonsense mediated decay (NMD) mechanism in case the sequence fusion results in formation of a nonsense codon in the new reading frame.
Trans-splicing between RNAs of remote genes occurs not that often in humans. Many apparent chimeric RNA molecules are artifacts that arise in the course of genome rearrangements or template switching by reverse transcription [108]. Nevertheless, the presence of chimeric RNAs generated by trans-splicing in human cells has been proved. In particular, in normal human mammary gland cells chimeric transcripts of genes located on different chromosomes have been detected [109]. Interchromosomal trans-splicing has been registered between an exogenous bacterial RNA and endogenous human RNA [110]. In the transcriptome of human pluripotent embryonic stem cells (both natural and induced), several lincRNAs originating from trans-splicing characterized by high intercellular content have been detected [111]. Suppression of expression of these RNAs with short RNAs (shRNAs) was accompanied by loss of the ability to support pluripotency by the cells. In general, the existence of the mechanism of intergenic splicing considerably widens the coding potential of the mammalian genome and increases the diversity of its proteome [112-114].
Another well-known phenomenon indicating the global functional significance of introns is the intron-mediated enhancement (IME) of transcription. The presence of introns in genes stimulated their expression in representatives of remote taxonomic groups, including animals and plants, which indicates the fundamental importance of the phenomenon [91]. In the absence of introns, many genes with intact promoters are not being expressed at all in vivo. As a rule, introns located in 5′- or 3′-UTRs, which differ considerably by their ability to stimulate transcription, influence expression the most. The ability of introns to provide efficient recombination of allelic protein encoding genes during meiosis is considered a factor accelerating protein evolution [115]. Prolonged sequences of introns increase the probability of recombination between mutant allelic exons and create proteins with new combinations of mutations, the functionality of which is further tested by the organism.
Introns are the site of localization of many regulatory elements of DNA. In addition to common sequences necessary for splicing progression (5′- and 3′-terminal splicing sites, branching site, polypyrimidine tract, intron enhancers, and splicing silencers [116]), introns can contain sequences regulating transcription (alternative promoters and transcription terminators; transcription enhancers and silencers), as well as miRNA and lncRNA genes. The rate of splicing as such can be a determining factor in regulation of gene expression. In particular, the rare U12-type introns, splicing of which involves spliceosomes containing the U12 snRNA, are removed from human pre-mRNA slower than the common U2 class introns, which is accompanied by a considerable suppression of biosynthesis of relevant proteins [117]. In many cases, regulatory functions of introns spread over a certain class of genes and even individual genes.
As follows from the above-mentioned facts, introns perform multiple important functions in regulation of gene expression. It is not surprising therefore, that mutations in introns are often accompanied by severe pathologic consequences. This is an additional indication of the important functional role of these noncoding sequences. Mutational substitutions of nucleotides impairing splicing through changing of donor or acceptor splicing sites occur particularly often [118]. Mutations in polypyrimidine tract and branching points are rare. In general, intron mutations constitute ~10% to the total of human pathogenic mutations known by now and are accompanied by various rearrangements of mature mRNAs.
Transcription enhancers. Enhancers are sequences regulating transcription located both closely to or at a considerable distance (up to several Mb) from promoters of genes they regulate. In some cases, they provide interchromosomal transfer of signals in trans (for example, see [119]). In terms of their size, typical enhancers (sequence length is <1 kb), as a rule located close to housekeeping genes, and superenhancers (3-50 kb) primarily located close to the key genes controlling ontogenesis (see [120] for review) are distinguished.
The major proteins that interact functionally with enhancers are transcription factors, which can be both activators and repressors [121]. In particular, the global transcription coactivator protein acetyl transferase p300, subunits of the Mediator complex, DNA-binding protein 7 of the chromodomain helicase, cohesin, CTCF, and RNA polymerase II are often associated with active enhancers in vivo [122]. Besides, histone H3 contacting the enhancers exhibits typical modifications: mono- and/or dimethylation of Lys at position 4 (H3K4me1 and H3K4me2), as well as acetylation of Lys27 (H3K27ac), while trimethylation of H3K4me3 typical for promoters is at insignificant levels [5, 123-125]. In addition, sequences of active enhancers exhibit hypersensitivity to DNase I (HS), which is typical of open chromatin state. Such molecular markers of enhancers create the prerequisites for their identification in a functioning genome. Patterns of histone modifications in enhancer chromatin of cells in different tissues correlate strictly with tissue-specific gene expression [126]. Evaluation with chromatin immunoprecipitation by anti-p300 protein antibodies and NGS technology showed that the total number of enhancers in the human genome reaches 106, and their sequences can make up to 3% of all nucleotides in the genome [126, 127]. As indicated by the data obtained by the ENCODE consortium, in the human genome enhancers occur each 3-30 kb [5]. Therefore, the number of enhancers in the human genome exceeds considerably the number of promoters; approximately half of enhancers are located inside genes [123].
Enhancers are characterized by modular structure: they contain several (sometimes over 10) short, 6-12-bp-long, transcription factor binding sites (motifs) organized in clusters [128]. Enhancers produce the major contribution to the provision of tissue-specific gene transcription and development of multicellular organisms by mediating the interaction between trans-acting protein transcription factors and regulatory sequences in promoters. The interaction of transcription factors with enhancers is under strict control and as such, without auxiliary proteins, can proceed without apparent functional consequences [129]. The modular composition of enhancers provides for functional joining of transcription factors in various combinations, which widens considerably the regulatory possibilities of enhancers. The same promoter can be under the effect of various regulatory elements of enhancer in different tissues and at different times.
Some enhancers are present in the genome in two or more distal copies that possess the same or considerably overlapping activities with respect to the gene they regulate. As a rule, the primary enhancer is located close to the target gene and other enhancers are distant from the gene, but often close to other genes. Such remote enhancers that cannot be distinguished from the primary enhancer by their properties are termed shadow enhancers [130]. First discovered in drosophila [131], shadow enhancers function in the human genome as well, locating, for example, in the introns of the GLI3 transcription factor gene [132] or upstream from the renin gene [132a]. The apparent functional redundancy of such enhancers increases the stability of the system to mutational changes and, in a number of cases, to extreme environmental factors and provides for accurate fulfillment of the organism’s early development program as well [130].
The stimulating effect of enhancers on transcription performed by RNA polymerase II does not depend upon their orientation in DNA molecules or their position relative to the promoter [133]. They are equally efficient in performing their functions being located both up- or downstream from a gene, inside it, or at a considerable distance (up to several hundred kb) from it, which is one of the criteria used to experimentally detect enhancers. Independence from the positioning in a DNA molecule is true for entire enhancers, but not their individual modules.
According to the modern perception, the answer to the question on molecular mechanisms underlying the regulatory signal transduction from enhancers to promoters at large distances is the looping out of DNA regions located between the enhancers and the promoters of genes and direct interaction of both regulatory elements [134]. The model is convincingly supported by multiple experiments using high-throughput techniques of analysis of chromatin conformation in the interphase nucleus based on the chromosome conformation capture (3C) technique. According to the 3C technique, interactions between chromosome regions are determined by introduction of cross-links into the interacting chromatin followed by DNA restriction, ligation of fragments brought together, and sequencing of the ligation products [135]. Transcription factors and their complexes are the major participants in the interaction between enhancers and promoters. These nucleoprotein complexes termed enhanceosomes also contain cohesin, which is involved besides in holding sister chromatids of chromosomes together during mitosis and meiosis (see [125] for review).
The main coactivator of transcription, that is, the CBP protein, also interacts with enhancers and recruits RNA polymerase II, transcribing most of the protein-encoding genes in eukaryotes to enhancers [136]. In accordance, many enhancers were found recently to perform functions of promoters from which RNA polymerase II synthesizes lncRNAs termed eRNAs [137-140]. Synthesis of eRNA can proceed in a single direction (1D-eRNA) or in two directions (2D-eRNA), and the transcripts are capped at their 5′-ends. Unidirectional transcripts are polyadenylated, which is not typical for most bidirectional transcripts, the main representatives of eRNA [126]. It gradually becomes clear that eRNAs play an important role in the initiation of formation and stabilization of promoter-enhancer loops (see [138, 140] for reviews). This is supported by the data on impairment of enhancer functioning resulting from directed destruction of eRNA by artificial siRNAs or antisense oligonucleotides [141, 142]. Since enhancer transcription, in turn is regulated by various signals, this creates a new level of global transcription regulation in eukaryotes.
The most prominent role of enhancers in the organism’s development program executed by embryonic stem cells is fulfilled through their preparation for activation via interaction with transcription factor proteins that maintain the enhancers in the open state ready for binding of other factors (competent state). The appearance of new transcription factors in cells leads to replacement of the old ones, activation of transcription of relevant genes, and progression of cell differentiation in the desired direction [143]. In addition to transcription factors, proteins typically comprising complexes that suppress transcription can be detected on some enhancers; here, these proteins adopt the unusual role of activators. The recent discovery of the Janus-faced repressors-activators of transcription is an important achievement of modern studies of transcription in eukaryotes (see [144] for review).
A remarkable property of some enhancers is their high evolutionary and functional conservation. For example, almost half of the 167 studied ultraconserved human enhancer sequences supported tissue-specific expression of a reporter gene in embryogenesis of transgenic mice and fish [145, 146].
The exceptional importance of enhancers in maintenance of tissue-specific gene expression patterns at various stages of individual development implies severe functional consequences of mutational changes in enhancers. Indeed, numerous mutations leading to the development of oncological diseases, deafness, systemic lupus erythematosus, multiple sclerosis, Crohn’s disease, and other pathological conditions in humans have been described [147].
Locus control regions. Many gene clusters differentially expressed during development of an organisms and in different tissues are coordinately regulated by locus control regions (LCR) (reviewed in [148, 149]). This type of cis-acting positive regulatory elements are similar to enhancers in their effects, but in contrast to enhancers provide for stable transcription of the controlled transgenes independently of the position of their integration site; the level of transcription controlled by LCR also depends on the number of the transgene copies in the genome. Expression of transgenes stably integrated in the genome is known to be dependent on the site of integration in the chromosome (the so-called position effect variegation); their transcription is suppressed upon incorporation into heterochromatized regions of chromosomes [150]. LCR located close to a promoter provide for functionality of the regulated gene independently of its position in the chromosome. This indicates that there should be insulators (see below) among LCR functional modules, which has been proven experimentally [151].
LCRs are viewed as complex enhancers composed of several enhancer modules. The distance from LCRs to the promoters they regulate can be of several dozens of kb. Therefore, it is typical of LCRs to possess several (up to 10) HS sites marking individual enhancers. Similar to common enhancers, these sequences are marked with acetylated forms of histone H3 typical for the open chromatin state. Besides, in the active state LCRs and genes they regulate are brought together and interact directly with each other, which is accompanied by looping out of DNA separating these sequences. Moreover, interchromosomal interactions between promoters and LCRs have been revealed; for example, in murine T-helpers, promoter of the interferon-γ gene in the TH1 locus located in chromosome 10 interacts with the LCR of the TH2 locus located in chromosome 11 [152]. Intergenic transcription initiated in LCRs and accompanied by generation of lncRNA imparts LCRs with even greater functional similarity to common enhancers [153]. Emergence of these noncoding transcripts correlates with structural changes in chromatin, DNA demethylation, and acetylation of histones and can play an important role in regulation of these processes. You will find more details on lncRNA in the review below.
In addition to the direct involvement in gene activation, enhancers (including LCRs) play an important role in maintenance of spatial organization of chromatin in interphase nuclei. For example, actively transcribed genes often move beyond their chromosome territories within chromatin loops; particularly, this process is influenced by the LCR of the β-globin locus [154]. When cells of the erythroid lineage differentiate, the β-globin locus migrates from the nucleus periphery to its center and associates with transcription factories or other self-assembling substructures, such as speckles involved in splicing, which is accompanied by activation of gene expression, and the process requires the LCR [155, 156]. The Eµ enhancer is necessary for migration of the IgH locus from the nucleus periphery to its center [157]. In our opinion, the necessity for correct localization of genetic loci being expressed within the interphase nuclei indicates another common function of intergenic noncoding sequences in the eukaryotic genome: they provide for the required mobility of the loci inside the nucleus, preserving the system of connected loci as a whole.
Chromatin insulators. Regulatory sequences termed insulators play the key role in maintenance of intranuclear spatial organization of the eukaryotic genome and formation of topologically associated domains (TADs), the length of which lies within 1 Mb. TADs are universal structural elements of spatial organization of human and animal genomes in the interphase nuclei, and their location in the genome is highly conserved [158]. Besides, multiple intragenomic contacts emerging under the effect of insulators are accompanied by spatial rapprochement of promoters and enhancers, which is necessary for their functioning. Insulators prevent the nonspecific cross effects of enhancers and silencers on promoters by isolating functional domains of chromatin from each other (reviewed in [159, 160]). Two main mechanisms are employed in the effect of insulators: (1) blocking of enhancer effects and (2) creation of a barrier on the way of heterochromatin spreading over the adjacent euchromatin regions of chromosomes. In genetic engineering experiments, the first mechanism is manifested through inactivation of a transgene by an insulator placed between the enhancer and the promoter, and the second, in protection of the transgene flanked by insulators from position effect variegation. Besides, the ability of insulators to govern specific rapprochement of enhancers and promoters accompanied by activation of relevant genes, as well as involvement of insulators in demarcation of borders between chromatin regions that are in different epigenetic states, has been demonstrated recently. In vertebrates, the range of influence of insulators is not limited to transcription. For example, recently, their involvement in regulation of V(D)J recombination in immunoglobulin loci has been demonstrated [160].
