Rat Liver Sinusoidal Surface N-Linked Glycoproteomic Analysis by Affinity Enrichment and Mass Spectrometric Identification
Jianglin Li1#, Jun Gao2#, Miao Jiang1#, Jia Chen1, Zhonghua Liu1, Ping Chen1*, and Songping Liang1*
1Key Laboratory of Protein Chemistry and Developmental Biology of the Ministry of Education, College of Life Sciences, Hunan Normal University, Changsha, 410081, P. R. China; E-mail: chenp@hunnu.edu.cn; liangsp@hunnu.edu.cn2Qingyuan City People’s Hospital of Jinan University, Qingyuan, Guangdong, 511518, P. R. China
# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received April 8, 2014; Revision received November 11, 2014
Glycosylation in liver is one of the most biologically important protein modifications. It plays critical roles in many physiological and pathological processes by virtue of its unique location at the blood–tissue interface, including angiogenesis, liver cancer, cirrhosis, and fibrosis. To analyze glycosylation of plasma membrane proteins in liver sinusoidal endothelial cells (LSEC), N-glycopeptides of the LSEC surface were enriched using a filter-assisted sample preparation-based lectin affinity capture method and subsequently identified with mass spectrometry. In total, 225 unique N-glycosylation sites on 152 glycoproteins were identified, of which 119 (53%) sites had not previously been determined experimentally. Among the glycoproteins, 53% were classified as plasma membrane proteins and 47 (31%) as signaling proteins and receptors. Moreover, 23 cluster of differentiation antigens with 49 glycopeptides were detected within the membrane glycoproteins of the liver sinusoidal surface. Furthermore, bioinformatics analysis revealed that the majority of identified glycoproteins have an impact on processes of LSEC. Therefore, N-glycoproteomic analysis of the liver sinusoidal surface may provide useful information on liver regeneration and facilitate liver disease diagnosis.
KEY WORDS: liver sinusoidal endothelial surface, N-glycoproteomic, N-glyco-FASP, mass spectrometryDOI: 10.1134/S0006297915030025
Abbreviations: CAM, cell adhesion molecule; CD, cluster of differentiation; ECM, extracellular matrix; EGFR, epidermal growth factor receptor; ER, endoplasmic reticulum; FASP, filter-aided sample preparation-based method; GO, gene ontology annotation; GRAVY, grand average hydropathy; KEGG, Kyoto Encyclopedia of Genes and Genomes; LC-MS/MS, liquid chromatography-tandem mass spectrometry; LSEC, liver sinusoidal endothelial cells; MS, mass spectrometry; NPM, plasma membrane pellet from the Nycodenz density centrifugation; PM, plasma membrane; SPM, LSEC PM fraction after sucrose density centrifugation; TMD, transmembrane domain.
Analysis of plasma membrane proteins and their posttranslational
modifications is an important route for the identification of disease
markers and potential drug targets [1, 2]. Protein glycosylation is one of the major
posttranslational modifications with significant effects on protein
folding, conformation distribution, stability, and activity. It is
estimated that more than half of all mammalian proteins are
glycosylated [3]. N-Glycosylation is by far the
most common cell surface modification, and it occurs at asparaginyl
residues within the consensus sequence N-!P-S/T (where !P is any amino
acid except proline) [1]. This modification is
involved in many cellular functions, including cell–cell and
receptor–ligand interactions, immune response, apoptosis, and
disease pathogenesis [4]. N-Linked glycosylation
predominantly occurs on membrane proteins, which are traditionally
difficult to analyze using proteomic methods. Notably, most mammalian
membrane proteins are extensively glycosylated [5].
Glycosylation of membrane proteins plays a key role in biological
processes such as cell adhesion, recognition, and signal transduction
[6]. The disruption and mutation of glycosylation
sites are basic features of oncogenesis and tumor progression.
Considerable attention has recently focused on the growing field of
glycoproteomics to determine the biological roles of protein
glycosylation and its relationship with specific diseases. Several
researchers have employed mass spectrometric methods to characterize
glycopeptides or glycans attached to certain glycosylation sites. To
date, extensive studies have been performed on body fluids such as
plasma and saliva to identify biomarkers of diseases [6]. Decades of research have shown that structural
changes in the glycan structures of serum proteins are an indication of
liver damage [7]. Protein glycosylation has been
focal in the search for an objective target feature in liver diseases
[6, 8]. A few reports on
membrane glycoproteins in liver tissue are documented in the
literature. A literature search revealed that only one study to date
has focused on the rat liver membrane; in that work, Lee et al. focused
on whole liver membrane glycoproteome, and they identified 335
potential N-linked glycosylation sites on 424 non-redundant membrane
proteins [9]. As we know, the liver contains
multiple cell types, including liver sinusoidal endothelial cells
(LSEC), stellate cells, Kupffer cells, biliary epithelial cells, and
hepatocytes, and therefore the membrane is extremely complex. LSECs
between the blood and vessel wall, namely, the luminal side of the
plasma membrane of vascular endothelial cells, are composed of a single
layer of flattened endothelial cells that line blood vessels and are
critical in various physiological and pathological processes [10-12]. Due to the hydrophobic
nature of plasma membrane (PM) proteins, mass-spectrometric analysis is
a difficult task. LSEC PM constitutes only 15.2% of all PM. In this
study, the LSEC PM was enriched and its N-glycoproteome was identified
by mass spectrometry [9]. Recent technological
developments in sample preparation, together with improvements in
mass-spectrometric analysis, have facilitated analysis of these
proteins and their posttranslational modifications [1, 10, 12].
A filter-aided sample preparation (FASP)-based method, in which
glycopeptides were enriched via lectin affinity purification on the
surface of a filter, is well suited for this purpose. According to this
method, most membrane proteins could be effectively solubilized in
sodium dodecyl sulfate (SDS), which could be removed easily in the
following washing step [1, 11,
13, 14]. In this study, we
successfully applied glycopeptide enrichment with an N-glyco-FASP for
N-glycoproteomic analysis of liver sinusoidal surface accessible from
the vasculature. In total, 225 N-glycosylation sites on 152
glycoproteins were identified. Among these, 23 cluster of
differentiation (CD) antigens with 49 glycopeptides were detected
within the membrane glycoproteins of LSEC. Many of the identified
glycoproteins, such as CD13, CD147, CD82, epidermal growth factor
receptor (EGFR), and integrin β1, are associated with
physiologically and pathologically significant events. Our data might
provide clues for identification of valuable candidate drug targets and
biomarkers from rat liver sinusoidal surface.
MATERIALS AND METHODS
An overview of the analytical strategy in this study is illustrated in Fig. S1 (see Supplement to this paper on the journal website http://protein.bio.msu.ru/biokhimiya).
In vivo membrane density perturbation technique. Animal housing and all experimental procedures were conducted according to the requirements of the Provisions and General Recommendations of Chinese Experimental Animal Administration Legislation. The study was approved by the local ethics committee and was conducted according to the principles expressed in the Helsinki Declaration. The silica nanoparticle subfraction method was used for LSEC plasma membrane isolation [15-17] except the in vivo membrane density perturbation steps were carried out according to our previous report [17]. A schematic view of the separation steps is shown in Fig. 1a. Briefly, the rats were anesthetized with 10% chloral hydrate (n = 6). The liver was perfused with colloidal silica suspension and then covered with polyacrylic acid. The membrane density perturbation procedures were repeated once to make a denser luminal membrane. After homogenization, crude LSEC PM (SPM) was obtained from the bottom of a sucrose density centrifugation with membrane density higher than 69% sucrose solution. Then the SPM from five rat livers were pooled. To purify further the LSEC membranes, the SPM was then sedimented through 70% Nycodenz density centrifugation. The plasma membrane pellet from the Nycodenz density centrifugation (NPM) was washed with lysis buffer twice. The NPM proteins were then recovered from the silica coating by solubilized in 2% SDS sample loading buffer, sonicated 5 times for 10 s, and boiled for 10 min. The solubilized LSEC PM protein was recovered from the supernatant after centrifugation at 15,000g for 30 min. The protein concentration was determined with an RCDC protein assay kit (Bio-Rad, USA).
