REVIEW: How Membrane Surface Affects Protein Structure
V. E. Bychkova*, L. V. Basova, and V. A. Balobanov
Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: bychkova@vega.protres.ru; balobanov@phys.protres.ru; uralm62@rambler.ru* To whom correspondence should be addressed.
Received August 4, 2014; Revision received September 29, 2014
The immediate environment of the negatively charged membrane surface is characterized by decreased dielectric constant and pH value. These conditions can be modeled by water–alcohol mixtures at moderately low pH. Several globular proteins were investigated under these conditions, and their conformational behavior in the presence of phospholipid membranes was determined, as well as under conditions modeling the immediate environment of the membrane surface. These proteins underwent conformational transitions from the native to a molten globule-like state. Increased flexibility of the protein structure facilitated protein functioning. Our experimental data allow understanding forces that affect the structure of a protein functioning near the membrane surface (in other words, in the membrane field). Similar conformational states are widely reported in the literature. This indicates that the negatively charged membrane surface can serve as a moderately denaturing agent in the cell. We conclude that the effect of the membrane field on the protein structure must be taken into account.
KEY WORDS: anionic phospholipid membranes, simple alcohols as a model, globular proteins, apo- and holomyoglobins, apo- and holocytochromes c, cytochrome b5, human α-lactalbumin, conformational changes, non-native protein states, membrane–protein interactions pathways, membrane fieldDOI: 10.1134/S0006297914130045
Abbreviations: apoCyt c, apoform of cytochrome c; apoHLA, Ca2+-free form of human α-lactalbumin; apoMb, apomyoglobin; CD, circular dichroism; Cp.exc, partial excess heat capacity; Cyt b5, cytochrome b5; DPPC, 1,2-dipalmitoyl-3-phosphatidylcholine; DPPG, 1,2-dipalmitoyl-3-phosphatidylglycerol; ER, endoplasmic reticulum; GuHCl, guanidinium hydrochloride; holoCyt c, cytochrome c with bound heme; holoMb, myoglobin with bound heme; I, intermediate state; IFlmax, fluorescence intensity at fluorescence spectrum maximum; iPrOH, isopropanol; –LUV, anionic large bilayer vesicles; MeOH, methanol; MG, molten globule state; –MIC, micelles of anionic lysoPhL; N, native state; Na-P, mixture of Na-phosphates; NMR, nuclear magnetic resonance; PhL, phospholipid; POPG, 1-palmitoyl-2-oleylphosphatidylglycerol; Pr, protein; PS, phosphatidylserine; RBP, retinol-binding protein; –SUV, anionic small bilayer vesicles; tBuOH, tret-butanol; U, completely unfolded state; UV, ultraviolet; ΔH, enthalpy change; εeff, effective dielectric constant of a medium; λFlmax, wavelength of fluorescence spectrum maximum; θ, ellipticity.
A little history. In 1981 a new state of the protein
molecule – an intermediate state between the native and
completely unfolded states – was discovered in the Laboratory of
Protein Physics, Institute of Protein Research (Pushchino, Russia).
This compact state with pronounced secondary structure and a
fluctuating tertiary structure [1, 2] was later called a molten-globule state. This state
was observed in vitro on decreasing pH of the solution, at
moderate concentrations of both strong denaturing reagents and some
salts, and frequently after heat denaturation of proteins [3-5]. Naturally the question arose
whether this state could be realized in the cell.
Analysis of the literature data published at that time showed that in some processes occurring in the cell the state of proteins is not an obviously native one, i.e. the proteins have no rigid tertiary structure. It was noticed that upon protein translocation through the mitochondrial membrane, competent to translocation are the states observed just after the biosynthesis and also after dilution of the solution of a protein unfolded with a strong denaturing reagent [6-8]. If for some period of time these proteins remained without membranes, through which they should be translocated, they became incompetent to translocation. In addition, it was noted that the membrane should have a negative charge. Another example is penetration of toxins into the cell. Colicin A undergoes conformational transformation close to the membrane surface; as a result, the hydrophobic helical hairpin is embedded in the membrane, entraining the remaining part of the protein. Several proteins are united, thus forming a pore in the membrane, which causes cell death. This is the toxic action of colicins [9, 10].
NON-NATIVE STATE OF PROTEINS IN CELLS
Conditions in the cell. Let us analyze what conditions can be seen in the cell. As known, neither extremely low pH, strong denaturing reagents, nor high temperatures are observed in the cell. The formed endocytic vesicles contain proton ATPases in their membrane, the work of which decreases pH to 6.0 in endosomes. Such a decrease in pH is sufficient to release some substances transferred by receptors whose structure performs a conformational transition at pH 6.0. The further maturation of endosomes results in the formation of lysosomes, where pH is already 4.5. It is at this pH that lysosomal proteases hydrolyze the proteins designated for degradation in lysosomes. Lower pH values are practically not observed in cells. The temperature of 37°C is far from the heat denaturation temperature of cellular proteins.
However the content of the cell is striking because of the presence of various membranes: a highly developed network of endoplasmic reticulum (ER) and the existence of a great number of organelles including mitochondria. The composition of cell membranes is very complicated. Their bilayer structure contains neutrally and negatively charged phospholipids, and the internal part of the bilayer consists of hydrophobic lipid units and has a low dielectric constant (ε). Membranes also include membrane proteins, the protruding loops of which (the cytoplasmic surface) are often charged negatively, receptors, phosphoinositides, and other components [11]. It is estimated that the content of negatively charged phospholipids can make up to 30% of the total amount of phospholipids in the membrane.
Conditions on the interphase. Taking into account the complicated composition of membranes and the negative charge on the external side of the membrane [12, 13], it was suggested that the membrane surface can affect the structure of proteins functioning in its vicinity. This effect may consist of at least two components. As mentioned above, on one hand, the membrane surface has some total negative charge, and on the other hand the internal part of the membrane has low dielectric constant. That is why the external side of the membrane is the boundary of two phases: an aqueous phase, where ε is about 80, and a hydrophobic phase with ε of about 2-4. As long ago as in 1982, Landau and Lifshits [14] noted that at the interphase ε may be equal to the half-sum of the components, i.e. it should be about 40. It is obvious that such conditions differ greatly from the aqueous environment in the cytoplasm.
CHARGED SURFACE OF MEMBRANES AS A DENATURING REAGENT IN THE
CELL
As knowledge on the non-native states of different proteins both in the cell and in vitro accumulated, the question arose as to what may be the denaturing agent in the cell, where there are no extreme denaturing conditions. Therefore, of great interest is the analysis of situations when under the action of any internal factors within the cell a protein with an initial native structure is transformed to the state similar to the denatured state. Some conditions in the cell such as moderately low pH (4.5-5.0) in lysosomes or negatively charged membrane surfaces as well as cytoskeleton elements can provide for rather denatured conditions for proteins.
It was shown in the team of Schatz [6-8] that partial protein unfolding on the membrane surface can have physiological significance. They demonstrated that this partial unfolding prior to translocation is explained by negative charges on the membrane surface. Indeed, as long ago as in 1979 it was found [15] and corroborated later [16] that in accord with simple electrostatic theory, the membrane surface with a high electrostatic potential can attract protons. This leads to a local decrease in pH by at least 2 units at a distance of 5-15 Å from the membrane surface. It should be mentioned that it is low pH makes that enables formation of the molten-globule state for many proteins in vitro [5, 17, 18]. As noted above, to perform translocation trough the membrane proteins should have the molten-globule state, and the membrane per se promotes the protein transition to the molten-globule state due to its negatively charged surface. The pH value close to the mitochondrial membrane may additionally decrease because of the release of protons from the respiratory system in the intermembrane space via the pores of the outer membrane [19]. But this decrease in pH (of about 2 units) is generally insufficient for acid denaturation of proteins. That is why we proposed [3] the existence of additional denaturing effect on the membrane surface – the local decrease in the effective dielectric constant close to the membrane surface. This may enhance the electrostatic interactions facilitating the local low pH to transform proteins to the molten-globule state. Calculations [14, 20] revealed that approaching the interface of the two media (for example, aqueous and organic ones) the ε value in the aqueous medium decreases reaching in the limit a value almost twice lower than that of bulk of water. Actually, the ε value of the aqueous environment of a protein molecule is constant and equal to ~80 at 20°C, but at high electrostatic potentials or local high concentrations of solvated ions it may change. Therefore, it is necessary to take into consideration the dependence of ε of the medium on the distance to the charged hydrophobic surface. Theoretical estimations of this dependence were made and experimental data were obtained with the use of fluorescence probes sensitive to the polarity of the environment. They demonstrate that pH in the vicinity of a charged hydrophobic surface is much lower than the pH of bulk water [21].
MODELING CONDITIONS IN THE VICINITY OF THE MEMBRANE
SURFACE
Conditions in the immediate vicinity of the membrane surface can be easily modeled. To this end, water–alcohol mixtures can be used to vary pH and ε. Investigations of the conformational state of a protein under these conditions reveal changes in the protein molecule if it is in the immediate vicinity of a membrane surface. It is evident that in addition to this, it is necessary to study also the influence of phospholipid membranes on protein structure. Let us analyze both approaches.
Water–alcohol mixtures as a model for the effect of membrane field on the structure of a protein. At moderately low pH values, water–alcohol mixtures are a simple and concurrently reliable model applicable for testing our assumption that the denaturing action of membranes is explained not only by a local decrease in pH, but also by the influence of the hydrophobic part of the membrane becoming apparent in the decrease of the dielectric constant of the medium [3, 5]. The choice of alcohol mixtures as an acceptable model is based on the fact that the denaturing action of alcohols mixable with water (like MeOH, PrOH, further alcohols) is determined largely by decreased ε value rather than by specific properties of individual alcohols [22, 23]. There are no systematic studies of this subject in the literature, and only a few different proteins have been investigated. Therefore, in the following section attention will focus on experimental results of the authors of this review. For a more complete understanding of the experimental material, we will briefly describe the methods used.
Methods for testing changes in protein structure. Tertiary structure. Changes in the tertiary structure of proteins are tested using scanning microcalorimetry and circular dichroism in the near UV region as well as fluorescence and nuclear magnetic spectroscopy. It is often found that with an increase in alcohol concentration, the heat absorption peak on the curve of heat absorption dependence on temperature disappears, i.e. the native structure and the tight packing of protein side groups are disturbed. This process is also seen in the change in CD spectra in the near UV region. A clearly expressed spectrum for the protein native form changes and practically disappears at a certain content of alcohol and pH, which is a test for protein denaturation and is observed under any denaturing action on the protein. It should be noted that at the same time the high intensity of CD spectra in the far UV region is retained. Upon protein denaturation, the NMR spectra lose their characteristic features in the high-field region, and the bands in the range of aromatic residues are shifted.
For proteins containing Trp, the position of the maximum of the fluorescence spectrum shifts from 320 nm characteristic of the native protein to 340 nm which is characteristic of the intermediate state for these proteins on other methods of denaturation. However, in no case the position of the Trp residue fluorescence has reached 350-360 nm characteristic of a completely unfolded state of the protein.
For proteins containing heme, absorption spectroscopy is also used. This allows tracing conformational changes in the heme region because absorption of the heme is very sensitive to the changes in its environment. The intensity and position of the spectrum maximum allow judging the influence of a denaturant on the structure of the heme environment.
Secondary structure. Changes in the secondary structure were tested by CD spectra in the far UV region. The content of α-helix increases at a high concentration of alcohol in the mixture. It is interesting that in such conditions no unfolding of the protein molecule is observed, and the denatured state is highly helical, in contrast to the high concentration of strong denaturants where the polypeptide chain is unfolded. It was shown for a large number of globular proteins that at moderate concentrations of alcohols, an intermediate state is formed. This indicates the presence of at least two stages (or three states) in the process of protein denaturation with alcohols. The first was initially described as “unfolding or partial unfolding of the native structure”, and the other as a transition to another denatured conformation characterized by a higher content of helical structure. It is worth mentioning that in earlier papers [22, 23] water–alcohol mixtures were not considered as possible models for analysis of the influence of the membrane field on the protein structure.
Methods as HPLC, macroscopic diffusion, and limited proteolysis were used to decide whether the protein is in unbound state in solution.
Choice of objects for study. To test our assumption, it was necessary to choose particular proteins. Our choice was based on the preference of proteins that are positioned and function near the membrane. We chose holo- and apocytochromes (holoCyt c and apoCyt c, respectively), plasma (RBP) and cellular (CRBP I) retinol-binding proteins, cytochrome b5 (Cyt b5), apomyoglobin (apoMb), and ubiquitin. The experiments were performed in water–alcohol mixtures at moderately low pH values which, as was assumed, could model the environment of the membrane surface.
Cytochrome c. Cyt c functions close to the internal mitochondrial membrane, transferring an electron from cytochrome-c-reductase to cytochrome-c-oxidase. Studies of the conformational behavior of this protein with CD and fluorescence spectroscopies revealed the existence of two stages (three states) of its denaturation with MeOH. It was demonstrated that in water–alcohol mixture (0.5 M NaCl in the presence of 40% methanol, pH 4.0), holoCyt c transforms into a state whose properties are similar to the molten-globule state of the protein in aqueous medium (pH 2.0, 0.5 M NaCl) [24]. The CD spectrum of this protein in the near UV region loses its specific features, which is evidence of the loss of tight packing of side groups, i.e. rigid tertiary structure. But in accord with the CD spectrum in the far UV region, which is similar to the analogous Cyt c spectrum in the molten globule (MG) state under aqueous conditions, the protein retains its secondary structure (Fig. 1a). Measurements of hydrodynamic dimensions of the protein under these conditions by following macroscopic diffusion give a Stokes radius similar to that for the molten-globule state. The state of Cyt c denatured with alcohol (depending on the content of MeOH) is highly helical, in contrast to the state of the protein unfolded with strong denaturants.
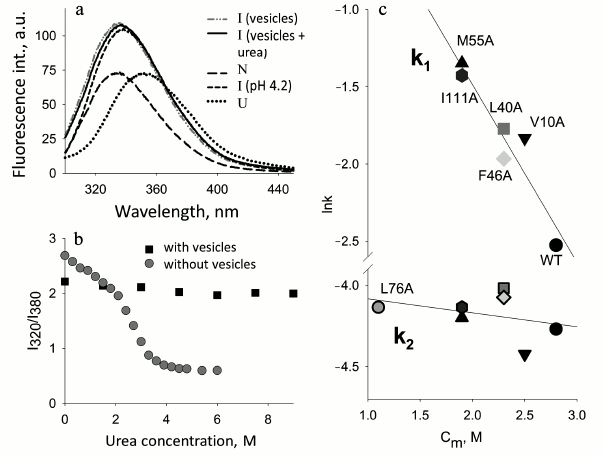
Fig. 1. Changes in parameters of proteins on their denaturation by alcohols. a) CD spectra in the far UV region of apo- and holoCyt c in aqueous solution and in the presence of 40% tBuOH and MeOH, respectively; apoCyt c is in an unfolded form in water, and holoCyt c in 0.5 M NaCl is in the native state: the measurements were performed in 5 mM Na-P buffer, pH 4.0 (adapted from [34]). b) Changes in the fluorescence intensity, the CD spectra in the far UV region at 220 nm, and absorbance at 412 nm (Soret band) for the soluble fragment of Cyt b5 depending on iPrOH concentration at pH 6.5 (adapted from [30]). c) Changes in the CD spectra in the far UV region of sperm whale apoMb depending on MeOH concentration, shown by numbers near the curves; c = 1 mg/ml. d) Changes in fluorescence intensity of sperm whale apoMb depending on MeOH concentration at pH 5.7 (adapted from [30]).
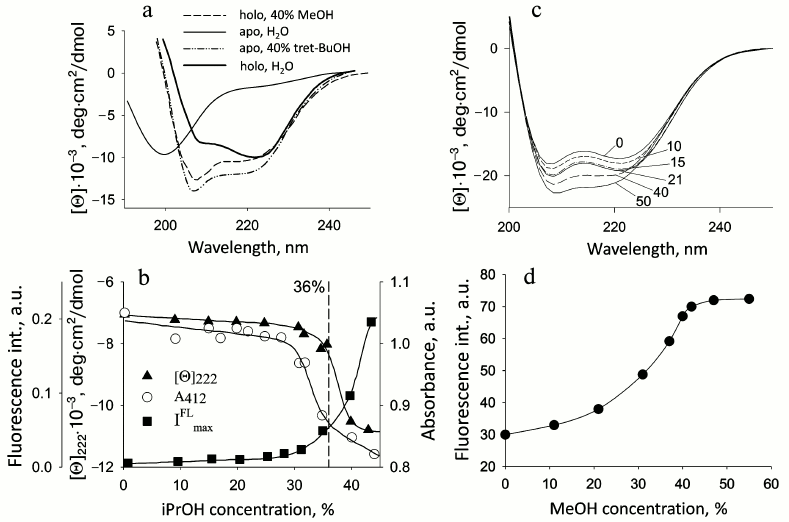
Apocytochrome c. ApoCyt c is a precursor of the functional holoprotein. It is synthesized in the cytoplasm on free ribosomes and then is transferred to the surface of mitochondria to be translocated to the intermembrane space. The protein becomes helical on the outer mitochondrial membrane and penetrates into it. In the intermembrane space, heme ligase binds the heme to apoCyt c. In water–alcohol mixtures, apoCyt c that in an aqueous environment is in the state of a random coil transforms into highly helical but rather compact state, the specific viscosity being 9.0 (unpublished data, [25]). The effect of a number of organic solvents on the properties of this protein was studied: they included alcohols with different length of the aliphatic chain (MeOH, EtOH, iPrOH, tBuOH), substituted alcohol (2,2,2-trifluoroethanol), and other organic solvents (dioxane, tetrahydrofuran, acetonitrile, and acetone). CD spectra in the far UV region showed that addition of alcohols in increasing concentration resulted in changes in both their shape and intensity. The presence of an isodichroic point (about 204 nm) in all the spectra indicates that in these systems disordered conformations are transformed into a conformation with a large amount of helix. Other organic solvents also cause an increase in ellipticity. But only for alcohols the denaturation curves coincide if the dependence of the fraction of denatured molecules is plotted as a function of dielectric constant (ε) of the water–alcohol mixture [22, 23]. This indicates the independence of changes in protein structure from the physical properties of a specific alcohol. Inasmuch as apoCyt c has no tertiary structure in aqueous medium, addition of alcohol would result in decreasing ε from 80 to 40-50 and structuring the unfolded polypeptide chain. Of importance is that in its structural properties the described alcohol-induced state of apoCyt c is similar to the state of this protein that is formed on the outer mitochondrial membrane surface and which is competent for translocation through the membrane [8]. CD spectra in the far UV region for Cyt c and its apoform in water–alcohol mixtures are given in Fig. 1a.
Water-soluble fragment of cytochrome b5. Cyt b5 functions as a water-soluble protein [26, 27] that reduces metHb in erythrocytes and metMb in other cells. With its amphipathic structure, Cyt b5 is a peripheral membrane protein from the outer layer of the ER membrane and consists of a polar N-terminal catalytic domain and nonpolar C-terminal region of the polypeptide chain embedded in the lipid bilayer of the membrane that is an anchor for the protein. The water-soluble domain functions near the ER membrane, interacting with numerous partners. Studies of the water-soluble domain under model conditions of water–methanol mixtures at MeOH concentrations varying from 40 to 60% and pH 7.2 demonstrated that the structure of this fragment undergoes a transition during which the rigid tertiary structure is lost but the secondary structure is preserved [29]. We performed an additional study of the conformational state of Cyt b5 in the presence of different concentrations of iPrOH at pH 6.5 to restrict the transition interval. The results revealed that in the presence of 36% iPrOH the heme environment becomes non-native, and no characteristic heat absorption peak is observed, whereas the secondary structure and compactness persist (see further Table 2). In other words, the addition of alcohol leads to transition of the protein structure from native to intermediate state (unpublished data, [30]). Figure 1b shows denaturation curves of Cyt b5 upon addition of iPrOH.
