REVIEW: Cytochrome bd Protects Bacteria against Oxidative and Nitrosative Stress: A Potential Target for Next-Generation Antimicrobial Agents
V. B. Borisov1*, E. Forte2, S. A. Siletsky1, M. Arese2, A. I. Davletshin1,3, P. Sarti2,4, and A. Giuffrè4
1Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119991 Moscow, Russia; fax: (495) 939-3181; E-mail: bor@genebee.msu.su2Department of Biochemical Sciences and Istituto Pasteur – Fondazione Cenci Bolognetti, Sapienza University of Rome, I-00185 Rome, Italy
3Institute of Oriental and Classical Studies, Russian State University for the Humanities, 125993 Moscow, Russia
4CNR Institute of Molecular Biology and Pathology, I-00185 Rome, Italy
* To whom correspondence should be addressed.
Received December 22, 2014; Revision received January 23, 2015
Cytochrome bd is a terminal quinol oxidase of the bacterial respiratory chain. This tri-heme integral membrane protein generates a proton motive force at lower efficiency than heme-copper oxidases. This notwithstanding, under unfavorable growth conditions bacteria often use cytochrome bd in place of heme-copper enzymes as the main terminal oxidase. This is the case for several pathogenic and opportunistic bacteria during host colonization. This review summarizes recent data on the contribution of cytochrome bd to bacterial resistance to hydrogen peroxide, nitric oxide, and peroxynitrite, harmful species produced by the host as part of the immune response to microbial infections. Growing evidence supports the hypothesis that bd-type oxidases contribute to bacterial virulence by promoting microbial survival under oxidative and nitrosative stress conditions. For these reasons, cytochrome bd represents a protein target for the development of next-generation antimicrobials.
KEY WORDS: antimicrobial agents, bacteria, oxidative stress, nitrosative stress, reactive oxygen and nitrogen species, respiratory chain, terminal oxidase, virulenceDOI: 10.1134/S0006297915050077
Abbreviations: k, observed rate constant; Ki, apparent inhibition constant; ONOO–, peroxynitrite; TMPD, N,N,N′,N′-tetramethyl-p-phenylenediamine.
GENERAL PROPERTIES
Cytochrome bd is a quinol:O2 oxidoreductase of the prokaryotic respiratory chain [1-3] that has not yet been identified in eukaryotic organisms [4]. The enzyme catalyzes the four-electron reduction of molecular oxygen to water using quinols as electron donors [5, 6]. The energy released in the redox reaction is stored in the form of a transmembrane electrical potential difference [7] through a molecular mechanism that has not been fully elucidated. It is assumed that the membrane potential is mainly created by the vectorial movement of protons through a proton transfer pathway that runs from the cytoplasm to the active site located on the opposite, periplasmic side of the membrane [8-12]. It has been found that, unlike heme-copper oxidases, cytochrome bd does not function as a proton pump [8-15]. Thus, it works with a lower energy efficiency as compared to heme-copper respiratory enzymes. For cytochrome bd, the H+/e– ratio (the number of protons transported across the membrane upon the transfer of one electron) is equal to 1, whereas for most heme-copper oxidases is equal to 2 [12, 16-19].
Cytochrome bd oxidases have been identified in both harmless and pathogenic bacteria, such as Mycobacterium tuberculosis [20], Klebsiella pneumoniae [21], Shigella flexneri [22], Listeria monocytogenes [23], Streptococcus [24], Brucella [25, 26], Salmonella [27, 28], and members of the Bacteroides class [29]. In these pathogens, a positive correlation between virulence and the expression level of cytochrome bd was noted [30].
Cytochrome bd expression is enhanced under unfavorable growth conditions, for example at low oxygen tension, in the presence of poisons (cyanide) [31] or uncouplers (protonophore) [32] in the environment, upon alkalization of the medium [31], or at high temperature [33]. In nitrogen-fixing bacteria, cytochrome bd contributes to protect nitrogenase against oxygen inactivation [34-36]. Cytochrome bd oxidase was also found to extend the oxygen concentration range at which anoxygenic phototrophic bacteria can grow [37]. Moreover, cytochrome bd-I from Escherichia coli participates in the regulation of disulfide bond formation during protein folding [38], as well as in heme biosynthesis (namely, at the level of the protoporphyrinogen IX oxidase enzyme) [39].
The three-dimensional structure of cytochrome bd is still unknown. The enzyme exhibits no homology with any heme-copper or alternative cyanide-resistant terminal oxidase [1, 7, 40, 41]. The enzymes from E. coli and Azotobacter vinelandii are mainly isolated as stable oxygenated complexes [42-44]. This is probably due to the high affinity of the enzyme for oxygen [45, 46]. Cytochrome bd-I from E. coli has been studied in detail. Until recently, the enzyme had been assumed to be composed of only two different integral membrane polypeptides, subunits I (CydA, 57 kDa) and II (CydB, 43 kDa). However, more recently it was shown that cytochrome bd contains an additional (4 kDa) polypeptide, CydX [47, 48]. This small polypeptide was suggested to be the third subunit of the oxidase, as it is required for maintenance of the enzyme activity and stabilization of the heme prosthetic groups [47-49].
Cytochrome bd has no copper, but it contains three redox-active hemes: the low-spin heme b558 and the high-spin hemes b595 and d [50, 51]. Heme b558 is located on subunit I, whereas hemes b595 and d are probably located at the interface between subunits I and II [52]. According to current views, the three hemes are all located closer to the outer (periplasmic) side of the membrane [53]. The hexacoordinate heme b558 is likely involved in quinol oxidation, whereas heme d binds molecular oxygen, being directly involved in its four-electron reduction to H2O. The role of the pentacoordinate heme b595 is still unclear; some authors suggested that its function is to mediate the electron transfer between heme b558 and heme d [54, 55]. According to other researchers, heme b595 represents a second redox center capable of reacting with oxygen [56, 57]. Finally, the data obtained with the enzymes from E. coli and A. vinelandii suggest that heme b595 may participate in the reduction of oxygen, forming together with heme d a di-heme oxygen-reducing center, similarly to the heme/Cu binuclear center in heme-copper oxidases [9, 58-68]. On the other hand, cytochrome bd from Geobacillus thermodenitrificans revealed no significant interaction between hemes b595 and d, at variance from cytochrome bd-I from E. coli [69]. This may reflect substantial differences in the arrangement of the active center between the two enzymes.
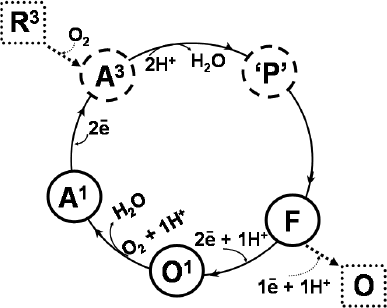
It is widely accepted that, during the catalytic cycle (Fig. 1), cytochrome bd undergoes the following transitions:
A1 → A3 → 'P' → F → O1 → A1,
where A1 and A3 are the heme d ferrous oxygenated forms of the enzyme with one (b5583+b5953+d2+–O2) and three (b5582+b5952+d2+–O2) electrons, respectively; 'P' is a short-lived state originally proposed to be a peroxo intermediate of heme d; F is the intermediate with ferryl heme d (b5583+b5953+d4+=O2–), and O1 is the one-electron-reduced form of the enzyme with ferric hemes d and b595 (b5582+b5953+d3+–OH). Under steady-state conditions, the A1 and F forms of the enzyme predominate, being the main catalytic intermediates [70]. Consistently, these intermediates are detected in preparations of the isolated and membrane-bound enzyme. Under the same conditions, a small fraction of the O1 intermediate is also detected [70]. A3 and 'P' are short-lived species that at room temperature can only be detected by “fast kinetic” methods [10, 11]. The existence of the 'P' intermediate was first reported by Belevich et al. [10]. The 'P' compound is possibly a ferryl intermediate, but with a π-cation radical on the porphyrin ring of heme d and one electron on heme b558 (b5582+b5953+d*4+=O2–) [71]. The fully oxidized (O, b5583+b5953+d3+–OH) and fully reduced (R3, b5582+b5952+d2+) forms (Fig. 1) most likely are not intermediates of the catalytic cycle [70-72], but can be obtained artificially. It is worth noting that the ferryl complex of cytochrome d, very similar spectroscopically to the bona fide catalytic intermediate F [10, 11], can be obtained by adding an excess of hydrogen peroxide to the enzyme, either “air-oxidized” or treated with a lipophilic oxidant [8, 73-75]. In the latter case, the reaction with hydrogen peroxide is quite fast, proceeding at an observed second-order rate constant k of 600 M–1·s–1 [74].