Fig. 1. Purification schematic and quality identification of sinusoidal endothelial surface plasma membrane from rat liver. a) Work flow of enrichment of sinusoidal endothelial surface plasma membrane from rat liver. b) SDS-PAGE image of crude plasma membrane, SPM, and NPM. c) Purity identification of SPM and NPM determined by Western blotting. Equal protein amounts from each fraction were loaded. SPM, the PM fraction from sucrose density centrifugation; NPM, the PM fraction from Nycodenz density centrifugation. Na+/K+-ATPase and flotillin-1, plasma membrane markers; Golgi 58, marker of Golgi; prohibitin, marker of mitochondria.
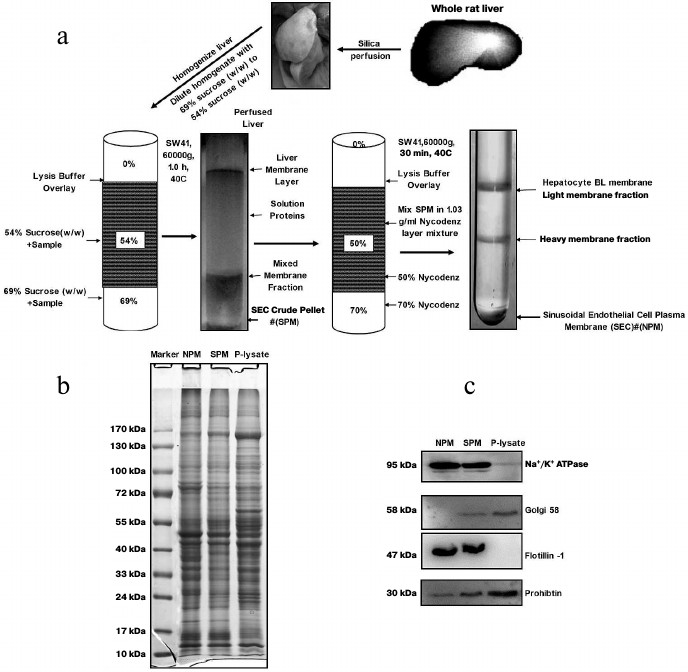
SDS-PAGE and immunoblotting analysis. For protein separation and purity verification, the protein samples were mixed with 4× SDS loading buffer and subjected to SDS-PAGE. After completion of electrophoresis, the gel was visualized by Coomassie brilliant blue G250 (Bio-Rad) staining or transferred to PVDF membrane (Merck Millipore, Germany) for further western blotting analysis. Na+/K+-ATPase antibody (BD Bioscience, USA) and flotillin-1 antibody (Abcam, UK) were used to detect the plasma membrane. Golgi 58 antibody (Abcam) was used for marker of Golgi. Prohibitin antibody was used to detect mitochondria. After exposure to Hyperfilm ECL (GE Healthcare, Amersham, USA), images were analyzed using Quantity One 1D-Analysis software (Bio-Rad).
Filter aided sample preparation. Approximately 0.5 mg LSEC PM protein was subjected to the FASP procedure as previously reported [1, 13]. Briefly, approximately 0.5 mg protein was mixed with 50 µl of lysis buffer (0.1 M Tris-HCl, 0.1 M DTT, 4% SDS) and incubated at 95°C for 3 min. The mixture was then centrifuged at 15,000g for 15 min, and the resulting supernatant was mixed with 200 µl 8 M urea in 0.1 M Tris-HCl, pH 8.5 (UA solution). The mixed sample was loaded into a 3-kDa Microcon filtration device (Merck Millipore) and centrifuged at 14,000g for a minimum of 20 min so that the remaining volume was less than 10 µl. The concentrates were then diluted in the devices with 200 µl of solution and centrifuged twice. After centrifugation, the concentrates were mixed with 100 µl 50 mM iodoacetamide in UA solution, incubated in darkness at room temperature for 30 min, and then centrifuged for 20 min. The concentrate was diluted with 200 µl of 8 M urea in 0.1 M Tris-HCl, pH 8.5, and concentrated again. This step was repeated twice. The samples were then diluted with 100 µl 40 mM NH4HCO3 and concentrated twice. After concentrating, 5 µg trypsin (Promega, USA) in 100 µl 40 mM NH4HCO3 (pH 7.8) was added for digestion overnight at 37°C. The resulting peptides were collected by centrifugation of filter units with 50 µl binding buffer (20 mM Tris-HCl, 0.5 M NaCl, 1 mM CaCl2, 1 mM MnCl2, pH 7.3) for 20 min. This step was repeated three times.
Lectin enrichment and de-glycosylation. Approximately 250 µg of digested peptides were mixed with lectin solution containing a combination of concanavalin A, wheat germ agglutinin, and Ricinus communis agglutinin (Sigma Aldrich, USA), resulting in mixtures of peptides and lectins with a mass proportion of 1 : 2. The mixtures were transferred to new YM-30 filter units (Merck Millipore). After 1 h incubation at room temperature, the unbound peptides were eluted by centrifugation. The captured peptides were washed followed by deglycosylation with peptide-N-glycosidase F (Sigma Aldrich). After incubation for 12 h at 37°C, the deglycosylated peptides were eluted. The deglycosylated peptides were purified on ZipTips C18 and dried by vacuum centrifugation.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. The peptide mixtures were analyzed using a linear ion trap-Orbitrap hybrid mass spectrometer (LTQ-Orbitrap XL; Thermo Fisher Scientific, USA) equipped with an ESI nano spray source. The digested peptides were injected into an Easy LC system (Proxeon, Denmark) with a pre-column (2 cm, ID 100 µm, 5 µm, C18). Peptides were eluted from a C18 column (10 cm, ID 75 µm, 3 µm, C18) with a linear gradient of 5-40% solvent B (99.9% acetonitrile with 0.1% formic acid) for 80 min with constant flow of 200 nl/min. The mass spectrometer was set so that each full MS scan (m/z 350-1800) in profile mode was acquired in the orbitrap with a resolution of 100,000 at m/z 400, followed by five MS/MS scans in the ion trap on the five most intense ions from the MS spectrum with dynamic exclusion setting: a repeat count of 2, a repeat duration of 30 s, and an exclusion duration of 90 s. Xcalibur 2.0.7 (Thermo Fisher Scientific) was used to control both the EASY n-LC and the Orbitrap MS/MS.
Protein identification and data analysis. Mascot (v. 2.2.06; Matrix Science, UK) was used for protein identification. Trypsin was chosen as the enzyme, and the number of missed cleavages was set to 1. The peptide charge was set to 2+, 3+, and 4+, and the peptide tolerance and MS/MS tolerance were 15 ppm and 0.5 Da, respectively. The IPI rat database (v. 3.68; 79,898 sequences; 42,135,824 residues) and a decoy database constructed as described by our laboratory group were appended to the target database. Variable modifications were carbamidomethyl for cysteines, oxidation for methionines, and an asparagine to aspartic acid conversion of +0.984 Da. The maximum number of missed cleavage sites was two. Spectra with a Mascot score >30 (significance threshold p < 0.05) and a valid glycosylation consensus sequence N-X-S/T (X is not proline) were considered for further manual evaluation. The UniProt system (http://www.uniprot.org/uniprot) was used for glycosylation data evaluation and protein function analysis. Identification was validated using a 1% false discovery rate assessed by reverse database searching, which was applied to non-redundant protein lists. A minimum of two unique peptide matches identified in at least two replicates was used to validate protein identifications. The grand average hydropathy (GRAVY) values for identified proteins and peptides were analyzed using the ProtParam program available at http://tw.expasy.org/tools/protparam.html. Putative transmembrane domains (TMDs) for identified proteins were mapped using the TMHMM 2.0 program based on transmembrane hidden Markov model (http://www.cbs.dtu.dk/services/TMHMM) by submitting the FASTA files. The information on membrane location was derived from UniProt, gene ontology annotation (GO), and the Swiss-Prot database. The N-glycosites were compared with the Swiss-Prot database and further recognized as “Known”, “Probable”, “By similarity”, “Potential”, and “Unknown”. UniProt and David Bioinformatics Resources 6.7 [18, 19] (http://david.abcc.ncifcrf.gov/home.jsp) were used to evaluate the enrichment of N-linked glycoproteins. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was used to evaluate the pathways of the detected N-glycoproteins [20].