In chromatin, insulators are often marked by specific proteins: CTCF transcription factor, which is a highly conserved protein with 11 zinc finger-like domains, and/or the TFIIIC transcription factor of RNA polymerase III, which is a multisubunit protein interacting with the B box of the promoter of tRNA genes (reviewed in [161]). Both proteins contact the cohesin complex, which, when bound to DNA, stabilizes the interaction of remote chromatin regions with each other upon formation of bases of chromatin domain loops. Therefore, sequences of insulators in animals may contain the CTCF binding site, which is a degenerate sequence 50-bp-long, or a tRNA gene.
Using the ChIP technique followed by NGS, up to 30,000 CTCF binding sites, and therefore, potential insulators, were found in human genome; 43% of them were localized in intergenic regions, 7% in 5′-UTR, 3% in exons, 29% in introns of genes, 2% in 3′-UTRs, and 16% in proximity of TSSs [162]. Using the same approach, several thousands of short sequences that interacted with TFIIIC were found in the human genome in addition to promoters of tRNA genes [163]. These additional sequences named extra TFIIIC (ETC) possessed the ability to bind TFIIIC independently of RNA polymerase III promoter containing sequences of A and B boxes. In contrast to these promoters, ETC sites contained either B box or the conserved GC-rich 16-nucleotide motif.
S/MAR sequences. Along with the insulators, DNA sequences that provide for loop base attachment to nuclear matrix (scaffold/matrix attachment regions, S/MARs) are another element playing an important role in spatial organization of functional domains of chromosomes in interphase nuclei. These AT-rich (>70%) DNA fragments remain associated with nuclear matrix after nucleus extraction by detergent or high ionic strength buffers [164] and are present in both genes and intergenic regions, close to insulators, enhancers, cis-acting regulatory elements, and replication origins. Besides, an S/MAR often marks the borders between the condensed and decondensed chromatin [165]. In the process of cell differentiation, S/MAR-mediated intranuclear rearrangement of chromatin loops that contact with the nuclear matrix occurs and is associated with changes in gene expression profiles [166, 167]. Studies of S/MAR did not reveal any consensus sequences in them, which, supposedly, can be explained by recognition of the features of spatial but not primary DNA structure by the matrix proteins interacting with the S/MARs. Recently, using hybridization on biochips, 453 S/MARs with average length of ~5 kb were mapped in a 30-Mb DNA sequence of HeLa cells representing 1% of the human genome [168]. Most S/MARs localized close to the terminal sequences of genes being expressed and were associated with RNA polymerase II and the transcription factor binding sites. At the same time, approximately 40% of S/MARs were located in intergenic regions or in proximity of inactive genes and could represent the classical border sequences or be involved in the total spatial organization of the genome. The distance between the neighboring S/MARs in HeLa cells is 80-90 kb [169] and can vary in different parts of the genome [168].
Noncoding RNAs
Modern analysis of the mammalian transcriptome by high-throughput methods using NGS demonstrated that a considerable part of the genome is represented by noncoding (nc) RNAs. All RNAs not encoding proteins belong to this group, which includes both well-studied rRNAs, tRNAs, small nuclear (sn) and small nucleolus (sno) RNAs, and the recently discovered short and long ncRNAs, the function of most of which is not known yet. This latter group of ncRNAs will be discussed in the current section of the review.
Short noncoding RNAs. Short noncoding RNAs discovered in mammals are usually divided into three classes: microRNA (miRNA), short interfering RNA (siRNA), and RNA interacting with PIWI (piRNA) (see [170, 171] for reviews). RNAs of all three classes are single-stranded oligoribonucleotides ~22 nt long that realize their regulatory effect through formation of specific complexes with mRNA targets based on complementarity differing by their biogenesis.
In humans, >3000 miRNA are known today; they are coded by genes spread over the genome and are transcribed by RNA polymerase II (less often, by RNA polymerase III) with the formation of miRNA precursors (pri-miRNA) having one or several stem-loop structural elements with the stem carrying the miRNA sequence. The miRNA genes can be located in both the intergenic regions of the genomic DNA, often (50% of all genes) in clusters, and in introns (40%) and noncoding exons (10%) of protein or RNA coding genes [172]. Intergenic miRNAs are transcribed from promoters of their own genes, while intron miRNAs are most often transcribed from promoters of genes where they are located and in ~1/3 of cases, from their own promoters [173]. An interesting kind of intron miRNA is the mirtrons – the first stages of biogenesis of such miRNAs result from splicing [174]. Besides, miRNAs can be encompassed within longer noncoding RNAs, e.g. snoRNAs [175]. Transcription from self promoters provides for additional possibilities for regulation of miRNA expression using the classical mechanisms. Pri-miRNA synthesized by RNA polymerase II are capped and polyadenylated.
Processing of pri-miRNAs is continued in the nucleus using the protein complex named the Microprocessor, the main components of which are the Drosha (possessing RNase III activity) and DGCR8 proteins and helicases p68 and p72. Their activity results in formation of pre-miRNA retaining the stem-loop structure, which is exported from the nucleus into the cytoplasm through the nuclear pore complex. In the cytoplasm, the stem is cleaved by an RNase III called Dicer with the formation of miRNA duplex, one strand of which is represented by the miRNA, and the other by an inactive miRNA* (passenger strand). Processing of pre-miRNA can proceed with deviations leading to formation of many isoforms of miRNA (isomirs) differing by size and primary structure and consequently functional activity [176].
In the process of formation of RNA-induced silencing complex (miRISC), miRNA duplex is enrolled into the AGO protein complex, which recognizes the 5′-monophosphate of the miRNA strand with the preference for A and U as a 5′-terminal nucleotide. The miRISC formation is completed with the replacement of the passenger strand and its degradation [177]. Supposedly, the strand of miRNA duplex, the 5′-end of which is less stably bound with the complementary strand than the 3′-end, is chosen as the miRNA (the so-called asymmetry rule). Realization of such mechanism of miRNA strand choice should be very sensitive to changes in its primary structure resulting from introduction of alternative nucleotides in SNPs or under the effect of mutations.
Mature miRISC interacts with mRNA target, which the encompassed miRNA is complementary to (at least partially). In approximately half of the studied complexes, one prerequisite for such an interaction is the presence of a seed fragment 2-7 nt long in the 5′-terminal part of miRNA completely complementary to mRNA [178]. However, noncanonical interactions between miRNA and mRNA [179] in the seed region of the complex, as well as the cooperatively interacting multiple seed regions [180] were discovered recently; together with the established regulatory effect of individual miRNAs on many mRNA targets, these new findings indicate the flexibility of recognition of mRNA targets by miRISC complex. Regions of mRNA interaction with miRNA are often located in 3′-UTR, but they were observed in 5′-UTR and coding fragments as well.
The main mechanism of the regulatory effect of miRNA is suppression of translation of mRNA through its decapping and deadenylation followed by degradation. Besides, interaction of mRNA with miRISC can be accompanied by inhibition of protein synthesis at the level of initiation or elongation. In rare cases, stimulation of translation under the effect of miRNA has been noted. Recently, involvement of nuclear miRNA in positive and negative regulation of transcription through direct interaction with promoters (see [181] for review) has been discovered. Besides, all stages of miRNA biogenesis and interactions with their targets are additionally regulated (reviewed in [170, 182]). Altogether, these facts indicate that miRNA is a key regulator of gene expression in mammals, and their genes make up a considerable fraction of the noncoding sequences of the genome.
Biogenesis of endogenous siRNAs differs from that of miRNAs insignificantly [183]. After joining of RNA with its antisense partner in the cytoplasm, the dsRNA becomes a substrate for Dicer, and the siRNA is further introduced into the RISC complex, where it implements its effect on the mRNA target. The main source of endogenous siRNAs in mammals is the bidirectional transcripts of transposons and overlapping regions of mRNAs and antisense RNA genes formed upon convergent or divergent transcription or transcription of pseudogenes.
As follows from their name, when the RISC complex is formed small piRNAs interact with PIWI proteins comprising the Piwi subfamily of Argonaute proteins and being expressed primarily in germ line cells, although PIWI proteins have been detected in embryonic stem cells and cells of various somatic tissues (reviewed in [172, 184, 185]). The length of piRNAs is 26-31 nt and the total number of their types reaches 1,000,000. The main function of piRNAs is the prevention of transcription of transposons and their mobilization. In contrast to small RNAs discussed above, piRNA precursors are single-stranded, and Dicer is not involved in their processing. Two pathways (primary and secondary) of piRNA biogenesis have been described. When the primary pathway is realized, piRNA precursors 1-100-kb-long are transcribed by RNA polymerase II from the gene clusters. The transcripts are antisense sequences with respect to RNA of transposons. Many enzymes involved in piRNA biogenesis have not been identified precisely. Presumably, after the synthesis of the precursor, it is fragmented by an endonuclease and the formed RNA fragments with 5′-terminal U interact with PIWI proteins; then, they are shortened from the 3′-end to the final size and methylated. The nucleoprotein complexes are transported to the nucleus, where they bind the growing mRNA strands of transposons being transcribed, attract relevant proteins, and trigger methylation or heterochromatization of DNA, which leads to suppression of MGE transcription. The secondary pathway of piRNA biogenesis provides for their amplification from the mRNA template of transposons via the ping-pong mechanism.
Ubiquitous participation of miRNAs in regulation gene expression implies their important role in the development of pathological processes. Indeed, studies of polymorphisms and mutations in miRNAs, their targets, enzymes, and auxiliary proteins involved in their processing and regulatory effects elucidated the traces of these pervasive small molecules in all studied pathological processes occurring in the organism [186].
Long noncoding RNAs. Long noncoding RNAs (lncRNA) comprise ncRNAs over 200-nt-long and represent the most numerous class of ncRNA, with the total number of genes encoding lncRNA reaching 10,000 in humans [187]. This recently compiled catalog contains over 15,000 transcripts; functions of most of them are not known. Based on the location of lncRNA sequences in the genome with respect to coding genes, lncRNAs are divided into long intergenic noncoding RNAs (lincRNA) and intragenic ones (antisense, intronic, exonic, and overlapping). Most lncRNAs are independent transcription units synthesized by RNA polymerase II, 40% of them having polyadenylation signal. Most lncRNAs (98%) are subject to splicing at canonical sites (GT/AG) and, as a rule, contain only two exons. Approximately a quarter of lncRNAs undergo alternative splicing and exist in several isoforms. Fifty eight percent of lncRNAs are small molecules (200-950 nt), 40% are 950-4800-nt-long, and 2% are represented by transcripts of even larger size. The largest size has been registered for the product of the single-exon gene NEAT1 (22,700 nt), which is involved in formation of nuclear paraspeckles. A lower level of expression and more pronounced tissue specificity of transcription is typical of lncRNA genes in comparison with usual genes. Also, pronounced nuclear localization has been reported for lncRNAs.
Known functions of lncRNAs are exceptionally diverse and affect all stages of gene expression (reviewed in [188, 189]). Most nuclear lncRNAs recruit chromatin-modifying proteins to relevant genetic loci [190]. The result may be repression or activation of genes of the locus through DNA or histone modification accompanied by heterochromatization of chromatin or changes in its conformation. Besides, as described above, nuclear lncRNA are involved in dose compensating inactivation of the X chromosome, imprinting establishment, and regulation of activity of enhancers and other elements of the genome.
In cytoplasm, lncRNAs can change the stability of mRNA and suppress or activate its translation. These activities of lncRNA are often implemented via complementary interaction with the target mRNA. As noted above, antisense lncRNAs can govern siRNA formation. Transcripts of pseudogenes act as miRNA traps that regulate expression of relevant genes. Such lncRNAs were termed competing endogenous RNAs (ceRNAs); an interesting kind of ceRNA is the recently discovered and widely represented circular RNAs (circRNA) [191, 192]. Linear ceRNAs possess low stability, while circRNAs are more stable and contain regulatory sites for miRNA binding involved in regulated inactivation of circRNA.
Pseudogenes
A pseudogene is a copy of a gene that has lost its ability to produce a functional protein. Depending on their origin, pseudogenes are classified as processed or unprocessed. The latter, in turn, are divided into unitary and duplicated pseudogenes (see [193-195] for reviews). A separate group is formed by nuclear mitochondrial (NUMT) pseudogenes [196].
Processed pseudogenes result from incorporation of the products of reverse transcription of mRNA of relevant genes in a new fragment of the genome. Then, various damaging mutations promote loss of the coding potential of these genes. The distinctive feature of processed pseudogenes is the absence of introns and the presence of poly-A tract at their 3′-ends. Often, processed pseudogenes contain no promoters and their expression involves other regulatory elements. For example, pseudogenes located in introns of other genes use the transcription apparatus of the host gene. Approximately 10% of genes possess processed pseudogenes. Such pseudogenes are most typical of the housekeeping genes.