Fig. 1. Catalytic cycle of cytochrome bd. Solid arrows show the catalytic reaction pathway. Dotted arrows indicate transitions that are not part of the catalytic cycle.
INTERACTION WITH HYDROGEN PEROXIDE
(H2O2)
A large body of evidence suggests that cytochrome bd contributes to bacterial resistance against the oxidative stress induced by hydrogen peroxide. Escherichia coli cytochrome bd-defective mutants are extremely sensitive to H2O2 [33]. Accordingly, the expression level of cytochrome bd in E. coli increases upon exposure to the peroxide [76]. Korshunov and Imlay [77], using an E. coli strain devoid of some antioxidant enzymes (the KatG and KatE catalases and the NADH-peroxidase Ahp), showed that, upon a sudden switch from anaerobic to aerobic growth conditions, a bd-type enzyme reduces the formation of intracellular H2O2. They suggested that cytochrome bd accomplishes this function indirectly, by diverging reducing equivalents from fumarate reductase, a key H2O2-generator [77].
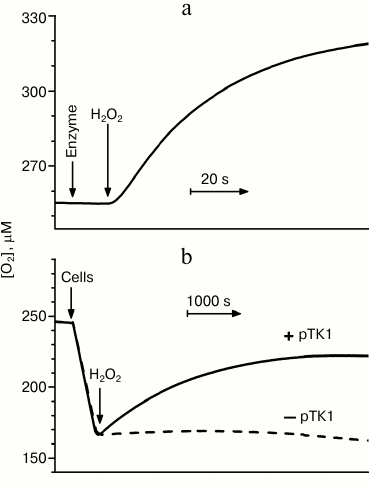
Recently, we found that cytochrome bd-I from E. coli is able also to directly decompose H2O2 [78, 79]. In all likelihood, for this purpose the enzyme can use two different mechanisms, exhibiting both catalase [78] and peroxidase [79] activity. A high catalase activity of cytochrome bd-I was described by Borisov et al. [78]. This activity was assessed polarographically by measuring the rate of O2 formation upon addition of H2O2 to the enzyme [78]. The activity was observed with both the isolated solubilized enzyme (Fig. 2a) and in cells of the catalase-deficient E. coli UM2 strain overexpressing cytochrome bd-I (solid line in Fig. 2b).
The reaction of H2O2 with the isolated enzyme was studied in more detail [78]. It turned out that the oxidase exhibits notable catalase activity not only in the “air-oxidized” state (Fig. 2a), but also in turnover with O2 (in the presence of ubiquinol-1 and an excess of dithiothreitol, serving as the reducing system). In turnover, the catalase and oxygen reductase activities were found not to compete with each other, suggesting that different active sites are responsible for these activities in the enzyme. Upon O2 depletion and consequent full reduction of the enzyme, the catalase activity disappears. The activity thus depends on the redox state of cytochrome bd-I.
As expected, the reaction rate was found to be proportional to the concentration of the oxidase, as well as to the concentration of H2O2 up to 0.5 mM. At higher H2O2 concentrations, the reaction rate tends to saturate, possibly due to a partial inactivation of the enzyme. It was shown that the reaction product (O2) does not inhibit the catalase activity of the enzyme, as almost identical rates have been measured under aerobic (~255 µM O2) and microaerobic (3-15 µM O2) conditions. The reaction proceeds with the formation of about half a mole of O2 per mole of H2O2, whereas no O2 generation was observed in control experiments (i.e. with the thermoinactivated enzyme, in the absence of the enzyme, or without the substrate).
Fig. 2. Catalase activity of cytochrome bd-I from E. coli. a) Effect of addition of 100 µM H2O2 to the isolated solubilized enzyme. b) Effect of addition of 235 µM H2O2 to catalase-deficient E. coli cells of the UM2 strain (devoid of the KatG and KatE catalases). The cells, when overexpressing cytochrome bd (due to the presence of the pTK1 plasmid carrying the operon encoding cytochrome bd-I), show catalase activity (solid line, +pTK1). In the absence of cytochrome bd-I overexpression, the cells do not exhibit a notable catalase activity (dashed line, –pTK1). The experimental details are given in Borisov et al. [78].
A number of experiments have been carried out with different inhibitors to gain insight into the nature of the site responsible for the observed catalase activity. The lack of effects by N-ethylmaleimide rules out that thiol groups of the protein are responsible for the catalase reaction. The involvement of a quinol binding site was also excluded as the reaction proved to be insensitive to antimycin A. Inhibitors targeting the reduced heme d, such as NO and CO, did not affect the catalase activity, thus arguing against a participation of this heme in the reaction. The lack of effects by NO, an effective inhibitor of bona fide catalases [78, 80], allowed us to exclude that the observed catalase activity was due to contaminant catalases in the cytochrome bd preparations.
The molecular mechanism at the basis of the catalase activity of cytochrome bd is unclear. The catalase activity of the enzyme is three orders of magnitude more sensitive to cyanide than the oxygen reductase one [78], pointing to the participation of a heme group in the catalase activity. As the oxygen reductase activity is directly linked to heme d and this activity does not compete with the catalase reaction, heme d should not be involved in the latter reaction. On this basis, it was tentatively suggested that the site responsible for the catalase activity was the high-spin heme b595 [78]. It was found that cyanide at a concentration completely inhibiting the catalase activity causes only small changes in the absorption spectrum of the “air-oxidized” enzyme, accounting for a binding of this ligand to maximally 4% of the hemes b. Therefore, in the preparations of the solubilized cytochrome bd-I, only a small fraction of the enzyme is endowed with the catalase activity (with a high apparent turnover number of at least 3250 s–1) [78]. It is important to emphasize that the observed catalase activity is not an artifact of the isolation/purification procedure and is not due to the contamination by a bacterial bona fide catalase, as cytochrome bd-I, when overexpressed, shows a notable catalase activity in intact cells devoid of the KatG and KatE catalases (solid line in Fig. 2b). Thus, one can conclude that in E. coli cytochrome bd-I can have a protective role against oxidative stress, in addition to the bacterial catalases.
Cytochrome bd-I from E. coli was shown to be endowed also with a peroxidase activity in the presence of different substrates, such as guaiacol, ferrocene, benzohydroquinone, and potassium ferrocyanide [79]. The guaiacol peroxidase activity was investigated in more detail. In particular, inhibitors of the oxygen reductase activity of the enzyme, such as cyanide, pentachlorophenol, and 2-n-heptyl 4-hydroxyquinoline-N-oxide proved to inhibit the peroxidase activity of E. coli cytochrome bd-I to a similar extent [79]. It is assumed that guaiacol donates electrons to cytochrome bd-I via a quinol binding site, and the reduction of H2O2 occurs in the oxygen-reducing center. Although the peroxidase activity of the solubilized enzyme towards guaiacol appeared to be rather low (the apparent turnover number was ~4 s–1), such an activity in vivo, i.e. with the physiological electron donors (such as quinols), may be much higher, thereby contributing to H2O2 detoxification in the bacterial cell.
The molecular mechanism through which the peroxide-utilizing activities of cytochrome bd-I are regulated in the E. coli cell is still unknown, as well as whether bd-type oxidases from other pathogenic bacteria are endowed with such activities. In this regard, it is interesting to note that disruption of the cytochrome c maturation system in Mycobacterium tuberculosis, the causative agent of tuberculosis, was reported to lead to a significant increase in both the expression of cytochrome bd and bacterial resistance to H2O2 [81]. This observation suggests that in M. tuberculosis the bd-type oxidase plays a role in protecting the pathogen against oxidative stress, by metabolizing H2O2 through a catalase and/or a peroxidase activity [82].