RESULTS
Isolation of LSEC PM from vasculature of rat liver. We employed an in vivo membrane density perturbation strategy previously applied in our laboratory to isolate liver sinusoidal PM. The purity of LSEC PM was determined by immunoblotting [17]. As shown in Fig. 1a, after homogenization, the liver sinusoidal membrane was separated on a benchtop centrifuge. Due to the cationic colloidal silica coating, the volume of pellet (containing P-lysate) from centrifugation became large. To reduce experimental costs and ensure biocompatibility with the lysis buffer system, sucrose density gradient centrifugation (with the highest density at 1.35 g/ml) was applied for the second separation. Since the volume of SPM was reduced significantly in the second preparation, a more expensive and appropriate gradient medium, Nycodenz, was employed for the third separation to further purify the membrane fraction (NPM). The final pellet was washed three times in lysis buffer, and the purity of SPM and NPM assessed. The protein staining patterns of the homogenate, SPM, and NPM were examined using SDS-PAGE, as shown in Fig. 1b. Several bands showed significantly different intensities. All P-lysate, SPM fractions, and NPM preparations were detected using Western blotting. The SPM fraction was virtually free of Golgi and mitochondrial contamination. Additionally, endoplasmic reticulum (ER) contamination was greatly reduced. Moreover, PM markers were greatly enriched, as evident from Na+/K+-ATPase and flotillin-1 activity. NPM was significantly purer than SPM after the third purification, as shown from Western blotting with antibodies against Golgi 58, Na+/K+-ATPase, and flotillin-1 (Fig. 1c).
N-glyco-FASP method. Analysis of membrane proteins presents a considerable challenge owing to their hydrophobic nature and low abundance. To resolve this problem, different strategies for the digestion and identification of membrane proteins have been reported [21]. Among them, the filter-aided sample preparation (FASP)-based method is particularly suitable for analyzing insoluble proteins, in particular plasma membrane proteins, since complete protein solubilization is achieved in SDS while allowing gel-free analysis [1, 13]. Zielinska and coworkers further developed a FASP-based N-linked glycopeptide capture method (N-glyco-FASP) involving binding to lectins on the top of a filter, and mapped 6367 N-glycosylation sites on 2352 proteins in four mouse tissues and blood plasma [1]. In the current study, we applied this procedure to collect LSEC PM proteins and achieve better digestion efficiency. The work flow diagram of the enrichment strategy of LSEC PM N-glycopeptides is outlined in Fig. S1 (see Supplement). Moreover, the eluted peptides are clean and suitable for lectin enrichment. The technique features efficient exchange of SDS for urea in a centrifugal ultrafiltration unit, followed by protein digestion and isolation of peptides, which are eluted with high purity for LC-MS/MS. For improving the recovery rate of peptides, filters were rinsed with 50 µl binding buffer three times. Efficient digestion was followed by the process of N-glycopeptide enrichment. For capture of all three classes of N-glycosylated peptides in LSEC PM, we used the N-glyco-FASP method based on a multilectin affinity strategy for glycopeptide enrichment [22]. Lectins do not need to be coupled to a solid support since they are retained by the filter, and therefore any lectin or mixture of lectins can be employed. We selected ConA that binds to mannose, wheat germ agglutinin that binds to sialic acid, N-acetylglucosamine and Ricinus communis agglutinin, which captures galactose modified at the 3-0 position (e.g. with sialic acid or another galactose) as well as terminal galactose. LSEC PM were independently prepared in triplicate and measured three times via single LC MS/MS runs with 165-min gradients. Additionally, N-glycosylation sites required no additional fractionation. Interestingly, the number of identified N-glycosylation sites did not increase significantly with repeated measurements and additional fractionation [1]. The methods were successfully applied to LSEC PM samples, leading to the identification of 277 N-glycosites in 162 glycoproteins. However, only 231 N-glycosites of 152 glycoproteins were annotated in the UniProt database (details of identified glycoproteins and glycopeptides are provided in Table S1 of the Supplement).
To address concerns of biological reproducibility, experiments were conducted on three biological replicates. The reproducibility of the method for the glycoproteomic study of LSEC PM was evaluated. One hundred fifty two non-redundant proteins were identified with 89, 95, and 103 glycoproteins in subsequent experiments. The reproducibility was 63.0% on average, with separate identification ratios of 58.6% (89/152), 62.5% (95/152), and 67.8% (103/152) (details are described in Table S6 of the Supplement).
Mapping of LSEC PM N-glycoproteins. The N-glyco-FASP method was further applied for rat liver sinusoidal surface glycoproteomic analysis. As shown in Table S1 (Supplement), 277 N-glycosylation sites on 260 unique glycopeptides were identified, corresponding to 162 N-glycoproteins. To enhance further identification reliability, only proteins recognized according to the UniProt database (version of July 2013) were approved. Consequently, 148 N-glycoproteins were annotated, among which 46 proteins were identified with more than two glycopeptides and 102 with one glycopeptide (Table S2 of the Supplement). Glycosylation sites were compared with the UniProt database. In total, 231 unique N-glycosites were recognized, and only seven sites were documented in UniProt based on experimental data. Moreover, 231 sites were annotated as “Potential” or “By similarity” in view of no experimental evidence. The remaining 224 sites identified in this study were not referred to in the UniProt database. To our knowledge, the other 127 (55%) N-glycoproteins have not hitherto been reported in rats (Fig. 2).
Fig. 2. Classification of N-glycosites identified in rat liver sinusoidal surface plasma membrane. N-Glycosites were compared with the Swiss-Prot database and further classified as “Known”, “By similarity”, “Potential”, and “Unknown”. “By similarity” and “Potential” are two types of non-experimental qualifiers indicating that the information provided is not based on experimentally proven findings in the Swiss-Prot database. The term ‘‘Potential’’ indicates logical or conclusive evidence that the given annotation could apply. ‘‘By similarity’’ is added to proven data for whole or partial protein, and transferred to other protein family members within a certain taxonomic range dependent on the biological event or characteristic (http://au.expasy.org/sprot/userman.html#non_experimental).
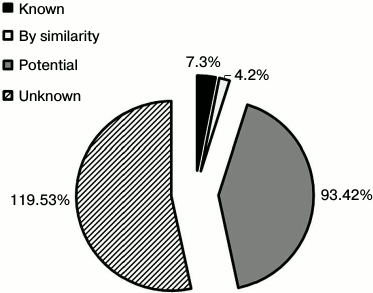
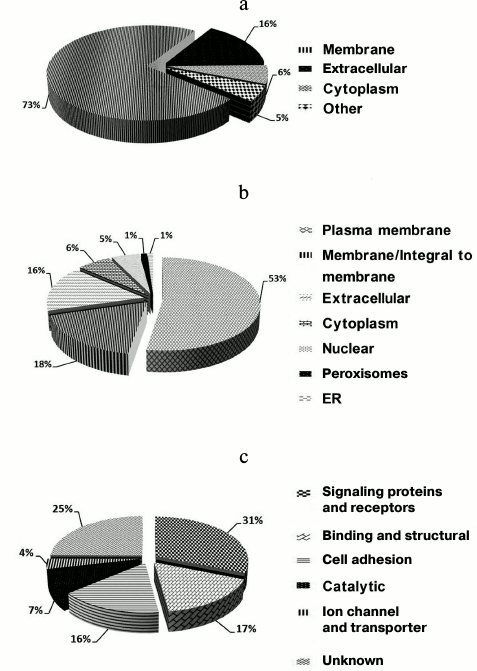