Apomyoglobin. ApoMb is a precursor of the holoprotein that functions as a carrier and donor of oxygen in cells. It binds the heme near the negatively charged outer mitochondrial membrane (the heme is synthesized on the inner mitochondrial membrane and then is transported to the outer membrane). Thus, the structure of apoMb can be influenced by the membrane field. Properties of sperm whale apoMb in modeled conditions of water–methanol mixtures at pH 5.2 were studied using methods of CD in the far and near UV regions, fluorescence, microcalorimetry, and gel chromatography. At pH 5.2 in the presence of 30% MeOH, no heat absorption peak is observed, which is evidence of disturbance of the protein tertiary structure. However, the secondary structure is preserved, and according to the fluorescence and gel-filtration data the protein remains compact [30]. Thus, under these conditions the protein structure becomes similar to the structure of the MG state in buffer solution (Fig. 1 (c and d) and Table 2 (see further)).
Similar results were obtained in [31] in the studies of horse apoMb in water–MeOH mixtures.
Retinol-binding protein. Blood plasma retinol-binding protein (RBP) transports retinol (vitamin A) and transfers its ligand to the membrane or to a corresponding receptor. Cellular retinol-binding protein (CRBP I) transfers vitamin A within the cell from its membrane to corresponding enzymes that modify retinol, adapting it to a form accessible for the cell. For RBP under moderately denaturing conditions (pH 8.5, 5 mM Na-P buffer and 55% MeOH, 37°C), it is possible to model both the transfer of retinol (vitamin A) and the changes in the protein structure [32]. In this case, concurrently with retinol release, the protein itself undergoes a conformational transition to a state similar to the MG state in aqueous medium at low pH. The release of retinol and denaturation of RBP at physiological temperature were investigated in dependence on the concentration of MeOH upon changes in pH from 2.0 to 8.5. As a result, a diagram of retinol release and RBP denaturation was plotted in coordinates of pH — concentration of MeOH — εeff (effective dielectric constant of the medium). Figure 2 shows spectra of different forms of RBP and the diagram as well as the dependence of the fraction of native RBP molecules versus the concentration of MeOH. It should be noted that RBP is a very stable protein and it releases retinol only at pH values from 3.5 to 2.0, so at pH > 3.5 we have the native holoprotein. The intermediate state observed in water–MeOH mixtures has a CD spectrum in the far UV region similar to that for the MG state at pH 2.0 (Fig. 2) [33]. This state has no rigid tertiary structure, but it remains compact almost as the native protein [34]. Thus, it is the first time that the process of retinol release from its complex with RBP was modeled in a simple artificial system (a water–alcohol mixture at moderately low pH values).
Fig. 2. Conformational behavior of RBP depending on MeOH concentration. a) Comparison of the CD spectra in the far UV region in a MG state (pH 2.0, in the absence of MeOH) with the spectra of RBP at pH 3.5 in 16% MeOH and at pH 8.5 in 55% MeOH (in both cases after the release of retinol). The CD spectrum in the far UV region of native apoRBP is shown for comparison. b) Relative fraction of RBP molecules binding retinol and RBP native molecules depending on MeOH concentration: 1) change in intensity of fluorescence at 460 nm (band of fluorescence intensity maximum of bound retinol); 2) according to band at 325 nm associated with retinol ellipticity; 3, 4) fraction of native molecules according to changes in ellipticity at [θ]220 for apo- and holoforms of RBP, respectively; 5) according to molar ellipticity of RBP in near UV region [θ]280 for apoRBP. c) Diagram of retinol release and RBP denaturation depending on pH and MeOH concentration at 37°C. εeff is the average value of the dielectric constant of MeOH–water mixtures. Two curves limit the region, in which retinol release and denaturation of RBP are observed, and delimit the area of the native and denatured states of RBP molecules (adapted from [34]).
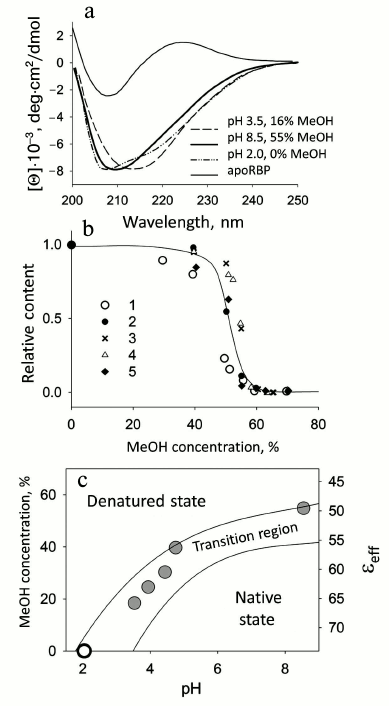
RBP is a characteristic representative of a whole class of transport proteins involved in the transfer of retinol and its derivatives. The mechanism of the release of hydrophobic ligands by other proteins of this class has not been studied in detail. However, taking into account the structural similarity of these proteins and the way of ligand binding, it can be proposed that the mechanism of release of these ligands will also have features in common with the mechanism of retinol release. An example of this is cellular CRBP I, for which comparable data were obtained [35].
Ubiquitin. Analogous studies were performed for ubiquitin, whose function is modification of proteins intended for degradation. It was demonstrated for this protein that the intermediate state observed at pH 2.0 and 60% methanol is almost as compact as the native state (intrinsic viscosity being 3.9), has a high content of secondary structure, but does not reveal cooperative temperature melting, i.e. is in the MG state (unpublished data). It is probable that a similar state is observed in this protein upon binding of its C-terminal glycine to different protein substrates, which may promote denaturation of its C-terminal part [36].
Other proteins. In addition to the above-described proteins, there are also some proteins for which similar studies in water–alcohol solutions have been made.
For human apolipoprotein H (ApoH), it was found that a change in the ε value (with addition of 57% methanol) resulted in the formation of α-helical structure in this β-structural protein, and under these conditions the CD spectra in the far UV region resemble those in the presence of phospholipid vesicles [37].
Modeling of conformational changes in the N-terminal binding domain of human apolipoprotein E (ApoE) in water–propanol solutions at pH 7.0 revealed that at 30% iPrOH the protein tertiary structure changes, though not as strongly as upon binding to vesicles. But under such conditions the protein is nearly as compact as the native one. When pH is decreased to 4.5 and 30% iPrOH is added, more significant changes occur both in the tertiary and secondary structures of the protein; these changes are similar to the changes caused by membranes, and simultaneously the Stokes radius becomes larger [38].
Babu and Douglas [39] studied equilibrium denaturation of holoMb at pH 4.0. When MeOH (35-40% alcohol) is added at this pH, the tertiary structure of the protein disappears and an intermediate state is formed whose secondary structure resembles the structure of the native protein. Under such conditions the heme still may be bound, but when the concentration of methanol exceeds 50% the protein releases the heme. A further increase in alcohol content results in the formation of a highly helical state of apoMb without the heme.
The analysis of changes in the structure of the C-terminal fragment of angiotensin II receptor (AT1A) in the presence of phospholipid membranes and in solutions of water–methanol mixtures by surface plasmon resonance showed that the fragment binds to anion phospholipids, and structures with a high content of α-helix are formed in the solution. The combination of hydrophobic and electrostatic interactions may be of importance for the function of this receptor [40].
Thus, we conclude that under model conditions (water–alcohol mixtures at moderately low pH), changes in the structural properties of proteins can be observed. In this case the proteins transform from the native state to intermediate states analogous in their properties to the MG state in aqueous solution.
The next stage was investigation of protein in the presence of phospholipid membranes at neutral pH, a condition approximating that in living cell.
CONFORMATIONAL STATE OF GLOBULAR PROTEINS IN THE PRESENCE OF
PHOSPHOLIPID MEMBRANES
Only scattered data on the influence of the membrane surface on protein structure, especially at physiological pH, are available at present. Taking into account the deficiency of data on the structure of proteins in the presence of membranes and the hypothesis on the denaturing action of the membrane surface, we performed a systematic investigation of the influence of negatively charged phospholipid membranes on the conformational state of globular proteins at neutral pH characteristic of the cytoplasm.
Choice of objects for study. To study conformational changes in proteins, we chose several proteins that are near the membrane during their functioning but do not bind to it. The latter is an important condition because it is known that direct interaction of proteins with the membrane can change their conformational state both upon insertion into the membrane and upon translocation through it [41].
Such proteins include the nearly neutral apo- and holoMb and negatively charged Cyt b5. For comparison, we chose negatively charged human lactalbumin (HLA), which in the course of biosynthesis binds to the internal side of the ER membrane. Positively charged proteins were excluded because their interaction with the negatively charged membrane is trivial. HoloMb and Cyt b5 are stable proteins. Under aqueous conditions, their denaturation occurs either at low pH of about 2.0 or at temperatures near 70-80°C. Therefore, to attain a substantial effect of membranes, we used 100% negatively charged phospholipid membranes from POPG and membranes with a lower content of these phospholipids. To maintain constant content of phospholipids, zwitterion POPC or DPPC were added. The investigations were conducted at neutral pH values (6.2 and 7.2). As stated above, in the presence of membranes a local decrease in pH by 2-4 units can take place near their surface (depending on the conditions) [16]. So, at pH 7.2 the expected decrease in this value by 2 units can give pH 5.2. Under such conditions, the pH-induced unfolding the protein still retains its native state. Another situation was observed if the experiments were done at pH 6.2. In this case in the presence of membranes the pH value near their surface can be 4.2 or lower, which can lead to transition to the MG state. Hence the conformational behavior of the protein will change. Comparison of the experimental data at the two pH values shows clearly whether near the membrane surface (i.e. on the interphase) the effective pH value decreases at neutral pH of solution. The decrease may be caused, as mentioned above, by the presence of the electrostatic potential of negatively charged phospholipids and low dielectric constant of the medium within the membrane bilayer that is on the interphase.
Myoglobin. Myoglobin carries oxygen in the cells of skeletal muscles and myocardium. It has a higher affinity to oxygen than hemoglobin and can bind an oxygen molecule at its low partial pressure near the capillary walls, thus oxymyoglobin (oxyMb) is formed. The high affinity to oxygen provides for oxygen storage and its transport from the sarcolemma to mitochondria. The lipophilic oxygen molecule can be dissolved in the lipid bilayer, and by free diffusion it can penetrate to the intermembrane space of mitochondria. Myoglobin can bind oxygen due to the presence of the heme. One of the six coordination bonds of the iron ion is occupied by the nitrogen atom of His93 (helix F) and another serves for binding the oxygen molecule, His64 (helix E) being located nearby [42]. Distal His, propionic chains, and residues of Thr, Val, and Phe form a hydrophobic “ligand pocket”, which together with the heme located within it and proximal His are included in the active center of myoglobin. The oxygen molecule interacts with the iron of the heme, but the iron oxidation state does not change. This is possible because a medium with low ε is formed in the hydrophobic heme pocket. However, the heme is located deep in the myoglobin molecule, and the oxygen molecule cannot easily reach it. It is presumed that fluctuations of amino acid side chains form a transport channel along which oxygen moves to the heme. Indeed, it was demonstrated that at least His64 is involved in this process. Thus, it is suggested that the binding of oxygen is explained by the mobility of side chains within the protein molecule, but this suggestion has not been proven experimentally.
In the cell, there are two forms of myoglobin: apo- and holoMb. Being a precursor of holoMb (as mentioned above), apoMb acquires the heme on the surface of the mitochondrial membrane [43]. Upon binding of the heme, a part of the polypeptide chain, especially helix F, is structured [44], which makes the protein more stable. Many researchers have thoroughly studied the structural and thermodynamic characteristics of both apoMb [44-53] and holoMb [54-57]. Despite the absence of the heme, apoMb also retains the hydrophobic core [58] and the tertiary structure characteristic of holoMb [59]. According to thermodynamic criteria, both holoMb and its apoform can undergo cooperative transitions upon heating, these being accompanied by noticeable changes in enthalpy and heat capacity.
The holo- and apoforms of myoglobin have similar tertiary structure. So it can be expected that negatively charged mitochondrial membranes would affect the structure of both forms in a similar way, but the amplitude of the effects would depend on the stability of the forms. Thermal melting is typical of both forms. In the presence of phospholipids, the mentioned temperature transition disappears, but for apoMb this occurs at PhL/Pr ratio of 25 : 1, while for holoMb this ratio is 200 : 1 [60]. The absence of the melting peak is evidence of disturbance of the tight packing of side groups.
Disturbance of tertiary structure of myoglobin in the presence of phospholipid membranes. Detailed studies of holoMb and apoMb have been done previously [60, 61]. The changes in the shape and intensity of CD spectra in the near UV region yield information on the disturbance of the protein tertiary structure in the presence of negatively charged vesicles from POPG, whose molar ratios (PhL/Pr) were varied from 25 : 1 to 200 : 1 at pH 7.2. At molar ratio PhL/Pr = 25 : 1 (pH 7.2), the spectrum of holoMb is very similar in all parameters to that of the native protein. An increase in the concentration of phospholipids leads to a decrease in the molar ellipticity value, retaining the shape of the spectra, which shows that the number of protein molecules with a rigid tertiary structure is reduced. With molar ratios of PhL/Pr = 200 : 1 at pH 7.2 and 25 : 1 at pH 6.2, all specific features of the spectrum disappear, which is evidence of the loss of tight packing of side groups in the presence of a high content of negatively charged phospholipid vesicles [60].
The CD spectrum of apoMb in the near UV region in the native state has less specific features and lower ellipticity than the holoform. With molar ratio PhL/Pr = 25 : 1 at pH 7.2, the spectrum shape in the range of 250-320 nm changes noticeably and the ellipticity value becomes close to zero [61].
The absorption spectrum of holoMb in the native state (N) has a well pronounced maximum at 409 nm (a Soret band), while in the intermediate state (I) there is almost no absorption of the heme, which is comparable to its value for the completely unfolded protein (Fig. 3c). Absorption spectra of holoMb in the Soret band in the presence of negatively charged vesicles were obtained at pH 6.2 with molar ratios of PhL/Pr varying from 25 : 1 to 100 : 1 (Fig. 3c). When vesicles are added to the native proteins, the intensity of the absorption band decreases and concurrently its width increases. Besides, a shoulder appears at 380 nm, which is evidence of accumulation of non-native protein. Consequently, the effect of vesicles leads to conformational changes in the structure of the myoglobin molecule in the vicinity of the heme. A decrease in pH from 7.2 to 6.2 causes more pronounced conformational changes in holoMb. Indeed, the intensity of the Soret band at pH 6.2 is changed at molar ratio PhL/Pr = 25 : 1, whereas a similar change in the intensity of the Soret band at pH 7.2 occurs only at the ratio of 200 : 1.
Fig. 3. Conformational behavior of myoglobin under different conditions: a, b) sperm whale apoMb; c-e) sperm whale holoMb. a) CD spectra in the far UV region for apoMb in the presence of POPG phospholipid membranes: 1, 2) spectra at pH 7.2 and a molar ratio of PhL/Pr = 25 : 1 and 100 : 1, respectively; 3) spectrum at pH 6.2 and PhL/Pr = 50 : 1; dotted lines show the spectra of the protein in the native state (N) at pH 7.2 and in the molten globule state (MG) at pH 4.2, 10 mM NaAc. b) Change in the intensity of the Trp fluorescence spectrum of apoMb at pH 7.2 and PhL/Pr ratios of 25 : 1 (1) and 100 : 1 (2), respectively, and pH 6.2 and PhL/Pr ratio of 50 : 1 (3); excitation wavelength is 293 nm; spectra for the N, MG, and fully unfolded in 6 M GuHCl (U) states are shown for comparison. c) Absorption spectra of holoMb at pH 6.2 in the presence of POPG vesicles at PhL/Pr ratio 25 : 1 (1), 50 : 1 (2), and 100 : 1 (3). Spectra of the protein in the N and intermediate at pH 3.6 (I) states without the vesicles are given for comparison (adapted from [30]). d) Changes in the Trp fluorescence intensity spectrum of holoMb at pH 6.2 and PhL/Pr ratios of 25 : 1 (1), 50 : 1 (2), and 100 : 1 (3), respectively; excitation wavelength is 293 nm; spectra for states N, intermediate at pH 3.6 (I), and completely unfolded in 6 M GuHCl (U) are shown for comparison (adapted from [30]). e) Trypsin digestion of holoMb at pH 7.2 in the native state (I) and in the presence of the vesicles with PhL/Pr ratio 25 : 1 (II) and 200 : 1 (III). Numbers near the lanes indicate the time of incubation with trypsin. Vertical lane between samples I and II indicate molecular weight markers (adapted from [30]).
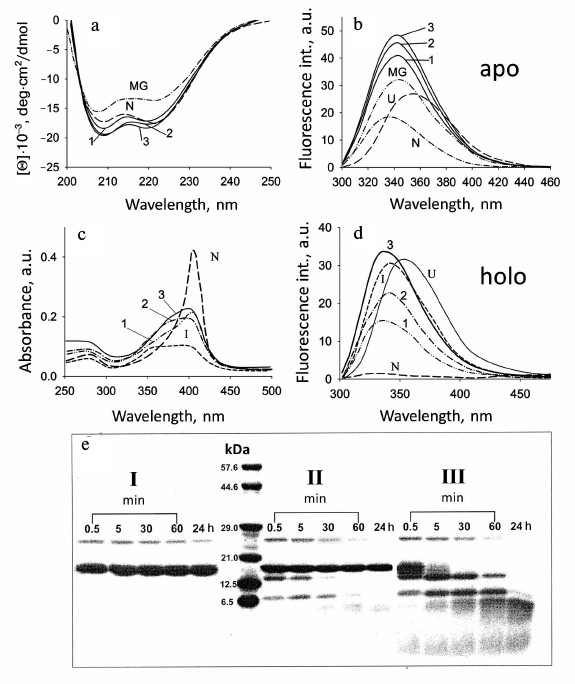
Both apoMb and holoMb contain two Trp residues in helix A (in positions 7 and 14), but in the native state the fluorescence of these residues is quenched strongly by amino acid residues surrounding them and by the heme in the holoprotein. This allows observing conformational changes in the protein using intrinsic Trp fluorescence. Complete unfolding of holoMb in 6 M GuHCl causes enhancement of the fluorescence intensity and evokes a long-wavelength shift of the spectral maximum to 355 nm (U, Fig. 3d). At pH 4.2 [57], the maximum of fluorescence of myoglobin Trp is at 340 nm and is intermediate between those of the native and completely unfolded states. Figure 3d shows for comparison the fluorescence spectrum of holoMb at pH 3.6 corresponding to the intermediate state (I). Fluorescence spectra of holoMb in the presence of negatively charged phospholipid membranes were recorded over a wide range of molar ratios of PhL/Pr at pH 7.2 and 6.2 (Fig. 3d). With a minimal molar ratio of PhL/Pr = 25 : 1 at pH 6.2, a small rise in the fluorescence intensity is observed, the maximum being shifted to 338 nm. An increase in the PhL concentration at constant protein concentration results in further enhance of the fluorescence intensity of Trp. With the ratio of PhL/Pr = 200 : 1 the fluorescence amplitude becomes close to the fluorescence amplitude specific for the protein in the intermediate and completely unfolded states, but no shift of the fluorescence maximum to 355 nm occurs. When pH decreases to 6.2, already at molar ratio PhL/Pr = 25 : 1 the intensity becomes half of this value for the denatured protein in aqueous solution, and at PhL/Pr = 100 : 1 it is even somewhat higher than that for the protein in a completely unfolded state (Fig. 3d). The position of the fluorescence maximum at all molar ratios of PhL/Pr and pH is the same, 338 nm, which is close to the position of the fluorescence maximum for the protein in an intermediate state. The position of the maximum of the Trp fluorescence spectrum at 338 nm is usually explained by incomplete exposure of Trp to water, i.e. it shows the presence of rather compact structure.
Using spectral methods, it has been established that at pH 7.2 and 6.2 negatively changed phospholipid membranes cause conformational changes in apo- and holoforms of myoglobin. ApoMb completely loses its rigid tertiary structure already at the molar ratio PhL/Pr = 25 : 1 at pH 7.2, the environment of Trp residues being different from the native one (Fig. 3b). Due to the presence of the heme, holoMb loses the tight packing of side groups only at molar ratio PhL/Pr = 200 : 1 at pH 7.2 and 25 : 1 at pH 6.2. In this case both Trp and the heme lose their native environment. These results show that negatively charged phospholipid vesicles cause changes in the N conformation of protein molecule leading to disturbance of its tertiary structure. In its properties, this state is similar to the intermediate state of the forms in an aqueous buffer, but it is not identical to the MG, though it reveals basic properties of the latter. A similar state is also observed for apoMb and holoMb in water–alcohol mixtures at high concentrations of methanol [31, 39].