Duplicated pseudogenes are formed in the course of tandem duplication or crossing over. Further mutations make these copies of genes functionally inactive. Such pseudogenes maintain the intron-exon structure. In contrast to processed pseudogenes, which can be located in various parts of the genome, duplicated pseudogenes are located at the same chromosomes as the precursor genes. Unitary pseudogenes result from damaging mutations of a single copy of the initial gene. Such pseudogenes have no functionally active parent gene.
NUMT pseudogenes are fragments of mitochondrial DNA (mtDNA) incorporated into various parts of the nuclear genome. Such pseudogenes were detected in various eukaryotic organisms, including humans (286 pseudogenes) [197]. It has been found that sequences of all mitochondrial genes are represented in the human nuclear genome and are evenly spread over the chromosomes. NUMT pseudogenes can represent individual mitochondrial genes and fragments of mtDNA encompassing two or more adjacent genes. Many NUMT pseudogenes have various mutations: substitutions, insertions, deletions, and duplications. Mechanisms of mtDNA penetration in the nuclear genome have not been studied completely. Supposedly, damaged by various endogenous and exogenous factors, mtDNA can be removed from mitochondria and, through the cytoplasm, reach the nucleus, where they incorporate into the nuclear DNA. According to many researchers, incorporation of mtDNA fragments into the nuclear genome can occur in the process of reparation of double-stranded DNA breaks through nonhomologous end joining (NHEJ). For some genes, e.g. the human PCNA gene, more than one pseudogene has been described [198].
For a long period, pseudogenes have been considered functionally inactive evolutionary shatters of genes. However, studies of the last decade demonstrated the important regulatory role of pseudogenes. Above all, many pseudogenes are being transcribed [199]. The generated RNAs realize their regulatory functions through several mechanisms. Recently, it has been shown that owing to the high homology of the PTENP1 pseudogene transcript and the parent PTEN gene, they can compete for the regulatory miRNAs that bind sites located in 3′-UTR of the genes [200]. The concept of functional interaction of genes and their transcribed pseudogenes at the level of mRNA has been defined in general terms, and the idea of competing endogenous mRNAs (ceRNA) was introduced.
The mechanism described above is implemented if transcription of a pseudogene results in a sense RNA strand with respect to mRNA of the parent gene. However, there are pseudogenes that are sources of antisense transcripts. Such RNAs form duplexes with transcripts of the precursor genes and can be cleaved by relevant enzymatic systems forming endogenous short interfering RNAs (siRNAs) [201, 202]. The siRNAs are involved in regulation of expression of precursor genes via the RNA interference mechanism (see the section on miRNAs above). For example, siRNA emerging from DNA duplex formed by RNA of the OCT4-pg5 pseudogene and antisense lncRNA of the OCT4 gene mediates suppression of the gene expression [203].
As noted above, processed pseudogenes can be located in various parts of the genome: in intergenic regions, exons, and introns of other genes. In the latter case, the sequence of the pseudogene may become a new exon in the host gene. Such process of a new exon emerging was termed exonization. Transcription of the pseudogene within the host gene can result in a chimeric mRNA. The protein translated from such mRNA will be different from the protein product of the host gene [195]. That is, in the process of exonization, functions of both pseudogene and host gene change.
The human genome contains ~18,000 pseudogenes, and approximately 2/3 of them are processed (http://www.pseudogene.org [204]). Most of the pseudogenes are associated with a limited number of families of actively transcribed genes. For example, genes of 79 human ribosomal proteins are associated with 20% of all human pseudogenes, and the gene encoding glyceraldehyde 3-phosphate dehydrogenase has 62 pseudogenes [205]. Such a great excess in the number of pseudogenes over their parent genes is explained by a flash of activity of retrotransposons in the human genome. On the other hand, taking into account the functionally active state of many pseudogenes, one may assume that they could be directly involved in regulation of expression of these actively transcribed genes. In general, studies of the recent years have raised the curtain over this vast class of genomic sequences that have long been considered noncoding and revealed the diversity of regulatory effects they produce on gene expression.
Repeated Sequences
Repeated sequences occupy most of the mammalian genome [206]. Except for centromere and telomere sequences, the functions of the repeats remain a mystery. Progress in understanding of the role of repeated sequences in functioning of the genome became apparent only when new high-throughput methods of DNA and RNA sequence and spatial structure analysis were developed and the first global studies of the genome and transcriptome were conducted.
Mobile genetic elements. Mobile genetic elements (MGE), or transposons, are moderately repeated DNA sequences in the eukaryotic genome. They make up 45% of the human genome and 40% of the murine genome. The name derives from their ability to change position in the genome of somatic cells and germ line cells. In addition to true MGEs, multiple fragments and copies thereof inactivated by mutations occur in the genome. Since complete MGEs contain genes providing for their mobility and survival in the genome, we refer these sequences to noncoding ones arbitrarily in this review, reflecting our poor knowledge of the major functions of transposons in eukaryotic cells.
According to molecular mechanisms that MGEs employ for their translocation in genomic sequences, they are divided into two big classes, that is, retroelements (class I) and DNA transposons (class II) [206]. For their mobilization, retroelements utilize mechanisms based on reverse transcription. Depending on the structural features and replication mechanisms, retroelements are divided into LTR-containing elements (retrotransposons and endogenous retroviruses) and non-LTR retroelements (long interspersed elements, LINE, and short interspersed elements, SINE) [207, 208]. Translocation of these MGEs in the genome occurs via transcription, synthesis of cDNA using the generated RNA template employing reverse transcriptase, and integration of the cDNA into a new genetic locus (copy-and-paste mechanism). Such transpositions result in an increasing number of copies of the retroelements in the genome.
Retrotransposons resemble exogenous viruses in terms of their structure and replication mechanisms. A particular feature of their structure is the presence of long terminal repeats (LTR) containing sequences involved in regulation of transcription and replication. LINEs, otherwise called long retroposons, possess the same genes as retrotransposons, but have no LTRs. Nevertheless, they have promoters of RNA polymerase II, which transcribes LINE genes. Since retrotransposons and LINEs possess all they need for translocations in the genome, they are called autonomous retrotransposons. SINEs (or short retroposons) are not autonomous; their transposition requires protein products of expression of autonomous genes of the transposons. They contain close to their 5′-end an internal promoter of RNA polymerase III, which transcribes them.
In contrast to retrotransposons, DNA transposons translocate in the genome through a cut-and-paste mechanism involving transposase, an enzyme belonging to the class of recombinases [206]. This results in excision of the transposon accompanied by duplication of a short nucleotide sequence in the old integration site and insertion of the copy into a new genome site, typically close to the old one.
The extremely high abundance of MGEs among genomic sequences evidences their importance in eukaryotic genome evolution and allows considering them as molecular endosymbionts of eukaryotic cells [209, 210].
Lately, it became clear that MGE could produce considerable influence on expression of common genes in the eukaryotic genome. First, promoters of retroelements and the associated regulatory sequences are involved in processes of common gene expression. Global analysis of retrotransposon expression in genomes of the human and mouse using the CAGE approach revealed ~275,000 and ~44,000 TSS in repeated sequences of the genomes respectively, which made ~31 and ~18% to all known TSSs of these organisms, although the levels of their activity were much lower than those of TSSs in common genes [40]. Transcription initiated at repeated sequences is of tissue-specific nature. For example, up to 30% of all TSSs of human embryonic tissues are detected in repeated sequences (16% in retrotransposons, 10% in satellites, and 5% in simple repeats). LINE sequences produce the major contribution to these RNA syntheses. Transcription of simple repeats clearly dominates in approximately half of the studied tissues. Utilization of ~35% of all TSSs associated with retrotransposons is regulated in ontogenesis. Analysis of LTRs in murine retrotransposons belonging to the VL30 family also revealed a distinct tissue-specific transcription that does not occur in brain, hypothalamus, or embryonic tissues. Synthesis of most of messenger and antisense transcripts revealed in the work was initiated at previously unknown promoters.
Pervasive transcription of repeated sequences influences the transcriptome of protein encoding genes [40]. It turned out that in mouse, 144 promoters of retrotransposons or their fragments, and 576 analogs in humans, are used as alternatives upon transcription of known genes. In addition, retrotransposons occurring in 3′-UTRs of over a quarter of genes are expressed, decreasing the level of the transcription. Bidirectional transcription accompanied by synthesis of both sense and antisense RNAs often starts within retrotransposon sequences and, as discussed above, provides the maintenance of the epigenome and establishment of borders between functional domains of chromosomes. Cases of inclusion of transposon enhancers and insulators into transcriptional networks of humans, other animals, and plants have been reported (see recent review [211]). Transposons are active in normal brain tissue of mammals and can influence its metabolism [212]. An LTR is used as alternative promoter for the murine erythroid transcription factor gene Pu.1, playing an important functional role in erythropoiesis [213]. Many retrotransposons are activated by demethylation and expressed in early embryogenesis of mammals, where they can provide for zygotic induction of gene expression in a developing embryo [214]. Indeed, experimental suppression of transcription of endogenous viruses and LINE1 was accompanied by decrease in the embryo’s competence for development [215]. Therefore, the results of recent studies support the earlier proposed hypothesis on the important role of MGEs in phylogenesis and ontogenesis in eukaryotes; however, their global significance for eukaryotic genome is yet to be elucidated.
Telomeres and centromeres. Simple repeats comprising telomeric and centromeric regions of chromosomes performing important biological functions make up a considerable fraction of noncoding sequences of the mammalian genome.
Telomeres are specialized genetic loci organized into big nucleoprotein complexes located at the ends of eukaryotic chromosomes providing for their robust replication and stability [216]. The existence of linear chromosomes in cells requires that at least two interconnected problems be solved. First, reproduction of linear DNA molecules in a series of cell generations without the engagement of specialized molecular mechanisms would inevitably result in under-replication of chromosomal ends and decrease in the size of the molecules in each cell cycle. Indeed, since the replicative DNA polymerases perform DNA synthesis only in the 5′→3′ direction, they cannot fill in the single strand break that would emerge upon removal of the last 5′-terminal RNA primer. Second, chromosome ends should be protected from erroneous recognition by reparation systems; otherwise, they would join the chromosomes at their ends with each other. The solution of both problems is provided by the telomeres.
Mammalian telomere DNA (tDNA) is built from tandems of repeated hexanucleotides TTAGGG, its total length in humans is 10-15 kb, and it can be as long as 20-50 kb in rodents. Mainly double-stranded, tDNA contains a G-rich 3′-extending strand that acts as a primer for telomerase. Single-stranded end can exist at least in two alternative conformations, forming the so-called t-loops and G-quadruplexes [217, 218]. A t-loop-like structure is formed with the participation of protein factors upon incorporation of the single-stranded end between tDNA chains of the closest TTAGGG repeats. G-quadruplexes are formed by stacks of four G residues held together in the same plane by Hoogsteen base pairing. G-quadruplexes have been detected in vivo and, presumably, can limit elongation of tDNA by telomerase.
A protein complex of six polypeptides called shelterin is formed on the tDNA. It preserves chromosome ends from fusion, which has been proven experimentally by inactivation of individual complex components [219]. Another important component of the telomere complex is the telomerase, a reverse transcriptase that, together with the auxiliary proteins, provides the formation and maintenance of tDNA [220]. An integral part of telomerase is the telomerase RNA (TR) – an integral part of the catalytic subunit (TERT), which uses TR as a template in tDNA synthesis. The single-stranded 3′-end of tDNA acts as a primer. TR not only functions as a template, but it also directs the assembly of the additional dyskerin complex that provides for TR stability and telomerase functioning in vivo. Human telomerase is able to add over 100 nt to telomeres per cell cycle. Single-stranded tDNA thus formed is made double-stranded by replicative DNA polymerases.
While TR is synthesized in all cell types, TERT expression is strictly regulated in the course of ontogenesis and is typical of germ line cells and embryonic stem cells, and not of somatic cells. Therefore, each division of somatic cells results in shortening of tDNA ends due to the above-mentioned problem of replication of DNA ends, which ultimately leads to cessation of cell division. An important role in regulation of telomerase activity is played by the CST protein trimer (CTC1–STN1–TEN1), which limits telomerase processivity [221]. Despite heterochromatization, tDNA is transcribed by RNA polymerase II from subtelomeric, telomeric sequences forming capped, and polyadenylated lncRNAs 0.1-9-kb-long termed telomeric repeat containing RNAs (TERRA) [222]. Only the non-polyadenylated TERRA turned out to be associated with chromatin. TERRA is partially complementary to TR and can interact directly with TERT and suppress telomerase activity. Besides, it is involved in heterochromatization of telomeric chromatin [223].
The association between telomeres and cell proliferation is clearly observed in oncological diseases. In 90% of tumors, telomere activity is increased, which promotes immortalization of malignant cells through elongation of tDNA. In humans, tDNA length is an inherited characteristic and, probably, can influence life duration, as well as reproductive functions, of an individual [224]. Multiple pathological states connected with telomerase and telomerase complex dysfunctions, which are poorly classified due to their high heterogeneity, have been termed telomeropathies [225].