Accordingly, the 224 (97%) N-glycosylation sites were either listed as “Potential” sites or not identified in the UniProt database. Table S2 (Supplement) provides details of the identified N-linked glycosylation sites and their parent glycoproteins. Reliable identification of peptides and N-glycosylation sites is based on the following principles: identified peptides must have a Mascot ion score above 30 (p < 0.05) and their expected value must be evaluated. Second, glycoproteins need at least one peptide containing the consensus N-!P-[S/T]-!P (where !P is not proline) N-glycosylation motif, and all identified glycopeptides require an asparagine to aspartic acid deamidation site with a mass increase of 0.984 Da. Finally, all MS/MS spectra for each identified glycopeptide must be manually checked. An example MS/MS spectrum of the N-linked glycopeptide, TYCSSELVSN#CTQK, from the precursor of CD164 is presented in Fig. S2 (see Supplement). The mass difference of 115 Da between fragments y4 and y5 confirms that conversion of N (asparagine) to D (aspartic acid) occurs at Asn97. Details of all the identified N-glycosites are provided in Table S1 (Supplement). These data could potentially be used for glycoprotein database completion and provide valuable information for further in-depth study of N-glycosylation in rat liver. Physicochemical characteristics (Fig. S3 in the Supplement) were employed to evaluate the bias of our sample preparation and separation methods. Overall, 28% (42) proteins with molecular weight (MW) above 100 kDa and 6% (9) proteins with pI above 9.0 were identified, as shown in Fig. S3a. When the calculated pI values were plotted against molecular weight on a logarithmic scale (Fig. S3b), over 40% of these proteins fell outside the typical limits of protein resolution with routine 2-DE (MW < 100 kDa; pI ranging from 4.5 to 8.5). The identified glycoproteins of LSEC PM were analyzed based on the calculated GRAVY values, and TMDs were predicted to assess the efficacy of the developed protocol for membrane protein identification (Table S1 in the Supplement). As the affinity approach in this study uses non-gel-based separation, it is particularly suitable for purification of highly hydrophobic integral membrane proteins. Several proteins with membrane-spanning domains were detected: 37% (55) of the glycoproteins had TMDs and 10% (15) were predicted to contain 2-11 TMDs (Fig. S3c). In addition, six glycoproteins with no TMDs, including CD54, were predicted as membrane proteins via database annotation, and one predicted as extracellular. These results further support the effectiveness of the method in successfully identifying integral membrane proteins containing multiple TMDs with no bias against complex multi-spanning proteins. The GRAVY index of identified glycoproteins shows a broad scattering of values ranging from hydrophilic proteins, such as RT1 class I, CE4 (–0.830), to extremely hydrophobic proteins, such as similar to epithelial membrane protein 1 (1.010). However, both proteins localize on plasma membrane and contain TMDs, despite the significant deviation in their GRAVY values. GRAVY values tended to be positive, and proteins were predicted as hydrophobic. However, in the region with 0-2 TMDs, GRAVY values were distributed in the range from –0.4 to 0.4. According to Santoni et al. [23], it is difficult to determine whether a protein is hydrophobic or hydrophilic based on the distribution of GRAVY values within this range. We classified the subcellular locations of N-glycoproteins according to Swiss-UniProt and GO annotation. Among the 59% (95) GO cellular component-annotated proteins, 73% (69) belonged to membrane-associated locations. Overall, 16% (15 proteins) were classified as extracellular, 6% (6) were located in the cytoplasm, and 5% (5) were mainly in the ER and proteasomes (Fig. 3a). Among the membrane proteins, 53% (50) localized to the plasma membrane and 16% (15) to extracellular (Fig. 3b). The molecular functions of glycoproteins identified in the current study were classified based on the GO database and literature surveys (Fig. 3c). Approximately, 40 (27%) of the N-glycoproteins had no annotated functions and were therefore classified as “uncharacterized” or “hypothetical”. Overall, 18% (26 proteins) were classified as cell adhesion molecular, 18% (27) with binding and structural activities, and 8% (12) with catalytic activity, due to ambiguous annotations of the GO database. Interestingly, 38% (50 proteins) were classified as signaling proteins and receptors with potential roles in a variety of cell signaling pathways. Further details of cellular localization are presented in Table S1 (Supplement). The cell surface membrane proteins identified included cell adhesion proteins, cell surface receptors, and many solute carrier transporters.
Fig. 3. Subcellular and functional distribution of glycoproteins determined with affinity enrichment and identification: a) total identified glycoproteins; b) membrane glycoproteins; c) functional distribution of the identified glycoproteins. All proteins were categorized using the UniProt database and GO annotation.
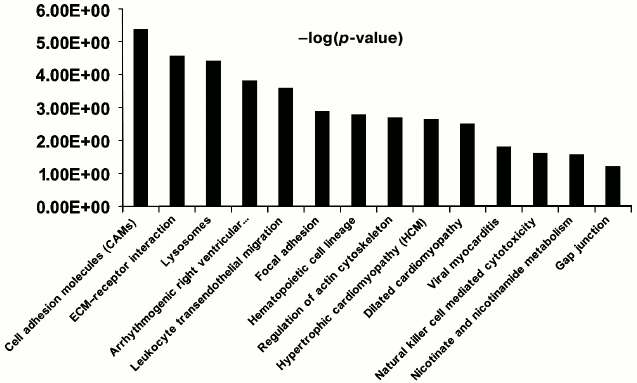
Functional characterization of membrane N-glycoproteins of rat liver sinusoidal surface. N-Glycoproteins were subjected to further bioinformatics analysis. While LSEC PM glycoproteins were greatly enriched, this did not represent the entire signaling cascade. Rather, the procedure was designed to capture N-linked glycoproteins, many of which are involved in LSEC-related signaling. DAVID analysis indicated significant enrichment for LSEC-related KEGG pathways (Fig. 4; Tables S4 and S5 in the Supplement).
Fig. 4. Significant KEGG pathways involved in protein–protein interaction (PPI) networks of 150 N-linked glycoproteins of LSEC PM. The p-value threshold in the corresponding KEGG pathways is shown.
LSECs act as a filter between the lumen of the hepatic sinusoid and surrounding hepatocytes. A major role of LSEC is to minimize barriers for the bidirectional transfer of small or soluble substrates between blood and extracellular space of Disse while excluding larger circulating particles, such as blood cells, platelets, and chylomicrons. This physiological role is achieved by the presence of numerous transcellular pores in LSECs known as fenestrations [24, 25]. The diameter and number of fenestrations are altered by various liver diseases, diabetes mellitus, and old age, and they are influenced by cytokines and hormones [26]. Alterations in the size and number of fenestrations influences the hepatic trafficking of lipoproteins [27], clearance of pharmaceutical agents [25, 28], liver regeneration and angiogenesis, and interactions between lymphocytes and hepatocytes [29, 30]. In the physiological and pathological processes of liver cancer and angiogenesis, LSEC first needs to adhere to the extracellular matrix and surrounding stromal cells via cell adhesion molecules (CAMs). CAMs are associated with liver angiogenesis progression and metastasis and have been proposed as effective biomarkers [31-33]. In our study, F11 receptor, RT1 class I, CE14, activated leukocyte cell adhesion molecule, cadherin 2, cadherin 5, endothelial cell adhesion molecule, integrin αM, integrin β1 (fibronectin receptor β), integrin β2, intercellular adhesion molecule 1 (CD54), and intercellular adhesion molecule 2 were classified as CAMs (the table). Integrins are important adhesion molecules that regulate tumor and endothelial cell survival, proliferation, and migration. Interestingly, integrin β1, integrin α5, and fibronectin 1 form complexes in angiogenesis of liver regeneration, and integrin α5β1 appears to play a critical role during angiogenesis [34]. Integrins are transmembrane glycoproteins involved in cell–matrix and cell–cell interactions [35], and thus they play critical roles in adhesion, cytoskeletal remodeling, and cell motility. Five integrin proteins were identified in our analysis, including integrin αM, integrin α1 (CD49a), integrin β1 (CD29), integrin β2, and integrin α5 [36]. Integrins act as important adhesion molecules that regulate endothelial cell and tumor survival, proliferation, and migration. A study of integrin glycosylation would therefore be not only beneficial in understanding liver physiology mechanisms, such as angiogenesis, but also liver disease diagnosis and treatment, for instance, distinguishing the stage of liver fibrosis, cirrhosis, and hepatocellular carcinoma carcinogenesis [6, 7].