Fluorescence spectra of apoMb in the presence of negatively charged vesicles at different molar ratios of PhL/Pr (pH 7.2) are given Fig. 3b. The spectrum of apoMb in the native state differs substantially from the fluorescence spectrum of holoMb in the absence of vesicles, which is connected with the lack of heme as a quencher. At the same time, other amino acid residues in the spatial environment of tryptophan residues continue quenching its fluorescence. Upon transition from the N state to the MG state, the fluorescence intensity is enhanced, a shift of the fluorescence maximum from 332 to 340 nm being observed, while upon protein unfolding with urea the spectral maximum shifts to 355 nm. In the presence of vesicles, already at molar ratio PhL/Pr = 25 : 1 (pH 7.2) the fluorescence intensity increases drastically to a value exceeding that for both the native and intermediate states of the protein. The maximum of the apoMb fluorescence in the presence of vesicles is at 340 nm, which is characteristic of the MG state and varies greatly from this value for the protein in a completely unfolded state. At high molar ratios of PhL/Pr (pH 7.2) as well as at the molar ratio PhL/Pr = 50 : 1 (pH 6.2), the fluorescence intensity increases insignificantly, which may be connected with the effect of the hydrophobic layer of the membrane on the environment of Trp. Thus, in the presence of vesicles apoMb also loses the rigid native environment of Trp residues.
Retention of myoglobin secondary structure in the presence of phospholipid vesicles. Regardless of the significant changes in tertiary structure caused by addition of phospholipids, the secondary structure of myoglobins does not alter noticeably. The CD spectrum of holoMb in the native state has the shape typical of α-helical proteins with two clear minima at 208 to 220 nm. Upon addition of large amounts of phospholipids, the CD spectra of holoMb change more significantly, the absolute value of ellipticity at 208 nm becoming somewhat larger than that at 220 nm. So, the spectra of the protein in the presence of vesicles become similar to those of the protein in the intermediate state, the latter in turn having the shape described for the MG state of other proteins [62]. The shape of the spectra of holoMb in the presence of phospholipid vesicles at pH 6.2 reminds that of the CD spectra in the far UV region at high concentration of alcohol [39]. High concentrations of PhL at pH 6.2 do not evoke noticeable changes in the spectrum shape, i.e. the secondary structure changes little if at all. It should be emphasized that CD spectra of holoMb obtained in the presence of vesicles differ greatly from the spectrum of the completely unfolded protein (U). Thus, the helical secondary structure of holoMb in the presence of a negatively charged membrane is preserved almost completely, though it differs from the structure characteristic of the native protein.
However, these changes are not critical because the spectra also differ greatly from the spectrum of the protein completely unfolded in solution. As in the case of holoMb, the changes in the apoMb structure take place at pH 6.2 at lower concentrations of phospholipids than at pH 7.2 (see Fig. 3a).
Therefore, the secondary structure of the holo- and apoforms of myoglobin in the presence of negatively charged PhL vesicles preserves its helical nature but differs from that typical of the N and U states of the protein. Small changes in the shape of the spectra are probably caused by conformational rearrangements in the protein three-dimensional structure under these conditions.
Myoglobin denaturation by phospholipid vesicles leads to interaction of the protein with the surface of vesicles. As follows from the above, negatively charged PhL vesicles at pH 7.2 and 6.2 induce conformational changes in apo- and holoforms of myoglobin leading to disturbance of the tertiary structure. The properties of the new state are similar to those of the I state of these forms in aqueous solution but are not identical to the properties of the MG state. A similar state for apoMb and holoMb is found in water–alcohol mixtures at high concentrations of methanol [31, 39].
As known, in the intermediate state the proteins have a propensity to interact with the membranes, but the efficiency of this interaction depends on some factors, in particular, on the protein stability. Methods of HPLC, macroscopic diffusion, and high-resolution 1H-NMR were used to determine the localization of non-native protein molecules. On account of strongly differing dimensions, elution volumes of an isolated protein and the protein bound to phospholipid vesicles differ greatly. This permits analyzing the association of the protein with phospholipids. With molar ratio PhL/Pr = 25 : 1 (pH 7.2) and the use of the gel-filtration chromatography, a peak is observed in the range of the native protein elution, i.e. the main part (~75%) of the protein remains unbound, though about a quarter of the protein is eluted in the peak corresponding to phospholipid vesicles [60]. At higher PhL/Pr molar ratios, the fraction of free protein decreases simultaneously with an increase in the portion of the protein bound to the phospholipid vesicles. The peak of the free protein disappears completely at pH 7.2 when the molar ratio of PhL/Pr is 200 : 1. At pH 6.2, a great part (~70%) of the bound protein is revealed at the ratio of PhL/Pr = 25 : 1, while at 50 : 1 the elution profile has only one protein peak eluted together with the vesicles.
Elution profiles of apoMb in the presence of vesicles for molar PhL/Pr ratios of 25 : 1 and 50 : 1 at pH 7.2 show that even at the lowest PhL/Pr ratio, the protein is completely bound to phospholipid vesicles [61].
Consequently, in accord with the gel-chromatography data, both forms of myoglobin are bound to vesicles. ApoMb is completely bound to vesicles at all molar ratios of PhL/Pr at both pH values. It should be noted that according to estimations of the surface areas of vesicles and the protein under the chosen conditions, the vesicle concentration in the system three times exceeds the number of protein molecules that can be bound to the surface of the vesicles.
HoloMb interacts with vesicles much more weakly than its apoform (see Table 2). The extent of the protein binding to the vesicles correlates with the portion of the denatured protein tested by CD in the near UV region [60]. As known, in the N state the protein does not interact with membranes [3, 5]; that is why only the conformational transition of myoglobin to the intermediate state with a larger hydrophobic surface results in the binding of the protein to the vesicles.
To test the conformational state of unbound holoMb observed at low concentrations of phospholipid vesicles, high-resolution 1H-NMR was used. Dispersion of chemical shifts shows that holoMb at pH 7.2 has a rigid tertiary structure [60]. Characteristic high- and low-field resonances of holoMb only insignificantly overlap with the signals of PhL vesicles (POPG), which allows estimating the state of the protein having specific resonances in these regions.
The changes observed in the NMR spectra of these proteins were connected mainly with the disappearance of high-field signals at 0-(–2) ppm characteristic of tight packing of side groups and also with changes in the spectrum at 6-8 ppm corresponding to the state of aromatic groups. Estimations of the integral area in the range of the CH2-group resonances of phospholipids showed that an increase in the area under the curve corresponds to the same decrease of the area in the range of protein resonances. This fact also corroborates the binding of the protein to the membrane. In this case some changes occur in the spectrum of vesicles as well. This may indicate to some distortion in the packing of phospholipids in the membrane bilayer [60, 61].
The data of gel chromatography show that at molar ratio PhL/Pr = 25 : 1 (pH 7.2) the greater part (~75%) of the protein in solution is in the unbound state. The dimensions of the protein and vesicle molecules differ greatly, which permits using macroscopic diffusion for determination of the protein and vesicle dimensions in a mixed system [63]. The diffusion coefficients were measured for holoMb not bound with vesicles at molar ratio PhL/Pr = 25 : 1 (pH 7.2) as well as for vesicles in the absence of the protein and the protein in the native state without vesicles. The data are listed in Table 1. At molar ratio PhL/Pr = 200 : 1 (pH 7.2) no free (unbound) protein can be revealed. This result is completely compatible with the gel-chromatography data. When estimating the area under the diffusion curve for unbound myoglobin at molar ratio PhL/Pr = 25 : 1 (pH 7.2), it was found that on average it is 25% smaller than that for the native protein. Thus, the data on macroscopic diffusion are in good agreement with the gel-chromatography data.
The existence or absence of molecules of native holoMb in the presence of vesicles was checked also using limited proteolysis. Native holoMb with a rigid tertiary structure (independent of the incubation time) is not prone to the action of protease (I, Fig. 3e). At molar ratio PhL/Pr = 25 : 1 (pH 7.2), cleavage of about 25% of all protein molecules is revealed just from the first minute of incubation of the mixture with trypsin (II, Fig. 3e). As the time of incubation grows, all the protein fragments formed during the first minute are completely cleaved and there remain only protein molecules inaccessible to the protease. Consequently, under these conditions this part of the protein (~75%) has tertiary structure similar to the native one, which complies well with the data obtained with spectral methods. Under complete binding at molar ratio PhL/Pr = 200 : 1 (pH 7.2), the first minute of incubation is characterized by cleavage of all protein molecules, and the band in the range of electrophoretic motility of the native protein is not found at all (III, Fig. 3e), which is evidence of the absence of protein molecules with rigid tertiary structure among those bound to vesicles [30].
Summarizing the results obtained by gel chromatography, macroscopic diffusion, and 1H-NMR, we conclude that at low PhL/Pr ratio (pH 7.2) approximately one third of the protein molecules have no rigid tertiary structure and are bound to vesicles. The remaining protein molecules are free and have properties characteristic of the native state. At high ratio PhL/Pr = 200 : 1 (pH 7.2) and molar ratio PhL/Pr = 70 : 1 (pH 6.2), myoglobin has no rigid structure and is completely bound to vesicles, thus leading to distortion of the phospholipid packing in the bilayer (see the above NMR data).
Stability of myoglobin in the presence of phospholipid vesicles. To characterize the conformational stability of the unbound (free) protein in the presence of PhL, scanning microcalorimetry was used. The temperature melting of holoMb was performed in the presence of vesicles from unsaturated PhL (T = 271K) for molar PhL/Pr ratios of 25 : 1, 50 : 1, 200 : 1 (pH 7.2), and 25 : 1 (pH 6.2). According to the spectral data and the data of gel filtration and diffusion, at PhL/Pr = 25 : 1 (pH 7.2), the greater part of the protein is not bound and has structure similar to the native structure. The rest of the holoMb has no rigid tertiary structure, and as known, proteins in a partially denatured state I have no peaks of thermal melting. That is why in this case it is possible to trace only the free protein melting. The thermal denaturation of holoMb in the native state proceeds by the “all-or-none” principle [64], because ΔHcal ≈ ΔHeff. At molar ratio PhL/Pr = 25 : 1 (pH 7.2), we have a protein heat absorption peak that differs greatly from the native protein peak. The difference is in the shift of the peak (by about 20°C) to lower temperatures (as compared to the native protein, 357K), the calorimetric enthalpy value decreasing from 500 kJ/mol (for the native protein) to 280 kJ/mol. The effective enthalpy is 370 kJ/mol under these conditions, thus the ratio of the calorimetric and effective enthalpies is 0.76. This means that in the presence of vesicles the cooperative region of the protein is smaller than in the native protein. It is important that the transitions observed are irreversible. It can be assumed that such a change in the heat absorption peak indicates destabilization of the native protein, the reason for which is the dependence of the stability of the native protein molecules on pH, ionic strength, and stabilization of denatured protein molecules due to binding with vesicles. At molar ratio PhL/Pr = 50 : 1 and higher (up to 200 : 1) (pH 7.2) and also at all molar ratios at pH 6.2, heat absorption curves of holoMb are absent, which is typical of protein having no rigid tertiary structure. However according to the CD data in the near UV region, at molar ratio PhL/Pr = 50 : 1 and pH 7.2, some of the protein retains the tight packing of side groups, but this does not conflict with the calorimetric data because the temperature itself is an additional denaturing factor.
Table 1. Parameters of holoMb under
different conditions obtained by macroscopic diffusion
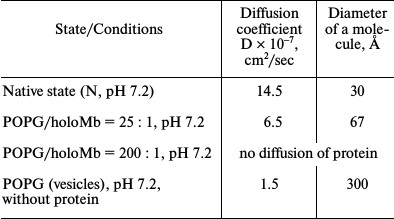
Thus, in the presence of vesicles some molecules of free holoMb at pH 7.2 have a native state, which, however, become less stable relative to the denatured state. Changes in the parameters of the gel-to-liquid-crystal transition of saturated phospholipids show the interaction of holoMb with vesicles, which results in the distortion of the regular packing of phospholipids in the bilayer. This agrees with the 1H-NMR data.
In the case of apoMb, the heat absorption peak of the native protein in the presence of vesicles from unsaturated PhL is absent already at molar ratio PhL/Pr = 25 : 1 and pH 7.2. This is evidence of the disturbance of the protein tertiary structure and is compatible with the data obtained using other methods. The fact that there is no heat absorption peak in the temperature range of apoMb melting seems to demonstrate that the sample has no native protein molecules, and a change in the shape of melting curves of vesicles in the presence of the protein may indicate the interaction of apoMb with phospholipid vesicles disturbing the bilayer structure. Most likely, this interaction is from partial immersion of the protein into the bilayer due to which the packing of PhL is changed. At pH 6.2 and all the above mentioned PhL/Pr ratios, the results are similar to those at pH 7.2.
Stability of the protein and interaction with membranes. In the Laboratory of Protein Physics (Institute of Protein Research, Russian Academy of Sciences) a detailed study of apoMb folding was done. The object of the investigation was the kinetics of folding of mutant proteins with substitutions both in the conserved and non-conserved positions important for folding [52, 53]. To study the influence of the protein stability on its interaction with membranes, a number of mutant apoMb were chosen. It was found that the negatively charged surface of membranes can play a dual role: the polypeptide chain of apoMb unfolded with urea folds on the membrane surface and acquires features of a structure characteristic of the MG state. On the other hand, native apoMb in the presence of the same membranes loses its N structure and transforms into an analogous state similar to the MG (Fig. 4) [65]. Analysis of the kinetics of interaction of native apoMb with the surface of vesicles demonstrated that when the content of negatively charged PhL drops from 100 to 20%, the interaction remains practically the same. But if the vesicles are formed of zwitterionic phospholipid POPC, the protein does not interact with the vesicles at all. At the same time, addition of one mole of urea increases fluctuations in the apoMb structure and restores the interaction of the protein and vesicles. The study of the conformational behavior of mutant proteins in the presence of negatively charged membranes revealed that their binding with the membranes depends significantly on the stability of the proteins. Figure 4 shows the dependence of the logarithm of the rate of the first stage of the mutant apoMb−PhL vesicles interaction on cm value for each mutant. It is seen that there is a distinct dependence: the more stable the protein structure, the lower the interaction.
Fig. 4. Effect of protein stability on protein interactions with phospholipid membranes (adapted from [65]). a) Changes in Trp fluorescence spectra upon addition of apoMb in the N and urea-unfolded U states to a solution of mixed vesicles (POPG–POPC) at 20% negatively charged POPG. b) The protein urea denaturation curves in the absence and presence of vesicles measured by Trp fluorescence intensities at 320 and 380 nm. The kinetics of the interaction of apoMb mutant forms with vesicles monitored by changes in the Trp fluorescence intensity at 335 nm. c) Dependence of the logarithms of constants k1 and k2 characterizing stages I and II of the protein interaction with vesicles depending on cm (urea, M), the middle of the transition between N and I for each mutant (from curves of protein denaturation by urea). Designations: ▲ M55A; □ L40A; ▼ V10A; ⌂ I111A; ◊ F46A; ● WT; ○ L76A.
Phospholipid membranes facilitate the release of oxygen from oxymyoglobin. As known, myoglobin binds oxygen and transports it from the sarcolemma to mitochondria. The mechanism of oxygen release is still not clear. It also remains unclear what stimulates this process. It has been shown recently that at physiological pressure, oxygen can be released from oxyMb only upon its direct contact with the mitochondrial surface, the reaction rate being independent of the protein concentration [66]. Our experiments on the influence of model membranes on the structure of myoglobin demonstrated that in the presence of a negatively charged membrane the protein tertiary structure can be disturbed. This suggests that in the presence of mitochondria the structure of a myoglobin molecule is destabilized due to which the affinity to oxygen diminishes drastically. Thus the kinetics of oxyMb autooxidation in the presence of model membranes (vesicles) was studied by absorption spectroscopy. The absorption spectra of oxyMb in the absence and presence of vesicles are shown in Fig. 5 (a and c). The absorption spectrum of the oxy form of myoglobin is characterized by clearly pronounced peaks at 543 and 581 nm, which disappear in the met form where the heme ligand is water. The met form is revealed by the appearance of maxima at 505 and 634 nm. The transition of the oxy form of myoglobin to its met form in the absence of vesicles at room temperature occurs slowly and takes several days, while at on increase in temperature to the physiological value (37°C) the process accelerates, however not so much as to result in fast release of oxygen. Addition of vesicles leads to faster oxygen release, and at molar ratio PhL/Pr = 25 : 1 (pH 7.2) at 37°C it occurs in 2.5 h, and at 22°C it is much slower (up to a day and a half). A 4-fold increase in the concentration of phospholipids (PhL/Pr = 100 : 1) accelerates the oxygen release 2-fold, whereas at molar ratio PhL/Pr = 200 : 1 oxyMb molecules acquire the met-form just after mixing with vesicles (not shown). These data demonstrate that the presence of membranes facilitates the release of oxygen. Similar data for oxyMb in the presence of other vesicles were reported in [67]. The described effect can be associated with the disturbance of the tertiary structure of the oxyMb molecule revealed by other methods for holoMb. The initial oxygen release occurs only in the myoglobin molecules which denature and bind to the membrane. It can be proposed that the slow phase corresponds to oxygen release by free protein molecules. It should be noted that oxygen release from oxyMb molecules not bound to vesicles occurs much faster in the presence of vesicles than from this protein in their absence, which is indirect evidence of destabilization of the structure of Mb molecules not bound to vesicles. At molar ratio PhL/Pr = 200 : 1 (pH 7.2) the protein is completely bound to vesicles (data of other methods); therefore, after mixing of the protein and vesicles, fast release of oxygen takes place and it is substituted by water, i.e. the met-state is produced.
Fig. 5. Kinetics of the ligand release in the presence of phospholipid membranes. a-d) OxyMb at 22°C (a, b) and 37°C (c, d). e, f) RBP at 22°C. a) Absorption spectra of oxyMb at various times (indicated by numbers near the curves) after addition of vesicles, the molar ratio of PhL/Pr = 25 : 1, pH 7.2. b) Kinetics of autooxidation oxyMb in the absence (○) and in the presence of vesicles at ratios 25 : 1 (▲) and 100 : 1 (●). c) OxyMb absorption spectra at varied times after addition of vesicles shown near the spectra, pH 7.2. d) Kinetics of oxygen release in the absence (○) and in the presence of vesicles at molar ratio PhL/Pr = 25 : 1 (▼) (adapted from [30]). e) CD spectra in the far UV region of RBP in the native state and in the presence of mixed vesicles (DOPC/DOPG = 1 : 1) at molar ratio PhL/Pr = 500 : 1 at different pH values, 22°C. Changes in the spectrum of the N state reflect protein denaturation. f) Kinetics of RBP denaturation in the presence of vesicles under conditions similar to those in Fig. 5e. Retinol release occurs simultaneously with protein denaturation (kinetics of retinol release is very fast in the beginning) (unpublished data).
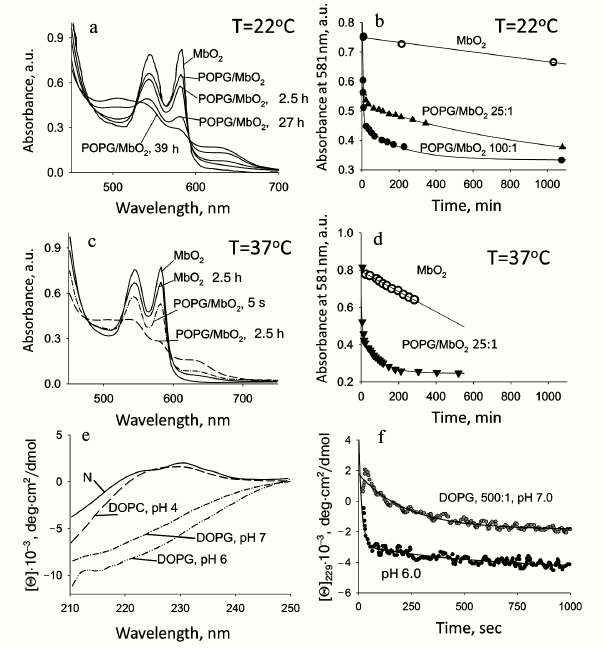
An analogous effect is observed upon vitamin A (retinol) delivery to the membrane. The kinetics of RBP denaturation and retinol release at pH 7.2 in the presence of membranes is sharply accelerated, and a decrease in pH to 6.0 results in a greater effect (Fig. 5, e and f). It should be emphasized that retinol can be released from RBP only upon disturbance of its structure. That is why the retinol release proceeds concurrently with the protein denaturation, but with a higher rate.