INTERACTION WITH NITRIC OXIDE (NO)
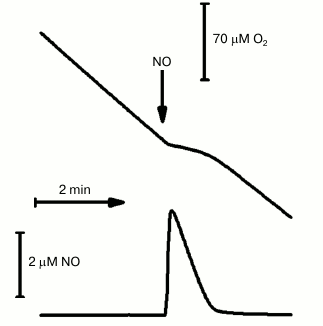
NO is produced by the host cell as part of the immune response to microbial infections. Interestingly, in some bacteria (E. coli [83], Staphylococcus aureus [84], M. tuberculosis [20], Desulfovibrio gigas [85], Bacillus subtilis [86]) NO induces expression of cytochrome bd-encoding genes. In this regard, investigating the interaction of cytochrome bd with NO is of particular interest. It was found that NO effectively inhibits the oxygen reductase activity of the bd-oxidases from E. coli and A. vinelandii [87]. At [NO] > 0.5 µM, a fast and complete inhibition of the activity of these enzymes is achieved. Cytochrome bd-I from E. coli was investigated in more detail. It was shown that following the removal of the added (<1 µM) NO from the solution, the oxygen reductase activity of the enzyme is recovered quickly and completely [87]. In contrast, at higher (micromolar) concentrations of NO, a small (<15%) irreversible inhibition of the enzyme was observed (Fig. 3, see also Borisov et al. [88]). For cytochrome bd-I from E. coli, at [O2] = 70 µM the value of the apparent inhibition constant (Ki) for NO is equal to 100 nM [87], close to the Ki value determined for the mitochondrial cytochrome c oxidase under similar experimental conditions [89]. After increasing the concentration of O2 in solution (up to 1 mM), the observed Ki value for NO appeared to be significantly higher (230 nM) [87]. This finding suggests a competition between NO and O2 binding to reduced unliganded heme d:
Fed2+ + NO → Fed2+–NO.
Since the rate constants for the binding of NO and O2 to ferrous heme d are likely similar [2], at low [NO]/[O2] ratio the onset of the inhibition is expected to be slow. However, this is not the case, as the enzyme is inhibited by NO rather quickly even at [NO]/[O2] ~ 0.005 [87]. This is probably due to the fact that NO can “trap” not only the unliganded ferrous heme d, but also some catalytic intermediates of the enzyme that are unreactive towards molecular oxygen. Accordingly, we have recently shown that the O2-unreactive A1 and F intermediates, prevailing under steady-state conditions [70], are able to react with NO [75, 90]. Upon interacting with the A1 intermediate, NO displaces O2 from heme d, eventually yielding the heme iron nitrosyl complex. In this reaction, the rate-limiting step is the dissociation of O2 from heme d (k = 78 s–1) [90]:
Fed2+–O2 → Fed2+ + O2,
Fed2+ + NO → Fed2+–NO.
Fig. 3. Inhibition by NO of the isolated solubilized cytochrome bd-I from E. coli. The O2 and NO traces were recorded in parallel. The O2-reductase activity of the enzyme was sustained with the reducing substrates 10 mM ascorbate and 0.5 mM TMPD. NO added, 4 µM; cytochrome bd-I, 100 nM. The experimental details are given in Borisov et al. [88].
Cytochrome bd apparently is not endowed with a NO reductase activity (i.e. it is unable to reduce NO to N2O) [87]. The reaction of nitric oxide with the F intermediate leads to the formation of a complex of the fully oxidized enzyme with nitrite bound at the ferric heme d. The reaction is quite fast (k ~ 105 M–1·s–1, for cytochrome bd from A. vinelandii [75]) and likely proceeds according to the following mechanism:
Fed4+=O2– + NO → Fed3+–NO2–.
In this reaction, NO is oxidatively degraded into the much less toxic nitrite. The reaction might therefore be physiologically relevant as a defense mechanism against NO.
Considering the interaction of NO with the different forms of cytochrome bd, it is worth mentioning that this ligand also reacts with heme d in the fully oxidized enzyme with a rate constant k ~ 102 M–1·s–1, forming a heme d nitrosyl adduct [91]:
Fed3+ + NO → Fed3+–NO ↔ Fed2+–NO+.
Importantly, after exhaustion of NO in the medium, the recovery of cytochrome bd-I activity occurs much faster than in the case of the mitochondrial cytochrome c oxidase [2, 87]. This happens because in the fully reduced isolated bacterial enzyme NO dissociates from heme d at a much higher rate than from heme a3 in the mitochondrial enzyme (k = 0.133 s–1 [90] versus k = 0.0035 s–1 [92]). The high rate of NO dissociation from cytochrome bd-I [90] has been also confirmed in intact E. coli cells [93]. Another important observation made on the isolated cytochrome bd-I is that the rate constant of the dissociation of NO from the completely reduced enzyme (k = 0.133 s–1) is significantly higher than koff from the one-electron-reduced enzyme (k = 0.036 s–1) [90]. This suggests that the redox state of the hemes b (most likely heme b595) affects the stability of the ferrous nitrosyl heme d complex, the rate of NO dissociation being maximal when heme b595 is in the reduced state. This unique ability of cytochrome bd to rapidly dissociate NO from the active site may explain why under specific conditions this particular oxidase is preferentially expressed in place of a heme-copper enzyme. The faster NO dissociation is indeed expected to speed the recovery of bacterial respiration from NO inhibition, thus conferring to the microorganism a higher resistance to nitrosative stress. Interestingly, it has been recently found that the bd-type terminal oxidase from Shewanella oneidensis also makes this Gram-negative facultative anaerobe more resistant to nitrite and NO under aerobic growth conditions [94, 95].
INTERACTION WITH PEROXYNITRITE (ONOO–)
In response to bacterial infection, cells of the mammalian immune system produce both nitric oxide (NO) and superoxide anion at high concentrations by activating the NO synthases and NADPH oxidase, respectively. As a result of the diffusion-controlled reaction of NO with superoxide anion, peroxynitrite (ONOO–) is formed. Peroxynitrite is a highly reactive toxic compound, which gives rise to both oxidative and nitrosative stress in bacteria [96]. Upon penetrating inside the bacterial cell, ONOO– can cause protein modifications [97, 98], lipid oxidation [99], and DNA damage [100].
As mentioned above, some pathogenic bacteria express cytochrome bd as the terminal oxidase of the respiratory chain during host infection [6]. It is therefore important to know (i) the extent of bd-type terminal oxidase sensitivity to ONOO–, and (ii) the benefits, in terms of resistance to oxidative and nitrosative stress, granted to a pathogen expressing a bd-type rather than a heme-copper oxidase, such as cytochrome c oxidase. Previously, Cooper et al. studied in detail the interaction of cytochrome c oxidase isolated from mitochondria with ONOO–, and they reported that ONOO– causes the irreversible inhibition of the purified cytochrome c oxidase [101-103].