Representative glycoproteins and N-glycosylation sites identified from
the LSEC PM
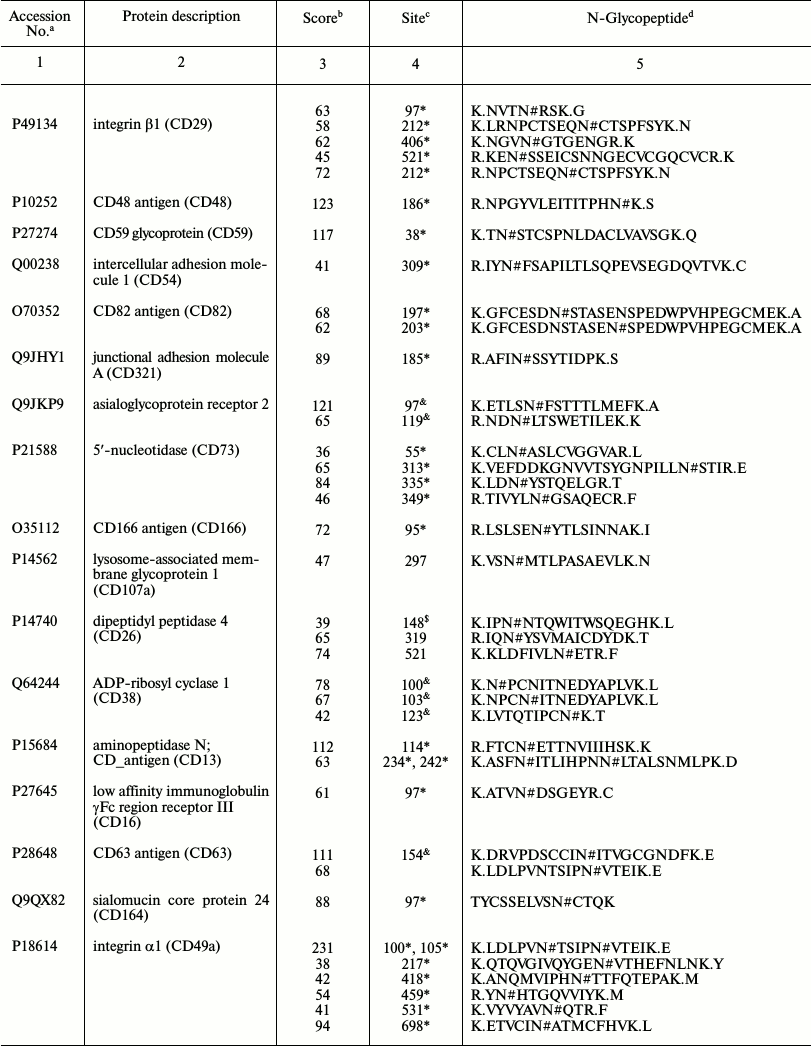
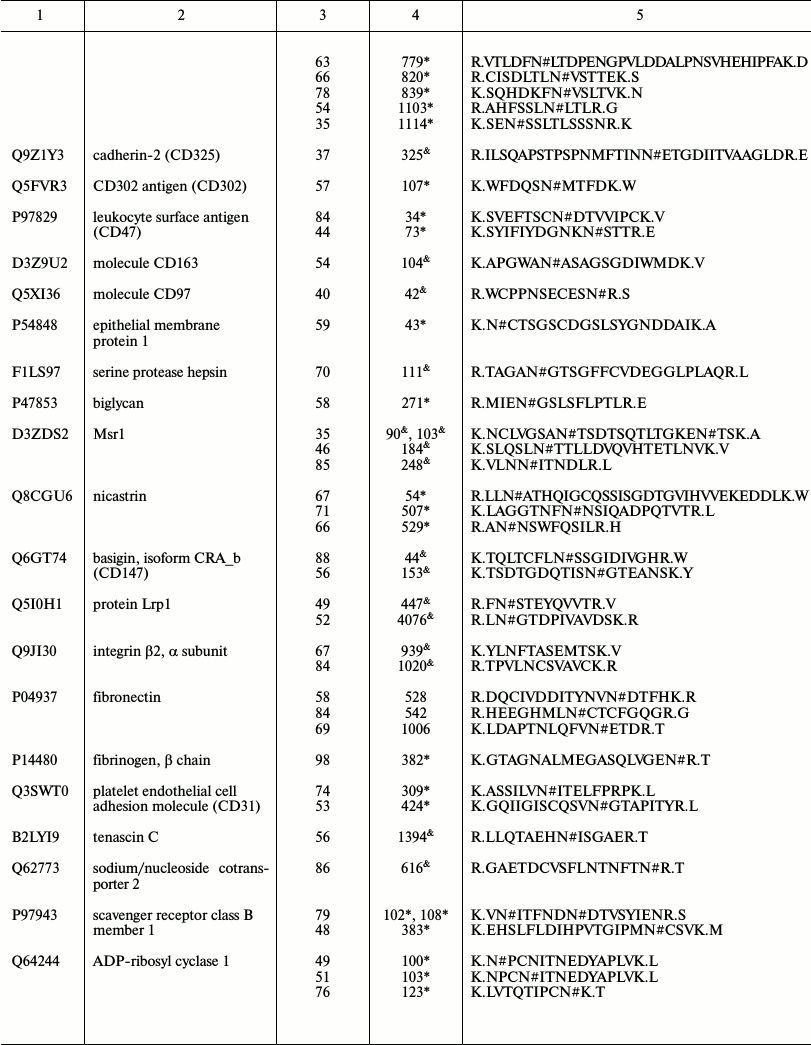
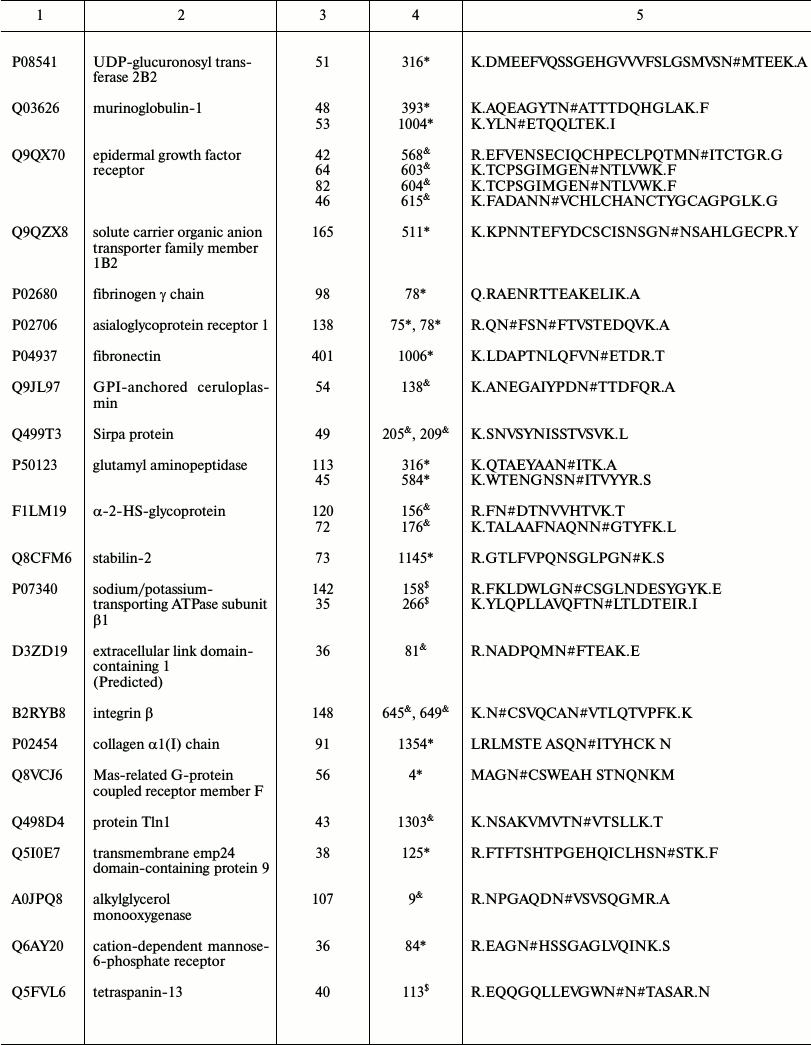

a Accession number from the UniProt database.
b The highest Mascot for the identified.
c The glycosylation sites are marked with &
Unknown, * Potential, $ By similarity. The N-glycosylation
sites were compared with the UniProt database and further recognized as
Known, Probable, By similarity, Potential, and Unknown. Probable, By
similarity, and Potential are three types of non-experimental
qualifiers indicated that the information given is not based on
experimentally proven findings in Swiss-Prot database. The term
‘‘Potential’’ indicates that there is some
logical or conclusive evidence that the given annotation could apply.
The term ‘‘Probable’’ is stronger than the
qualifier “Potential” and there must be at least some
experimental evidence. ‘‘By similarity’’ is
added to facts that were proven for a protein or part of it, and which
is then transferred to other protein family members within a certain
taxonomic range.
d Glycosylation sites are marked with # according to the
UniProt database. All the glycosylation proteins were identified at
least from two experimental data.