The examples of oxyMb and RBP undoubtedly show that the presence of the negatively charged surface of membranes leads to larger fluctuations in the protein structure, which in addition to the fluctuations caused by the temperature increase to 37°C contribute to the functions of these proteins.
Water-soluble fragment of cytochrome b5. Features of disturbance of native interactions in tertiary structure of cytochrome b5 in the presence of phospholipid vesicles. Cyt b5 is a peripheral membrane protein [28, 68, 69] that is spontaneously imbedded into the outer layer of the ER membrane in the posttranslational period.
The water-soluble domain of Cyt b5 functions near the ER membrane and interacts with numerous partners including the NADH-Cyt b5 reductase [70, 71] and NADPH cytochrome P450 reductase [72, 73]. It receives one electron from the reductases and transfers it to other proteins that are components of the electron transfer system (such as cytochrome P450). These proteins catalyze desaturation of fatty acids [74, 75], hydroxylation of steroids, and xenobiotic transformation of drugs and different natural products [70, 72, 76]. Erythrocytes have a natural water-soluble form of Cyt b5 (without a hydrophobic domain) involved in the reduction of metMb [70, 77] and metHb [26, 78]. It is believed that Cyt b5 deprived of the hydrophobic domain cannot form a complex with cytochrome P450, and hydrophobic interactions play a more significant role than electrostatic interactions upon complex formation [79].
The structure of the catalytic fragment of Cyt b5 was studied by X-ray analysis [80] and homo- [81, 82] and hetero-NMR [83, 84]. The X-ray analysis revealed that the sequence of the catalytic domain has a large number of negatively charged amino acid residues in the region of the heme that determine the charge of the fragment [80]. Some are involved in the interaction with cytochromes [74, 85] and globins [26, 77, 86], and others are engaged in the interaction with reductases [71, 77, 87, 88]. The most of the negatively charged residues are on the protein surface and localized near the heme, and just these residues are involved in interactions of Cyt b5 with partner proteins in redox reactions. On the contrary, the part of the protein that is facing the membrane is strongly depleted of negatively charged amino acid residues. Studies on the conformational state of the water-soluble fragment of Cyt b5 near the membrane surface are extremely important for understanding the mechanism of its functioning. Structural investigations of Cyt b5 were performed both in the presence of vesicles from negatively charged unsaturated phospholipids and in the presence of vesicles from a mixture of saturated uncharged (DPPC) and unsaturated (POPG) phospholipids at pH 7.2 and 5.5. It is worth mentioning that at pH 5.5, Cyt b5 is still in its native state, being insignificantly destabilized due to the pH dependence of the native state stability [89].
Features of tertiary structure of cytochrome b5 in the presence of phospholipid vesicles. Heat denaturation reveals changes in the protein tertiary structure under different conditions. The heat absorption curves for Cyt b5 at molar PhL/Pr ratios varying from 50 : 1 to 200 : 1 at pH 7.2 and 5.5 show that there is no significant shift of the protein melting temperature even at pH 5.5. However, with a decrease in pH, an approximately 2-fold decrease in the calorimetric enthalpy occurs, which may be connected with destabilization of the protein structure [89]. The ratio of the calorimetric and effective enthalpies also changes, decreasing to 0.5, which suggests association and probably dimerization of the protein in the presence of vesicles.
The data of [89] suggest that at PhL/Pr = 50 : 1, a few of the protein molecules are in the denatured state and make no contribution to the common calorimetric enthalpy, but only the protein molecules that have retained their tertiary structure are melted. At PhL/Pr = 100 : 1 and higher (pH 7.2), a protein heat absorption peak is absent, and hence the rigid tertiary structure of the protein is already disturbed. It should be noted that an increase in temperature does by itself destabilize the protein, and together with vesicles their denaturing action on the protein structure is even stronger. At pH 5.5 and all ratios of PhL/Pr, it is impossible to observe heat melting of the protein, which is evidence that with a decrease in pH the addition of vesicles results in disturbance of the tight packing of protein side groups.
The gel-to-liquid-crystal phase transition of saturated phospholipids (DPPG) is revealed at 312K (39°C), which makes it possible to trace the process. Calorimetric measurements using vesicles from saturated phospholipids were made at molar ratios of PhL/Pr = 100 : 1 and pH 7.2 and 5.5. At PhL/Pr = 100 : 1 and pH 7.2, no changes were revealed in the parameters of the temperature transition of the vesicles, which may indicate only electrostatic interaction of the protein with the surface of the vesicles without disturbance of the phospholipid packing in the bilayer. At pH 5.5 and the same molar ratio, the melting peak of the vesicles varies not only in the values of heat capacity and enthalpy, but also shifts to lower temperatures; hence, the protein interacts more intensively with the vesicles.
At pH 7.2, Cyt b5 in the native state has a rigid asymmetric environment of aromatic amino acids resulting in the appearance of a pronounced CD spectrum in the near UV region. The spectrum consists of characteristic bands that are completely absent in the spectrum obtained for the protein in the intermediate MG state [89]. The shape of the CD spectrum in the near UV region for Cyt b5 in the presence of a heat absorption peak is very close to that of the native protein. Such a spectrum demonstrates the presence of tertiary interactions in the major part of protein molecules. When the concentration of phospholipids (pH 7.2) increases, the native protein features in the CD spectra in the near UV region disappear, showing the disturbance of the tight packing of its side groups. At pH 5.5 for all studied PhL/Pr molar ratios, the CD spectra in the near UV region lose all specific features of the native protein spectrum. This means that under such conditions Cyt b5 interacts with phospholipid vesicles and in this state has no tight packing of its side groups. These data are compatible with the results obtained by microcalorimetry.
Absorption spectra of Cyt b5 in the characteristic band of the heme are sensitive to changes in its environment. At PhL/Pr = 50 : 1 and pH 7.2, the absorption spectrum has no distinct changes and is close to the native one. A four-fold or higher increase in the concentration of phospholipids results in the following: widening of the Soret band, decrease in the absolute value of the peak, and shift of the maximum to shorter wavelength with an additional shoulder at 380 nm characteristic of the denatured protein. At pH 5.5, already at PhL/Pr = 25 : 1 more marked changes occur in the Soret band. These changes indicate that in the presence of vesicles the environment of the Cyt b5 heme differs from that typical of the native state in solution, which indicates denaturation of the protein on the whole. In this case, the absorption spectra in the presence of vesicles differ greatly from the protein spectrum at pH 3.0 when the heme is released from the hydrophobic pocket.
The amplitude of the intrinsic Trp fluorescence of Cyt b5 in the native state is quite low, a weakly expressed maximum at 335 nm being characteristic of the fluorescence spectrum, which may be caused, as in holoMb, by quenching of the single Trp residue by the heme and the surrounding amino acid residues. At PhL/Pr = 50 : 1 and pH 7.2, the fluorescence intensity increases insignificantly, i.e. Trp is still strongly affected by the heme and has a rather rigid environment. When the concentration of phospholipids increases to ratio PhL/Pr = 500 : 1 at pH 7.2, the intensity of Trp fluorescence grows remarkably, but only insignificantly exceeding that for this protein in the MG state. At pH 5.5 and PhL/Pr = 50 : 1, the fluorescence intensity is somewhat higher than at PhL/Pr = 500 : 1 and pH 7.2. The intensity of the fluorescence spectra for other ratios at pH 5.5 is the same and only slightly exceeds that for PhL/Pr = 50 : 1. However, under these conditions the fluorescence intensity is only 75% of the value for the completely unfolded state. Thus the fluorescence spectroscopy data show that in the presence of phospholipids the distance and mutual orientation of the heme and Trp are changed. The wavelength at the fluorescence maximum is 340 nm for all molar PhL/Pr ratios, which is close to the value for the native state of Cyt b5. This suggests a rather hydrophobic environment of Trp in the denatured protein.
Summarizing the data obtained in the studies of the tertiary structure of Cyt b5 in the presence of negatively charged phospholipid vesicles, we conclude that in the presence of phospholipids the tight packing of the protein side groups is disturbed. Cyt b5 has proved to be more stable to the denaturing action of negatively charged vesicles than holoMb, which completely loses its tertiary structure at lower value of the PhL/Pr ratio. This difference in the interaction of the proteins with vesicles could be connected with both the difference in their total charge and their structural organization.
Secondary structure of cytochrome b5 is preserved in the presence of phospholipid membranes. CD spectra in the far UV region were analyzed for Cyt b5 under the same conditions as for measuring the absorption and fluorescence. The CD spectrum of Cyt b5 in the N state has one well-expressed minimum at 220 nm and a shoulder at 208 nm. In the MG state [89] at pH 3.0, the shape of the spectrum changes and the shoulder at 208 nm turns into a clearly expressed minimum. For molar ratio PhL/Pr = 50 : 1 at pH 7.2, changes in the CD spectrum are insignificant, and all parameters resemble the spectrum of the protein in the native state. The shape of the spectra of Cyt b5 in the presence of high concentrations of PhL at pH 7.2 become similar to that of the spectrum of the protein in the MG state, and they vary greatly from the spectrum of the unfolded protein. Consequently, at pH 7.2 the secondary structure of Cyt b5 in the presence of vesicles changes slightly, and the change in the shape of the spectra is associated with conformational rearrangements of the protein molecule. Decrease in pH to 5.5 leads to an increase in the absolute value of ellipticity at 220 nm already at PhL/Pr = 50 : 1, as compared to the spectrum of the MG protein, though the shape of the spectra is similar. Increase in the concentration of vesicles results in a still higher amplitude of the CD spectrum. A comparable change in the CD spectra in the far UV region is observed for this protein at higher concentrations of MeOH [29]. This effect can be explained by increased stability of hydrogen bonds inside the polypeptide chain when the protein gets into a more hydrophobic environment, i.e. in a medium with a lower ε value. We suggest that the content of the secondary structure increases due to partial immersion of the protein molecule in the bilayer, where the influence of the hydrophobic part of the membrane is rather strong. This agrees with the scanning microcalorimetry data. Thus, Cyt b5 in the presence of charged phospholipid vesicles as well as in the MG state has a pronounced secondary structure, which, however, differs from that of the native structure.
Interaction of cytochrome b5 with phospholipid membranes. As known, the native water-soluble fragment of this protein does not interact with the membrane, so the binding to the membrane is also indirect evidence of denaturation of the protein. The elution profiles obtained during gel filtration of mixtures of proteins and phospholipid vesicles have two peaks: one in the place of elution of the free protein and the other in the place of elution of vesicles, in this case in the void volume. At PhL/Pr = 50 : 1 and pH 7.2, the area of the peak corresponding to the free protein diminishes slightly (by about 10%). An increase in the molar concentration of phospholipids results in a decrease of the peak height in the range of native Cyt b5 elution, and concurrently the peak area increases in the range of vesicle elution.
The binding of protein molecules at pH 7.2 occurs only at molar ratio PhL/Pr = 500 : 1, though the area of the vesicle surface is many times larger (about 20-fold) than the area of the surface of all protein molecules. For pH 5.5, the propensity for changes in the value of elution peaks is similar to that at pH 7.2, but complete binding of the protein with vesicles proceeds at a much lower concentration of phospholipids (PhL/Pr = 200 : 1). Thus, for Cyt b5 the interaction with vesicles is also observed, but for this a higher molar concentration of phospholipids than that for holoMb is required. Destabilization of the protein caused by decreasing pH to 5.5 results in protein transition from the native state to the intermediate state and the consequent binding with the membrane at lower molar PhL/Pr ratio than is required at pH 7.2.
Interaction of Cyt b5 with mixed vesicles. The lipid composition of biological membranes includes both negatively charged phospholipids and neutral phospholipids, and this is true for the ER membrane. Therefore, the effect of vesicles from a mixture (50 : 50) of saturated uncharged (DPPC) and unsaturated negatively charged (POPG) phospholipids on the conformational state of Cyt b5 was studied. Scanning microcalorimetry was used to assess the state of the tertiary structure of the protein. For the native protein, the heat absorption peak is observed at 340K (67°C), and for the vesicles at 300K (27°C). The melting peaks of the protein and vesicles do not overlap, which permits reliable observation of changes in each process independently. At PhL/Pr = 25 : 1 and pH 7.2, the protein melting temperature in the maximum is practically unchanged, but in this case the calorimetric enthalpy decreases twice as compared to the native protein. A similar change in the curve of temperature transition was observed for Cyt b5 in the presence of vesicles from negatively charged phospholipids but at PhL/Pr = 50 : 1. As to mixed phospholipids at the same molar ratio, only a small protein melting peak is observed with a maximum at 343K, which shows that a large part of the protein molecules have already lost their tertiary structure. The other protein molecules preserve the tight packing of side groups. Thus, the disruption of the tertiary structure of Cyt b5 in the presence of vesicles from the mixture of uncharged and charged phospholipids was more efficient than with the use of only negatively charged unsaturated phospholipids. The mixture of saturated and unsaturated phospholipids forms a bilayer whose influence, despite the double decrease in negative charge of the membrane surface, is more efficient for this protein.
Hence both the neutrally charged myoglobin and negatively charged Cyt b5 respond to the influence of the charged surface of PhL membrane on their tertiary structure, which changes their conformational state.
α-Lactalbumin. Lactalbumin (HLA) is a well-studied globular protein found in the milk of humans and other mammals. HLA is a modifier of the enzyme galactosyltransferase and causes the formation of a lactose-synthesized enzyme complex that catalyzes the final step in the biosynthesis of lactose during lactation. The 3D structure of α-HLA has been determined to resolution 1.7 Å [90]. The HLA molecule contains four α-helices and a triple antiparallel β-sheet. The protein structure is stabilized by four disulfide bridges. HLA is an acidic protein with pI 4.4, and at neutral pH it has a charge of –8.
Conformational transitions in the structure of the protein under the influence of different denaturants in aqueous solutions are well studied [1, 2, 91]. One of the most important features of the protein is its high affinity for calcium ions, which stabilize the protein [92].
HLA is a water-soluble protein, but after biosynthesis in its apoform it is associated with the ER inner layer membrane. However, the conformation of the membrane-bound protein state is not known. The binding of Ca2+ only leads to the following: a change in the protein conformation, acquiring of the native functional conformation, overcoming of the ER quality control system, and subsequent exocytosis.
Conformational changes in α-lactalbumin structure in the presence of phospholipid vesicles. HLA is a secretory protein. The calcium-free form of HLA was chosen for study just because it interacts with the membrane after biosynthesis. Structural changes in the protein in the presence of vesicles were studied by scanning microcalorimetry, CD in the far UV region, and high-resolution 1NMR.
Heat absorption curves for apoHLA (in the presence of 10 mM EDTA) were obtained in the absence of vesicles at pH 6.2 and in the presence of a mixture of PhL vesicles, as well as for free vesicles from a mixture of the same PhL (Fig. 6a). Free vesicles have their own melting peak with a maximum at 300K. The maximum of the protein melting peak is at 311K, so the protein peak overlaps with the vesicle melting peak. The calorimetric enthalpy value of protein melting is 168 kJ/mol, while for vesicles it is only about 23 kJ/mol. The protein heat absorption peak is absent in the presence of vesicles, but the melting peak of vesicles shifts to higher temperatures. The observed shift of the heat absorption curve of vesicles indicates a change in their properties due to the interaction with the protein.
Fig. 6. Conformational behavior of the Ca2+-free form of human lactalbumin (apoHLA) in the presence of phospholipid membranes. a) Dependence of excess partial heat capacity Cp.exc on temperature in the presence of mixed vesicles of zwitterionic DPPC and charged POPG (1 : 1) for ratio PhL/Pr = 25 : 1, pH 6.2. The heat absorption curve for the native state of the protein at pH 6.2 in the absence of vesicles is shown for comparison. b) CD spectra in the far UV region of apoHLA in the presence of vesicles (conditions are same as in Fig. 6a). Spectra of the protein in the N, MG (pH 2.0), and U (6 M GuHCl) states are shown for comparison. c) High resolution 1H-NMR spectra of the apoHLA native state (spectrum 1) and in the presence of vesicles (spectrum 2) under conditions similar to those in Fig. 6a; 3) spectrum of vesicles without the protein (adapted from [30]).
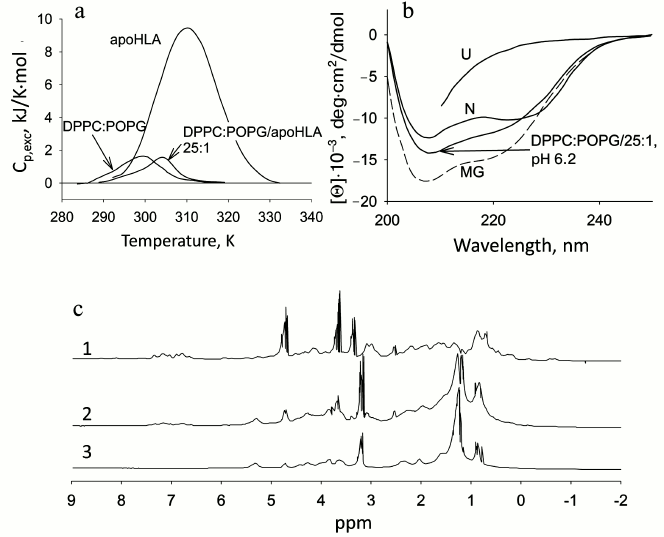
Changes in Trp fluorescence spectra are similar to those for apoMb. As the intensity of the fluorescence peak is increasing, the position of the spectral maximum shifts to 340 nm, which corresponds to that of the protein in the MG state. Such changes indicate the HLA denaturation in the presence of vesicles, but it is difficult to judge the compactness of the protein because we cannot exclude the influence of the hydrophobic layer of the membrane on Trp fluorescence.
CD spectra in the far UV region of HLA are shown in Fig. 6b. As seen, the shape of the spectrum of the protein in the presence of vesicles changes, the ellipticity at 208 nm increases, and, thereby, the spectrum becomes more similar to the spectrum of the protein in the MG state. In general, this pattern of changes suggests a restructuring of secondary structure elements due to interaction of the protein with vesicles. However, complete destruction of the secondary structure is not observed, as the spectrum of the protein in the presence of vesicles is not similar to that of the unfolded form.
The NMR spectrum of the native protein has a number of well-resolved secondary chemical shifts, which almost completely disappear upon addition of PhL vesicles. Only features characteristic of the free vesicles are left (Fig. 6c). It should be noted that the NMR spectrum of the protein in the presence of vesicles differs significantly from that of free vesicles in the region of vesicle resonances (2-0.5 ppm), in particular, there is an increase in the integrated area under the curve. Resonances characteristic of the native protein do not appear under these conditions. This fact suggests that the protein binds to the surface of the vesicles, and this leads to a slight disturbance of the regular packing of phospholipids in the bilayer.
Conformational states and thermodynamics of the binding of HLA to membranes were studied in Chenal’s group [93]. It was shown that the conformation of the protein associated with the membrane surface is very sensitive to external factors such as pH, concentration of Ca2+, and charge and curvature of the membrane. The anchoring of the protein in the membrane is limited to amphipathic α-helices.
Thus, the calcium-free form of human HLA in the presence of PhL vesicles undergoes disturbance of the tertiary structure while maintaining the secondary structure. This indicates that after biosynthesis, the calcium-free form of HLA associated with the membrane is in the intermediate state rather than the native state.