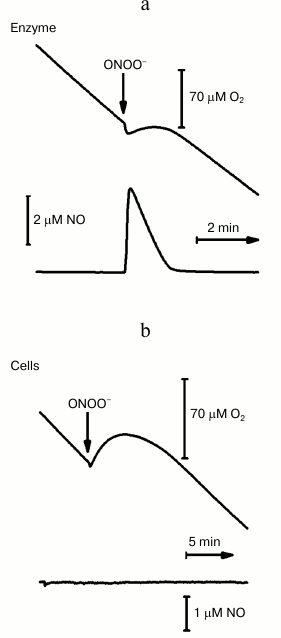
Recently, we investigated the effect of ONOO– on the oxygen reductase activity of cytochrome bd-I from E. coli [88] and found that upon adding ONOO– to the isolated solubilized enzyme in turnover with O2 (with an excess of the reductants ascorbate and TMPD), two events can be observed: a transient stop of the oxygen consumption and the formation of NO (Fig. 4a). For example, upon addition of 50 µM ONOO–, a release of 4 µM NO was observed. Once the NO disappears from the solution by reacting with O2 and the ferryl complex of cytochrome bd-I, the oxygen consumption activity of the enzyme resumes. Detailed analysis of the oxygen consumption rates measured before and after addition of ONOO– showed that, if the concentration of NO formed following the addition of ONOO– is greater than 1 µM, the enzymatic activity does not return to its initial level. Virtually identical results have been obtained in control experiments upon adding authentic NO instead of ONOO– (Fig. 3). The latter result suggests that the small irreversible inhibition observed after addition of high ONOO– concentrations has to be attributed to NO rather than to ONOO–. The maximum irreversible inhibition (~15%) has been observed at 6 µM NO, either exogenously added or produced following the addition of 100 µM ONOO– [88]. It is important to emphasize that, upon addition of ONOO– to cytochrome bd-I overexpressing E. coli cells, a rapid and reversible stop of the oxygen consumption is also observed (Fig. 4b). In this case, however, following the addition of high concentrations of ONOO– to the cells, the extent of the irreversible inhibition was very small (<5%), in agreement with the finding that no noticeable amounts of NO are produced (Fig. 4b) under these experimental conditions. This led us to conclude that the oxygen reductase activity of cytochrome bd-I, as isolated or in cells, is not inhibited by ONOO– per se [88]. It is interesting to note that in both cases (isolated enzyme or cells) the addition ONOO– not only leads to a temporary stop of the oxygen consumption, but also to a short-term formation of a small amount of O2 (see the increase in the oxygen trace in Fig. 4). Since H2O2 may be a contaminant of the commercial preparations of ONOO– and/or a secondary product of ONOO– decay, we assume that the observed evolution of O2 is a consequence of the NO-insensitive catalase activity of cytochrome bd-I [78, 88].
Fig. 4. Effect of peroxynitrite on the oxygen consumption rate by cytochrome bd-I from E. coli. a) ONOO– (50 µM) was added to the isolated solubilized enzyme (100 nM) in turnover with O2 and excess reductants (10 mM ascorbate and 0.5 mM TMPD). b) ONOO– (80 µM) was added to the respiring (due to endogenous substrates) E. coli cells (of the GO105 strain devoid of cytochrome bo3) overexpressing the bd-I oxidase (due to the presence of the pTK1 plasmid carrying the operon encoding this enzyme). The experimental details are given in Borisov et al. [88].
We have also directly measured the peroxynitrite-detoxifying activity of cytochrome bd-I isolated from E. coli by stopped-flow absorption spectroscopy. In these experiments, the ONOO– concentration was monitored over time measuring the absorption at 310 nm. A number of control experiments (without the enzyme or the reducing substrates or ONOO–) was also carried out, whose results were taken into account to calculate the rate constant of the enzymatic ONOO– decomposition by cytochrome bd-I in turnover with O2 and excess ascorbate and TMPD reductants. As expected, this observed rate constant was found to increase linearly with the concentration of the enzyme [88]. Moreover, upon increasing the concentration of TMPD from 150 to 300 µM, at each cytochrome bd-I concentration tested, a faster decay of ONOO– was observed, consistent with an increase in the apparent turnover number of the ONOO–-detoxifying activity of the enzyme, from 7 to 10 moles ONOO–/mole enzyme per second [88].
All in all, we have found that (i) differently from mitochondrial cytochrome c oxidase, cytochrome bd-I from E. coli is not inactivated by ONOO– up to a concentration of 100 µM; (ii) the bd-I enzyme, in turnover with oxygen and the reducing substrates, is capable of metabolizing ONOO– quite rapidly, thus acting as a detoxifying agent for this highly reactive toxic compound. To our knowledge, this has been the first time that the kinetics of ONOO– decomposition by a terminal oxidase was directly measured [88].
PHARMACOLOGICAL PREPARATIONS – SPECIFIC INHIBITORS OF
ENERGY METABOLISM IN PATHOGENIC BACTERIA
To combat pathogenic microorganisms, a number of antibiotics with different modes of action have been used for many decades. However, in recent years there has been a significant increase in the resistance (including multidrug resistance) of pathogenic microorganisms to antimicrobial drugs, particularly to antibiotics that in the past were highly effective. As a result, the infectious diseases caused by such resistant pathogens are not amenable to standard treatment, thus leading to prolonged illness and increased risk of death. Hence, there is an urgent need for the development of new antimicrobial drugs acting through novel mechanisms. In this regard, the enzymes playing a central role in the energy metabolism represent suitable targets for novel antibacterial compounds.
Cytochrome bd oxidase is emerging as one of such targets. As indicated above, the enzyme is able to significantly increase the resistance of a microbial cell to oxidative and nitrosative stress, thereby presumably helping a bacterial pathogen to evade the host immune defense. Since cytochrome bd is found only in bacteria, selective inhibitors of bd-type oxidases should not have negative effects on the host energy metabolism.
The development of next-generation drugs by the world pharmaceutical industry is only in its infancy. The first next-generation drug targeting the energy metabolism of a bacterial pathogen is bedaquiline (diarylquinoline). The bactericidal effect of this anti-tuberculosis drug approved by the U.S. Food and Drug Administration (U.S. FDA) is due its ability to selectively inhibit the F1Fo-ATP synthase of Mycobacterium tuberculosis [104]. The inhibition of ATP synthesis leads to disruption of the energy production system and, eventually, to the death of the microbial cell. However, compared to other frontline anti-tuberculosis drugs, such as isoniazid, bedaquiline kills the M. tuberculosis cells rather slowly [105]. Recently, we raised the hypothesis that cytochrome bd could confer to M. tuberculosis cells higher resistance to oxidative stress, thanks to its ability to degrade hydrogen peroxide [82]. Following our work, Berney et al. reported that the rate at which bedaquiline kills the M. tuberculosis cells increases remarkably, if the bd-type terminal oxidase is knocked-out (by replacing the cydA gene with a hygromycin cassette by using specialized transduction) [106]. This is consistent with the observation that in M. tuberculosis cells treated with bedaquiline the expression levels of cytochrome bd increase substantially [105].
Another drug targeting the respiratory chain of M. tuberculosis is compound Q203, based on imidazo[1,2-α]pyridine [107]. Its action is based on the inhibition of the bc1 respiratory complex [107]. Recently, the effect of a series of five different scaffolds, imidazo[1,2-α]pyridine derivatives, on clinical strains of M. tuberculosis was reported [108]. Although imidazo[1,2-α]pyridines completely inhibit the growth of most of the M. tuberculosis strains tested by inhibiting the bc1 complex, the laboratory-adapted strains H37Rv, CDC1551, and Erdman appeared to overcome this growth inhibition [108]. This was suggested to be due to an increase in the cytochrome bd expression levels. Indeed, deletion of this terminal oxidase in the H37Rv strain makes the mutant more sensitive to imidazo[1,2-α]pyridines [108].
In summary, it is hoped that the use of a specific inhibitor of cytochrome bd in combination with inhibitors of other enzymes of the energy metabolism (e.g. bedaquiline and/or imidazo[1,2-α]pyridines) will have a synergistic effect, thereby representing an innovative pharmacological strategy to fight bacterial pathogens.
The authors are grateful to Dr. A. A. Konstantinov and Dr. V. P. Skulachev for their interest in this work, useful discussions, and critical remarks.
The reported study was partially supported by the Russian Foundation for Basic Research (research projects No. 14-04-00153-a and 15-04-06266-a), and by Ministero dell’Istruzione, dell’Universita e della Ricerca of Italy (PNR-CNR Aging Program 2012-2014, FIRB RBIN06E9Z8 and PRIN 20107Z8XBW_005).
REFERENCES
1.Poole, R. K., and Cook, G. M. (2000) Redundancy of
aerobic respiratory chains in bacteria? Routes, reasons and regulation,
Adv. Microb. Physiol., 43, 165-224.