Our data indicated that the CD47 molecule, collagen (type VI, α2), procollagen (type VI, α3), the protein similar to integrin α-IIb precursor (CD41 antigen), fibronectin 1, and integrins are involved in the extracellular matrix (ECM) receptor interaction pathway (table). ECM is an important regulator of angiogenesis and vascular remodeling. Specifically, an activated leukocyte–cell adhesion molecule, ALCAM (CD166), has been identified as a cancer stem cell marker [37, 38]. Although not identified with KEGG analysis, several other proteins are associated with cell adhesion, including the CD97 antigen associated with increased motility and invasiveness [39] owing to the ability to break through endothelial cell barriers and attach to distant organs. Recent research has further revealed that CD97 is involved in angiogenesis [40] and leukocyte trafficking [41]. Cadherin 2 [42] and cadherin 5 [43], involved in integrin-mediated cell adhesion and fibronectin matrix remodeling in tumor cells, and basigin (also known as CD147 or extracellular matrix metalloproteinase inducer, EMMPRIN), are enriched on tumor cell surfaces. Basigin stimulates the production of collagenase, thus contributing to tumor progression [44, 45].
Cell proliferation, survival, migration, invasion, and organ development are also regulated by specialized focal adhesion complexes that anchor actin filaments to integrins. Integrins bind either to the RGD motif (fibronectin, ACTB) or to DGEA and GFOGER motifs (collagen). Downstream signaling, including focal adhesion kinase, enables reorganization of the actin cytoskeleton and allows cells to change their shape and migrate, a key process during metastasis. In addition to integrins and collagens, epidermal growth factor receptor (EGFR) was identified in this study. EGFR is a cell surface protein that binds to the epidermal growth factor implicated in focal adhesion, possibly mediated through two members of the tensin family [46]. Mutations leading to EGFR overexpression are involved in several cancers and act as targets for a number of anticancer therapies [47]. Rat EGFR has no reported glycosylation sites in the UniProt database, but in humans and mice, EGFR has been annotated as a glycoprotein, with tyrosine kinase activity in the intracellular region [48]. Three N-linked glycosylation sites in EGFR were identified (the table). Eleven glycosylation sites for this protein in humans are reported in the UniProt database, including two partially glycosylated modification sites that may be related to protein function [49, 50]. At present, no evidence of a correlation between glycosylation of EGFR and MDR (multidrug resistance) in cancers is available. However, Liu et al. demonstrated that glycosylation attenuates EGFR-mediated invasion of lung cancer cells. The interaction between EGF and its receptor ultimately leads to cell proliferation [51].
The most striking functional characteristic of LSECs is their high endocytic activity, leading to their designation as scavenger endothelial cells [52-54]. LSECs play a critical role in the removal of colloids and soluble macromolecular waste products from the systemic circulation, and they represent the largest endocytotic tissue in the body. Based on DAVID cluster analysis, scavenger receptor class B member 2, CD164, sialomucin, CD63, cathepsin L1, lysosomal-associated membrane protein 1,2, and mannose-6-phosphate receptor were classified as lysosome pathway components in KEGG.
DISCUSSION
Chronic liver disease is a serious health problem worldwide. Analysis of plasma membrane proteins and their posttranslational modifications is critical for the effective identification of disease markers and targets for drug treatment [2, 6, 55]. Glycosylation is a well-known posttranslational modification in many secreted proteins. Research over decades has shown that structural changes in the glycan structures of serum proteins represent an indication of liver damage [7]. Organelle proteomics, an accepted technique for cellular fraction analysis, has two main advantages, specifically, decreased complexity of the sample for analysis and provision of subcellular information on identified proteins [56].
In organelle proteomics, a defined cell type or homogeneous cell population is essential as good starting material. The liver is a large, complex organ containing multiple cell types, including LSECs, stellate cells, Kupffer cells, biliary epithelial cells, and hepatocytes [57]. Various receptors are present on the sinusoidal and hepatocyte surfaces, and proteins bind to these receptors via their carbohydrate moieties. In addition to changes in glycosylation patterns, alterations in receptor concentration and distribution occur in various chronic liver diseases (including cirrhosis, hepatocellular carcinoma (HCC), and alcoholic liver disease (ALD), leading to the accumulation of specific glycoproteins in the circulation [58, 59]. The role of glycosylation in liver fibrosis, its relationship with various liver pathologies (ALD, hepatitis B, bile-related diseases, and obesity), and role in HCC are of significant interest. Recently, several analyses have focused on protein glycosylation alterations in liver diseases. However, no N-glycoproteomics researchers have investigated the LSEC PM to date. A stereological study showed that liver endothelial cell PM constitutes only 15.2% of all PM. Considering the difficulties in isolation of homogeneous endothelial cells and impact of heterogeneous microcirculation on cells in vivo and in vitro, we applied the in vivo membrane density perturbation strategy developed previously in our laboratory to isolate LSEC PM [17]. Owing to their significant hydrophobicity, membrane proteins demonstrate low solubility and high tendency of aggregation. As a complementary method, N-glyco-FASP has the advantages of FASP for membrane glycoprotein analysis. In the current study, the N-glyco-FASP method was applied for surface glycoproteomic analysis of the LSEC plasma membrane. Consequently, 231 N-glycosylation sites were identified on 152 glycoproteins. Interestingly, among the identified membrane glycoproteins of the liver sinusoidal surface, 23 CD antigens with 49 glycopeptides (51 N-glycosylation sites) were detected. Among these, 127 (53%) N-glycosylation sites not previously determined experimentally were highlighted. We classified 53% of these glycoproteins as plasma membrane proteins and 50 (41%) as signaling proteins and receptors. The results clearly support the utility of N-glyco-FASP as an effective strategy for the identification of plasma membrane proteins and modifications in their glycosylation patterns.
The liver contains multiple cell types, and therefore the membrane is extremely complex. LSEC PM constitutes only 15.2% of all PM. In contrast to the study by Lee et al. [9], we targeted the PM of LSEC for enrichment. As a result, the complexity of the sample was significantly reduced [9]. Theoretically, our data represent a subset of that reported by the group of Lee, but only about 20 (9%) N-glycoproteins were identified from both studies. This finding confirms the significance of sample handling in proteomics research and validates the effectiveness of our strategy. On the other hand, the percentage similarity with the LSEC PM proteome was low in this study [9]. Comparison of the datasets indicated 40% (170/425) common proteins in the PM proteome of LSEC and this study (Table S7 in the Supplement). The unique protein percentage was 60% (255/425) in this study (identification information and specific annotations of all identified proteins are shown in Table S3 in the Supplement). In total, 162 N-glycoproteins were identified based on the IPI database. Surprisingly, only 99 (61.1%) N-glycoproteins were identified in this work, which may be attributed partly to differences in protein separation and identification, but mainly to different sample enrichment strategies. While our study material remained the same, the main aim of our study was different. We employed the N-glyco-FASP method for surface glycoproteomic analysis of the LSEC plasma membrane N-glycoproteome.
Our results showed that proteins within the N-glycoproteome of LSEC PM are diversely involved in structural, signaling, transporting, traffic, and adhesion functions, possibly encompassing almost all the biological processes involved in LSEC PM. Numerous researchers have suggested an important role of LSEC surface proteins under various physiological and pathological conditions, including hypoxia, cirrhosis, fibrosis, liver cancer and angiogenesis [24, 27, 60]. LSEC surface proteins have been previously shown to participate in these important pathological processes. Many of the identified CD antigens have important biological functions and are prospective drug targets and biomarkers. The other proteins analyzed in liver ECM included collagens, fibrinogen, fibronectin, and integrin (the table). ECM is mainly located at the interface between blood flow and endothelial cells. Although ECM comprises less than 3% of the relative areas of a normal liver section, quantitative, topographic, or qualitative modification of ECM proteins would rapidly affect liver structure and function [61]. Proteolytic processing of ECM molecules has either stimulatory or inhibitory effects on liver diseases, such as angiogenesis. ECM degradation by matrix metalloproteinases could promote angiogenesis by stimulating endothelial cell migration. These proteins include CD147, aminopeptidase N (CD13), and scavenger receptor class B member 2. The multifunctional cation-independent mannose-6-phosphate receptor in our list displays metalloproteinase activity. Since many known disease-related proteins have been identified using the colloidal coating strategy, we expected that this method could be applied to disease research on animal models.
Future studies using an affinity approach on the vascular endothelium surface will mainly aim to optimize and integrate the strategy with various experimental workflows. For example, the strategy could be used to discover new and promising liver disease biomarkers in combination with quantitative proteomic approaches, such as stable isotope labeling (for instance, iTRAQ, 18O labeling). This method might lead to the identification of a catalog of potential biomarkers correlated with cancer development as well as future drug targets via quantitative analysis of the differential expression of vascular endothelium surface glycoproteins under various conditions, for example, during different stages of liver regeneration and hepatocellular carcinoma.