Comparison of structural features of different proteins in the presence of phospholipid vesicles. The above data allow for a comparison of the effect of negatively charged phospholipid vesicles on the conformational state of weakly positively charged holo- and apomyoglobins and a negatively charged water-soluble fragment of Cyt b5 at neutral pH specific to the cytoplasm. All investigated proteins function near the membrane and do not bind to it, but can be affected by it. It was found that PhL vesicles have a denaturing effect on all of these proteins, and they have a general propensity to structural changes, in spite of the differences in the spatial structure of the proteins. The main result is that the tertiary structure of these proteins in the presence of vesicles is disturbed, whereas the regular secondary structure is preserved. This state is intermediate between the N and U states of proteins in aqueous solution, and its properties are close to the molten globule state. However, the observed intermediate state of the protein in the presence of phospholipid membranes cannot be called a typical state of MG, observed in aqueous conditions, as all these proteins bind to vesicles, forming a complex. This fact did prevented us from determining the size of the protein using available methods. The binding of proteins to vesicles occurs due to electrostatic interactions, and after immersion of the protein in the outer membrane layer hydrophobic interactions arise. If we consider the behavior of each protein in the presence of vesicles separately, a number of features related to the structural characteristics of the protein and the difference in the total charge will be revealed. ApoMb and the calcium-free form of HLA do not contain ligands that could stabilize the protein molecule, so their denaturation and binding to vesicles occur even at the lowest concentration used (PhL/Pr = 25 : 1). The secondary structure of these proteins in the presence of vesicles is changed slightly. HoloMb and Cyt b5 contain a noncovalently bound heme that leads to an increase in the protein stability, i.e. specific rigidity of the tertiary structure at neutral pH. However, despite the greater stability of the tertiary structure, the addition of negatively charged vesicles at pH 7.2 leads to an increase in the mobility of the side groups, and, ultimately, to disruption of their dense packing. In this case, the processes of denaturing and binding, unlike in the case of apoproteins, occur stepwise, i.e. at low concentrations of PhL only part of the protein molecules loose their close packing of side groups and are associated with vesicles. Gradually, with increasing concentration of PhL, a growing number of protein molecules are denatured until all of the protein molecules have an intermediate state, wherein the protein is associated with vesicles. For myoglobin at pH 7.2, all the molecules are completely bound to vesicles at molar ratio PhL/Pr = 200 : 1, and for Cyt b5 only at PhL/Pr = 500 : 1, although as estimated in such systems the surface area of the vesicles accessible for the binding exceeds the surface area of all the protein molecules. This can be explained by the difference in the total charge of the protein. This fact emphasizes the importance of electrostatic interactions in the binding of proteins with phospholipid membranes. Myoglobin molecules not bound with vesicles, which are present at lower PhL concentrations, are in the native state, which, however, becomes less stable relative to the denatured protein molecules associated with vesicles. This is explained by the fact that the phospholipid membranes have their “field” at a distance of 5-20 Å, due to the presence of negatively charged phospholipid heads, and the change in the dielectric constant of the medium from a value of about 40 at the interface to 80 in the bulk aqueous phase. In the case of Cyt b5, free protein molecules in the presence of vesicles retain native tertiary structure for a longer period, until collisions with the membrane result in a transition to an intermediate state competent for binding. Lowering the pH to 6.2 for myoglobin and 5.5 for Cyt b5 facilitates denaturation and the binding to the phospholipid membrane, as in this case holoMb changes on the beginning of its pH-induced transition and therefore should not undergo substantial changes in tertiary structure, and Cyt b5 at this lowered pH will be in the middle of its pH-dependent transition. Based on our data, effective lowering of the pH near the surface of the charged vesicles for apoMb is about three pH units in the used conditions. The dependence of holoMb and Cyt b5 properties on the PhL content in general resembles the dependence of their stability on pH.
COMPARISON OF CONFORMATIONAL STATES OF PROTEINS IN
WATER–ALCOHOL MIXTURES AND IN THE PRESENCE OF PHOSPHOLIPID
MEMBRANES
Comparative analysis of experimental data on the involvement of non-native states of a protein in a number of cellular processes led to a hypothesis about a functional role of the intermediate states of the protein [94]. In subsequent years, this hypothesis was confirmed. For example, it was experimentally shown that, besides the translocation of proteins across the membrane and ligand exchange near the membrane, a protein acquires the MG state after biosynthesis, and this state is recognized by chaperones. Proteins destined for degradation are modified, which leads to destabilization of their structure. Moreover, other important cellular processes also require a non-native state of the protein. However, the occurrence of the non-native protein state in a cell may lead to the formation of plaques and fibrils on the membrane surface that might result in the appearance of human diseases.
Estimation of the conformational state of different proteins in water–alcohol mixtures and in the presence of phospholipid membranes. The bilayer membrane has a complicated structure [12, 13, 95]. The results obtained for the retinol-binding protein [32] and Cyt c [24] showed that the change in a number of significant features of the protein structure observed in the presence of PhL membranes can be modeled by a moderate decrease of ε and pH. In particular, it has been shown that at moderately low concentrations of MeOH (16%) at pH 3.5, RBP releases retinol, and the protein undergoes a transition to an intermediate state similar to the MG state.
The effect of water–alcohol mixtures on the structure of horse apoMb was studied in [31], horse holoMb in [39], and negatively charged Cyt b5 in [29]. During the study of the conformational state of apoMb in water–methanol mixtures at moderately low pH (~30% MeOH at pH 5.2), an intermediate state was found that is characterized by the absence of rigid tertiary structure and retaining its compactness and nearly native secondary structure (see Table 2). At alcohol concentrations above 50%, a more structured denatured helical state is observed. Similar results were observed in the case of holoMb, which is transformed into an intermediate state of the MG type in 40% MeOH at pH 4.0 (see Table 2). The Cyt b5 soluble fragment was studied at pH 7.2 [29], and only at concentrations of 40 to 60% MeOH was a transition from the native to intermediate state observed.
We investigated the conformational state of Cyt b5 in the presence of various concentrations of iPrOH at pH 6.5 using absorption spectroscopy, Trp fluorescence, CD in the far UV region (Fig. 1b), and scanning microcalorimetry. The results showed that at 36% alcohol concentration the heme environment becomes non-native and no heat absorption peak is observed, whereas the secondary structure and compactness are preserved (see Table 2), i.e. the transition from the native protein structure into an intermediate state occurs.
Comparing the structural data obtained for various proteins in the presence of negatively charged vesicles and in water–alcohol mixtures, we can conclude that the propensity for conformational transitions is similar in both cases. Under the influence of both denaturants, under certain conditions proteins lose their rigid tertiary structure but retain secondary structure. It is difficult to judge the compactness of the protein molecule in the presence of vesicles, since proteins bind to the membrane. The main structural characteristics of the proteins studied under different conditions are shown in Table 2.
Our data on protein denaturation in the presence of vesicles are in agreement with the results obtained for the protein in water–alcohol mixtures at moderately low pH. These results support the hypothesis [3, 94] that membranes affect the structure of the protein due to enhanced electrostatic interactions near their surface, namely, through the combined action of lowering both pH and ε of the local environment. These results leave no doubt that the surface of the membrane can serve as a moderately denaturing agent in the cell.
POSSIBLE PATHWAYS OF MEMBRANE–PROTEIN INTERACTIONS
Translocation of proteins across membranes. The first indication that the non-native state of the protein is involved in the translocation process appeared in reports of Schatz’s group. They studied the translocation of proteins renatured from solutions of a strong denaturant [96-99]. It was found that the renatured proteins translocate faster and more efficiently than native precursors, and the translocation process is not inhibited by either low temperature or specific ligands.
In studies by the same group [6, 8], it was observed that the native precursor connected to the outer membrane surface of mitochondria is transformed into a state competent for transport. This state may be similar to the intermediate state in protein folding. They noted that “such a partial unfolding should have physiological significance”.
Further investigations of Schatz’s group clearly showed that denaturation of a protein is required for its translocation across the membrane. First, there is evidence that the stabilization of proteins by ligand binding inhibits translocation: thus, methotrexate associated with dihydrofolate reductase stabilizes its structure against proteolysis and prevents it from passing through the membrane [7, 96]; or the introduction of a disulfide bond in the precursor of dihydrofolate reductase, stabilizing the structure of the protein, blocks its import [100]. In addition, destabilization of a protein by urea, by point mutations, or by binding to the membrane surface dramatically accelerates the import of the protein into the mitochondria [6, 7, 98, 101]. Besides, it is shown in [102] that the stabilization of the structure of ribonuclease barnase by its inhibitor barstar completely inhibits the import of barnase into mitochondria. It was found that holoproteins or their precursors, for example, such as cytochromes c and b, are imported in their apoforms, just after biosynthesis, whereas heme binding inhibits their translocation [103]; the wild-type apoform of maltose-binding protein sensitive to proteolysis is exported quickly and efficiently [104].
Second, it was found that non-native proteins are translocated much more easily than their folded forms. Dihydrofolate reductase without C-terminal residues is translocated even in the absence of ATP [105]. The precursor of β-lactamase binds to GroEL immediately after biosynthesis, before the acquisition of the rigid structure, and can be translocated, but it loses this ability upon reaching its native state [106].
Third, it was shown that the proteins associated with the membrane are in a non-native state. Translocation of dihydrofolate reductase occurs in two steps: binding to the membrane, which is inhibited by salts, occurs in a partially unfolded form accessible to proteolysis, and the translocation proper is inhibited by methotrexate [96, 97]. Insertion of membrane proteins synthesized in the cytoplasm into the membrane occurs stepwise, passing through intermediate states [107, 108].
New data on the mechanism of protein translocation across a membrane have appeared, in particular, protein transport across the mitochondrial membrane has been studied in detail. Many proteins are transported from the cytosol into mitochondria via the TOM complex or channels with a limited diameter [109]. To go through such a channel, even the smallest proteins should be denatured [110, 111]. Disturbance of the initially folded protein structure can be caused by the mitochondrial membrane itself due to the presence of specific proteins (e.g. Hsp70) and the potential on the mitochondrial membrane [112, 113].
Table 2. Structural features of proteins in
the native state, in the presence of phospholipid vesicles at neutral
pH, and in water–alcohol mixtures at moderately low pH values
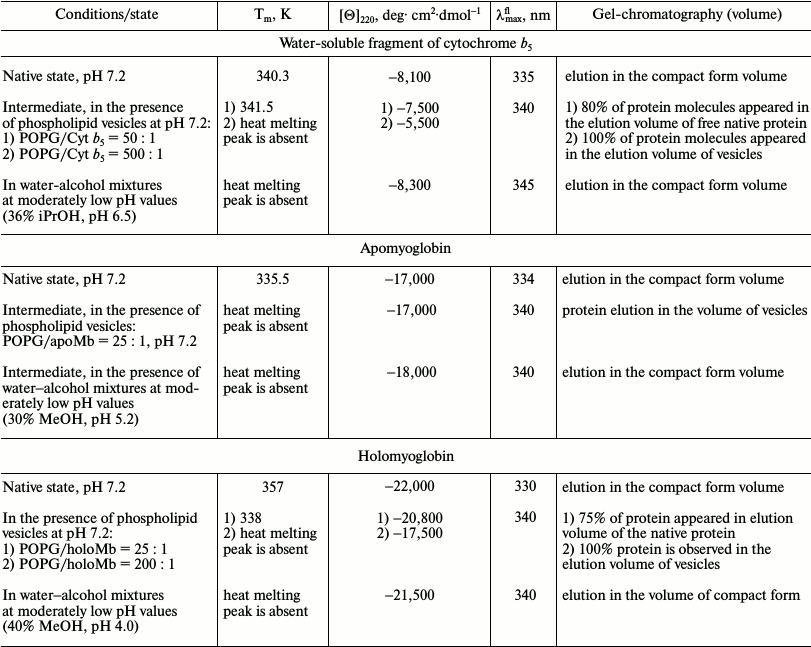
Translocation process. Studies of toxins indicate that they penetrate through the membrane in a non-native state. It was shown for the pore-forming domain of colicin A that its binding to the membrane and penetration into it increase sharply at lower pH values [9, 10]. On the other hand, it was found that this domain is transformed into the molten globule state at low pH, and this pH value coincides with the local pH at the membrane surface. The data allowed the authors to conclude that the local low pH at the membrane surface denatures the pore forming domain of the toxin to the molten globule state. It makes the structure of the domain more fluctuating or flexible and allows the hydrophobic hairpin, in the native state immersed in the hydrophobic core of the protein, to separate from the rest of the protein and penetrate into the membrane [9, 114]. Later it was shown that the binding of colicin A to negatively charged vesicles leads to a conformational change in the tertiary structure of the protein [115-118].
Other toxins having pore-forming domains, such as colicin E1 [119] and δ-toxin [120], behave similarly.
For diphtheria toxin (DT), for which the transition from the native to the molten globule state is observed at acidic pH [122], it was shown that denaturation facilitates its binding to the membrane [121]. It is well known that DT enters the cell through organelles with internal acidic pH. Under in vivo conditions, in lysosomes DT is exposed to acidic pH at which it is transformed into the molten globule state. When corresponding toxin domains are transferring to the cytoplasm, their renaturation from the MG state to the native active state occurs [121-124]. Similar data were obtained for exotoxin, which has homology and enzymatic activity analogous to DT [125], as well as for influenza hemagglutinin [126]. Later, in 2009, Chenal’s group extensively studied the penetration of the toxic domain of DT through the membrane using neutron reflectometry and solid-state NMR spectroscopy [127].
The A-domain of the dimer toxin ricin binds to negatively charged phospholipid vesicles, but it does not interact with neutral phosphatidylcholine vesicles. The binding is accompanied by conformational changes in ricin and destabilization of the lipid bilayer [128, 129]. On the interaction of the protein with the vesicles, the protein structure undergoes a transition to a partially unfolded state, as evidenced by its cleavage by trypsin and the loss of ability to bind adenine, i.e. to perform its function.
Denaturing effect of membranes is also revealed during penetration of staphylococcal α-toxin into a cell [130]. Penetration and translocation of the catalytic domain of the pathogenic bacterium Bordetella pertussis CyaA toxin across the membrane was studied in [129-131]. A long fragment (1-489 a.a.) that had the ability to bind to the membrane and destabilize the structure of the bilayer was discovered, while the shorter fragment of this protein (1-386 a.a.) did not have such properties. Destabilization of the protein structure near the membrane exposed the hydrophobic binding site and its binding to the membrane. This made it possible to transfer the hydrophobic catalytic domain through a hydrophilic extracellular environment into the hydrophobic environment of the cytoplasmic membrane.
These data confirm that protein rigid structure should be disturbed prior to interaction with the membrane. This is confirmed in the literature. Cholera and pertussis toxins are unstable relative to temperature, which allows them to be in a non-native conformation and to use the ER degradation system for penetration into the cytoplasm. Thermal destabilization of the tertiary structure of the cholera toxin catalytic subunit usually occurs prior to disordering of its secondary structure. Therefore, any structural stabilization of cholera toxin [132, 133] or pertussis toxin [134, 135] inhibits the transfer of the toxins from the ER to the cytoplasm and the cell toxicity. Thus, stabilization of the tertiary structure of the cholera toxin catalytic subunit by glycerol prevents its exit from the ER [132]. This work showed that the thermal instability of the cholera toxin plays a key role in the process of intoxication. This also applies to the S1 subunit of pertussis toxin [134]. The authors of [136] showed that the membrane surrounding of the neurotoxin facilitates the binding of the ligand to its receptor. The interaction of the neurotoxin with charged heads of phosphatidylserine (PS), surrounding the acetylcholine receptor, leads to the appearance of the specific topology of the toxin on the membrane surface, which promotes the blocking of the receptor.
Interactions of the P-type cardiotoxin from cobra venom with different phospholipid membranes were studied using 1H-NMR. It was found that the toxin binds to negatively charged vesicles (but not neutral ones) and penetrates into the bilayer, which leads to further destruction of the membrane [137].
Binding of non-native state of proteins to membranes. In the non-native state, hydrophobic groups are exposed to the surface of the protein. This leads to the binding of hydrophobic dyes, protein aggregation, and interaction with membranes. This is true for HLA, which undergoes a transition from the native to the molten globule state at acidic pH, and only after that it binds to the membrane bilayer and then partially buries into it [138]. In later studies it was shown using NMR that the binding of HLA to the membrane occurs due to interaction of hydrophobic residues of helices A and C with the lipid bilayer [139]. It also is established that apoMb is associated with large vesicles at acidic pH values, and the sequence of the fragment interacting with the membrane was determined [140].
It was found that anionic phospholipids can modulate the function of several enzymes: for example, for activation of hydrolytic activity of water-soluble ATPase SecA, its binding to the Escherichia coli inner membrane is required. For this action, a partially unfolded protein conformation has to be formed [141]. The modulating effect of negatively charged membranes was shown for peptides isolated from the C-terminal domain of apolipoprotein E (ApoE). These peptides are able to independently form lipoprotein complexes, and the authors suggested that the surface charge of ApoE can regulate its metabolism [142]. To regulate the activity and stability of tyrosine hydroxylase, only its weak binding to the membrane is required [143]. Similar conditions are necessary for activation of prothrombin [144]. The water-soluble dimer of acetylcholinesterase from Torpedo californica, while in the native state, does not interact with vesicles of phosphatidylcholine, whereas the transformation of the protein to the MG state leads to its binding to the membrane through a hydrophobic peptide of 5 kDa [145]. This amphiphilic peptide is placed horizontally on the surface of the membrane, and only its hydrophobic surface is immersed in the membrane. Acetylcholinesterase from Bungarus fasciatus easily undergoes thermal denaturation in the presence of membranes, but is not associated with them and remains in the denatured state in solution [146], i.e. in this case the membrane surface acts as a catalyst of denaturation.
The above examples of proteins were obtained under non-native conditions, when the intermediate states of proteins were formed prior to their interaction with membranes. An exception is acetylcholinesterase, for which it was found that the presence of membranes catalyzes its thermal denaturation at neutral pH [147].
It was shown for saposin C that its binding to negatively charged PS vesicles causes their fusion [148]. In the process of fusion of the vesicles, both protein–membrane and protein–protein interactions are involved. Vesicle fusion occurs due to immersion of the N- and C-terminal helices of the protein into the bilayer of different vesicles. This process is accompanied by a change in the conformation of the protein molecule, which, in turn, is able to interact with another protein molecule situated near the first one.
The authors of [149] note that in the presence of PG phospholipids in the membrane, the rate of folding and efficient yield of folded trimeric α-helical membrane protein diacylglycerol kinase increase. Substitution of PG for lyso-PG reduces the rate and efficiency of folding of this enzyme.
For small GTPase H-Ras, anchored to the membrane, it was shown that it forms active dimers due to protein–protein interactions (without involvement of the lipid) only on the membrane surface. In solution, the protein has exclusively monomeric form [150].
The process of forming transport vesicle shells is also associated with the membranes. Thus, the self-organization of clathrins and similar proteins (coat proteins) occurs solely on the membrane surface [151].
Interaction of cytochrome c with membranes. Studies of Cyt c and its apoform began long ago. In the literature, there is an example of functional structuring of apoCyt c due to the interaction of its initially unfolded chains with the negatively charged surface of micelles. First, the protein forms helices, then it is embedded into the membrane in this helical state and becomes able to bind the heme [152]. The study of the conformational state of apo- and holoCyt c in the presence of various membranes as well as the mechanism of interaction of these proteins with membranes was carried out in Pinheiro’s group for several years [153-158]. In the first papers of this series, they studied the structure and dynamics of the PhL bilayer, which contains bound Cyt c, using one-dimensional 31P-NMR [153]. It was shown that the binding of Cyt c with the membrane is a result of both destabilization of the protein molecule and disturbance of its tertiary structure. In its binding with the membrane, electrostatic interactions play an important role, since positively charged Cyt c interacts with negatively charged lipid heads, particularly cardiolipin, which is a major phospholipid of the inner mitochondrial membrane. Consequently, the specific interaction of the protein with the membrane depends on the type of PhL of which the bilayer is composed. The interaction of Cyt c to membranes was also studied by fluorescence and absorption spectroscopy using the stopped-flow technique and NMR [154, 158]. Following this work, the authors proposed a kinetic mechanism of Cyt c unfolding near the membrane surface. It was found that the initial binding of the protein to the membrane occurs via electrostatic interactions of positively charged groups of the protein with the negatively charged PhL head groups, after which an intermediate with structure similar to that of the native protein is formed. The next step is the disturbance of the tertiary structure and transition into a denatured state with helical secondary structure, again similar to the native protein structure. The kinetic studies were continued in investigations of the folding of apoCyt c in the presence of lipid micelles [155, 156]. As mentioned above, apoCyt c is the precursor of Cyt c, which functions in the intermembrane space of mitochondria. ApoCyt c is synthesized in the cytoplasm, and then it is translocated through the outer membrane of mitochondria and binds the heme in the intermembrane space. Using fluorescence spectroscopy and CD in the far-UV region, the authors described a possible mechanism of translocation of apoCyt c through the outer membrane of mitochondria. It was shown that the formation of the helical structure precedes the protein interaction with the membrane and its penetration into it [155].