Centromeres are the genetic loci of eukaryotic chromosomes controlling their separation into daughter cells in mitosis and meiosis (reviewed in [226, 227]). Each centromere is a site of assembly of a multiprotein complex called the kinetochore, which provides for chromosome attachment to microtubules and its migration along the mitotic spindle during karyokinesis. Centromeres of humans and other primates are formed by sequences of α-satellite DNA (satDNA) built from (head-to-tail) tandems of 171-bp-long monomers. Individual monomers exhibit 50-70% homology and are grouped into a new repeated unit, a higher order repeat (HOR) 1-3-kb-long, which repeats continuously and forms the centromere locus. The length of satDNA thus organized is 0.25-5 Mb. In total, centromere sequences make up ~5% of all sequences of the human genome.
Each chromosome contains a unique sequence of α-satellites, in which HOR multimers contain a unique number of monomers in tandem, which allows the differentiation of individual chromosomes. The total size of centromeres differs even in homologous chromosomes of one human, as well as between individuals. Also, polymorphism is typical of sequences of individual HOR monomers: some contain a specific element called CENP-B box that is recognized by a specific DNA-binding protein of centromeres, CENP-B. Other monomers are marked with SNPs.
The functional importance of human satDNA for activity of centromeres has been established in experiments on artificial chromosomes. In the process of bottom-down construction of X and Y chromosomes in hybrid human–mouse and human–hen cells, chromosome size progressively decreased upon homologous recombination with plasmid DNA containing telomeric sequences and markers for selection. In thus formed minimal constructs retaining the ability to segregate correctly into daughter cells, centromeres were built from satDNA. In the bottom-up approach, yeast and bacterial artificial chromosomes were integrated into synthetic or cloned satDNA that imparted the artificial chromosomes with the required properties. However, not all satDNA possessed the ability to form centromeres. Their functionality was manifested only in the presence of the native CENP-B box in them.
In light of the recently discovered pervasive transcription, it does not seem surprising that the entire centromeric locus, together with the pericentromeric regions, is being transcribed (reviewed in [228]). Transcripts, including the polyadenylated ones, are detected in both nucleus and cytoplasm. Pericentromeric satDNA is actively transcribed in embryogenesis and is involved in heterochromatization of these chromosomal regions in mice. Destruction of these transcripts using antisense technique leads to growth arrest [229]. Depression of satDNA transcription was registered in many human epithelial tumors; however, it remains unclear whether the transcription is the reason or the consequence of the tumorigenesis [230]. New data arrive showing participation of satDNA transcripts in the assembly and maintenance of kinetochores [231]. All these facts point out the important and multifaceted functions of simple repeating sequences of satDNA in the mammalian genome.
CONCLUSION
Sequences of human and animal genomes not encoding proteins are full with various regulatory elements and ncRNA genes (see table). The best-known and well-studied elements include the gene flanking cis-acting regulatory sequences: promoters and sequences representing 5′- and 3′-UTRs. Although they occupy only a small part of the genomes under discussion, their role in regulation of gene expression is extremely high. Many genes use alternative regulatory sequences for regulation; therefore, the total number of promoters and UTRs considerably exceeds the number of genes annotated in the genomes.
Content of known and proposed functional noncoding DNA sequences in the
human genome
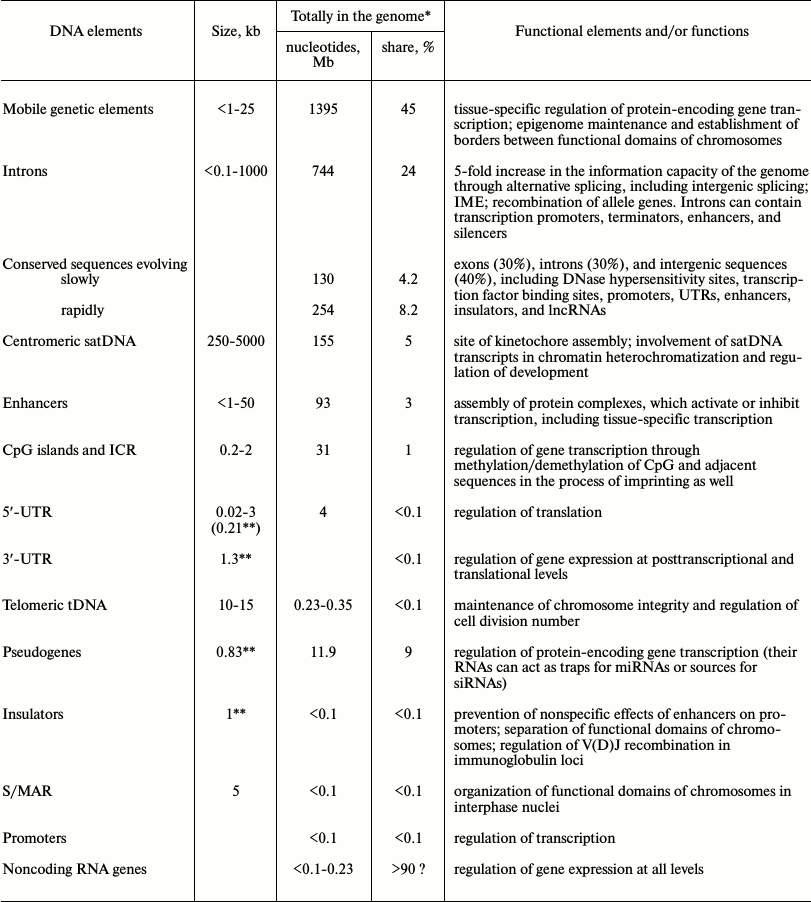
* The size of a haploid human genome is 3100 Mb.
** Average size.
Enhancers, locus control regions, insulators, and S/MAR sequences belong to a more massive group of regulatory elements of eukaryotic genomes. The share of enhancers, together with superenhancers, reaches 3% in the human genome. These elements play the key role in the establishment of highly ordered transcription in many tissues of an organism and in the process of ontogenesis. A distinctive feature of insulators and S/MAR sequences, although they represent a minor fraction of the human genome (<0.1%), is that they spatially and functionally organize prolonged (~1 Mb) chromosome domains expressing genes in interphase nuclei. The observed migration of the genetic loci from nucleus periphery to its center and back associated with their activation/deactivation indicate another rarely discussed function of noncoding sequences, which can make up a considerable share of these loci. In our opinion, evolutionary inclusion of noncoding sequences between the expressed genes imparts chromosomal genes with the flexibility required for maintenance of the dynamic intranuclear state of working genetic loci and their mobility within chromosomal territories. Such linker function imposes no strict requirements on the primary structure of these noncoding sequences, only the linear size is under the pressure of selection. If the hypothesis is true, hardly any intergenic linker sequences are significantly conserved, since their functions do not require it.
A considerable part (~1%) of the human genome is made of genomic CGI sites, which mark DNA methylation regions located in the intergenic fragments and close to 5′-termini of many genes, as well as inside the genes, and participate in regulation of transcription thereof. Taking into account sequences flanking CGIs (the so-called shores and shelves) that cover ~4 kb on either side of CGIs, their fraction in the genome can be higher. Recently, Prof. Romanov and colleagues developed a new concept that looks from an unexpected side on the role of DNA methylation in ontogenesis of eukaryotes [232]. They found that methylated CpG dinucleotides are nonrandomly included in various codons of gene exons in humans and other animals. Spontaneous deamination of 5-mC occurring during the individual’s lifetime is accompanied by formation of T and rising of mutations. Transformation of a sense codon into a stop-codon or codon encoding an amino acid unfavorable for the protein leads to inactivation of the protein or enzyme. Such codons were called by authors dangerous. According to the proposed model, the rate of dangerous codons is specific for organisms of different taxonomic groups and correlates negatively with individual lifespan, while DNA methylation patterns in dangerous codons represent a unique code of the aging of a biological species.
Pseudogenes represent another wide class of noncoding sequences, which until recently have been considered nonfunctional shatters of relevant genes. By now, it has been convincingly demonstrated that those pseudogenes that are transcribed in forward or reverse direction forming mRNAs or antisense RNAs are sources for ceRNAs and siRNAs involved in translational regulation of expression of genes they were generated by.
Recent studies of introns, which make up ~24% of all human genomic sequences, also revealed their various functions in regulation of gene expression. Alternative splicing and trans-splicing of mRNA precursors that were made possible by the presence of introns increase the informational capacity of the genome 5-fold. The global functional significance of introns also follows from the intron-mediated enhancement of gene expression (IME phenomenon). Besides, introns are the location sites for many regulatory DNA elements, such as alternative promoters and transcription terminators, transcription enhancers and silencers, and miRNA and lncRNA genes.
The recently discovered pervasive transcription covering 99% of human genome sequences opens new horizons in studies of the functional role of multiple ncRNAs it generates. It has already been found that short and long noncoding RNAs present among the transcripts produce regulatory effects at all levels of eukaryotic gene expression. Recently discovered regulatory networks involving cellular RNAs uncovered the top of a new unexplored iceberg of regulatory mechanisms governing eukaryotic gene expression.
Finally, repeated sequences that make up the major part of the mammalian genome no longer appear a desert in the landscape of the genome. Simple repeats forming centromeres and telomeres of chromosomes are transcribed and play an important role in functioning of these genetic loci and the cell as a whole. Transposons actively participate in regulation of expression of common genes and formation and maintenance of the epigenome and bordering barriers between functional domains of chromosomes.
Despite the diversity of functions of noncoding sequences in the eukaryotic genome that we demonstrated in this review, the importance of most of them for the living cell and the organism remains unclear. The previously established gene-protective function of noncoding sequences against endogenous chemical mutagens could be the primary function with respect to all others (see our review [14] and references therein). Indeed, modern eukaryotic organisms live and evolve in the atmosphere of oxygen. Reactive oxygen species formed in the process of living constantly destroy DNA integrity, causing up to 200,000 damages in each cell daily; without reparation, some of the damages can become mutations. Since noncoding sequences represent most of the mammalian genome, such mutations occur mainly in noncoding sequences without harmful consequences for cells and the entire organism. After the genome had evolutionarily gained its modern size and the definite ratio between coding and noncoding sequences, equilibrium was established in the organism, with the system of DNA reparation and gene protection by noncoding sequences providing for the acceptable level of endogenous mutagenesis compatible with life of a multicellular organism. At the same time, intragenomic expansion of noncoding sequences creates conditions for further evolving, accompanied with the emergence of new functions, most of which are yet to be established in the course of further studies.
Authors are grateful to Professor G. A. Romanov for critical review of the manuscript and valuable remarks.
This work was supported by the Russian Foundation for Basic Research (project No. 14-08-00801-A).
REFERENCES
1.Griffiths, P. E., and Stotz, K. (2006) Genes in the
postgenomic era, Theor. Med. Bioeth., 27, 499-521.
2.El-Hani, C. N. (2007) Between the cross and the
sword: the crisis of the gene concept, Genet. Mol. Biol.,
30, 297-307.
3.Boyle, A. P., Araya, C. L., Brdlik, C., Cayting,
P., Cheng, C., Cheng, Y., Gardner, K., Hillier, L.
W., Janette, J., Jiang, L., et al. (2014) Comparative
analysis of regulatory information and circuits across distant species,
Nature, 512, 453-456.
4.Dunham, I., Kundaje, A., Aldred, S. F., Collins, P.
J., Davis, C., Doyle, F., Epstein, C. B., Frietze, S., Harrow, J.,
Kaul, R., et al. (2012) An integrated encyclopedia of DNA elements in
the human genome, Nature, 489, 57-74.
5.The ENCODE Project Consortium, Birney, E.,
Stamatoyannopoulos, J. A., Dutta, A., Guigo, R., Gingeras, T. R.,
Margulies, E. H., Weng, Z., Snyder, M., Dermitzakis, E. T., Thurman, R.
E., et al. (2007) Identification and analysis of functional elements in
1% of the human genome by the ENCODE pilot project, Nature,
447, 799-816.
6.1000 Genomes Project Consortium, Abecasis, G. R.,
Altshuler, D., Auton, A., Brooks, L. D., Durbin, R. M., Gibbs, R. A.,
Hurles, M. E., McVean, G. A., et al. (2010) A map of human genome
variation from population-scale sequencing, Nature, 467,
1061-1073.
7.1000 Genomes Project Consortium, Abecasis, G. R.,
Auton, A., Brooks, L. D., DePristo, M. A., Durbin, R. M., Handsaker, R.
E., Kang, H. M., Marth, G. T., and McVean, G. A. (2012) An integrated
map of genetic variation from 1092 human genomes, Nature,
491, 56-65.
8.Roberts, N. J., Vogelstein, J. T., Parmigiani, G.,
Kinzler, K. W., Vogelstein, B., and Velculescu, V. E. (2012) The
predictive capacity of personal genome sequencing, Sci. Transl.
Med., 4, 135le5.
9.Gjuvsland, A. B., Vik, J., Beard, D. A., Hunter, P.
J., and Omholt, S. W. (2013) Bridging the genotype-phenotype gap: what
does it take? J. Physiol., 591, 2055-2066.
10.Van der Sijde, M. R., Ng, A., and Fu, J. (2014)
Systems genetics: from GWAS to disease pathways, Biochim. Biophys.
Acta, 1842, 1903-1909.