This work was supported by grants from the National Natural Science Foundation of China (81070353 and 31370817) and the Cooperative Innovation Center of Engineering and New Products for Developmental Biology of Hunan Province (20134486).
REFERENCES
1.Zielinska, D. F., Gnad, F., Wisniewski, J. R., and
Mann, M. (2010) Precision mapping of an in vivo N-glycoproteome
reveals rigid topological and sequence constraints, Cell,
141, 897-907.
2.Wisniewski, J. R. (2011) Tools for phospho- and
glycoproteomics of plasma membranes, Amino Acids, 41,
223-233.
3.Apweiler, R., Hermjakob, H., and Sharon, N. (1999)
On the frequency of protein glycosylation, as deduced from analysis of
the SWISS-PROT database, Biochim. Biophys. Acta (BBA)-General
Subjects, 1473, 4-8.
4.Varki, A. (1993) Biological roles of
oligosaccharides: all of the theories are correct, Glycobiology,
3, 97-130.
5.Gahmberg, C. G., and Tolvanen, M. (1996) Why
mammalian cell surface proteins are glycoproteins, Trends Biochem.
Sci., 21, 308-311.
6.Tian, Y., and Zhang, H. (2013) Characterization of
disease-associated N-linked glycoproteins, Proteomics,
13, 504-511.
7.Blomme, B., Van Steenkiste, C., Callewaert, N., and
Van Vlierberghe, H. (2009) Alteration of protein glycosylation in liver
diseases, J. Hepatol., 50, 592-603.
8.Scott, D. W., and Patel, R. P. (2013) Endothelial
heterogeneity and adhesion molecules N-glycosylation: implications in
leukocyte trafficking in inflammation, Glycobiology, 23,
622-633.
9.Lee, A., Kolarich, D., Haynes, P. A., Jensen, P.
H., Baker, M. S., and Packer, N. H. (2009) Rat liver membrane
glycoproteome: enrichment by phase partitioning and glycoprotein
capture, J. Proteome Res., 8, 770-781.
10.Wisniewski, J. R., Zougman, A., Nagaraj, N., and
Mann, M. (2009) Universal sample preparation method for proteome
analysis, Nat. Methods, 6, 359-362.
11.Wisniewski, J. R., Ostasiewicz, P., and Mann, M.
(2011) High recovery FASP applied to the proteomic analysis of
microdissected formalin-fixed paraffin-embedded cancer tissues
retrieves known colon cancer markers, J. Proteome Res.,
10, 3040-3049.
12.Zielinska, D. F., Gnad, F., Schropp, K.,
Wisniewski, J. R., and Mann, M. (2012) Mapping N-glycosylation sites
across seven evolutionarily distant species reveals a divergent
substrate proteome despite a common core machinery, Mol. Cell,
46, 542-548.
13.Wisniewski, J. R., Zougman, A., and Mann, M.
(2009) Combination of FASP and Stage Tip-based fractionation allows
in-depth analysis of the hippocampal membrane proteome, J. Proteome
Res., 8, 5674-5678.
14.Wisniewski, J. R., Zielinska, D. F., and Mann, M.
(2011) Comparison of ultrafiltration units for proteomic and
N-glycoproteomic analysis by the filter-aided sample preparation
method, Anal. Biochem., 410, 307-309.
15.Stolz, D. B., Ross, M. A., Salem, H. M., Mars, W.
M., Michalopoulos, G. K., and Enomoto, K. (1999) Cationic colloidal
silica membrane perturbation as a means of examining changes at the
sinusoidal surface during liver regeneration, Am. J. Pathol.,
155, 1487-1498.
16.Durr, E., Yu, J., Krasinska, K. M., Carver, L.
A., Yates, J. R., Testa, J. E., Oh, P., and Schnitzer, J. E. (2004)
Direct proteomic mapping of the lung microvascular endothelial cell
surface in vivo and in cell culture, Nat. Biotechnol.,
22, 985-992.
17.Li, X., Xie, C., Cao, J., He, Q., Cao, R., Lin,
Y., Jin, Q., Chen, P., Wang, X., and Liang, S. (2008) An in vivo
membrane density perturbation strategy for identification of liver
sinusoidal surface proteome accessible from the vasculature, J.
Proteome Res., 8, 123-132.
18.Huang, D. W., Sherman, B. T., Tan, Q., Kir, J.,
Liu, D., Bryant, D., Guo, Y., Stephens, R., Baseler, M. W., and Lane,
H. C. (2007) DAVID bioinformatics resources: expanded annotation
database and novel algorithms to better extract biology from large gene
lists, Nucleic Acids Res., 35, W169-W175.
19.Huang, D. W., Sherman, B. T., and Lempicki, R. A.
(2008) Systematic and integrative analysis of large gene lists using
DAVID bioinformatics resources, Nat. Protoc., 4,
44-57.
20.Aoki, K. F., and Kanehisa, M. (2005) Using the
KEGG database resource, Curr. Protoc. Bioinform., 1,
1.12; DOI: 10.1002/0471250953.
21.Josic, D., and Clifton, J. G. (2007) Mammalian
plasma membrane proteomics, Proteomics, 7, 3010-3029.
22.Qiu, R., and Regnier, F. E. (2005) Use of
multidimensional lectin affinity chromatography in differential
glycoproteomics, Anal. Chem., 77, 2802-2809.
23.Santoni, V., Kieffer, S., Desclaux, D., Masson,
F., and Rabilloud, T. (2000) Membrane proteomics: use of additive main
effects with multiplicative interaction model to classify plasma
membrane proteins according to their solubility and electrophoretic
properties, Electrophoresis, 21, 3329-3344.
24.Geraud, C., Schledzewski, K., Demory, A., Klein,
D., Kaus, M., Peyre, F., Sticht, C., Evdokimov, K., Lu, S., and
Schmieder, A. (2010) Liver sinusoidal endothelium: a
microenvironment-dependent differentiation program in rat including the
novel junctional protein liver endothelial differentiation-associated
protein-1, Hepatology, 52, 313-326.
25.Ganesan, L. P., Mohanty, S., Kim, J., Clark, K.
R., Robinson, J. M., and Anderson, C. L. (2011) Rapid and efficient
clearance of blood-borne virus by liver sinusoidal endothelium, PLoS
Pathog., 7, e1002281.
26.Mitchell, S. J., Huizer-Pajkos, A., Cogger, V.
C., McLachlan, A. J., Le Couteur, D. G., Jones, B., de Cabo, R., and
Hilmer, S. N. (2011) Age-related pseudocapillarization of the liver
sinusoidal endothelium impairs the hepatic clearance of acetaminophen
in rats, J. Gerontol. Ser. A: Biol. Sci. Med. Sci., 66,
400-408.
27.Reichen, J. (1999) The role of the sinusoidal
endothelium in liver function, Physiology, 14,
117-121.
28.Solaun, M. S., Mendoza, L., De Luca, M.,
Gutierrez, V., Lopez, M. P., Olaso, E., Sim, L., Kim, B., and
Vidal-Vanaclocha, F. (2002) Endostatin inhibits murine colon carcinoma
sinusoidal-type metastases by preferential targeting of hepatic
sinusoidal endothelium, Hepatology, 35, 1104-1116.
29.Ding, B.-S., Nolan, D. J., Butler, J. M., James,
D., Babazadeh, A. O., Rosenwaks, Z., Mittal, V., Kobayashi, H., Shido,
K., and Lyden, D. (2010) Inductive angiocrine signals from sinusoidal
endothelium are required for liver regeneration, Nature,
468, 310-315.
30.Gamble, J., Vadas, M., and McCaughan, G. (2011)
Sinusoidal endothelium is essential for liver regeneration,
Hepatology, 54, 731-733.
31.Kuwada, S. K., Kuang, J., and Li, X. (2005)
Integrin alpha5/beta1 expression mediates HER-2 down-regulation in
colon cancer cells, J. Biol. Chem., 280, 19027-19035.
32.Dingemans, A.-M. C., van den Boogaart, V., Vosse,
B. A., van Suylen, R.-J., Griffioen, A. W., and Thijssen, V. L. (2010)
Integrin expression profiling identifies integrin alpha5 and beta1 as
prognostic factors in early stage non-small cell lung cancer, Mol.