Relation between protein–membrane interactions and human diseases. A key event in the origin of prion diseases is the conversion of the normal cellular form of prion protein (PrP) into the toxic form, which is prone to formation of fibrils and plaques. A cause of protein misfolding could be the presence of membranes. It was shown experimentally that the β-structural form of the recombinant prion protein binds to anionic vesicles at pH 7.0, and much better at pH 5.0 [159]. This preserves a major part of the β-structure and the disordered regions. β-PrP forms diffuse aggregates upon binding to the membrane at pH 5.0 with the emergence of protofibrils. Moreover, denaturation of the PrP β-form occurs in the presence of planar membranes containing a mixture of DPPC, cholesterol, and sphingomyelin, with the subsequent formation of fibrils. The PrP α-helical form interacts with three types of membranes: negatively charged membrane, membrane containing neutral phospholipids, and planar membrane consisting of phosphatidylcholine (PC), sphingomyelin, and cholesterol [160]. Stronger binding occurs with the negatively charged membrane at pH 5.0, as compared to pH 7.0, due to both protonation of His and an increase in the charge of the protein (from +4 to +9), i.e. in this system electrostatic interactions play a key role in the binding.
The protein α-synuclein (α-Syn) might be a precursor in the formation of plaques in Alzheimer’s and Parkinson’s diseases. α-Syn is a small soluble protein that has a disordered secondary structure in aqueous solution. It was shown that α-Syn is able to bind to negatively charged phospholipid vesicles [161]. The disordered secondary structure of the protein is stabilized on its interaction with the membrane, and segments with helical structure appear. Furthermore, it was found that the removal of sterol from the membranes (which makes them more fluid) reduces the binding of α-Syn to the cytoplasmic membrane, leading to amplified vesicle association, and thus resulting in greater protein toxicity. Thus, a higher concentration of sterol in the plasma membrane could prevent development of synucleinopathy. This effect requires further investigation in connection with the widespread use of statins and their possible impact on membrane-bound proteins [162].
The role of PS-containing membranes and factor V(a) in the activation of prothrombin is discussed in [144]. It was found that both factors approximately equally accelerate this process, but PS-containing membranes significantly increase the advantage of the activation pathway via formation of one of the intermediates, mesothrombin, at a certain concentration of the membranes.
The τ-protein is capable of forming neurofibrils and plaques [163]. It was suggested that places of fibril and plaque formation, which contribute to such transformation of the protein in the cell, may contain negatively charged membranes.
The ability of membranes to influence protein functions was investigated for the antimicrobial polypeptide cecropin A, for which all stages of interaction (approaching of the polypeptide to the membrane, interaction with its surface, and penetration into the membrane) were investigated. It was found that the polypeptide acquires a well-defined secondary structure at the membrane surface, oriented parallel to its surface. Although the polypeptide immersed in the bilayer membrane increases slightly in size, its structure and orientation remain unchanged [164].
Interaction of amylin hormone with a membrane promotes the formation of pre-amyloid aggregates, the latter leading to the formation of amyloid fibrils in type 2 diabetes [165].
The main regulator of cell growth and metabolism, as well as cytoskeletal reorganization of TOR (Ser/Thr kinase target of rapamycin), undergoes a significant conformational change both in the presence of phospholipid membranes containing the regulator phosphatidic acid (PA) and without it. Dysregulation at TOR-signaling transfer affects the change in metabolism and causes neurological diseases. It was found that the deficiency of glucose leads to a decrease in intracellular pH from ~7.5 to ~6.0, which can promote changes in the protein structure [166].
Phospholipid binding modules are found in intracellular proteins such as kinases. These modules play a crucial role in the regulation of many protein kinases depending on their localization. For many of them, their membrane localization depends on the presence of PS phospholipids. Using X-ray analysis, for many modules structurally conserved binding sites with anionic phospholipids were identified. In this case, kinases bind other regulators, similar to septins, only when bound to the membrane surface. These facts are important for understanding defects in the action of kinases, so far as it is associated with various diseases such as Alzheimer’s disease, cancer, and autism [167].
The lipid domains play an important role in maintaining the myelin structure during transmission of nerve impulses. These domains regulate adsorption of MBP (myelin basic protein) on the membrane and the interaction with the lipid bilayers of myelin, which are in close contact. These interactions create conditions for an environment with a low dielectric constant through compact bilayers, which is essential for efficient nerve pulse propagation. Any disturbance of the structure of the myelin sheath and interactions within the system leads to neurological diseases, the most common being multiple sclerosis [166-168].
The examples briefly described above show that protein–membrane interactions are very important in the initiation of various diseases. Progress in this direction will help to find new ways to prevent their development.
New directions. In recent years, interest in studies of lactalbumin has renewed with regard to its ability to form a complex with oleic acid. This molecular complex is able to overcome twice the membrane barrier and destroy the DNA of cancer cells [107, 169-172]. Lactalbumin forms a complex only in the calcium-free form having a partially unfolded conformation like the MG state, and its modified form is able to bind with the membrane, both cellular and mitochondrial.
Increasing attention has been focused recently on what is the possible influence of cell membranes on proteins that are operating near or on their surface. This problem is important for studying the folding of integral membrane proteins and their penetration into the membrane [107, 173]. Such outer membrane proteins as OmpA [107] and OmpG [174] are synthesized as globular precursors, which undergo a conformational transition to the MG state near the membrane surface. Then, this altered state interacts with the membrane leading to penetration of this already intermediate structure into the membrane. However, the factors influencing the process of protein–membrane interactions are still not sufficiently understood.
Interaction of proteins with membranes is also evident in the choice of ways for protein delivery into cells [175]. Membrane proteins continue to be targets for the delivery of drugs into the cell, because they allow communication between neighboring cells and are involved in the transport of ions and substances through the cell membrane.
There is evidence of this in the literature. Membranes may have a regulatory effect. Delivery of transmitters requires fusion of synaptic vesicles with the plasma membrane. This is achieved by structuring under the influence of the membrane of the disordered cytoplasmic domain of synaptobrevin upon Ca2+-dependent formation of a SNARE-complex causing the formation of a tetra-helical complex. This complex combines three components of the SNARE-complex (syntaxin 1A, SNAP-25, and synaptobrevin-2) [176-178], where syntaxin and synaptobrevin are bound to the membrane by their C-terminal fragments. However, it was not clear how syntaxin is clustered on the membrane surface. The authors of [179] revealed that this fusion is facilitated by electrostatic interactions with the anionic lipid phosphatidylinositol-4,5-biphosphate (PIP2), which forms an extensive cluster that is necessary for the sorption of syntaxin. This work focuses on the fact that the electrostatic interaction between the protein and phospholipids may lead to the formation of microdomains of the membrane irrespective of other factors.
It was also noted that the partially denatured state of the acidic fibroblast growth factor interacts with phospholipid membranes, which is not observed for the native protein [180].
There is another aspect to be considered when analyzing the changes in the structure of the proteins upon their interaction with the membrane. It is the targeted delivery of proteins (such as α-interferon, insulin, or growth hormone) in the body by their oral intake. For successful solutions of this problem, it is necessary to take into account the biochemical and physiological processes that determine membrane permeability, transfer through the gastrointestinal tract, and the interaction with mucous membranes. For this purpose, special carriers have to be designed that are resistant to a certain type of actions (e.g. pH 1.0 in the stomach), but usually it is a compound that can cause changes in the structure of proteins. Often, within these carriers, the transported proteins are in a state similar to the MG state in solution [175]. Most often, the place of delivery is the small intestine, where the conditions for the proteins are far from native. The presence of a branched membrane surface [181] and the presence of aromatic bile acids create conditions for destabilizing the structure of proteins and their transition to an intermediate state. This creates a problem for the delivery and preservation of physiologically important properties of protein compounds [182-184].
EFFECT OF DIFFERENT TYPES OF PHOSPHOLIPID MEMBRANES ON PROTEIN
STRUCTURE
In the cell, there are a variety of membranes. They have highly dynamic nature with regard to their composition, shape, and packing [12, 13, 95]. Furthermore, for processes of apoptosis, differentiation, and development the cell must be able to reduce the size of the organelles and to create them again, i.e. during its life the cell uses different types of membranes depending on the momentary needs. On the other hand, the effect of different kinds of membrane on proteins must be different. Figure 7 schematically shows the most common types of phospholipid membranes used in vitro. This is large bilayer vesicles (LUV), whose diameter is 1000-2000 Å, and giant vesicles are still larger. Frequently small bilayer vesicles (SUV) are used whose dimensions are 300-500 Å. A micelle can be formed at low concentrations of PhL. These vesicles allow simulating both flat membrane and those similar in shape to a sphere or the rounded part of an ellipsoid observed in vivo.
Fig. 7. Diversity of phospholipid membranes. Fragments of (a) large bilayer membranes (LUV), (b) small bilayer membranes (SUV), (c) micelles of lysophospholipids (Micelles), and (d) miscellaneous proteins on a comparable scale are presented. Different kinds of membranes are recognized by curvature, distance between phosphate groups, and accessibility of the hydrophobic lipid region to the aqueous medium.
The choice of the form of vesicles and their constituent PhL in vitro is determined by the goal being pursued. Therefore, most often proteins are studied using only one type of vesicles. However, the two proteins – apoMb and Cyt c – were studied in the presence of different types of PhL membranes, and therefore it is possible to compare the results.
The case of apoMb. It was shown [61] that in the presence of anionic –SUV (molar ratio of PhL/Pr = 25 : 1) apoMb structure undergoes a conformational change at neutral pH 7.2 and 6.2. On the other hand, in [138] it was found that in the presence of anionic –LUV (molar ratio of PhL/Pr = 300 : 1) the interaction of apoMb with the membrane occurs only at pH 5.5. Moreover, in 1984 it was reported [43] that stabilization of –SUV with cholesterol slows hemin release 2-fold. The protein–membrane interaction is also influenced by the concentration of negatively charged PG phospholipid. The binding efficiency decreases with decreasing concentration of PG in mixed vesicles. At the same time, the rate of membrane–protein interaction slows, and the stability of the protein structure has a noticeable effect as well [65]. Additionally, POPG –SUV can unfold native apoMb and fold the protein unfolded by urea.
The case of Cyt c. It has been shown for this protein that small vesicles (–SUV), composed of POPG and DOPS, can lead to denaturation of Cyt c [152]. Collapsing of apoCyt c in the presence of DPPG –SUV was studied by FTIR and H/D exchange [185, 186]. In the presence of zwitterionic POPC SUV, no effect of membranes on the protein structure could be detected [185]. However, if lysoPhL were taken instead of PhL, a small effect of membranes on the protein structure was observed, leading to the binding and structuring of approximately 60% of the molecules [185, 186].
A special case is the anionic micelles (–MIC) of lysoPG. Using –MIC lysoPG leads to folding of unstructured apoCyt c into the helical state, reaching almost 100% helicity of the native holoCyt c in solution [150, 185, 186]. In this case the helices were arranged perpendicularly to the membrane surface. But more striking is that denaturation of stable holoCyt c was observed by CD, absorption in the Soret band, and fluorescence methods even in the presence of zwitterionic MIC lysoPC [155], which is not observed for PC vesicles.
Analyzing these data, we can say that the difference between the membranes used is only in the packing of phospholipids. Large vesicles can have sufficiently flat areas, where PhL have dense packing, and only the phosphate heads are exposed to water. Therefore, the effect will depend mainly on the charge of these heads, and the low dielectric constant of the lipid part will have little effect. Another case is small vesicles and to a greater extent micelles. In these membranes, the PhL packing is not very dense due to significant curvature of the surface. Therefore, the influence of the hydrophobic part is more important, and this will enhance the electrostatic effect of the phosphate heads. Therefore, the denaturing effect will be more significant, as observed in the above examples.
CONCLUSION
The abovementioned experimental data reliably confirm the hypothesis [3, 94] that the membrane surface affects protein structure due to increased electrostatic interactions near its surface, namely due to the combined action of the local lowering of pH and the dielectric constant of the medium near the membrane surface. This indicates that the membrane surface can serve as a moderately denaturing agent in the cell.
The review of the existing literature data and the experimental work of the authors emphasize the requirement to take into account the possible influence or direct interaction of proteins and membranes under certain conditions. This may be important when studying the behavior of proteins near a membrane, or the folding of membrane proteins, or the delivery of protein drugs. This may also be important when choosing the ways of drug delivery in the body. In the analysis of these problems, one should pay attention to detailed study of protein–lipid interactions that have been described exhaustively in recently published reviews [187-189]. Conversion of membranes depends on their interaction with proteins and includes changing of organelle forms [190] as well as deformation of vesicles during their transport [191-193]. Taking into account the role of the high concentration of proteins in the cell, contributing to a change in the curvature of the membranes [194, 195], all of the abovementioned emphasizes the importance of the topics discussed in this review.
The authors are grateful to T. B. Kuvshinkina for the help in preparing this article for publication.
This work was supported by the Russian Scientific Foundation (grant No. 14-24-00157).
REFERENCES
1.Dolgikh, D. A., Gilmanshin, R. I., Brazhnikov, E.
V., Bychkova, V. E., Semisotnov, G. V., Venyaminov, S. Yu., and
Ptitsyn, O. B. (1981) α-Lactalbumin: compact state with
fluctuating tertiary structure? FEBS Lett., 136,
311-315.
2.Dolgikh, D. A., Abaturov, L. V., Bolotina, I. A.,
Brazhnikov, E. V., Bushuev, V. N., Bychkova, V. E., Gilmanshin, R. I.,
Lebedev, Yu. O., Semisotnov, G. V., Tiktopulo, E. I., and Ptitsyn, O.
B. (1985) Compact state of a protein molecule with pronounced
small-scale mobility: bovine alpha-lactalbumin, Eur. Biophys.
J., 13, 109-121.
3.Bychkova, V. E., and Ptitsyn, O. B. (1993) The
molten globule in vitro and in vivo, Chemtracts
– Biochem. Mol. Biol., 4, 133-163.
4.Bychkova, V. E., and Ptitsyn, O. B. (1993) The
state of molten globules of protein molecules is more quickly becoming
a rule, rather than an exception, Biofizika, 38,
58-66.
5.Ptitsyn, O. B. (1995) Molten globule and protein
folding, Adv. Protein Chem., 47, 83-229.
6.Endo, T., and Schatz, G. (1988) Latent membrane
perturbation activity of a mitochondrial precursor protein is exposed
by unfolding, EMBO J., 7, 1153-1158.
7.Eilers, M., Endo, T., and Schatz, G. (1989)
Adriamycin, a drug interacting with acidic phospholipids, blocks import
of precursor proteins by isolated yeast mitochondria, J. Biol.
Chem., 264, 2945-2950.
8.Endo, T., Eilers, M., and Schatz, G. (1989) Binding
of a tightly folded artificial mitochondrial precursor protein to the
mitochondrial outer-membrane involves a lipid-mediated conformational
change, J. Biol. Chem., 264, 2951-2956.
9.Van der Goot, F. G., Gonzales-Manas, J. M., Lakey,
J. H., and Pattus, F. (1991) A molten-globule membrane-insertion
intermediate of the pore-forming domain of colicin A, Nature,
354, 408-410.
10.Van der Goot, F. G., Lakey, J. H., and Pattus, F.
(1992) The molten globule intermediate for protein insertion or
translocation through membranes, Trends Cell Biol., 2,
343-348.
11.Schmitt, S., Prokisch, H., Schlunck, T., Camp II,
D. G., Ahting, U., Waizenegger, T., Scharfe, C., Meitinger, T., Imhof,
A., Neupert, W., Oefner, P. J., and Rapaport, D. (2006) Proteome
analysis of mitochondrial outer membrane from Neurospora crassa,
Proteomics, 6, 72-80.
12.Engelman, D. M. (2005) Membranes are more mosaic,
than fluid, Nature, 438, 578-580.
13.Von Meer, G., Voelker, D. R., and Feigenson, G.
W. (2008) Membrane lipids: where they are and how they behave,
Nature Rev. Mol. Cell Biol., 9, 112-124.
14.Landau, L. D., and Lifshits, E. M. (1982)
Theoretical Physics, Vol. 8. Electrodynamics of Continuous
Media [in Russian], Nauka, Moscow, p. 60.
15.Eisenberg, M., Gresalfi, T., Riccio, T., and
McLaughlin, S. (1979) Absorption of monovalent cations to bilayer
membranes containing negative phospholipids, Biochemistry,
18, 5213-5223.
16.Prats, M., Teissie, J., and Toccane, J. F. (1986)
Lateral proton conduction at lipid–water interfaces and its
implications for the chemiosmotic-coupling hypothesis, Nature,
322, 756-758.
17.Ptitsyn, O. B. (1987) Protein folding: hypotheses
and experiments, J. Prot. Chem., 6, 273-293.
18.Ptitsyn, O. B. (1992) The molten globule state,
in Protein Folding (Creighton, T. E., ed.) New York, pp.
243-299.
19.Skulachev, V. P. (1988) Biomembrane
Energetics, Spring-Verlag, Berlin-Heidelberg.
20.Finkelstein, A. V., and Ptitsyn, O. B. (2002) in
Protein Physics, Academic Press, An Imprint of Elsevier Science,
Amsterdam-Boston-London-New York-Oxford-Paris-San Diego-San
Francisco-Singapore-Sydney-Tokyo, pp. 1-380.
21.Winitski, A. P., McLaughlin, A. C., McDaniel, R.
V., Eisenberg, M., and McLaughlin, S. (1986) An experimental test of
the discreteness-of-charge effect in positive and negative lipid
bilayers, Biochemistry, 25, 8206-8214.
22.Wilkinson, K. D., and Mayer, A. N. (1986)
Alcohol-induced conformational changes of ubiquitin, Arch. Biochem.
Biophys., 250, 390-399.
23.Dufour, E., Bertrand-Harb, C., and Haertle, T.
(1993) Reversible effects of medium dielectric constant on structural
transformation of β-lactoglobulin and its retinol binding,
Biopolymers, 33, 589-598.
24.Bychkova, V. E., Dujsekina, A. E., Klenin, S. I.,
Tiktopulo, E. I., Uversky, V. N., and Ptitsyn, O. B. (1996) Molten
globule-like state of cytochrome c under conditions simulating
those near the membrane surface, Biochemistry, 35,
6058-6063.
25.Dujsekina, A. E. (1998) Conformations of
Proteins in Mediums with Moderately Low Values of a Dielectric Constant
and pH: PhD Thesis [in Russian], Pushchino.
26.Poulos, T. L., and Mauk, A. G. (1983) Models for
complexes formed between cytochrome b5 and subunits
of methemoglobin, J. Biol. Chem., 258, 7369-7373.
27.Livingston, D. J., McLachlan, S. J., La Mar, G.
N., and Brown, W. D. (1985) Myoglobin–cytochrome
b5 interactions and the kinetic mechanism of
metmyoglobin reductase, J. Biol. Chem., 260,
15699-15707.
28.Spatz, L., and Strittmatter, P. (1971) A form of
cytochrome b5 that contains an additional hydrophobic
sequence of 40 amino acid residues, Proc. Natl. Acad. Sci. USA,
68, 1042-1046.
29.Wang, Z.-Q., Wang, Y-H., Quan, W., Wang, H.-H.,
Chunyu, L.-J., Xie, Yi, and Huang, Z.-X. (1999) Methanol-induced
unfolding and refolding of cytochrome b5 and its P40V
mutant monitored by UV-visible, CD, and fluorescence spectra, J.