11.Clark, M. B., Amaral, P. P., Schlesinger, F. J.,
Dinger, M. E., Taft, R. J., Rinn, J. L., Ponting, C.
P., Stadler, P. F., Morris, K. V., Morillon, A., et al.
(2011) The reality of pervasive transcription, PLoS Biol.,
9, e1000625.
12.Harmston, N., Baresic, A., and Lenhard, B. (2013)
The mystery of extreme non-coding conservation, Philos. Trans. R.
Soc. Lond. B. Biol. Sci., 368, 1471-2970.
13.Gregory, T. R. (2005) The C-value enigma in
plants and animals: a review of parallels and an appeal for
partnership, Ann. Bot., 95, 133-146.
14.Patrushev, L. I., and Minkevich, I. G. (2008) The
problem of the eukaryotic genome size, Biochemistry (Moscow),
73, 1519-1552.
15.Kellis, M., Wold, B., Snyder, M. P., Bernstein,
B. E., Kundaje, A., Marinov, G. K., Ward, L. D., Birney, E., Crawford,
G. E., Dekker, J., et al. (2014) Defining functional DNA elements in
the human genome, Proc. Natl. Acad. Sci. USA, 111,
6131-6138.
16.Ponting, C. P., and Hardison, R. C. (2011) What
fraction of the human genome is functional? Genome Res.,
21, 1769-1776.
17.Lunter, G., Ponting, C. P., and Hein, J. (2006)
Genome-wide identification of human functional DNA using a neutral
indel model, PLoS Comput Biol., 2, e5.
18.Chiaromonte, F., Weber, R. J., Roskin, K. M.,
Diekhans, M., Kent, W. J., and Haussler, D. (2003) The share of human
genomic DNA under selection estimated from human-mouse genomic
alignments, Cold Spring Harb. Symp. Quant. Biol., 68,
245-254.
19.Lindblad-Toh, K., Garber, M., Zuk, O., Lin, M.
F., Parker, B. J., Washietl, S., Kheradpour, P., Ernst, J., Jordan, G.,
Mauceli, E., et al. (2011) A high-resolution map of human evolutionary
constraint using 29 mammals, Nature, 478, 476-482.
20.Rands, C. M., Meader, S., Ponting, C. P., and
Lunter, G. (2014) 8.2% of the human genome is constrained: variation in
rates of turnover across functional element classes in the human
lineage, PLoS Genet., 10, e1004525.
21.Kapranov, P., Cawley, S. E., Drenkow, J.,
Bekiranov, S., Strausberg, R. L., Fodor, S. P., and Gingeras, T. R.
(2002) Large-scale transcriptional activity in chromosomes 21 and 22,
Science, 296, 916-919.
22.Rinn, J. L., Euskirchen, G., Bertone, P.,
Martone, R., Luscombe, N. M., Hartman, S., Harrison, P. M., Nelson, F.
K., Miller, P., Gerstein, M., Weissman, S., and Snyder, M. (2003) The
transcriptional activity of human chromosome 22, Genes Dev.,
17, 529-540.
23.Bertone, P., Stolc, V., Royce, T. E., Rozowsky,
J. S., Urban, A. E., Zhu, X., Rinn, J. L., Tongprasit, W., Samanta, M.,
Weissman, S., Gerstein, M., and Snyder, M. (2004) Global identification
of human transcribed sequences with genome tiling arrays,
Science, 306, 2242-2246.
24.Cheng, J., Kapranov, P., Drenkow, J., Dike, S.,
Brubaker, S., Patel, S., Long, J., Stern, D., Tammana,
H., Helt, G., et al. (2005) Transcriptional maps of 10 human
chromosomes at 5-nucleotide resolution, Science, 308,
1149-1154.
25.Johnson, J. M., Edwards, S., Shoemaker, D., and
Schadt, E. E. (2005) Dark matter in the genome: evidence of widespread
transcription detected by microarray tiling experiments, Trends
Genet., 21, 93-102.
26.Kapranov, P., Sementchenko, V. I., and Gingeras,
T. R. (2003) Beyond expression profiling: next generation uses of high
density oligonucleotide arrays, Brief Funct. Genom. Proteom.,
2, 47-56.
27.Wang, Z., Gerstein, M., and Snyder, M. (2009)
RNA-Seq: a revolutionary tool for transcriptomics, Nature Rev.
Genet., 10, 57-63.
28.Mutz, K. O., Heilkenbrinker, A., Lonne, M.,
Walter, J. G., and Stahl, F. (2013) Transcriptome analysis using
next-generation sequencing, Curr. Opin. Biotechnol., 24,
22-30.
29.Okoniewski, M. J., and Miller, C. J. (2006)
Hybridization interactions between probe sets in short oligo
microarrays lead to spurious correlations, BMC Bioinformatics,
7, 276.
30.Royce, T. E., Rozowsky, J. S., and Gerstein, M.
B. (2007) Toward a universal microarray: prediction of gene expression
through nearest-neighbor probe sequence identification, Nucleic
Acids Res., 35, e99.
31.Pareek, C. S., Smoczynski, R., and Tretyn A.
(2011) Sequencing technologies and genome sequencing, Appl.
Genet., 52, 413-435.
32.Schadt, E. E., Turner, S., and Kasarskis, A.
(2010) A window into third-generation sequencing, Hum. Mol.
Genet., 19, 227-240.
33.Costa, V., Angelini C., De Feis, I., and
Ciccodicola, A. (2010) Uncovering the complexity of transcriptomes with
RNA-seq, J. Biomed. Biotechnol., 2010, 853916.
34.Mortazavi, A., Williams, B. A., Mc Cue, K.,
Schaeffer, L., and Wold, B. (2008) Mapping and quantifying mammalian
transcriptomes by RNA-Seq, Nature Methods, 5,
621-628.
35.Harbers, M., and Carninci, P. (2005) Tag-based
approaches for transcriptome research and genome annotation, Nature
Methods, 2, 495-502.
36.Jacquier, A. (2009) The complex eukaryotic
transcriptome: unexpected pervasive transcription and novel small RNAs,
Nature Rev. Genet., 10, 833-844.
37.Kapranov, P., and St. Laurent, G. (2012) Dark
matter RNA: existence, function, and controversy, Front. Genet.,
3, 60.
38.Kapranov, P., St. Laurent, G., Raz, T., Ozsolak,
F., Reynolds, C. P., Sorensen, P. H., Reaman, G., Milos, P., Arceci, R.
J., Thompson, J. F., and Triche, T. J. (2010). The majority of total
nuclear-encoded non-ribosomal RNA in a human cell is “dark
matter” unannotated RNA, BMC Biol., 8, 149.
39.Shiraki, T., Kondo, S., Katayama,
S., Waki, K., Kasukawa, T., Kawaji, H., Kodzius,
R., Watahiki, A., Nakamura, M., Arakawa,
T., Fukuda, S., Sasaki, D., Podhajska, A., Harbers,
M., Kawai, J., Carninci, P., and Hayashizaki, Y. (2003)
Cap analysis gene expression for high-throughput analysis of
transcriptional starting point and identification of promoter usage,
Proc. Natl. Acad. Sci. USA, 100, 15776-15781.
40.Faulkner, G. J., Kimura, Y., Daub, C. O., Wani,
S., Plessy, C., Irvine, K. M., Schroder, K., Cloonan, N., Steptoe, A.
L., Lassmann, T., et al. (2009) The regulated retrotransposon
transcriptome of mammalian cells, Nature Genet., 41,
563-571.
41.Maston, G. A., Evans, S. K., and Green, M. R.
(2006) Transcriptional regulatory elements in the human genome,
Annu. Rev. Genom. Hum. Genet., 7, 29-59.
42.Lenhard, B., Sandelin, A., and Carninci, P.
(2012) Metazoan promoters: emerging characteristics and insights into
transcriptional regulation, Nature Rev. Genet., 13,
233-245.
43.Kadonaga, J. T. (2012) Perspectives on the RNA
polymerase II core promoter, Wiley Interdisc. Rev. Dev. Biol.,
1, 40-51.
44.Thomas, M. C., and Chiang, C. M. (2006) The
general transcription machinery and general cofactors, Crit. Rev.
Biochem. Mol. Biol., 41, 105-178.
45.Massari, M. E., and Murre, C. (2000)
Helix-loop-helix proteins: regulators of transcription in eucaryotic
organisms, Mol. Cell. Biol., 20, 429-440.
46.Nakahata, Y., Yoshida, M., Takano, A., Soma,
H., Yamamoto, T., Yasuda, A., Nakatsu, T.,
and Takumi, T. (2008) A direct repeat of E-box-like elements is
required for cell-autonomous circadian rhythm of clock genes, BMC
Mol. Biol., 9, 1.
47.Santoro, N., Johansson, N., and Thiele, D. J.
(1998) Heat shock element architecture is an important determinant in
the temperature and transactivation domain requirements for heat shock
transcription factor, Mol. Cell. Biol., 18,
6340-6352.
48.Perry, R. P. (2005) The architecture of mammalian
ribosomal protein promoters, BMC Evol Biol., 5, 15.
49.Pankratova, E. V. (2008) Alternative promoters
and the complexity of the mammalian transcriptome, Mol. Biol.
(Moscow), 42, 422-443.
50.Bestor, T. H. (2000) The DNA methyltransferases
of mammals, Hum. Mol. Genet., 9, 2395-2402.
51.Trinklein, N. D., Aldred, S. F., Hartman, S. J.,
Schroeder, D. I., Otillar, R. P., and Myers, R. M. (2004) An abundance
of bidirectional promoters in the human genome, Genome Res.,
14, 62-66.
52.Yang, M. Q., and Elnitski, L. L. (2008) Diversity
of core promoter elements comprising human bidirectional promoters,
BMC Genomics, 9 (Suppl. 2), S3.
53.Rachakonda, P. S., Hosen, I., de Verdeir, P. J.,
Fallah, M., Heidenreich, B., Ryk, C., Wiklund, N. P., Steineck, G.,
Schadendorf, D., Hemminki, K., and Kumar, R. (2013) TERT promoter
mutations in bladder cancer affect patient survival and disease
recurrence through modification by a common polymorphism, Proc.
Natl. Acad. Sci. USA, 110, 17426-17431.
54.Savinkova, L. K., Ponomarenko, M. P.,
Ponomarenko, P. M., Drachkova, I. A., Lysova, M. V.,
Arshinova, T. V., and Kolchanov, N. A. (2009) TATA box
polymorphisms in human gene promoters and associated hereditary
pathologies, Biochemistry (Moscow), 74, 117-129.
55.Mignone, F., Gissi, C., Liuni, S., and Pesole, G.
(2002) Untranslated regions of mRNAs, Genome Biol., 3
(3).
56.Zhang, T., Haws, P., and Wu, Q. (2004) Multiple
variable first exons: a mechanism for cell- and tissue-specific gene
regulation, Genome Res., 14, 79-89.
57.Iakova, P., Wang, G. L., Timchenko, L.,
Michalak, M., Pereira-Smith, O. M., Smith, J. R., and
Timchenko, N. A. (2004) Competition of CUGBP1 and calreticulin for the
regulation of p21 translation determines cell fate, EMBO J.,
23, 406-417.
58.Araujo, P. R., Yoon, K., Ko, D., Smith, A. D.,
Qiao, M., Suresh, U., Burns, S. C., and Penalva, L. O. F. (2012) Before
it gets started: regulating translation at the 5′ UTR, Comp.
Funct. Genom., 2012, 475731.
59.Wethmar, K., Smink, J. J., and Leutz, A. (2010)
Upstream open reading frames: molecular switches in (patho)physiology,
Bioessays, 32, 885-893.
60.Chatterjee, S., and Pal, J. K. (2009) Role of
5′- and 3′-untranslated regions of mRNAs in human diseases,
Biol. Cell, 101, 251-262.
61.Matoulkova, E., Michalova, E., Vojtesek, B., and
Hrstka, R. (2012) The role of the 3′ untranslated region in
posttranscriptional regulation of protein expression in mammalian
cells, RNA Biol., 9, 563-576.
62.Zhao, W., Blagev, D., Pollack, J. L., and Erle,
D. J. (2011) Toward a systematic understanding of mRNA 39 untranslated
regions, Proc. Am. Thorac. Soc., 8, 163-166.
63.Smith, R. W. P., Blee, T. K. P., and Gray, N. K.
(2014) Poly(A)-binding proteins are required for diverse biological
processes in metazoans, Biochem. Soc. Trans., 42,
1229-1237.
64.Chen, J., and Kastan, M. B. (2010)
5′-3′-UTR interactions regulate p53 mRNA translation and
provide a target for modulating p53 induction after DNA damage,
Genes Dev., 24, 2146-2156.
65.Sandberg, R., Neilson, J. R., Sarma, A., Sharp,
P. A., and Burge, C. B. (2008) Proliferating cells express mRNAs with
shortened 30 untranslated regions and fewer microRNA target sites,
Science, 320, 1643-1647.
66.Hitti, E., and Khabar, K. S. A. (2012) Sequence
variations affecting AU-rich element function and disease, Front.
Biosci., 17, 1846-1860.