2.Giuffrè, A., Borisov, V. B., Arese, M.,
Sarti, P., and Forte, E. (2014) Cytochrome bd oxidase and
bacterial tolerance to oxidative and nitrosative stress, Biochim.
Biophys. Acta, 1837, 1178-1187.
3.Borisov, V. B. (1996) Cytochrome bd:
structure and properties, Biochemistry (Moscow), 61,
565-574.
4.Gavrikova, E. V., Grivennikova, V. G., Borisov, V.
B., Cecchini, G., and Vinogradov, A. D. (2009) Assembly of a chimeric
respiratory chain from bovine heart submitochondrial particles and
cytochrome bd terminal oxidase of Escherichia coli,
FEBS Lett., 583, 1287-1291.
5.Borisov, V. B., and Verkhovsky, M. I. (2009) Oxygen
as acceptor, in EcoSal Plus – Cellular and Molecular Biology
of E. coli, Salmonella, and the Enterobacteriaceae; doi:
10.1128/ecosalplus.3.2.7 (http://www.asmscience.org/content/journal/ecosalplus/10.1128/ecosalplus.3.2.7)
(Stewart, V., ed.) ASM Press, Washington, DC, pp. 1-31.
6.Giuffrè, A., Borisov, V. B., Mastronicola,
D., Sarti, P., and Forte, E. (2012) Cytochrome bd oxidase and
nitric oxide: from reaction mechanisms to bacterial physiology, FEBS
Lett., 586, 622-629.
7.Borisov, V. B., Gennis, R. B., Hemp, J., and
Verkhovsky, M. I. (2011) The cytochrome bd respiratory oxygen
reductases, Biochim. Biophys. Acta, 1807, 1398-1413.
8.Jasaitis, A., Borisov, V. B., Belevich, N. P.,
Morgan, J. E., Konstantinov, A. A., and Verkhovsky, M. I. (2000)
Electrogenic reactions of cytochrome bd, Biochemistry,
39, 13800-13809.
9.Belevich, I., Borisov, V. B., Zhang, J., Yang, K.,
Konstantinov, A. A., Gennis, R. B., and Verkhovsky, M. I. (2005)
Time-resolved electrometric and optical studies on cytochrome bd
suggest a mechanism of electron-proton coupling in the di-heme active
site, Proc. Natl. Acad. Sci. USA, 102, 3657-3662.
10.Belevich, I., Borisov, V. B., and Verkhovsky, M.
I. (2007) Discovery of the true peroxy intermediate in the catalytic
cycle of terminal oxidases by real-time measurement, J. Biol.
Chem., 282, 28514-28519.
11.Borisov, V. B., Belevich, I., Bloch, D. A., Mogi,
T., and Verkhovsky, M. I. (2008) Glutamate 107 in subunit I of
cytochrome bd from Escherichia coli is part of a
transmembrane intraprotein pathway conducting protons from the
cytoplasm to the heme b595/heme d active site,
Biochemistry, 47, 7907-7914.
12.Borisov, V. B., Murali, R., Verkhovskaya, M. L.,
Bloch, D. A., Han, H., Gennis, R. B., and Verkhovsky, M. I. (2011)
Aerobic respiratory chain of Escherichia coli is not allowed to
work in fully uncoupled mode, Proc. Natl. Acad. Sci. USA,
108, 17320-17324.
13.Puustinen, A., Finel, M., Haltia, T., Gennis, R.
B., and Wikström, M. (1991) Properties of the two terminal
oxidases of Escherichia coli, Biochemistry, 30,
3936-3942.
14.Bertsova, Y. V., Bogachev, A. V., and Skulachev,
V. P. (1997) Generation of protonic potential by the bd-type
quinol oxidase of Azotobacter vinelandii, FEBS Lett.,
414, 369-372.
15.Kolonay, J. F., Jr., and Maier, R. J. (1997)
Formation of pH and potential gradients by the reconstituted
Azotobacter vinelandii cytochrome bd respiratory
protection oxidase, J. Bacteriol., 179,
3813-3817.
16.Siletsky, S. A., and Konstantinov, A. A. (2012)
Cytochrome c oxidase: charge translocation coupled to
single-electron partial steps of the catalytic cycle, Biochim.
Biophys. Acta, 1817, 476-488.
17.Siletsky, S. A. (2013) Steps of the coupled
charge translocation in the catalytic cycle of cytochrome c
oxidase, Front. Biosci., 18, 36-57.
18.Siletsky, S. A., Belevich, I., Soulimane, T.,
Verkhovsky, M. I., and Wikström, M. (2013) The fifth electron in
the fully reduced caa3 from Thermus
thermophilus is competent in proton pumping, Biochim. Biophys.
Acta, 1827, 1-9.
19.Siletsky, S. A., Belevich, I., Jasaitis, A.,
Konstantinov, A. A., Wikström, M., Soulimane, T., and Verkhovsky,
M. I. (2007) Time-resolved single-turnover of ba3
oxidase from Thermus thermophilus, Biochim. Biophys.
Acta, 1767, 1383-1392.
20.Shi, L., Sohaskey, C. D., Kana, B. D., Dawes, S.,
North, R. J., Mizrahi, V., and Gennaro, M. L. (2005) Changes in energy
metabolism of Mycobacterium tuberculosis in mouse lung and under
in vitro conditions affecting aerobic respiration, Proc.
Natl. Acad. Sci. USA, 102, 15629-15634.
21.Juty, N. S., Moshiri, F., Merrick, M., Anthony,
C., and Hill, S. (1997) The Klebsiella pneumoniae cytochrome
bd terminal oxidase complex and its role in microaerobic
nitrogen fixation, Microbiology, 143, 2673-2683.
22.Way, S. S., Sallustio, S., Magliozzo, R. S., and
Goldberg, M. B. (1999) Impact of either elevated or decreased levels of
cytochrome bd expression on Shigella flexneri virulence,
J. Bacteriol., 181, 1229-1237.
23.Larsen, M. H., Kallipolitis, B. H., Christiansen,
J. K., Olsen, J. E., and Ingmer, H. (2006) The response regulator ResD
modulates virulence gene expression in response to carbohydrates in
Listeria monocytogenes, Mol. Microbiol.,
61, 1622-1635.
24.Yamamoto, Y., Poyart, C., Trieu-Cuot, P.,
Lamberet, G., Gruss, A., and Gaudu, P. (2005) Respiration metabolism of
group B Streptococcus is activated by environmental heme and
quinone and contributes to virulence, Mol. Microbiol.,
56, 525-534.
25.Endley, S., McMurray, D., and Ficht, T. A. (2001)
Interruption of the cydB locus in Brucella abortus
attenuates intracellular survival and virulence in the mouse model of
infection, J. Bacteriol., 183, 2454-2462.
26.Loisel-Meyer, S., Jimenez de Bagues, M. P.,
Kohler, S., Liautard, J. P., and Jubier-Maurin, V. (2005) Differential
use of the two high-oxygen-affinity terminal oxidases of Brucella
suis for in vitro and intramacrophagic multiplication,
Infect. Immun., 73, 7768-7771.
27.Zhang-Barber, L., Turner, A. K., Martin, G.,
Frankel, G., Dougan, G., and Barrow, P. A. (1997) Influence of genes
encoding proton-translocating enzymes on suppression of Salmonella
typhimurium growth and colonization, J. Bacteriol.,
179, 7186-7190.
28.Turner, A. K., Barber, L. Z., Wigley, P.,
Muhammad, S., Jones, M. A., Lovell, M. A., Hulme, S., and Barrow, P. A.
(2003) Contribution of proton-translocating proteins to the virulence
of Salmonella enterica serovars Typhimurium,
Gallinarum, and Dublin in chickens and mice, Infect.
Immun., 71, 3392-3401.
29.Baughn, A. D., and Malamy, M. H. (2004) The
strict anaerobe Bacteroides fragilis grows in and benefits from
nanomolar concentrations of oxygen, Nature, 427,
441-444.