Prot. Chem., 18, 547-555.
30.Basova, L. V. (2004) Influence of a Membrane
Surface on the Conformational State of Globular Proteins: PhD
Thesis [in Russian], Pushchino.
31.Kamatari, Yu. O., Ohji, S., Konno, T., Seki, Ya.,
Soda, K., Kataoka, M., and Akasaka, K. (1999) The compact and expanded
denatured conformations of apomyoglobin in the methanol–water
solvent, Protein Sci., 8, 873-882.
32.Bychkova, V. E., Dujsekina, A. E., Ptitsyn, O.
B., Fantuzzi, A., and Rossi, G. L. (1998) Release of retinol and
denaturation of its plasma carrier, retinol-binding protein, Fold.
Des., 3, 285-291.
33.Bychkova, V. E., Berni, R., Rossi, G. L.,
Kutyshenko, V. P., and Ptitsyn, O. B. (1992) Retinol-binding protein is
in the molten globule state at low pH, Biochemistry, 31,
7566-7571.
34.Dujsekina, A. E., Bychkova, V. E., Fantuzzi, A.,
Rossi, G. L., and Ptitsyn, O. B. (1998) Release of hydrophobic ligand
from retinol-binding protein under conditions mimicked the membrane
field, Mol. Biol. (Moscow), 32, 133-140.
35.Torta, F., Dyuysekina, A. E., Cavazzini, D.,
Fantuzzi, A., Bychkova, V. E., and Rossi, G. L. (2004) Solvent-induced
ligand dissociation and conformational states of cellular
retinol-binding protein type I, Biochim. Biophys. Acta,
1703, 21-29.
36.Stockman, B. J., Euvrard, A., and Scahill, T. A.
(1993) Heteronuclear three-dimensional NMR spectroscopy of a partially
denatured protein: the A-state of human ubiquitin, J. Biomol.
NMR, 3, 285-296.
37.Wang, S. X., Sun, Y. T., and Sui, S. F. (2000)
Membrane-induced conformational changes in human apolipoprotein H,
Biochem. J., 348, Pt. 1, 103-106.
38.Clement-Collin, V., Leroy, A., Monteilhet, C.,
and Aggerbeck, L. P. (1999) Mimicking lipid-binding-induced
conformational changes in the human apolipoprotein E N-terminal
receptor binding domain effects of low pH and propanol, Eur. J.
Biochem., 264, 358-368.
39.Babu, K. R., and Douglas, D. J. (2000)
Methanol-induced conformation of myoglobin at pH 4.0,
Biochemistry, 39, 14702-14710.
40.Mozsolits, H., Unabia, S., Ahmad, A., Morton, C.
J., Thomas, W. G., and Aguilar, M. I. (2002) Electrostatic and
hydrophobic forces tether the proximal region of the angiotensin II
receptor (AT1A) carboxyl terminus to anionic lipids,
Biochemistry, 41, 7830-7840.
41.Bychkova, V. E. (1997) On functional role of
non-native proteins, Uspekhi Biol. Khim., 37, 49-99.
42.Perutz, M. F. (1989) Myoglobin and hemoglobin:
role of distal residues in reactions with haem ligands, Trends
Biochem. Sci., 14, 42-44.
43.Cannon, J. B., Kuo, F. S., Pasternack, R. F.,
Wong, N. M., and Muller-Eberhard, U. (1984) Kinetics of the interaction
of hemin liposomes with heme binding proteins, Biochemistry,
23, 3715-3721.
44.Hughson, F. M., Wright, P. E., and Baldwin, R. L.
(1990) Structural characterization of a partly folded apomyoglobin
intermediate, Science, 249, 1544-1548.
45.Griko, Yu. V., Privalov, P. L., Venyaminov, S.
Yu., and Kutyshenko, V. P. (1988) Thermodynamic study of the
apomyoglobin structure, J. Mol. Biol., 202, 127-138.
46.Postnikova, G. B., Komarov, Y. E., and Yumakova,
E. M. (1991) Fluorescence study of the conformational properties of
myoglobin structure. 1. pH-dependent changes of tryptophanyl
fluorescence in intact and chemically modified sperm whale
apomyoglobins, Eur. J. Biochem., 198, 223-232.
47.Jennings, P., and Wright, P. E. (1993) Formation
of a molten globule intermediate early in the kinetic folding pathway
of apomyoglobin, Science, 262, 892-896.
48.Gast, K., Damaschun, H., Misselwitz, R.,
Mueller-Frohne, M., Zirwer, D., and Damaschun, G. (1994) Compactness of
protein molten globules: temperature-induced structural changes of the
apomyoglobin folding intermediates, Eur. Biophys. J., 23,
297-305.
49.Nishii, I., Kataoka, M., and Goto, Y. (1995)
Thermodynamic stability of the molten globule states of apomyoglobin,
J. Mol. Biol., 250, 223-238.
50.Postnikova, G. B. (1999) Fluorescence study of
conformational transitions in the myoglobin structure, Biochemistry
(Moscow), 64, 267-286.
51.Nishimura, C., Dyson, H. J., and Wright, P. E.
(2006) Identification of native and non-native structure in kinetic
folding intermediates of apomyoglobin, J. Mol. Biol.,
355, 139-156.
52.Samatova, E. N., Katina, N. S., Balobanov, V. A.,
Melnik, B. S., Dolgikh, D. A., Bychkova, V. E., and Finkelstein, A. V.
(2009) How strong are side chain interactions in the folding
intermediate? Protein Sci., 18, 2152-2159.
53.Samatova, E. N., Melnik, B. S., Balobanov, V. A.,
Katina, N. S., Dolgikh, D. A., Semisotnov, G. V., Finkelstein, A. V.,
and Bychkova, V. E. (2010) Folding intermediate and folding nucleus for
I/N and U/I/N transitions in apomyoglobin: contributions by conserved
and non-conserved residues, Biophys. J., 98,
1694-1702.
54.Antonini, E., and Brunori, M. (1971) Hemoglobin
and myoglobin in their reactions with ligands, in Frontiers of
Biology (Neuberger, A., and Tatum, E. L., eds.) Vol. 21,
North-Holland Publishing Company, Amsterdam-London.
55.Bismuto, E., Colonna, G., Savy, F., and Irace, G.
(1985) Myoglobin structure and regulation of solvent accessibility of
heme pocket, Int. J. Pept. Protein Res., 26, 195-207.
56.Privalov, P. L., Griko, Yu. V., Venyaminov, S.
Yu., and Kutyshenko, V. P. (1986) Cold denaturation of myoglobin, J.
Mol. Biol., 190, 487-498.
57.Postnikova, G. B., Komarov, Y. E., and Yumakova,
E. M. (1991) Fluorescence study of the conformational properties of
myoglobin structure. 2. pH- and ligand-induced conformational changes
in ferric- and ferrous myoglobins, Eur. J. Biochem., 198,
233-239.
58.Griko, Y. V., and Privalov, P. L. (1994)
Thermodynamic puzzle of apomyoglobin unfolding, J. Mol. Biol.,
235, 1318-1325.
59.Cocco, M. J., Kao, Y. H., and Lecomte, J. T.
(1996) The native state of apomyoglobin described by proton NMR
spectroscopy: the A-B-G-H interface of wild-type sperm whale
apomyoglobin, Proteins, 25, 267-285.
60.Basova, L. V., Tiktopulo, E. I., Kutyshenko, V.
P., Klenin, S. I., Balobanov, V. A., and Bychkova, V. E. (2014)
Membrane-induced changes in the holomyoglobin tertiary structure:
interplay with function, Eur. Biophys. J., 43,
317-329.
61.Basova, L. V., Tiktopulo, E. I., Kashparov, I.
A., and Bychkova, V. E. (2004) The conformational state of apomyoglobin
in the presence of phospholipid vesicles at neutral pH, Mol. Biol.
(Moscow), 38, 323-332.
62.Vassilenko, K. S., and Uversky, V. N. (2002)
Native-like secondary structure of molten globules, Biochim.
Biophys. Acta, 1564, 168-177.
63.Tsvetkov, V. N., and Klenin, S. I. (1953)
Diffusion coefficients of polystyrene fractions in dichloroethane,
Dokl. Akad. Nauk SSSR, 88, 49-52.
64.Privalov, P. L. (1979) Stability of proteins:
small globular proteins, Adv. Protein Chem., 33,
167-241.
65.Balobanov, V. A., Ilyina, N. B., Katina, N. S.,
Kashparov, I. A., Dolgikh, D. A., and Bychkova, V. E. (2010) Kinetics
of interaction between apomyoglobin and phospholipid membrane, Mol.
Biol. (Moscow), 44, 708-717.
66.Postnikova, G. B., and Tselikova, S. V. (2005)
Myoglobin and mitochondria: kinetics of oxymyoglobin deoxygenation in
mitochondria suspension, Biofizika, 50, 297-306.
67.Postnikova, G. B., and Shekhovtsova, E. A. (2013)
Effect of artificial and natural phospholipid membranes on rate of
sperm whale oxymyoglobin autooxidation, Biochemistry (Moscow),
78, 267-272.
68.Funk, W. D., Lo, T. P., Mauk, M. R., Brayer, C.
D., MacGillivray, R. T., and Mauk, A. G. (1990) Mutagenic,
electrochemical, and crystallographic investigation of the cytochrome
b5 oxidation-reduction equilibrium: involvement of
asparagine 57, serine 64, and heme propionate 7, Biochemistry,
29, 5500-5508.
69.Mitoma, J., and Ito, A. (1992) The
carboxy-terminal 10 amino acid residues of cytochrome
b5 are necessary for its targeting to the endoplasmic
reticulum, EMBO J., 11, 4197-4203.
70.Hultquist, D. E., and Passon, P. G. (1971)
Catalysis of methemoglobin reduction by erythrocyte cytochrome
b5 and cytochrome b5 reductase,
Nature New. Biol., 229, 252-254.
71.Kawano, M., Shirabe, K., Nagai, T., and
Takeshita, M. (1998) Role of carboxyl residues surrounding heme of
human cytochrome b5 in the electrostatic interaction
with NADH-cytochrome b5 reductase, Biochem.
Biophys. Res. Commun., 245, 666-669.
72.Hildebrandt, A. G., and Estabrook, R. W. (1972)
Evidence for the participation of cytochrome b5 in
hepatic microsomal mixed function oxidation reactions, Arch.
Biochem. Biophys., 143, 66-79.
73.Yamazaki, H., Johnson, W. W., Ueng, Y-F.,
Shimada, T., and Guengerich, F. P. (1996) Lack of electron transfer
from cytochrome b5 in stimulation of catalytic
activities of cytochrome P450 3A4. Characterization of a reconstituted
cytochrome P450 3A4/NADPH-cytochrome P450 reductase system and studies
with apocytochrome b5, J. Biol. Chem.,
271, 27438-27444.
74.Strittmatter, P., Spatz, L., Corcoran, D.,
Rogers, M. J., Setlow, B., and Redline, R. (1974) Purification and
properties of rat liver microsomal stearyl coenzyme A desaturase,
Proc. Natl. Acad. Sci. USA, 71, 4565-4569.
75.Keyes, S. R., and Cinti, D. L. (1980) Biochemical
properties of cytochrome b5-dependent microsomal
fatty acid elongation and identification of products, J. Biol.
Chem., 255, 11357-11364.
76.Kozutsumi, Y., Kawano, T., Yamakawa, T., and
Suruki, A. (1998) Participation of cytochrome b5 in
CMP-N-acetylneuraminic acid hydroxylation in mouse liver cytosol, J.
Biochem., 108, 704-706.
77.Livingston, D. J., McLachlan, S. J., La Mar, G.
N., and Brown, W. D. (1985) Myoglobin–cytochrome
b5 interactions and the kinetic mechanism of
metmyoglobin reductase, J. Biol. Chem., 260,
15699-15707.
78.Gacon, G., Lostanlen, D., Labie, D., and Kaplan,
J.-C. (1980) Interaction between cytochrome b5 and
hemoglobin: involvement of β66 (E10) and β95 (FG2) lysyl
residues of hemoglobin, Proc. Natl. Acad. Sci. USA, 77,
1917-1921.
79.Usanov, S. A., Bendzko, P., Pfeil, W., Janig, G.
R., and Ruckpaul, K. (1983) The role of the hydrophobic fragment of
cytochrome b5 in interaction with cytochrome P-450,
Bioorg. Khim., 9, 450-461.
80.Mathews, F. S., Levine, M., and Argos, P. (1972)
Three-dimensional Fourier synthesis of calf liver cytochrome
b5 at 2.8 Å resolution, J. Mol. Biol.,
64, 449-464.
81.Veitch, N. C., Concar, D. W., Williams, R. J. P.,
and Whitford, D. (1988) Investigation of the solution structures and
mobility of oxidized and reduced cytochrome b5 by 2D
NMR spectroscopy, FEBS Lett., 238, 49-55.
82.Whitford, D. (1992) The identification of cation
binding domains on the surface of microsomal cytochrome
b5 using 1H NMR paramagnetic difference
spectroscopy, Eur. J. Biochem., 203, 211-223.
83.Guiles, R. D., Basus, V. J., Kuntz, I., and
Waskell, L. (1992) Sequence specific 1H and 15N
resonance assignments for both equilibrium forms of the soluble heme
binding domain of rat ferrocytochrome b5,
Biochemistry, 31, 11365-11375.
84.Muskett, F. W., Kelly, G. P., and Whitford, D.
(1996) The solution structure of bovine ferricytochrome
b5 determined using heteronuclear NMR methods, J.
Mol. Biol., 258, 172-189.
85.Mauk, A. G., Mauk, M. R., Moore, G. R., and
Northrup, S. H. (1995) Experimental and theoretical analysis of the
interaction between cytochrome c and cytochrome
b5, J. Bioenerg. Biomembr., 27,
311-330.
86.Stayton, P. S., Fisher, M. T., and Sligar, S. G.
(1988) Determination of cytochrome b5 association
reactions. Characterization of metmyoglobin and cytochrome P-450
binding to genetically engineered cytochrome b5,
J. Biol. Chem., 263, 13544-13548.
87.Stayton, P. S., Poulos, T. L., and Sligar, S. G.
(1989) Putidaredoxin competitively inhibits cytochrome
b5–cytochrome P-450cam association:
a proposed molecular model for a cytochrome P-450cam
electron-transfer complex, Biochemistry, 28,
8201-8205.
88.Lederer, F. (1994) The cytochrome
b5-fold: an adaptable module, Biochimie,
76, 674-692.
89.Basova, L. V., Ilyina, N. B., Vasilenko, K. S.,
Tiktopulo, E. I., and Bychkova, V. E. (2002) Conformational states of
the water-soluble fragment of cytochrome b5: pH-induced
denaturation, Mol. Biol. (Moscow), 36, 891-900.
90.Acharya, K. R., Ren, J. S., Stuart, D. I.,
Phillips, D. C., and Fenna, R. E. (1991) Crystal structure of human
alpha-lactalbumin at 1.7 Å resolution, J. Mol. Biol.,
221, 571-581.
91.Kuwajima, K., Nitta, K., Yoneyama, M., and Sugai,
S. (1976) Three-state denaturation of alpha-lactalbumin by guanidine
chloride, J. Mol. Biol., 106, 359-373.
92.Permyakov, E. A., Yarmolenko, V. V.,
Kalinichenko, L. P., Morozova, L. A., and Burstein, E. A. (1981)
Calcium binding to alpha-lactalbumin: structural arrangement and
association constant evaluation by means of intrinsic fluorescence
changes, Biochem. Biophys. Res. Commun., 100,
191-197.
93.Chenal, A., Vernier, G., Savarin, P., Bushmarina,
N. A., Geze, A., Guillain, F., Gillet, D., and Forge, V. (2005)
Conformational states and thermodynamics of alpha-lactalbumin bound to
membranes: a case study of the effects of pH, calcium, lipid membrane
curvature and charge, J. Mol. Biol., 349, 890-905.
94.Bychkova, V. E., Pain, R. H., and Ptitsyn, O. B.
(1988) The “molten globule” state is involved in the
translocation of proteins across membranes? FEBS Lett.,
238, 231-234.
95.Bigay, J., and Bruno, A. (2012) Curvature, lipid
packing, and electrostatics of membrane organelles: defining cellular
territories in determining specificity, Dev. Cell, 23,
886-895.
96.Eilers, M., and Schatz, G. (1986) Binding of a
specific ligand inhibits import of a purified precursor protein into
mitochondria, Nature, 322, 228-232.
97.Eilers, M., and Schatz, G. (1988) Protein
unfolding and energetics of protein translocation across biological
membranes, Cell, 52, 481-483.
98.Vestweber, D., and Schatz, G. (1988) Point
mutations destabilizing a precursor protein enhance its
posttranslational import into mitochondria, EMBO J., 7,
1147-1151.
99.Glick, B., and Schatz, G. (1991) Import of
proteins into mitochondria, Annu. Rev. Genet., 25,
21-44.
100.Vestweber, D., and Schatz, G. (1988) A chimeric
mitochondrial precursor protein with internal disulfide bridges blocks
import of authentic precursors into mitochondria and allows
quantitation of import sites, J. Cell Biol., 107,
2037-2043.
101.Eilers, M., Hwang, S., and Schatz, G. (1988)
Unfolding and refolding of a purified precursor protein during import
into isolated mitochondria, EMBO J., 7, 1139-1145.
102.Huang, S., Murphy, S., and Matouschek, A.
(2000) Effect of the protein import machinery at the mitochondrial
surface on precursor stability, Proc. Natl. Acad. Sci. USA,
97, 12991-12996.
103.Hay, R., Bohni, P., and Gasser, S. (1984) How
mitochondria import proteins, Biochim. Biophys. Acta,
779, 65-87.
104.Randall, L. L., and Hardy, S. J. S. (1986)
Correlation of competence for export with lack of tertiary structure of
the mature species. A study in vivo of maltose-binding protein
from Escherichia coli, Cell, 46, 921-928.
105.Verner, K., and Schatz, G. (1988) Protein
translocation across membranes, Science, 241,
1307-1313.
106.Bochkareva, E. S., Lissin, N. M., and
Girshovich, A. S. (1988) Transient association of newly synthesized
unfolded proteins with the heat-shock GroEL protein, Nature,
336, 254-257.
107.Kleinschmidt, J. H., and Tamm, L. K. (1996)
Folding intermediates of a beta-barrel membrane protein. Kinetic
evidence for a multi-step membrane insertion mechanism,
Biochemistry, 35, 12993-13000.
108.Booth, P. J., and Curnow, P. (2009) Folding
scene investigation: membrane proteins, Curr. Opin. Struct.
Biol., 19, 8-13.
109.Schwartz, M. P., and Matouschek, A. (1999) The
dimensions of the protein import channels in the outer and inner
mitochondrial membranes, Proc. Natl. Acad. Sci. USA, 96,
13086-13090.
110.Diekert, K., de Kroon, A. I. P. M., Ahting, U.,
Niggermeyer, B., Neupert, W., de Kruijff, B., and Lill, R. (2001)
Apocytochrome c requires the TOM complex for translocation
across the mitochondrial outer membrane, EMBO J., 20,
5626-5635.
111.Prakash, S., and Matouschek, A. (2004) Protein
unfolding in the cell, Trends Biochem. Sci., 29,
593-600.
112.Bolliger, L., Junne, T., Schatz, G., and
Lithgow, T. (1995) Acidic receptor domains on both sides of the outer
membrane mediate translocation of precursor proteins into yeast
mitochondria, EMBO J., 14, 6318-6326.
113.Huang, S., Ratliff, K. S., and Matouschek, A.
(2002) Protein unfolding by the mitochondrial membrane potential,
Nat. Struct. Biol., 9, 301-307.
114.Cavard, D., Sauve, P., Heitz, F., Pattus, F.,
Martinez, C., Dijkman, R., and Lazdunski, C. (1988) Hydrodynamic
properties of colicin A. Existence of a high-affinity lipid-binding
site and oligomerization at acid pH, Eur. J. Biochem.,
172, 507-512.
115.Muga, A., Gonzalez-Manas, J. M., Lakey, J. H.,
Pattus, F., and Surewicz, W. K. (1993) pH-dependent stability and
membrane interaction of the pore-forming domain of colicin A, J.