67.Halees, A. S., Hitti, E., Al-Saif, M., Mahmoud,
L., Vlasova-St. Louis, I. A., Beisang, D. J., Bohjanen, P. R., and
Khabar, K. (2011) Global assessment of GU-rich regulatory content and
function in the human transcriptome, RNA Biol., 8,
681-691.
68.Hentze, M. W., Muckenthaler, M. U., and Andrews,
N. C. (2004) Balancing acts: molecular control of mammalian iron
metabolism, Cell, 117, 285-297.
69.Chavatte, L., Brown, B. A., and Driscoll, D. M.
(2005) Ribosomal protein L30 is a component of the UGA-selenocysteine
recoding machinery in eukaryotes, Nature Struct. Mol. Biol.,
12, 408-416.
70.Park, E., and Maquat, L. E. (2013)
Staufen-mediated mRNA decay, Wiley Interdiscip. Rev. RNA,
4, 423-435.
71.Nguyen, A. (2000) Prothrombin G20210A
polymorphism and thrombophilia, Mayo Clin Proc., 75,
595-604.
72.Varol, N., Conac, E., Gurocak, S., and Sozen, S.
(2011) The realm of micro RNAs in cancers, Mol. Biol. Rep.,
38, 1079-1089.
73.Li, E., and Zhang, Y. (2014) DNA methylation in
mammals, Cold Spring Harb. Perspect. Biol., 6 (5).
74.Medvedeva, Y. A., Khamis, A. M., Kulakovskiy, I.
V., Ba-Alawi, W., Bhuyan, M. S. I., Kawaji, H., Lassmann, T., Harbers,
M., Forrest, A. R., and Bajic, V. B.; FANTOM consortium (2013) Effects
of cytosine methylation on transcription factor binding sites, BMC
Genom., 15, 119.
75.Stadler, M. B., Murr, R., Burger, L., Ivanek, R.,
Lienert, F., Scholer, A., van Nimwegen, E., Wirbelauer, C., Oakeley, E.
J., Gaidatzis, D., Tiwari, V. K., and Schubeler, D. (2011) DNA
binding factors shape the mouse methylome at distal regulatory regions,
Nature, 480, 490-495.
76.Bestor, T. H., and Bourchis, D. (2004) Transposon
silencing and imprint establishment in mammalian germ cells, Cold
Spring Harb. Symp. Quant. Biol., 69, 381-387.
77.Maunakea, A. K., Nagarajan, R. P., Bilenky, M.,
Ballinger, T. J., D’Souza, C., Fouse, S. D., Johnson, B.
E., Hong, C., Nielsen, C., Zhao, Y., Turecki, G.,
et al. (2010) Conserved role of intragenic DNA methylation in
regulating alternative promoters, Nature, 466,
253-257.
78.Plongthongkum, N., van Eijk, K. R., de Jong, S.,
Wang, T., Sul, J. H., Boks, M. P., Kahn, R. S., Fung, H.
L., Ophoff, R. A., and Zhang, K. (2014) Characterization of
genome–methylome interactions in 22 nuclear pedigrees, PLoS
ONE, 9 (7).
79.Paul, D. S., and Beck, S. (2014) Advances in
epigenome-wide association studies for common diseases, Trends Mol.
Med., pii: S1471-4914(14)00115-4.
80.Hackett, J. A., and Surani, M. A. (2012) DNA
methylation dynamics during the mammalian life cycle, Philos. Trans.
R. Soc. Lond. B. Biol. Sci., 368 (1609).
81.Illingworth, R. S., and Bird, A. P. (2009) CpG
islands – a “rough guide”, FEBS Lett.,
583, 1713-1720.
82.Irizarry, R. A., Ladd-Acosta, C., Wen, B., Wu,
Z., Montano, C., Onyango, P., Cui, H., Gabo, K., Rongione, M., Webster,
M., et al. (2009) The human colon cancer methylome shows similar hypo-
and hypermethylation at conserved tissue-specific CpG island shores,
Nature Genet., 41, 178-186.
83.Kozlenkov, A., Roussos, P., Timashpolsky, A.,
Barbu, M., Rudchenko, S., Bibikova, M., Klotzle, B., Byne, W., Lyddon,
R., Di Narzo, A. F., et al. (2014) Differences in DNA methylation
between human neuronal and glial cells are concentrated in enhancers
and non-CpG sites, Nucleic Acids Res., 42, 109-127.
84.Blackledge, N. P., Thomson, J. P., and Skene, P.
J. (2013) CpG island chromatin is shaped by recruitment of ZF-CxxC
proteins, Cold Spring Harb. Perspect. Biol., 5,
a018648.
85.Vinson, C., and Chatterjee, R. (2012) CG
methylation, Epigenomics, 4, 655-663.
86.Sharp, A. J., Stathaki, E., Migliavacca, E.,
Brahmachary, M., Montgomery, S. B., Dupre, Y., and Antonarakis, S. E.
(2011) DNA methylation profiles of human active and inactive X
chromosomes, Genome Res., 21, 1592-1600.
87.Barlow, D. P., and Bartolomei, M. S. (2014)
Genomic imprinting in mammals, Cold Spring Harb. Perspect.
Biol., 6, 018382.
87a.Wang, L., Zhang, J., Duan, J., Gao, X., Zhu, W., et al. (2014)
Programming and inheritance of parental DNA methylomes in mammals,
Cell, 157, 979-991.
88.Unoki, M., and Nakamura, Y. (2003). Methylation
at CpG islands in intron 1 of EGR2 confers enhancer-like activity,
FEBS Lett., 554, 67-72.
89.Rakyan, V. K., Down, T. A., Balding, D. J., and
Beck, S. (2011) Epigenome-wide association studies for common human
diseases, Nature Rev. Genet., 12, 529-541.
89a.Feber, A., Wilson, G. A., Zhang, L., Presneau, N., Idowu, B., Down,
T. A., Rakyan, V. K., Noon, L. A., Lloyd, A. C., Stupka, E.,
Schiza, V., Teschendorff, A. E., Schroth, G. P., Flanagan, A., and
Beck, S. (2011) Comparative methylome analysis of benign and malignant
peripheral nerve sheath tumors, Genome Res., 21,
515-524.
90.Dick, K., Nelson, C. P., Tsaprouni, L., Sandling,
J. K., Aissi, D., Wahl, S., Meduri, E., Morange, P. E., Gagnon, F.,
Grallert, H., Waldenberger, M., Peters, A., Erdmann, J.,
Hengstenberg, C., Cambien, F., Goodall, A. H., Ouwehand, W. H.,
Schunkert, H., Thompson, J. R., Spector, T. D., Gieger, C., Tregouet,
D. A., Deloukas, P., and Samani, N. J. (2014) DNA methylation and
body-mass index: a genome-wide analysis, Lancet, 383,
1990-1998.
91.Rose, A. B. (2008) Intron-mediated regulation of
gene expression, Curr. Top. Microbiol. Immunol., 326,
277-290.
92.Bang, M. L., Centner, T., Fornoff, F., Geach, A.
J., Gotthardt, M., McNabb, M., Witt, C. C., Labeit,
D., Gregorio, C. C., Granzier, H., and Labeit, S. (2001) The
complete gene sequence of titin, expression of an unusual approximately
700-kDa titin isoform, and its interaction with obscurin identify a
novel Z-line to I-band linking system, Circ. Res., 89,
1065-1072.
93.Grzybowska, E. A. (2012) Human intronless genes:
functional groups, associated diseases, evolution, and mRNA processing
in absence of splicing, Biochem. Biophys. Res. Commun.,
424, 1-6.
94.Lander, E. S., Linton, L. M., Birren, B.,
Nusbaum, C., Zody, M. C., Baldwin, J., Devon, K., Dewar, K., Doyle, M.,
FitzHugh, W., et al. (2001) Initial sequencing and analysis of the
human genome, Nature, 409, 860-921.
95.Venter, J. C., Adams, M. D., Myers, E. W., Li, P.
W., Mural, R. J., Sutton, G. G., Smith, H. O., Yandell, M., Evans, C.
A., Holt, R. A., et al. (2001) The sequence of the human genome,
Science, 291, 1304-1351.
96.Koonin, E. V. (2006) The origin of introns and
their role in eukaryogenesis: a compromise solution to the
introns-early versus introns-late debate? Biol Direct.,
1, 22.
97.Irimia, M., and Roy, S. W. (2014) Origin of
spliceosomal introns and alternative splicing, Cold Spring Harb.
Perspect. Biol., 6 (6).
98.Gilbert, W. (1987) The exon theory of genes,
Cold Spring Harb. Symp. Quant Biol., 52, 901-905.
99.Elliott, D. J. (2014) Illuminating the
transcriptome through the genome, Genes, 5, 235-253.
100.Djebali, S., Davis, C. A., Merkel, A., Dobin,
A., Lassmann, T., Mortazavi, A., Tanzer, A., Lagarde, J., Lin, W.,
Schlesinger, F., et al. (2012) Landscape of transcription in human
cells, Nature, 489, 101-108.
101.Harrison, P. M., Kumar, A., Lang, N., Snyder,
M., and Gerstein, M. (2002) A question of size: the eukaryotic proteome
and the problems in defining it, Nucleic Acids Res., 30,
1083-1090.
102.Amrani, N., Ganesan, R., Kervestin, S., Mangus,
D. A., Ghosh, S., and Jacobson, A. (2004) A faux 3′-UTR promotes
aberrant termination and triggers nonsense-mediated mRNA decay,
Nature, 432, 112-118.
103.Buckley, P. T., Lee, M. T., Sul, J. Y.,
Miyashiro, J. Y., Bell, T. J., Fisher, S. A., Kim, J., and Eberwine, J.
(2011) Cytoplasmic intron sequence retaining transcripts can be
dendritically targeted via ID element retrotransposons, Neuron,
69, 877-884.
104.Sorek, R., Shamir, R., and Ast, G. (2004) How
prevalent is functional alternative splicing in the human genome?
Trends Genet., 20, 68-71.
105.Roy, S. W., and Irimia, M. (2008) Intron
mis-splicing: no alternative? Genome Biol., 9, 208.
106.Lasda, E. L., and Blumenthal, T. (2011)
Trans-splicing, WIREs RNA, 2, 417-434.
107.Akiva, P., Toporik, A., Edelheit, S., Peretz,
Y., Diber, A., Shemesh, R., Novik, A., and Sorek, R. (2006)
Transcription-mediated gene fusion in the human genome, Genome
Res., 16, 30-36.
108.McManus, C. J., Duff, M. O., Eipper-Mains, J.,
and Graveley, B. R. (2010) Global analysis of trans-splicing in
Drosophila, Proc. Natl. Acad. Sci. USA, 107,
12975-12979.
109.Fang, W., Wei, Y., Kang, Y., and Landweber, L.
F. (2012) Detection of a common chimeric transcript between human
chromosomes 7 and 16, Biol. Direct., 7, 49.
110.Hu, G.-J., Chen, J., Zhao, X.-N., Xu, J.-J.,
Guo, D.-Q., Lu, M., Zhu, M., Xiong, Y., Li, Q., Chang, C. C., et al.
(2013) Production of ACAT1 56-kDa isoform in human cells via
trans-splicing involving the ampicillin resistance gene, Cell
Res., 23, 1007-1024.
111.Wu, C.-S., Yu, C.-Y., Chuang, C.-Y., Hsiao, M.,
Kao, C. F., Kuo, H. C., and Chuang, T. J. (2014) Integrative
transcriptome sequencing identifies trans-splicing events with
important roles in human embryonic stem cell pluripotency, Genome
Res., 24, 25-36.
112.Gingeras, T. R. (2009) Implications of chimeric
non-co-linear transcripts, Nature, 461, 206-211.
113.Frenkel-Morgenstern, M., Lacroix, V., Ezkurdia,
I., Levin, Y., Gabashvili, A., Prilusky, J., Del Pozo, A., Tress, M.,
Johnson, R., Guigo, R., and Valencia, A. (2012) Chimeras taking shape:
potential functions of proteins encoded by chimeric RNA transcripts,
Genome Res., 22, 1231-1242.
114.Greger, L., Su, J., Rung, J., Ferreira, P. G.,
Geuvadis consortium, Lappalainen, T., Dermitzakis, E. T., and Brazma,
A. (2014) Tandem RNA chimeras contribute to transcriptome diversity in
human population and are associated with intronic genetic variants,
PLoS ONE, 9 (8).
115.Fedorova, L., and Fedorov, A. (2003) Introns in
gene evolution, Genetica, 118, 123-131.
116.Wang, Z., and Burge, C. B. (2008) Splicing
regulation: from a parts list of regulatory elements to an integrated
splicing code, RNA, 14, 802-813.
117.Patel, A. A., McCarthy, M., and Steitz, J. A.
(2002) The splicing of U12-type introns can be a rate-limiting step in
gene expression, EMBO J., 21, 3804-3815.
118.Lewandowska, M. A. (2013) The missing puzzle
piece: splicing mutations, Int. J. Exp. Pathol., 6,
2675-2682.
119.Lomvardas, S., Barnea, G., Pisapia, D. J.,
Mendelsohn, M., Kirkland, J., and Axel, R. (2006) Interchromosomal
interactions and olfactory receptor choice, Cell, 126,
403-413.