30.Forte, E., Borisov, V. B., Konstantinov, A. A.,
Brunori, M., Giuffrè, A., and Sarti, P. (2007) Cytochrome
bd, a key oxidase in bacterial survival and tolerance to
nitrosative stress, Ital. J. Biochem., 56,
265-269.
31.Avetisyan, A. V., Bogachev, A. V., Murtasina, R.
A., and Skulachev, V. P. (1992) Involvement of a d-type oxidase
in the Na+-motive respiratory chain of Escherichia
coli growing under low
ΔμΗ conditions, FEBS
Lett., 306, 199-202.
32.Bogachev, A. V., Murtazina, R. A., Shestopalov,
A. I., and Skulachev, V. P. (1995) Induction of the Escherichia
coli cytochrome d by low ΔμH+
and by sodium ions, Eur. J. Biochem., 232,
304-308.
33.Wall, D., Delaney, J. M., Fayet, O., Lipinska,
B., Yamamoto, T., and Georgopoulos, C. (1992) arc-Dependent
thermal regulation and extragenic suppression of the Escherichia
coli cytochrome d operon, J. Bacteriol.,
174, 6554-6562.
34.Poole, R. K., and Hill, S. (1997) Respiratory
protection of nitrogenase activity in Azotobacter vinelandii –
roles of the terminal oxidases, Biosci. Rep., 17,
307-317.
35.Bertsova, Y. V., Demin, O. V., and Bogachev, A.
V. (2005) Respiratory protection of nitrogenase complex in
Azotobacter vinelandii, Uspekhi Biol. Khim., 45,
205-234.
36.Dincturk, H. B., Demir, V., and Aykanat, T.
(2011) Bd oxidase homologue of photosynthetic purple sulfur
bacterium Allochromatium vinosum is co-transcribed with a
nitrogen fixation related gene, Antonie van Leeuwenhoek,
99, 211-220.
37.Hassani, B. K., Steunou, A. S., Liotenberg, S.,
Reiss-Husson, F., Astier, C., and Ouchane, S. (2010) Adaptation to
oxygen: role of terminal oxidases in photosynthesis initiation in the
purple photosynthetic bacterium, Rubrivivax gelatinosus, J.
Biol. Chem., 285, 19891-19899.
38.Bader, M., Muse, W., Ballou, D. P., Gassner, C.,
and Bardwell, J. C. A. (1999) Oxidative protein folding is driven by
the electron transport system, Cell, 98, 217-227.
39.Mobius, K., Arias-Cartin, R., Breckau, D.,
Hannig, A. L., Riedmann, K., Biedendieck, R., Schroder, S., Becher, D.,
Magalon, A., Moser, J., Jahn, M., and Jahn, D. (2010) Heme biosynthesis
is coupled to electron transport chains for energy generation, Proc.
Natl. Acad. Sci. USA, 107, 10436-10441.
40.Van der Oost, J., deBoer, A. P. N., de Gier,
J.-W. L., Zumft, W. G., Stouthamer, A. H., and van Spanning, R. J. M.
(1994) The heme-copper oxidase family consists of three distinct types
of terminal oxidases and is related to nitric oxide reductase, FEMS
Microbiol. Lett., 121, 1-10.
41.Green, G. N., Fang, H., Lin, R.-J., Newton, G.,
Mather, M., Georgiou, C. D., and Gennis, R. B. (1988) The nucleotide
sequence of the cyd locus encoding the two subunits of the
cytochrome d terminal oxidase complex of Escherichia
coli, J. Biol. Chem., 263, 13138-13143.
42.Poole, R. K., Kumar, C., Salmon, I., and Chance,
B. (1983) The 650 nm chromophore in Escherichia coli is an
“Oxy-“ or oxygenated compound, not the oxidized form of
cytochrome oxidase d: a hypothesis, J. Gen.
Microbiol., 129, 1335-1344.
43.Kahlow, M. A., Loehr, T. M., Zuberi, T. M., and
Gennis, R. B. (1993) The oxygenated complex of cytochrome d
terminal oxidase: direct evidence for Fe-O2 coordination in
a chlorin-containing enzyme by resonance Raman spectroscopy, J. Am.
Chem. Soc., 115, 5845-5846.
44.Borisov, V. B., Smirnova, I. A.,
Krasnosel’skaya, I. A., and Konstantinov, A. A. (1994) Oxygenated
cytochrome bd from Escherichia coli can be converted into
the oxidized form by lipophilic electron acceptors, Biochemistry
(Moscow), 59, 437-443.
45.Belevich, I., Borisov, V. B., Konstantinov, A.
A., and Verkhovsky, M. I. (2005) Oxygenated complex of cytochrome
bd from Escherichia coli: stability and photolability,
FEBS Lett., 579, 4567-4570.
46.Belevich, I., Borisov, V. B., Bloch, D. A.,
Konstantinov, A. A., and Verkhovsky, M. I. (2007) Cytochrome bd
from Azotobacter vinelandii: evidence for high-affinity oxygen
binding, Biochemistry, 46, 11177-11184.
47.Van Orsdel, C. E., Bhatt, S., Allen, R. J.,
Brenner, E. P., Hobson, J. J., Jamil, A., Haynes, B. M., Genson, A. M.,
and Hemm, M. R. (2013) The Escherichia coli CydX protein is a
member of the CydAB cytochrome bd oxidase complex and is
required for cytochrome bd oxidase activity, J.
Bacteriol., 195, 3640-3650.
48.Hoeser, J., Hong, S., Gehmann, G., Gennis, R. B.,
and Friedrich, T. (2014) Subunit CydX of Escherichia coli
cytochrome bd ubiquinol oxidase is essential for assembly and
stability of the di-heme active site, FEBS Lett.,
588, 1537-1541.
49.Chen, H., Luo, Q., Yin, J., Gao, T., and Gao, H.
(2015) Evidence for requirement of CydX in function but not assembly of
the cytochrome bd oxidase in Shewanella oneidensis,
Biochim. Biophys. Acta, 1850, 318-328.
50.Lorence, R. M., Koland, J. G., and Gennis, R. B.
(1986) Coulometric and spectroscopic analysis of the purified
cytochrome d complex of Escherichia coli: evidence for
the identification of “cytochrome a1” as
cytochrome b595, Biochemistry, 25,
2314-2321.
51.Miller, M. J., Hermodson, M., and Gennis, R. B.
(1988) The active form of the cytochrome d terminal oxidase
complex of Escherichia coli is a heterodimer containing one copy
of each of the two subunits, J. Biol. Chem., 263,
5235-5240.
52.Newton, G., and Gennis, R. B. (1991) In
vivo assembly of the cytochrome d terminal oxidase complex
of Escherichia coli from genes encoding the two subunits
expressed on separate plasmids, Biochim. Biophys. Acta,
1089, 8-12.
53.Zhang, J., Barquera, B., and Gennis, R. B. (2004)
Gene fusions with β-lactamase show that subunit I of the
cytochrome bd quinol oxidase from E. coli has nine
transmembrane helices with the O2 reactive site near the
periplasmic surface, FEBS Lett., 561, 58-62.
54.Poole, R. K., and Williams, H. D. (1987) Proposal
that the function of the membrane-bound cytochrome
a1-like haemoprotein (cytochrome
b595) in Escherichia coli is a direct electron
donation to cytochrome d, FEBS Lett., 217,
49-52.
55.Hata-Tanaka, A., Matsuura, K., Itoh, S., and
Anraku, Y. (1987) Electron flow and heme-heme interaction between
cytochromes b-558, b-595 and d in a terminal
oxidase of Escherichia coli, Biochim. Biophys. Acta,
893, 289-295.
56.D’mello, R., Hill, S., and Poole, R. K.
(1996) The cytochrome bd quinol oxidase in Escherichia
coli has an extremely high oxygen affinity and two-oxygen-binding
hemes: implications for regulation of activity in vivo by oxygen
inhibition, Microbiology, 142, 755-763.
57.Rothery, R. A., Houston, A. M., and Ingledew, W.