Biol. Chem., 268, 1553-1557.
116.Lakey, J. H., Gonzalez-Manas, J. M., van der
Goot, F. G., and Pattus, F. (1992) The membrane insertion of colicins,
FEBS Lett., 307, 26-29.
117.Parker, M. W., Tucker, A. D., Tsernoglou, D.,
and Pattus, F. (1990) Insights into membrane insertion based on studies
of colicins, Trends Biochem. Sci., 15, 126-129.
118.Anderluh, G., and Lakey, J. H. (2008) Disparate
proteins use similar architectures to damage membranes, Trends
Biochem. Sci., 33, 482-490.
119.Merrill, A. R., Cohen, F. S., and Cramer, W. A.
(1990) On the nature of the structural change of the colicin-E1 channel
peptide necessary for its translocation competent state,
Biochemistry, 29, 5829-5836.
120.Li, J. D., Carroll, J., and Ellar, D. J. (1991)
Crystal structure of insecticidal delta-endotoxin from Bacillus
thuringiensis at 2.5-A resolution, Nature, 353,
815-821.
121.Zhao, J.-M., and London, E. (1988) Conformation
and model membrane interactions of diphtheria toxin fragment A,
J. Biol. Chem., 263, 15369-15377.
122.London, E. (1992) Diphtheria toxin: membrane
interaction and membrane translocation, Biochim. Biophys. Acta,
1113, 25-51.
123.Jiang, J. X., Abrams, F. S., and London, E. I.
(1991) Folding changes in membrane-inserted diphtheria toxin that may
play important roles in its translocation, Biochemistry,
30, 3857-3864.
124.Cabiaux, V., Brasseur, R., Wattiez, R.,
Falmagne, P., Ruysschart, J.-M., and Goormaghtigh, E. (1989) Secondary
structure of diphtheria toxin and its fragments interacting with acidic
liposomes studied by polarized infrared spectroscopy, J. Biol.
Chem., 264, 4928-4938.
125.Allured, V. S., Collier, R. J., Carroll, S. F.,
and McKay, D. B. (1986) Structure of exotoxin A of Pseudomonas
aeruginosa at 3.0-A resolution, Proc. Natl. Acad. Sci. USA,
83, 1320-1324.
126.Tsurudome, M., Gluck, R., Graf, R., Falchetto,
R., Schaller, U., and Brunner, J. (1992) Lipid interactions of the
hemagglutinin HA2 NH2-terminal segment during influenza
virus-induced membrane fusion, J. Biol. Chem., 267,
20225-20232.
127.Chenal, A., Prongidi-Fix, L., Perier, A.,
Aisenbrey, C., Vernier, G., Lambotte, S., Haertlein, M., Dauvergne, M.
T., Fragneto, G., Bechinger, B., Gillet, D., Forge, V., and Ferrand, M.
(2009) Deciphering membrane insertion of the diphtheria toxin T domain
by specular neutron reflectometry and solid-state NMR spectroscopy,
J. Mol. Biol., 391, 872-883.
128.Day, P. J., Pinheiro, T. J. T., Roberts, L. M.,
and Lord, J. M. (2002) Binding of ricin A-chain to negatively charged
phospholipids vesicles leads to protein structural changes and
destabilizes the lipid bilayer, Biochemistry, 41,
2836-2843.
129.Mayerhofer, P. U., Cook, J. P., Wahlman, J.,
Pinheiro, T. J. T., Moore, K. A. H., Lord, J. M., Johnson, A. E., and
Roberts, L. M. (2009) Ricin A chain insertion into endoplasmic
reticulum membranes is triggered by a temperature increase to
37oC, J. Biol. Chem., 284, 10232-10242.
130.Vecsey-Semjen, B., Moellby, R., and van der
Goot, F. G. (1996) Partial C-terminal unfolding is required for channel
formation by staphylococcal α-toxin, J. Biol. Chem.,
271, 8655-8660.
131.Karst, J. C., Baker, R., Devi, U., Swann, M.
J., Davi, M., Roser, S. J., Ladant, D., and Chenal, A. (2012)
Identification of a region that assists membrane insertion and
translocation of the catalytic domain of Bordetella pertussis
CyaA toxin, J. Biol. Chem., 287, 9200-9212.
132.Massey, S., Banerjee, T., Pande, A. H., Taylor,
M., Tatulian, S. A., and Teter, K. (2009) Stabilization of the tertiary
structure of the cholera toxin A1 subunit inhibits toxin dislocation
and cellular intoxication, J. Mol. Biol., 393,
1083-1096.
133.Taylor, M., Banerjee, T., Navarro-Garcia, F.,
Huerta, J., Massey, S., Burlingame, M., Pande, A. H., Tatulian, S. A.,
and Teter, K. (2011) A therapeutic chemical chaperone inhibits cholera
intoxication and unfolding/translocation of the cholera toxin A1
subunit, PLoS One, 6, e18825, doi:
10.1371/joural.pone.0018825.
134.Pande, A. H., Moe, D., Jamnadas, M., Tatulian,
S. A., and Teter, K. (2006) The pertussis toxin S1 is a thermally
unstable protein susceptible to degradation by the 20S proteasome,
Biochemistry, 45, 13734-13740.
135.Pande, A. H., Scaglione, P., Taylor, M., Nemec,
K. N., Tulhill, S., Moe, D., Holmes, R. K., Tatulian, S. A., and Teter,
K. (2007) Conformational instability of the cholera toxin A1
polypeptide, J. Mol. Biol., 374, 1114-1128.
136.Lesovoy, D. M., Bocharov, E. V., Lyukmanova, E.
N., Kosinsky, Y. A., Shulepko, M. A., Dolgikh, D. A., Kirpichnikov, M.
P., Efremov, R. G., and Arseniev, A. S. (2009) Specific membrane
binding of neurotoxin II can facilitate its delivery to acetyl choline
receptor, Biophys. J., 97, 2089-2097.
137.Dubovskii, P. V., Lesovoy, D. M., Dubinnyi, M.
A., Utkin, Y. N., and Arseniev, A. S. (2003) Interaction of the P-type
cardiotoxin with phospholipid membranes, Eur. J. Biochem.,
270, 2038-2046.
138.Kim, J. G., and Kim, H. M. (1989) Interaction
of alpha-lactalbumin with phospholipid vesicles as studied by
photoactivated hydrophobic labeling, Biochim. Biophys. Acta,
983, 1-8.
139.Halskau, O., Froystein, N. G., Muga, A., and
Martinez, A. (2002) The membrane-bound conformational of
α-lactalbumin studied by NMR-monitored 1H-exchange,
J. Mol. Biol., 321, 99-110.
140.Vernier, G., Chenal, A., Vitrac, H.,
Barumandzadhe, R., Montagner, C., and Forge, V. (2007) Interactions of
apomyoglobin with membranes: mechanism and effects on heme uptake,
Protein Sci., 16, 391-400.
141.Ahn, T., Kim, J-S., Lee, B-C., and Yun, C-H.
(2001) Effects of lipids on the interaction of SecA with model
membranes, Arch. Biochem. Biophys., 359, 14-20.
142.Pande, A. H., Tripathy, R. K., and Narkar, S.
A. (2009) Membrane surface charge modulates lipoprotein complex forming
capability of peptides derived from the C-terminal domain of
apolipoprotein E, Biochim. Biophys. Acta, 1788,
1366-1376.
143.Thorolfsson, M., Doskeland, A. P., Muga, A.,
and Martinez, A. (2002) The binding of tyrosine hydroxylase to anionic
lipid bilayers involves the N-terminal region of the enzyme, FEBS
Lett., 519, 221-226.
144.Weinreb, G. E., Mukhopadhyay, K., Majumder, R.,
and Lentz, B. R. (2003) Cooperative roles of factor V(a) and
phosphatidylserine-containing membranes as cofactors in prothrombin
activation, J. Biol. Chem., 278, 5679-5684.
145.Shin, I., Kreimer, D., Silman, I., and Weiner,
L. (1997) Membrane-promoted unfolding acetylcholinesterase: a possible
mechanism for insertion into the lipid bilayer, Proc. Natl. Acad.
Sci. USA, 94, 2848-2852.
146.Shin, I., Silman, I., Bon, C., and Weiner, L.
(1998) Liposome-catalyzed unfolding of acetylcholinesterase from
Bungarus fasciatus, Biochemistry, 37,
4310-4316.
147.Shin, I., Wachtel, E., Roth, E., Bon, C.,
Silman, I., and Weiner, L. (2002) Thermal denaturation of Bungarus
fasciatus acetylcholinesterase: is aggregation a driving force in
protein unfolding? Protein Sci., 11, 2022-2032.
148.Wang, Y., Grabowski, G. A., and Qi, X. (2003)
Phospholipid vesicle fusion induced by saposin C, Arch. Biochem.
Biophys., 415, 43-53.
149.Seddon, A. M., Lorch, M., Ces, O., Templer, R.
H., Macrae, F., and Booth, P. J. (2008) Phosphatidylglycerol lipids
enhance folding of an alpha helical membrane protein, J. Mol.
Biol., 380, 548-556.
150.Lin, W-C., Iversen, L., Tu, H-L., Rhodes, C.,
Christensen, S. M., Iwing, J. S., Hansen, S. D., Huang, W. Y. C., and
Groves, J. T. (2014) H-Ras forms dimers on membrane surfaces via a
protein–protein interface, Proc. Natl. Acad. Sci. USA,
111, 2996-3001.
151.Faini, M., Beck, R., Wieland, F. T., and
Briggs, J. A. G. (2013) Vesicle coats: structure, function, and general
principles of assembly, Trends Cell Biol., 23,
279-288.
152.De Jongh, H. H. J., Killian, J. A., and de
Kruijff, B. (1992) A water–lipid interface induces a highly
dynamic folded state in apocytochrome c and cytochrome c,
which may represent a common folding intermediate, Biochemistry,
31, 1636-1643.
153.Pinheiro, T. J. T., and Watts, A. (1994)
Resolution of individual lipids in mixed phospholipids membranes and
specific lipid–cytochrome c interactions by magic-angle
spinning solid-state phosphorus-31 NMR, Biochemistry, 33,
2459-2467.
154.Pinheiro, T. J. T., Elove, G. A., Watts, A.,
and Roder, H. (1997) Structural and kinetic description of cytochrome
c unfolding induced by the interaction with lipid vesicles,
Biochemistry, 36, 13122-13132.
155.Bryson, E. A., Rankin, S. E., Carey, M., Watts,
A., and Pinheiro, T. J. T. (1999) Folding of apocytochrome c in
lipid micelles: formation of alpha-helix precedes membrane insertion,
Biochemistry, 38, 9758-9767.
156.Rankin, S. E., Watts, A., Roder, H., and
Pinheiro, T. J. T. (1999) Folding of apocytochrome c induced by
the interaction with negatively charged lipid micelles proceeds via a
collapsed intermediate state, Protein Sci., 8,
381-393.
157.Sanghera, N., and Pinheiro, T. J. T. (2000)
Unfolding and refolding of cytochrome c driven by the
interaction with lipid micelles, Protein Sci., 9,
1194-1202.
158.Pinheiro, T. J. T., Cheng, H., Seeholzer, S.
H., and Roder, H. (2000) Direct evidence for the cooperative unfolding
of cytochrome c in lipid membranes from H-2H exchange kinetics,
J. Mol. Biol., 303, 617-626.
159.Sanghera, N., and Pinheiro, T. J. T. (2002)
Binding of prion protein to lipid membranes and implications for prion
conversion, J. Mol. Biol., 315, 1241-1256.
160.Critchley, P., Kazlauskaite, J., Eason, R., and
Pinheiro, T. J. T. (2004) Binding of prion proteins to lipid membranes,
Biochem. Biophys. Res. Commun., 313, 559-567.
161.Davidson, W. S., Jonas, A., Clayton, D. F., and
George, J. M. (1998) Stabilization of alpha-synuclein secondary
structure upon binding to synthetic membranes, J. Biol. Chem.,
273, 9443-9449.
162.Valastyan, J. S., Termine, D. J., and
Lindquist, S. (2014) Splice isoform and pharmacological studies reveal
that sterol depletion relocalizes alpha-synuclein and enhances its
toxicity, Proc. Natl. Acad. Sci. USA, 111, 3014-3019.
163.Chirita, C. N., Necula, M., and Kuret, J.
(2003) Anionic micelles and vesicles induce tau fibrillization in
vitro, J. Biol. Chem., 278, 25644-25650.
164.Silvestro, L., and Axelsen, P. H. (2000)
Membrane-induced folding of cecropin A, Biophys. J., 79,
1465-1477.
165.Hebda, J. A., Magzoub, M., and Miranker, A. D.
(2014) Small molecule screening in context: lipid-catalyzed amyloid
formation, Protein Sci., 23, 1341-1348; doi:
10.1002/pro.2518.
166.Rodriguez Camargo, D. C., Link, M. N., and
Dames, S. A. (2012) The FKBP-rapamycin binding domain of human TOR
undergoes strong conformational changes in the presence of membrane
mimetics with and without the regulator phosphatidic acid,
Biochemistry, 51, 4909-4921.
167.Moravcevic, K., Mendrola, J. M., Schmitz, K.
R., Wang, Y-H., Slochower, D., Janmey, P. A., and Lemmon, M. A. (2010)
Kinase associated-1 domains drive MARK/PAR1 kinases to membrane targets
by binding acidic phospholipids, Cell, 143, 966-977.
168.Lee, D. W., Banquy, X., Kristiansen, K.,
Kaufman, Y., Boggs, J. M., and Israelachvili, J. N. (2014) Lipid
domains control myelin basic protein adsorption and membrane
interactions between model myelin lipid bilayers, Proc. Natl. Acad.
Sci. USA, 111, E768-E775.
169.Kohler, C., Gogvadze, V., Hakansson, A.,
Svanborg, C., Orrenius, S., and Zhivotovsky, B. (2001) A folding
variant of human alpha-lactalbumin induces mitochondrial permeability
transition in isolated mitochondria, Eur. J. Biochem.,
268, 186-191.
170.Svensson, M., Fast, J., Mossberg, A. K.,
Dueringer, C., Gustafsson, L., Hallgren, O., Brooks, C. L., Berliner,
L., Linse, S., and Svanborg, C. (2003) Alpha-lactalbumin unfolding is
not sufficient to cause apoptosis, but is required for the conversion
to HAMLET (human alpha-lactalbumin made lethal to tumor cells),
Protein Sci., 12, 2794-2804.
171.Hallgren, O., Aits, S., Brest, P., Gustafsson,
L., Mossberg, A. K., Wullt, B., and Svanborg, C. (2008) Apoptosis and
tumor cell death in response to HAMLET (human alpha-lactalbumin made
lethal to tumor cells), Adv. Exp. Med. Biol., 606,
217-240.
172.Pettersson-Kastberg, J., Mossberg, A. K.,
Trulsson, M., Yong, Y. J., Min, S., Lim, Y., O’Brien, J. E.,
Svanborg, C., and Mok, K. H. (2009) Alpha-lactalbumin, engineered to be
nonnative and inactive, kills tumor cells when in complex with oleic
acid: a new biological function resulting from partial unfolding, J.
Mol. Biol., 394, 994-1010.
173.Booth, P. J., and Curnow, P. (2009) Folding
scene investigation: membrane proteins, Curr. Opin. Struct.
Biol., 19, 8-13.
174.Liang, B., and Tamm, L. K. (2007) Structure of
outer membrane protein G by solution NMR spectroscopy, Proc. Natl.
Acad. Sci. USA, 104, 16140-16145.
175.Milstein, S. J., Leipold, H., Sarubbi, D.,
Leone-Bay, A., Mlynek, G. M., Robinson, J. R., Kasimova, M., and
Freire, E. (1998) Partially unfolded proteins efficiently penetrate
cell membranes – implications for oral drug delivery, J.
Control Release, 53, 259-267.
176.Kweon, D.-H., Kim, C. S., and Shin, Y.-K.
(2003) Regulation of neuronal SNARE assembly by membrane, Nat.
Struct. Biol., 10, 440-447.
177.Bowen, M., and Brunger, A. T. (2006)
Conformation of the synaptobrevin transmembrane domain, Proc. Natl.
Acad. Sci. USA, 103, 8378-8383.
178.Ellena, J. F., Liang, B., Wiktor, M., Stein,
A., Cafiso, D. S., Jahn, R., and Tamm, L. K. (2009) Dynamic structure
of lipid-bound synaptobrevin suggests a nucleation-propagation
mechanism for trans-SNARE complex formation, Proc. Natl. Acad. Sci.
USA, 106, 20306-20311.
179.Van den Bogaart, G., Meyenberg, K., Risselada,
H. J., Amin, H., Willig, K. I., Hubrich, B. E., Dier, M., Hell, S. W.,
Grubmueller, H., Diederichsen, U., and Jahn, R. (2011) Membrane protein
sequestering by ionic protein–lipid interactions, Nature,
479, 552-555.
180.Mach, H., and Middaugh, C. R. (1995)
Interaction of partially structured states of acidic fibroblast growth
factor with phosphor-lipid membranes, Biochemistry, 34,
9913-9920.
181.Ugolev, A. M. (1967) Physiology and
Pathology of Membrane (Contact) Digestion [in Russian], Nauka,
Leningrad.
182.Stroll, B. R., Leipold, H. R., Milstein, S.,
and Edwards, D. A. (2000) A mechanistic analysis of carrier-mediated
oral delivery of protein therapeutics, J. Control. Release,
64, 217-228.
183.Lennernas, H. (2007) Modeling gastrointestinal
drug absorption requires more in vivo biopharmaceutical data:
experience from in vivo dissolution and permeability studies in
humans, Curr. Drug Metab., 8, 645-657.
184.Wawrezinieck, A., Pean, J. M., Wuethrich, P.,
and Benoit, J. P. (2008) Oral bioavailability and drug/carrier
particulate systems, Med. Sci. (Paris), 24, 659-664.
185.Rankin, S. E., Watts, A., and Pinheiro, T. J.
(1998) Electrostatic and hydrophobic contributions to the folding
mechanism of apocytochrome c driven by the interactions with
lipid, Biochemistry, 37, 12588-12595.
186.Bryson, E. A., Rankin, S. E., Goormaghtigh, E.,
Ruysschaert, J. M., Watts, A., and Pinheiro, T. J. (2000) Structure and
dynamics of lipid-associated states of apocytochrome c, Eur.
J. Biochem., 267, 1390-1396.
187.McLaughlin, S., and Murray, D. (2005) Plasma
membrane phosphoinositide organization by protein electrostatics,
Nature, 438, 605-611.
188.Li, L., Shi, X., Guo, X., Li, H., and Xu, C.
(2014) Ionic protein–lipid interactions at plasma membrane: what
can the charge do? Trends Biochem. Sci., 39, 130-140.
189.Derganc, J., Antonny, B., and Copic, A. (2013)
Membrane bending: the power of protein imbalance, Trends Biochem.
Sci., 38, 576-584.
190.Shibata, Y., Hu, J., Kozlov, M. M., and
Rapoport, T. A. (2009) Mechanism shaping the membranes of cellular
organelles, Annu. Rev. Cell Dev. Biol., 25, 329-354.
191.Zimmerberg, J., and Kozlov, M. M. (2006) How
proteins produce cellular membrane curvature, Nature Rev. Mol. Cell
Biol., 7, 9-19.
192.Sens, P., Johannes, L., and Bassereau, P.
(2008) Biophysical approaches to protein-induced membrane deformations
in trafficking, Curr. Opin. Cell Biol., 20, 476-482.
193.Copic, A., Latham, C. F., Horlbeck, M. A.,
D’Arcangelo, J. G., and Miller, E. A. (2012) ER cargo properties
specify a requirement for COPII coat rigidity mediated by Sec 13p,
Science, 335, 1359-1362.
194.Leventis, R., and Silvius, J. R. (2010)
Quantitative experimental assessment of macromolecular crowding effects
at membrane surfaces, Biophys. J., 99, 2125-2133.
195.Stachowiak, J. C., Hayden, C. C., and Sasaki,
D. Y. (2010) Steric confinement of proteins on lipid membranes can
drive curvature and tubulation, Proc. Natl. Acad. Sci. USA,
107, 7781-7786.