Cancer, 9, 1476-4598.
33.Wang, J. L., Ren, K. F., Chang, H., Jia, F., Li,
B. C., Ji, Y., and Ji, J. (2013) Direct adhesion of endothelial cells
to bioinspired poly(dopamine) coating through endogenous fibronectin
and integrin alpha5/beta1, Macromol. Biosci., 13,
483-493.
34.Welti, J., Loges, S., Dimmeler, S., and
Carmeliet, P. (2013) Recent molecular discoveries in angiogenesis and
antiangiogenic therapies in cancer, J. Clin. Invest.,
123, 3190-3200.
35.Priglinger, C. S., Szober, C. M., Priglinger, S.
G., Merl, J., Euler, K. N., Kernt, M., Gondi, G., Behler, J., Geerlof,
A., Kampik, A., Ueffing, M., and Hauck, S. M. (2013) Galectin-3 induces
clustering of CD147 and integrin-beta1 transmembrane glycoprotein
receptors on the RPE cell surface, PLoS One, 8.
36.Gu, J., Isaji, T., Sato, Y., Kariya, Y., and
Fukuda, T. (2009) Importance of N-glycosylation on alpha5/beta1
integrin for its biological functions, Biol. Pharm. Bull.,
32, 780-785.
37.Weidle, U. H., Eggle, D., Klostermann, S., and
Swart, G. W. (2010) ALCAM/CD166: cancer-related issues, Cancer
Genom. Proteom., 7, 231-243.
38.Yan, M., Yang, X., Wang, L., Clark, D., Zuo, H.,
Ye, D., Chen, W., and Zhang, P. (2013) Plasma membrane proteomics of
tumor spheres identify CD166 as a novel marker for cancer stem-like
cells in head and neck squamous cell carcinoma, Mol. Cell.
Proteom., 12, 3271-3284.
39.Safaee, M., Clark, A. J., Ivan, M. E., Oh, M. C.,
Bloch, O., Sun, M. Z., Oh, T., and Parsa, A. T. (2013) CD97 is a
multifunctional leukocyte receptor with distinct roles in human cancers
(review), Int. J. Oncol., 43, 1343-1350.
40.Wang, T., Ward, Y., Tian, L., Lake, R., Guedez,
L., Stetler-Stevenson, W. G., and Kelly, K. (2005) CD97, an adhesion
receptor on inflammatory cells, stimulates angiogenesis through binding
integrin counterreceptors on endothelial cells, Blood,
105, 2836-2844.
41.Hamann, J., Veninga, J. H. H., de Groot, D. M.,
Visser, L., Hofstra, C. L., Tak, P. P., Laman, J. D., Boots, A. M., and
van Eenennaam, H. (20011) CD97 in leukocyte trafficking, Adv. Exp.
Med. Biol., 706, 128-137.
42.Jung, M.-Y., Park, S.-Y., and Kim, I.-S. (2007)
Stabilin-2 is involved in lymphocyte adhesion to the hepatic sinusoidal
endothelium via the interaction with alpha M/beta 2 integrin,
J. Leukoc. Biol., 82, 1156-1165.
43.Mertz, A. F., Che, Y., Banerjee, S., Goldstein,
J. M., Rosowski, K. A., Revilla, S. F., Niessen, C. M., Marchetti, M.
C., Dufresne, E. R., and Horsley, V. (2013) Cadherin-based
intercellular adhesions organize epithelial cell–matrix traction
forces, Proc. Natl. Acad. Sci. USA, 110,
842-847.
44.Guo, H., Li, R., Zucker, S., and Toole, B. P.
(2000) EMMPRIN (CD147), an inducer of matrix metalloproteinase
synthesis, also binds interstitial collagenase to the tumor cell
surface, Cancer Res., 60, 888-891.
45.Tang, W., Chang, S. B., and Hemler, M. E. (2004)
Links between CD147 function, glycosylation, and caveolin-1, Mol.
Biol. Cell., 15, 4043-4050.
46.Katz, M., Amit, I., Citri, A., Shay, T.,
Carvalho, S., Lavi, S., Milanezi, F., Lyass, L., Amariglio, N., and
Jacob-Hirsch, J. (2007) A reciprocal tensin-3-cten switch mediates
EGF-driven mammary cell migration, Nature Cell Biol., 9,
961-969.
47.Seshacharyulu, P., Ponnusamy, M. P., Haridas, D.,
Jain, M., Ganti, A. K., and Batra, S. K. (2012) Targeting the EGFR
signaling pathway in cancer therapy, Exp. Opin. Therap. Targets,
16, 15-31.
48.Mottolese, M., Natali, P., Coli, A., Bigotti, G.,
Benevolo, M., Cione, A., Raus, L., and Carapella, C. (1997) Comparative
analysis of proliferating cell nuclear antigen and epidermal growth
factor receptor expression in glial tumors: correlation with
histological grading, Anticancer Res., 18, 1951-1956.
49.Sato, C., Kim, J.-H., Abe, Y., Saito, K.,
Yokoyama, S., and Kohda, D. (2000) Characterization of the
N-oligosaccharides attached to the atypical Asn-X-Cys sequence of
recombinant human epidermal growth factor receptor, J. Biochem.,
127, 65-72.
50.Wu, S.-L., Kim, J., Hancock, W. S., and Karger,
B. (2005) Extended Range Proteomic Analysis (ERPA): a new and sensitive
LC-MS platform for high sequence coverage of complex proteins with
extensive post-translational modifications comprehensive analysis of
beta-casein and epidermal growth factor receptor (EGFR), J. Proteome
Res., 4, 1155-1170.
51.Liu, Y.-C., Yen, H.-Y., Chen, C.-Y., Chen, C.-H.,
Cheng, P.-F., Juan, Y.-H., Chen, C.-H., Khoo, K.-H., Yu, C.-J., and
Yang, P.-C. (2011) Sialylation and fucosylation of epidermal growth
factor receptor suppress its dimerization and activation in lung cancer
cells, Proc. Natl. Acad. Sci. USA, 108,
11332-11337.
52.Smedsrod, B., De Bleser, P., Braet, F.,
Lovisetti, P., Vanderkerken, K., Wisse, E., and Geerts, A. (1994) Cell
biology of liver endothelial and Kupffer cells, Gut, 35,
1509-1516.
53.Smedsrod, B., Melkko, J., Araki, N., Sano, H.,
and Horiuchi, S. (1997) Advanced glycation end products are eliminated
by scavenger-receptor-mediated endocytosis in hepatic sinusoidal
Kupffer and endothelial cells, Biochem. J., 322,
567-573.
54.Seternes, T., Sorensen, K., and Smedsrod, B.
(2002) Scavenger endothelial cells of vertebrates: a nonperipheral
leukocyte system for high-capacity elimination of waste macromolecules,
Proc. Natl. Acad. Sci. USA, 99, 7594-7597.
55.Chandler, K., and Goldman, R. (2013) Glycoprotein
disease markers and single protein omics, Mol. Cell. Proteom.,
12, 836-845.
56.Yates Iii, J. R., Gilchrist, A., Howell, K. E.,
and Bergeron, J. J. (2005) Proteomics of organelles and large cellular
structures, Nature Rev. Mol. Cell Biol., 6, 702-714.
57.Racanelli, V., and Rehermann, B. (2006) The liver
as an immunological organ, Hepatology, 43, S54-S62.
58.Sawamura, T., Nakada, H., Hazama, H., Shiozaki,
Y., Sameshima, Y., and Tashiro, Y. (1984) Hyperasialoglycoproteinemia
in patients with chronic liver diseases and/or liver cell carcinoma.
Asialoglycoprotein receptor in cirrhosis and liver cell carcinoma,
Gastroenterology, 87, 1217-1221.
59.Burgess, J. B., Baenziger, J. U., and Brown, W.
R. (1992) Abnormal surface distribution of the human asialoglycoprotein
receptor in cirrhosis, Hepatology, 15, 702-706.
60.Braet, F., and Wisse, E. (2002) Structural and
functional aspects of liver sinusoidal endothelial cell fenestrae: a
review, Comp. Hepatol., 1, 1.
61.Bedossa, P., and Paradis, V. (2003) Liver
extracellular matrix in health and disease, J. Pathol.,
200, 504-515.
Supplementary TABLES S1-S5 (MS Excel)
Supplementary TABLE S6 (MS Excel)
Supplementary FIGS S1-S3 (PDF)