J. (1987) The respiratory chain of anaerobically grown Escherichia
coli: reactions with nitrite and oxygen, J. Gen.
Microbiol., 133, 3247-3255.
58.Hill, J. J., Alben, J. O., and Gennis, R. B.
(1993) Spectroscopic evidence for a heme–heme binuclear center in
the cytochrome bd ubiquinol oxidase from Escherichia
coli, Proc. Natl. Acad. Sci. USA, 90, 5863-5867.
59.Tsubaki, M., Hori, H., Mogi, T., and Anraku, Y.
(1995) Cyanide-binding site of bd-type ubiquinol oxidase from
Escherichia coli, J. Biol. Chem., 270,
28565-28569.
60.Borisov, V., Arutyunyan, A. M., Osborne, J. P.,
Gennis, R. B., and Konstantinov, A. A. (1999) Magnetic circular
dichroism used to examine the interaction of Escherichia coli
cytochrome bd with ligands, Biochemistry, 38,
740-750.
61.Vos, M. H., Borisov, V. B., Liebl, U., Martin,
J.-L., and Konstantinov, A. A. (2000) Femtosecond resolution of
ligand–heme interactions in the high-affinity quinol oxidase
bd: a di-heme active site? Proc. Natl. Acad. Sci. USA,
97, 1554-1559.
62.Borisov, V. B., Sedelnikova, S. E., Poole, R. K.,
and Konstantinov, A. A. (2001) Interaction of cytochrome bd with
carbon monoxide at low and room temperatures: evidence that only a
small fraction of heme b595 reacts with CO, J.
Biol. Chem., 276, 22095-22099.
63.Borisov, V. B., Liebl, U., Rappaport, F., Martin,
J.-L., Zhang, J., Gennis, R. B., Konstantinov, A. A., and Vos, M. H.
(2002) Interactions between heme d and heme
b595 in quinol oxidase bd from Escherichia
coli: a photoselection study using femtosecond spectroscopy,
Biochemistry, 41, 1654-1662.
64.Arutyunyan, A. M., Borisov, V. B., Novoderezhkin,
V. I., Ghaim, J., Zhang, J., Gennis, R. B., and Konstantinov, A. A.
(2008) Strong excitonic interactions in the oxygen-reducing site of
bd-type oxidase: the Fe-to-Fe distance between hemes d
and b595 is 10 Å, Biochemistry,
47, 1752-1759.
65.Rappaport, F., Zhang, J., Vos, M. H., Gennis, R.
B., and Borisov, V. B. (2010) Heme–heme and heme–ligand
interactions in the di-heme oxygen-reducing site of cytochrome
bd from Escherichia coli revealed by nanosecond
absorption spectroscopy, Biochim. Biophys. Acta, 1797,
1657-1664.
66.Borisov, V. B., and Verkhovsky, M. I. (2013)
Accommodation of CO in the di-heme active site of cytochrome bd
terminal oxidase from Escherichia coli, J. Inorg.
Biochem., 118, 65-67.
67.Siletsky, S. A., Zaspa, A. A., Poole, R. K., and
Borisov, V. B. (2014) Microsecond time-resolved absorption spectroscopy
used to study CO compounds of cytochrome bd from Escherichia
coli, PLoS One, 9, e95617; doi:
95610.91371/journal.pone.0095617.
68.Borisov, V. B. (2008) Interaction of
bd-type quinol oxidase from Escherichia coli and carbon
monoxide: heme d binds CO with high affinity, Biochemistry
(Moscow), 73, 14-22.
69.Arutyunyan, A. M., Sakamoto, J., Inadome, M.,
Kabashima, Y., and Borisov, V. B. (2012) Optical and magneto-optical
activity of cytochrome bd from Geobacillus
thermodenitrificans, Biochim. Biophys. Acta, 1817,
2087-2094.
70.Borisov, V. B., Forte, E., Sarti, P., and
Giuffrè, A. (2011) Catalytic intermediates of cytochrome
bd terminal oxidase at steady-state: ferryl and oxy-ferrous
species dominate, Biochim. Biophys. Acta, 1807,
503-509.
71.Paulus, A., Rossius, S. G., Dijk, M., and de
Vries, S. (2012) Oxoferryl-porphyrin radical catalytic intermediate in
cytochrome bd oxidases protects cells from formation of reactive
oxygen species, J. Biol. Chem., 287,
8830-8838.
72.Yang, K., Borisov, V. B., Konstantinov, A. A.,
and Gennis, R. B. (2008) The fully oxidized form of the cytochrome
bd quinol oxidase from E. coli does not participate in
the catalytic cycle: direct evidence from rapid kinetics studies,
FEBS Lett., 582, 3705-3709.
73.Borisov, V. B., Gennis, R. B., and Konstantinov,
A. A. (1995) Interaction of cytochrome bd from Escherichia
coli with hydrogen peroxide, Biochemistry (Moscow),
60, 231-239.
74.Borisov, V., Gennis, R., and Konstantinov, A. A.
(1995) Peroxide complex of cytochrome bd: kinetics of generation
and stability, Biochem. Mol. Biol. Int., 37,
975-982.
75.Borisov, V. B., Forte, E., Sarti, P., Brunori,
M., Konstantinov, A. A., and Giuffrè, A. (2006) Nitric oxide
reacts with the ferryl-oxo catalytic intermediate of the
CuB-lacking cytochrome bd terminal oxidase, FEBS
Lett., 580, 4823-4826.
76.Lindqvist, A., Membrillo-Hernandez, J., Poole, R.
K., and Cook, G. M. (2000) Roles of respiratory oxidases in protecting
Escherichia coli K12 from oxidative stress, Antonie Van
Leeuwenhoek, 78, 23-31.
77.Korshunov, S., and Imlay, J. A. (2010) Two
sources of endogenous hydrogen peroxide in Escherichia coli,
Mol. Microbiol., 75, 1389-1401.
78.Borisov, V. B., Forte, E., Davletshin, A.,
Mastronicola, D., Sarti, P., and Giuffrè, A. (2013) Cytochrome
bd oxidase from Escherichia coli displays high catalase
activity: an additional defense against oxidative stress, FEBS
Lett., 587, 2214-2218.
79.Borisov, V. B., Davletshin, A. I., and
Konstantinov, A. A. (2010) Peroxidase activity of cytochrome bd
from Escherichia coli, Biochemistry (Moscow), 75,
428-436.
80.Brown, G. C. (1995) Reversible binding and
inhibition of catalase by nitric oxide, Eur. J. Biochem.,
232, 188-191.
81.Small, J. L., Park, S. W., Kana, B. D., Ioerger,
T. R., Sacchettini, J. C., and Ehrt, S. (2013) Perturbation of
cytochrome c maturation reveals adaptability of the respiratory
chain in Mycobacterium tuberculosis, MBio, 4,
e00475-00413.
82.Forte, E., Borisov, V. B., Davletshin, A.,
Mastronicola, D., Sarti, P., and Giuffrè, A. (2013) Cytochrome
bd oxidase and hydrogen peroxide resistance in Mycobacterium
tuberculosis, MBio, 4, e01006-01013.
83.Pullan, S. T., Gidley, M. D., Jones, R. A.,
Barrett, J., Stevanin, T. M., Read, R. C., Green, J., and Poole, R. K.
(2007) Nitric oxide in chemostat-cultured Escherichia coli is
sensed by Fnr and other global regulators: unaltered methionine
biosynthesis indicates lack of S nitrosation, J. Bacteriol.,
189, 1845-1855.
84.Richardson, A. R., Dunman, P. M., and Fang, F. C.
(2006) The nitrosative stress response of Staphylococcus aureus
is required for resistance to innate immunity, Mol.
Microbiol., 61, 927-939.
85.Machado, P., Felix, R., Rodrigues, R., Oliveira,
S., and Rodrigues-Pousada, C. (2006) Characterization and expression
analysis of the cytochrome bd oxidase operon from
Desulfovibrio gigas, Curr. Microbiol., 52,
274-281.