120.Mousavi, K., Zare, H., Koulnis, M., and
Sartorelli, V. (2014) The emerging roles of eRNAs in transcriptional
regulatory networks, RNA Biol., 11, 106-110.
121.Bulger, M., and Groudine, M. (2010) Enhancers:
the abundance and function of regulatory sequences beyond promoters,
Dev Biol., 339, 250-257; doi:
10.1016/j.ydbio.2009.11.035.
122.Zentner, G. E., Tesar, P. J., and Scacheri, P.
C. (2011) Epigenetic signatures distinguish multiple classes of
enhancers with distinct cellular functions, Genome Res.,
21, 1273-1283.
123.Heintzman, N. D., Stuart, R. K., Hon, G., Fu,
Y., Ching, C. W., Hawkins, R. D., Barrera, L. O., Van Calcar, S., Qu,
C., Ching, K. A., et al. (2007) Distinct and predictive chromatin
signatures of transcriptional promoters and enhancers in the human
genome, Nature Genet., 39, 311-318.
124.Visel, A., Blow, M. J., Li, Z., Zhang, T.,
Akiyama, J. A., Holt, A., Plajzer-Frick, I., Shoukry, M., Wright, C.,
Chen, F., et al. (2009) ChIP-seq accurately predicts tissue-specific
activity of enhancers, Nature, 457, 854-858.
125.Ong, C. T., and Corces, V. G. (2011) Enhancer
function: new insights into the regulation of tissue-specific gene
expression, Nature Rev. Genet., 12, 283-293.
126.Heintzman, N. D., Hon, G. C., Hawkins, R. D.,
Kheradpour, P., Stark, A., Harp, L. F., Ye, Z., Lee, L. K., Stuart, R.
K., Ching, C. W., et al. (2009) Histone modifications at human
enhancers reflect global cell-type-specific gene expression,
Nature, 459, 108-112.
127.Cho, K. W. Y. (2012) Enhancers: WIRE review,
WIREs Dev. Biol., 1, 469-478.
128.Spitz, F., and Furlong, E. E. M. (2012)
Transcription factors: from enhancer binding to developmental control,
Nature Rev. Genet., 13, 613-626.
129.Maston, G. A., Landt, S. G., Snyder, M., and
Green, M. R. (2012) Characterization of enhancer function from
genome-wide analyses, Annu. Rev. Genom. Hum. Genet., 13,
29-57.
130.Barolo, S. (2012) Shadow enhancers: frequently
asked questions about distributed cis-regulatory information and
enhancer redundancy, Bioessays, 34, 135-141.
131.Hong, J. W., Hendrix, D. A., and Levine, M. S.
(2008) Shadow enhancers as a source of evolutionary novelty,
Science, 321, 1314.
132.Abbasi, A. A., Paparidis, Z., Malik, S., Bangs,
F., Schmidt, A., Koch, S., Lopez-Rios, J., and Grzeschik, K. H. (2010)
Human intronic enhancers control distinct subdomains of Gli3 expression
during mouse CNS and limb development, BMC Devel. Biol.,
10, 44.
132a.Zhou, X., and Sigmund, C. D. (2008) The chorionic enhancer is
dispensable for regulated expression of the human renin gene, Am. J.
Physiol. Regul. Integr. Comp. Physiol., 294, 279-287.
133.Blackwood, E. M., and Kadonaga, J. T. (1998)
Going the distance: a current view of enhancer action, Science,
281, 60-63.
134.Krivega, I., and Dean, A. (2012) Enhancer and
promoter interactions – long distance calls, Curr. Opin.
Genet. Devel., 22, 79-85.
135.Lieberman-Aiden, E., van Berkum, N. L.,
Williams, L., Imakaev, M., Ragoczy, T., Telling, A., Amit, I., Lajoie,
B. R., Sabo, P. J., Dorschner, M. O., et al. (2009) Comprehensive
mapping of long-range interactions reveals folding principles of the
human genome, Science, 326, 289-293.
136.Kim, T.-K., Hemberg, M., Gray, J. M., Costa, A.
M., Bear, D. M., Wu, J., Harmin, D. A., Laptewicz, M., Barbara-Haley,
K., Kuersten, S., et al. (2010) Widespread transcription at neuronal
activity-regulated enhancers, Nature, 465, 182-187.
137.Orom, U. A., Derrien, T., Beringer, M.,
Gumireddy, K., Gardini, A., Bussotti, G., Lai, F., Zytnicki, M.,
Notredame, C., Huang, Q., Guigo, R., and Shiekhattar, R. (2010) Long
noncoding RNAs with enhancer-like function in human cells, Cell,
143, 46-58.
138.Orom, U. A., and Shiekhattar, R. (2013) Long
noncoding RNAs usher in a new era in the biology of enhancers,
Cell, 154, 1190-1193.
139.Kowalczyk, M. S., Hughes, J. R., Garrick, D.,
Lynch, M. D., Sharpe, J. A., Sloane-Stanley, J. A., McGowan, S. J., De
Gobbi, M., Hosseini, M., Vernimmen, D., et al. (2012) Intragenic
enhancers act as alternative promoters, Mol. Cell, 45,
447-458.
140.Lam, M. T. Y., Li, W., Rosenfeld, M. G., and
Glass, C. K. (2014) Enhancer RNAs and regulated transcriptional
programs, Trends Biochem. Sci., 39, 170-182.
141.Li, W., Notani, D., Ma, Q., Tanasa, B., Nunez,
E., Chen, A. Y., Merkurjev, D., Zhang, J., Ohgi, K., and Song, X.
(2013) Functional roles of enhancer RNAs for estrogen-dependent
transcriptional activation, Nature, 498, 516-520.
142.Melo, C. A., Drost, J., Wijchers, P.
J., van de Werken, H., de Wit, E., Oude Vrielink, J.
A., Elkon, R., Melo, S. A., Leveille, N., Kalluri,
R., de Laat, W., and Agami, R. (2013) eRNAs are required for
p53-dependent enhancer activity and gene transcription, Mol.
Cell, 49, 524-535.
143.Xu, J., Watts, J. A., Pope, S. D., Gadue, P.,
Kamps, M., Plath, K., Zaret, K. S., and Smale, S. T. (2009)
Transcriptional competence and the active marking of tissue-specific
enhancers by defined transcription factors in embryonic and induced
pluripotent stem cells, Genes Dev., 23, 2824-2838.
144.Reynolds, N., O’Shaughnessy, A., and
Hendrich, B. (2013) Transcriptional repressors: multifaceted regulators
of gene expression, Development, 140, 505-512.
145.Pennacchio, L. A., Ahituv, N., Moses, A. M.,
Prabhakar, S., Nobrega, M. A., Shoukry, M., Minovitsky, S., Dubchak,
I., Holt, A., Lewis, K. D., et al. (2006) In vivo enhancer
analysis of human conserved non-coding sequences, Nature,
444, 499-502.
146.Navratilova, P., Fredman, D., Hawkins, T. A.,
Turner, K., Lenhard, B., and Becker, T. S. (2009) Systematic
human/zebrafish comparative identification of cis-regulatory
activity around vertebrate developmental transcription factor genes,
Dev Biol., 327, 526-540.
147.Herz, H.-M., Hu, D., and Shilatifard, A. (2014)
Enhancer malfunction in cancer, Mol. Cell, 53,
859-866.
148.Dean, A. (2006) On a chromosome far, far away:
LCRs and gene expression, Trends Genet., 22, 38-45.
149.Liang, S., Moghimi, B., Yang, T. P.,
Strouboulis, J., and Bungert, J. (2008) Locus control region mediated
regulation of adult β-globin gene expression, J. Cell
Biochem., 105, 9-16.
150.Yankulov, K. (2013) Dynamics and stability:
epigenetic conversions in position effect variegation, Biochem. Cell
Biol., 91, 6-13.
151.Farrell, C. M., West, A. G., and Felsenfeld, G.
(2002) Conserved CTCF insulator elements flank the mouse and human
β-globin loci, Mol. Cell. Biol., 22, 3820-3831.
152.Spilianakis, C. G., Lalioti, M. D., Town, T.,
Lee, G. R., and Flavel, R. A. (2005) Interchromosomal associations
between alternatively expressed loci, Nature, 435,
637-645.
153.Gribnau, J., Diderich, K., Pruzina, S.,
Calzolari, R., and Fraser, P. (2000) Intergenic transcription and
developmental remodeling of chromatin subdomains in the human
β-globin locus, Mol. Cell, 5, 377-386.
154.Noordermeer, D., Branco, M. R., Splinter, E.,
Klous, P., van Ijcken, W., Swagemakers, S., Koutsourakis, M., van der
Spek, P., Pombo, A., and de Laat, W. (2008) Transcription and chromatin
organization of a housekeeping gene cluster containing an integrated
beta-globin locus control region, PLoS Genet., 4,
e1000016.
155.Ragoczy, T., Bender, M. A., Telling, A., Byron,
R., and Groudine, M. (2006) The locus control region is required for
association of the murine beta-globin locus with engaged transcription
factories during erythroid maturation, Genes Dev., 20,
1447-1457.
156.Brown, J. M., Green, J., das Neves, R. P.,
Wallace, H. A., Smith, A. J., Gray, N., Taylor, S., Wood, W. G., Higgs,
D. R., et al. (2008) Association between active genes occurs at nuclear
speckles and is modulated by chromatin environment, J. Cell
Biol., 182, 1083-1097.
157.Guo, C., Gerasimova, T., Hao, H., Ivanova, I.,
Chakraborty, T., Selimyan, R., Oltz, E. M., and Sen, R. (2011) Two
forms of loops generate the chromatin conformation of the
immunoglobulin heavy-chain gene locus, Cell, 147,
332-343.
158.Dixon, J. R., Selvaraj, S., Yue, F., Kim, A.,
Li, Y., Shen, Y., Hu, M., Liu, J. S., and Ren, B. (2012) Topological
domains in mammalian genomes identified by analysis of chromatin
interactions, Nature, 485, 376-380.
159.Van Bortle, K., and Corces, V. G. (2013) The
role of chromatin insulators in nuclear architecture and genome
function, Curr. Opin. Genet. Dev., 23, 212-218.
160.Chetverina, D., Aoki, T., Erokhin, M.,
Georgiev, P., and Schedl, P. (2013) Making connections: insulators
organize eukaryotic chromosomes into independent cis-regulatory
networks, Bioessays, 36, 163-172.
161.Van Bortle, K., and Corces, V. G. (2012) tDNA
insulators and the emerging role of TFIIIC in genome organization,
Transcription, 3, 277-284.
162.Jothi, R., Cuddapah, S., Barski, A., Cui, K.,
and Zhao, K. (2008) Genome-wide identification of in vivo
protein–DNA binding sites from ChIP-Seq data, Nucleic Acids
Res., 36, 5221-5231.
163.Moqtaderi, Z., Wang, J., Raha, D., White, R.
J., Snyder, M., Weng, Z., and Struhl, K. (2010) Genomic binding
profiles of functionally distinct RNA polymerase III transcription
complexes in human cells, Nature Struct. Mol. Biol., 17,
635-640.
164.Mirkovitch, J., Mirault, M. E., and Laemmli, U.
K. (1984) Organization of the higher order chromatin loop: specific DNA
attachment sites on nuclear scaffold, Cell, 39,
223-232.
165.Gerasimova, T. I., Byrd, K., and Corces, V. G.
(2000) A chromatin insulator determines the nuclear localization of
DNA, Mol. Cell, 6, 1025-1035.
166.Iarovaia, O. V., Akopov, S. B., Nikolaev, L.
G., Sverdlov, E. D., and Razin, S. V. (2005) Induction of transcription
within chromosomal DNA loops flanked by MAR elements causes an
association of loop DNA with the nuclear matrix, Nucleic Acids
Res., 33, 4157-4163.
167.Shaposhnikov, S. A., Akopov, S. B., Chernov, I.
P., Thomsen, P. D., Joergensen, C., Collins, A. R., Frengen,
E., and Nikolaev, L. G. (2007) A map of nuclear matrix attachment
regions within the breast cancer loss-of-heterozygosity region on human
chromosome 16q22.1, Genomics, 89, 354-361.
168.Keaton, M. A., Taylor, C. M., Layer, R. M., and
Dutta, A. (2011) Nuclear scaffold attachment sites within ENCODE
regions associate with actively transcribed genes, PLoS ONE,
6 (3).
169.Jackson, D. A., Dickinson, P., and Cook, P. R.
(1990) The size of chromatin loops in HeLa cells, EMBO J.,
9, 567-571.
170.Libri, V., Miesen, P., van Rij, R. P., and
Buck, A. H. (2013) Regulation of microRNA biogenesis and turnover by
animals and their viruses, Cell Mol. Life Sci., 70,
3525-3544.
171.Hirose, T., Mishima, Y., and Tomari, Y. (2014)
Elements and machinery of non-coding RNAs: toward their taxonomy,
EMBO Rep., 15, 489-507.
172.Kim, V. N., Han, J., and Siomi, M. C. (2009)
Biogenesis of small RNAs in animals, Nature Rev. Mol. Cell
Biol., 10, 126-139.
173.Monteys, A. M., Spengler, R. M., Wan,
J., Tecedor, L., Lennox, K. A., Xing, Y.,
and Davidson, B. L. (2010) Structure and activity of putative
intronic miRNA promoters, RNA, 16, 495-505.
174.Westholm, J. O., and Lai, E. C. (2011)
Mirtrons: microRNA biogenesis via splicing, Biochimie,
93, 1897-1904.
175.Scott, M. S., and Ono, M. (2011) From snoRNA to
miRNA: dual function regulatory non-coding RNAs, Biochimie,
93, 1987-1992.
176.Neilsen, C. T., Goodall, G. J., and Bracken, C.
P. (2012) IsomiRs – the overlooked repertoire in the dynamic
microRNAome, Trends Genet., 28, 544-549.
177.Kawamata, T., and Tomari, Y. (2010) Making
RISC, Trends Biochem. Sci., 35, 368-376.
178.Elkayam, E., Kuhn, C-D., Tocilj, A., Haase, A.
D., Greene, E. M., Hannon, G. J., and Joshua-Tor, L. (2012) The
structure of human Argonaute-2 in complex with miR-20a, Cell,
150, 100-110.
179.Helwak, A., Kudla, G., Dudnakov, T., and
Tollervey, D. (2013) Mapping the human miRNA interactome by CLASH
reveals frequent noncanonical binding, Cell, 153,
654-665.
180.Broderick, J., Salomon, W. E., Ryder, S. P.,
Aronin, N., and Zamore, P. D. (2011) Argonaute protein identity
and pairing geometry determine cooperativity in mammalian RNA
silencing, RNA, 17, 1858-1869.
181.Salmanidis, M., Pillman, K., Goodall, G., and
Bracken, C. (2014) Direct transcriptional regulation by nuclear
microRNAs, Int. J. Biochem. Cell Biol., 54, 304-311.
182.Stroynowska-Czerwinska, A., Fiszer, A., and
Krzyzosiak, W. J. (2014) The panorama of miRNA-mediated mechanisms in
mammalian cells, Cell. Mol. Life Sci., 71, 2253-2270.
183.Okamura, K., and Lai, E. C. (2008) Endogenous
small interfering RNAs in animals, Nature Rev. Mol. Cell Biol.,
9, 673-678.
184.Luteijn, M. J., and Ketting, R. F. (2013)
PIWI-interacting RNAs: from generation to transgenerational
epigenetics, Nature Rev. Genet., 14, 523-534.
185.Ross, R. J., Weiner, M. M., and Lin, H. (2014)
PIWI proteins and PIWI-interacting RNAs in the soma, Nature,
505, 353-359.
186.Sayed, D., and Abdellatif, M. (2011) MicroRNAs
in development and disease, Physiol. Rev., 91,
827-887.
187.Derrien, T., Johnson, R., Bussotti, G., Tanzer,
A., Djebali, S., Tilgner, H., Guernec, G., Martin, D., Merkel, A.,
Knowles, D. G., et al. (2012) The GENCODE v7 catalog of human long
noncoding RNAs: analysis of their gene structure, evolution, and
expression, Genome Res., 22, 1775-1789.
188.Fatica, A., and Bozzoni, I. (2014) Long
non-coding RNAs: new players in cell differentiation and development,
Nature Rev. Genet., 15, 7-21.
189.Batista, P. J., and Chang, H. Y. (2013) Long
noncoding RNAs: cellular address codes in development and disease,
Cell, 152, 1298-1307.
190.Khalil, A. M., Guttman, M., Huarte, M., Garber,
M., Raj, A., Rivea Morale, D., Thomas, K., Presser, A., Bernstein, B.
E., van Oudenaarden, A., et al. (2009) Many human large intergenic
noncoding RNAs associate with chromatin-modifying complexes and affect
gene expression, Proc. Natl. Acad. Sci. USA, 106,
11667-11672.
191.Hansen, T. B., Jensen, T. I., Clausen, B. H.,
Bramsen, J. B., Finsen, B., Damgaard, C. K., and Kjems, J. (2013)
Natural RNA circles function as efficient microRNA sponges,
Nature, 495, 384-388.
192.Memczak, S., Jens, M., Elefsinioti,
A., Torti, F., Krueger, J., Rybak, A., Maier,
L., Mackowiak, S. D., Gregersen, L. H., Munschauer,
M., Loewer, A., Ziebold, U., Landthaler, M., Kocks,
C., le Noble, F., and Rajewsky, N. (2013) Circular RNAs are a
large class of animal RNAs with regulatory potency, Nature,
495, 333-338.
193.Pink, R. C., Wicks, K., Caley, D. P., Punch, E.
K., Jacobs, L., and Carter, D. R. (2011) Pseudogenes: pseudo-functional
or key regulators in health and disease, RNA, 17,
792-798.
194.Yang, W., and Wang, X. (2013) Pseudogenes:
pseudo or real functional elements? J. Genet. Genom., 40,
171-177.
195.Poliseno, L. (2012) Pseudogenes: newly
discovered players in human cancer, Sci. Signal., 5
(242).
196.Gaziev, A. I., and Shaikhaev, G. O. (2010)
Nuclear mitochondrial pseudogenes, Mol. Biol. (Moscow),
44, 405-417.
197.Tourmen, Y., Baris, O., Dessen, P., Jacques,
C., Malthiery, Y., and Reynier, P. (2002) Structure and chromosomal
distribution of human mitochondrial pseudogenes, Genomics,
80, 71-77.
198.Stoimenov, I., and Lagerqvist, A. (2012) The
PCNA pseudogenes in the human genome, BMC Res. Notes, 5
(87).
199.Muro, E. M., Mah, N., and Andrade-Navarro, M.
A. (2011) Functional evidence of posttranscriptional regulation by
pseudogenes, Biochimie, 93, 1916-1921.
200.Poliseno, L., Salmena, L., Zhang,
J., Carver, B., Haveman, W. J., and Pandolfi, P. P.
(2010) A coding-independent function of gene and pseudogene mRNAs
regulates tumor biology, Nature, 465, 1033-1038.
201.Tam, O. H., Aravin, A. A., Stein, P., Girard,
A., Murchison, E. P., Cheloufi, S., Hodges, E., Anger,
M., Sachidanandam, R., Schultz, R. M., and Hannon, G. J.
(2008) Pseudogene-derived small interfering RNAs regulate gene
expression in mouse oocytes, Nature, 453, 534-538.
202.Watanabe, T., Totoki, Y., Toyoda, A., Kaneda,
M., Kuramochi-Miyagawa, S., Obata, Y., Chiba, H., Kohara,
Y., Kono, T., Nakano, T., Surani, M. A., Sakaki,
Y., and Sasaki, H. (2008) Endogenous siRNAs from naturally formed
dsRNAs regulate transcripts in mouse oocytes, Nature,
453, 539-543.
203.Hawkins, P. G., and Morris, K. V. (2010)
Transcriptional regulation of Oct4 by a long noncoding RNA antisense to
Oct4-pseudogene 5, Transcription, 1, 165-175.
204.Zhang, Z., and Gerstein, M. (2004) Large-scale
analysis of pseudogenes in the human genome, Curr. Opin. Genet.
Dev., 14, 328-335.
205.Liu, Y. J., Zheng, D., Balasubramanian, S.,
Carriero, N., Khurana, E., Robilotto, R., and Gerstein, M. B. (2009)
Comprehensive analysis of the pseudogenes of glycolytic enzymes in
vertebrates: the anomalously high number of GAPDH pseudogenes
highlights a recent burst of retrotranspositional activity, BMC
Genom., 10, 480.
206.Lopez-Flores, I., and Garrido-Ramos, M. A.
(2012) The repetitive DNA content of eukaryotic genomes, Genome
Dyn., 7, 1-28.
207.Wicker, T., Sabot, F., Hua-Van, A., Bennetzen,
J. L., Capy, P., Chalhoub, B., Flavell, A., Leroy, P., Morgante, M.,
Panaud, O., Paux, E., SanMiguel, P., and Schulman, A. H. (2007) A
unified classification system for eukaryotic transposable elements,
Nature Rev. Genet., 8, 973-982.
208.Kramerov, D. A., and Vassetzky, N. S. (2005)
Short retroposons in eukaryotic genomes, Int. Rev. Cytol.,
247, 165-221.
209.Orgel, L. E., Crick, F. H. C., and Sapienza, C.
(1980) Selfish DNA, Nature, 288, 645-646.
210.Georgiev, G. P. (1984) Mobile genetic elements
in animal cells and their biological significance, Eur. J.
Biochem., 145, 203-220.
211.De Souza, F. S. J., Franchini, L. F., and
Rubinstein, M. (2013) Exaptation of transposable elements into novel
cis-regulatory elements: is the evidence always strong? Mol.
Biol. Evol., 30, 1239-1251.
212.Abrusan, G. (2006) Somatic transposition in the
brain has the potential to influence the biosynthesis of metabolites
involved in Parkinson’s disease and schizophrenia, Biol.
Direct., 7, 41.
213.Upton, K. R., and Faulkner, G. J. (2014) Blood
from “junk”: the LTR chimeric transcript Pu.2
promotes erythropoiesis, Mobile DNA, 5 (15).
214.Smith, Z. D., Chan, M. M., Mikkelsen, T. S.,
Gu, H., Gnirke, A., Regev, A., and Meissner, A. (2012) A unique
regulatory phase of DNA methylation in the early mammalian embryo,
Nature, 484, 339-344.
215.Beraldi, R., Pittoggi, C., Sciamanna, I.,
Mattei, E., and Spadafora, C. (2006) Expression of LINE-1 retroposons
is essential for murine pre-implantation development, Mol. Reprod.
Dev., 73, 279-287.
216.Nandakumar, J., and Cech, T. R. (2013) Finding
the end: recruitment of telomerase to the telomere, Nature Rev. Mol.
Cell Biol., 14, 69-82.
217.Williamson, J. R., Raghuraman, M. K., and Cech,
T. R. (1989) Monovalent cation-induced structure of telomeric DNA: the
G-quartet model, Cell, 59, 871-880.
218.Griffith, J. D., Comeau, L., Rosenfield, S.,
Stansel, R. M., Bianchi, A., Moss, H., and de Lange, T. (1999)
Mammalian telomeres end in a large duplex loop, Cell, 97,
503-514.
219.Sfeir, A., and de Lange, T. (2012) Removal of
shelterin reveals the telomere end-protection problem, Science,
336, 593-597.
220.Zvereva, M. I., Shcherbakova, D. M., and
Dontsova, O. A. (2010) Telomerase: structure, functions, and activity
regulation, Biochemistry (Moscow), 75, 1563-1583.
221.Chen, L. Y., and Lingner, J. (2013) CST for the
grand finale of telomere replication, Nucleus, 4,
277-282.
222.Azzalin, C. M., Reichenbach, P., Khoriauli, L.,
Giulotto, E., and Lingner, J. (2007) Telomeric repeat containing RNA
and RNA surveillance factors at mammalian chromosome ends,
Science, 318, 798-801.
223.Cifuentes-Rojas, C., and Shippen, D. E. (2012)
Telomerase regulation, Mutat. Res., 730, 20-27.
224.Eisenberg, D. T., Hayes, M. G., and Kuzawa, C.
W. (2012) Delayed paternal age of reproduction in humans is associated
with longer telomeres across two generations of descendants, Proc.
Natl. Acad. Sci. USA, 109, 10251-10256.
225.Holohan, B., Wright, W. E., and Shay, J. W.
(2014) Telomeropathies: an emerging spectrum disorder, J. Cell
Biol., 205, 289-299.
226.Plohl, M., Mestrovic, N., and Mravinac, B.
(2014) Centromere identity from the DNA point of view,
Chromosoma, 123, 313-325.
227.Aldrup-MacDonald, M. E., and Sullivan, B. A.
(2014) The past, present, and future of human centromere genomics,
Genes, 5, 33-50.
228.Gent, J. I., and Dawe, R. K. (2012) RNA as a
structural and regulatory component of the centromere, Annu. Rev.
Genet., 46, 443-453.
229.Probst, A. V., Okamoto, I., Casanova, M., El
Marjou, F., Le Baccon, P., and Almouzni, G. (2010) A strand-specific
burst in transcription of pericentric satellites is required for
chromocenter formation and early mouse development, Dev. Cell,
19, 625-638.
230.Ting, D. T., Lipson, D., Paul, S., Brannigan,
B. W., Akhavanfard, S., Coffman, E. J., Contino,
G., Deshpande, V., Iafrate, A. J., Letovsky,
S., Rivera, M. N., Bardeesy, N., Maheswaran, S.,
and Haber, D. A. (2011) Aberrant overexpression of satellite
repeats in pancreatic and other epithelial cancers, Science,
331, 593-596.
231.Chan, F. L., Marshall, O. J., Saffery, R., Kim,
B. W., Earle, E., Choo, K. H., and Wong, L. H. (2012) Active
transcription and essential role of RNA polymerase II at the centromere
during mitosis, Proc. Natl. Acad. Sci. USA, 109,
1979-1984.
232.Romanov, G. A., Suhoverov, V. S., and
Vanyushin, B. F. (2015) Epigenetic mutagenesis as a program for
age-related protein disfunction and ageing, Russ. J. Develop.
Biol., 46, in press.