86.Moore, C. M., Nakano, M. M., Wang, T., Ye, R. W.,
and Helmann, J. D. (2004) Response of Bacillus subtilis to
nitric oxide and the nitrosating agent sodium nitroprusside, J.
Bacteriol., 186, 4655-4664.
87.Borisov, V. B., Forte, E., Konstantinov, A. A.,
Poole, R. K., Sarti, P., and Giuffrè, A. (2004) Interaction of
the bacterial terminal oxidase cytochrome bd with nitric oxide,
FEBS Lett., 576, 201-204.
88.Borisov, V. B., Forte, E., Siletsky, S. A.,
Sarti, P., and Giuffrè, A. (2015) Cytochrome bd from
Escherichia coli catalyzes peroxynitrite decomposition,
Biochim. Biophys. Acta, 1847, 182-188.
89.Mason, M. G., Nicholls, P., Wilson, M. T., and
Cooper, C. E. (2006) Nitric oxide inhibition of respiration involves
both competitive (heme) and noncompetitive (copper) binding to
cytochrome c oxidase, Proc. Natl. Acad. Sci. USA,
103, 708-713.
90.Borisov, V. B., Forte, E., Sarti, P., Brunori,
M., Konstantinov, A. A., and Giuffrè, A. (2007) Redox control of
fast ligand dissociation from Escherichia coli cytochrome
bd, Biochem. Biophys. Res. Commun., 355,
97-102.
91.Borisov, V. B., Forte, E., Giuffrè, A.,
Konstantinov, A., and Sarti, P. (2009) Reaction of nitric oxide with
the oxidized di-heme and heme-copper oxygen-reducing centers of
terminal oxidases: different reaction pathways and end-products, J.
Inorg. Biochem., 103, 1185-1187.
92.Sarti, P., Giuffrè, A., Forte, E.,
Mastronicola, D., Barone, M. C., and Brunori, M. (2000) Nitric oxide
and cytochrome c oxidase: mechanisms of inhibition and NO
degradation, Biochem. Biophys. Res. Commun., 274,
183-187.
93.Mason, M. G., Shepherd, M., Nicholls, P., Dobbin,
P. S., Dodsworth, K. S., Poole, R. K., and Cooper, C. E. (2009)
Cytochrome bd confers nitric oxide resistance to Escherichia
coli, Nat. Chem. Biol., 5, 94-96.
94.Fu, H., Chen, H., Wang, J., Zhou, G., Zhang, H.,
Zhang, L., and Gao, H. (2013) Crp-dependent cytochrome bd
oxidase confers nitrite resistance to Shewanella oneidensis,
Environ. Microbiol., 15, 2198-2212.
95.Zhang, H., Fu, H., Wang, J., Sun, L., Jiang, Y.,
Zhang, L., and Gao, H. (2013) Impacts of nitrate and nitrite on
physiology of Shewanella oneidensis, PLoS One, 8,
e62629.
96.Ferrer-Sueta, G., and Radi, R. (2009) Chemical
biology of peroxynitrite: kinetics, diffusion, and radicals, ACS
Chem. Biol., 4, 161-177.
97.McLean, S., Bowman, L. A., Sanguinetti, G., Read,
R. C., and Poole, R. K. (2010) Peroxynitrite toxicity in Escherichia
coli K12 elicits expression of oxidative stress responses and
protein nitration and nitrosylation, J. Biol. Chem.,
285, 20724-20731.
98.Lindemann, C., Lupilova, N., Muller, A.,
Warscheid, B., Meyer, H. E., Kuhlmann, K., Eisenacher, M., and
Leichert, L. I. (2013) Redox proteomics uncovers
peroxynitrite-sensitive proteins that help Escherichia coli to
overcome nitrosative stress, J. Biol. Chem., 288,
19698-19714.
99.Rubbo, H., Trostchansky, A., and O’Donnell,
V. B. (2009) Peroxynitrite-mediated lipid oxidation and nitration:
mechanisms and consequences, Arch. Biochem. Biophys.,
484, 167-172.
100.Salgo, M. G., Bermudez, E., Squadrito, G. L.,
and Pryor, W. A. (1995) Peroxynitrite causes DNA damage and oxidation
of thiols in rat thymocytes, Arch. Biochem. Biophys.,
322, 500-505.
101.Sharpe, M. A., and Cooper, C. E. (1998)
Interaction of peroxynitrite with mitochondrial cytochrome oxidase.
Catalytic production of nitric oxide and irreversible inhibition of
enzyme activity, J. Biol. Chem., 273,
30961-30972.
102.Cooper, C. E., and Davies, N. A. (2000) Effects
of nitric oxide and peroxynitrite on the cytochrome oxidase
Km for oxygen: implications for mitochondrial
pathology, Biochim. Biophys. Acta, 1459, 390-396.
103.Cooper, C. E., Davies, N. A., Psychoulis, M.,
Canevari, L., Bates, T. E., Dobbie, M. S., Casley, C. S., and Sharpe,
M. A. (2003) Nitric oxide and peroxynitrite cause irreversible
increases in the Km for oxygen of mitochondrial
cytochrome oxidase: in vitro and in vivo studies,
Biochim. Biophys. Acta, 1607, 27-34.
104.Andries, K., Verhasselt, P., Guillemont, J.,
Gohlmann, H. W., Neefs, J. M., Winkler, H., Van Gestel, J., Timmerman,
P., Zhu, M., Lee, E., Williams, P., de Chaffoy, D., Huitric, E.,
Hoffner, S., Cambau, E., Truffot-Pernot, C., Lounis, N., and Jarlier,
V. (2005) A diarylquinoline drug active on the ATP synthase of
Mycobacterium tuberculosis, Science, 307,
223-227.
105.Koul, A., Vranckx, L., Dhar, N., Gohlmann, H.
W., Ozdemir, E., Neefs, J. M., Schulz, M., Lu, P., Mortz, E., McKinney,
J. D., Andries, K., and Bald, D. (2014) Delayed bactericidal response
of Mycobacterium tuberculosis to bedaquiline involves remodeling
of bacterial metabolism, Nat. Commun., 5,
3369.
106.Berney, M., Hartman, T. E., and Jacobs, W. R.,
Jr. (2014) A Mycobacterium tuberculosis cytochrome bd
oxidase mutant is hypersensitive to bedaquiline, MBio, 5,
e01275-01214.
107.Pethe, K., Bifani, P., Jang, J., Kang, S.,
Park, S., Ahn, S., Jiricek, J., Jung, J., Jeon, H. K., Cechetto, J.,
Christophe, T., Lee, H., Kempf, M., Jackson, M., Lenaerts, A. J., Pham,
H., Jones, V., Seo, M. J., Kim, Y. M., Seo, M., Seo, J. J., Park, D.,
Ko, Y., Choi, I., Kim, R., Kim, S. Y., Lim, S., Yim, S. A., Nam, J.,
Kang, H., Kwon, H., Oh, C. T., Cho, Y., Jang, Y., Kim, J., Chua, A.,
Tan, B. H., Nanjundappa, M. B., Rao, S. P., Barnes, W. S., Wintjens,
R., Walker, J. R., Alonso, S., Lee, S., Kim, J., Oh, S., Oh, T.,
Nehrbass, U., Han, S. J., No, Z., Lee, J., Brodin, P., Cho, S. N., Nam,
K., and Kim, J. (2013) Discovery of Q203, a potent clinical candidate
for the treatment of tuberculosis, Nat. Med., 19,
1157-1160.
108.Arora, K., Ochoa-Montano, B., Tsang, P. S.,
Blundell, T. L., Dawes, S. S., Mizrahi, V., Bayliss, T., Mackenzie, C.
J., Cleghorn, L. A., Ray, P. C., Wyatt, P. G., Uh, E., Lee, J., Barry,
C. E., 3rd, and Boshoff, H. I. (2014) Respiratory flexibility in
response to inhibition of cytochrome c oxidase in
Mycobacterium tuberculosis, Antimicrob. Agents
Chemother., 58, 6962-6965.