REVIEW: Electron Transfer in Photosystem I Containing Native and Modified Quinone Acceptors
A. Yu. Semenov1,2*, A. A. Petrova1, M. D. Mamedov1, and V. A. Nadtochenko2
1A.N. Belozersky Institute of Physical-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: +7 (495) 939-3181; E-mail: semenov@genebee.msu.ru2N.N. Semenov Institute of Chemical Physics, Russian Academy of Sciences, 119991 Moscow, Russia; fax: +7 (499) 137-8357
* To whom correspondence should be addressed.
Received January 27, 2015; Revision received March 8, 2015
The pigment–protein complex of photosystem I (PS I) catalyzes light-driven oxidation of plastocyanin or cytochrome c6 and reduction of ferredoxin or flavodoxin in oxygenic photosynthetic organisms. In this review, we describe the current state of knowledge of the processes of excitation energy transfer and formation of the primary and secondary ion-radical pairs within PS I. The electron transfer reaction involving quinone cofactor in the A1 site and its role in providing asymmetry of electron transport as well as interaction with oxygen and ascorbate in PS I are discussed.
KEY WORDS: photosystem I, reaction center, electron transfer, asymmetry, primary reactions, quinone acceptors, interaction with oxygen and ascorbateDOI: 10.1134/S0006297915060024
Abbreviations: A0A and A0B, primary electron acceptor chlorophylls in A and B branches; A1A and A1B, quinone molecules – secondary electron acceptors in A and B branches; Chl, chlorophyll; Chl1A/Chl1B, Chl2A/Chl2B, and Chl3A/Chl3B are the first, second, and third molecules of chlorophyll in symmetrical branches of redox cofactors A and B in PS I; Cl2NQ, 2,3-dichloro-1,4-naphthoquinone; P700, dimer of chlorophyll – primary electron donor; PhQ, phylloquinone; PQ, plastoquinone; PS I, photosystem I; RC, reaction center.
STRUCTURE OF PHOTOSYSTEM I COMPLEXES
Pigment–protein complex of photosystem I (PS I) is one of the crucial enzymes in the photosynthetic electron transfer chain in thylakoid membranes of oxygenic organisms [1]. PS I complex catalyzes the light-driven reaction of electron transfer from peripheral donor proteins plastocyanin and cytochrome c6 to ferredoxin and flavodoxin.
The three-dimensional structure of the trimeric form of PS I from the thermophilic cyanobacterium Synechococcus elongatus has been obtained by X-ray structural analysis with 2.5 Å resolution [2]. Each monomer has molecular weight ~330 kDa, contains one copy of 12 protein subunits (nine transmembrane and three peripheral) and ~130 cofactors (96 chlorophyll (Chl) a, 22 β-carotene, three [4Fe-4S] clusters, two phylloquinone (PhQ), and four lipid molecules). It should be noted that PS I also contains 201 water molecules and a calcium ion. Two transmembrane subunits PsaA and PsaB compose C2-symmetrical heterodimeric core complex containing most of the electron transfer cofactors.
The electron transfer chain of PS I consists of chlorophyll dimer P700 (Chl1A/Chl1B), A0 (pairs of Chl molecules designated as Chl2A/Chl3A and Chl2B/Chl3B), A1 (phylloquinone molecules designated as A1A/A1B), and iron–sulfur clusters FX, FA, and FB (Fig. 1).
Fig. 1. General view of electron transfer cofactors and electron transfer in PS I.
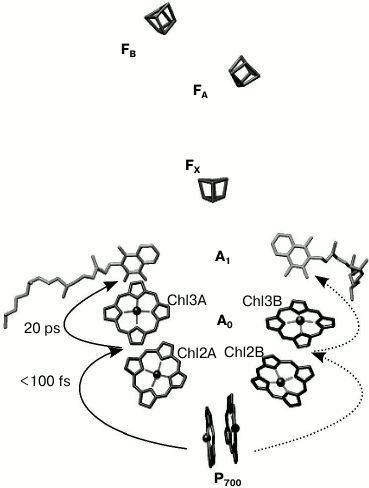
It is known that electron transfer in PS I occurs through both branches of the redox cofactors from P700 to FX, but the degree of asymmetry of this transfer and the factors that cause it are not clear [3, 4]. In addition, to date the kinetics of the primary charge separation and the nature of the primary electron donor and acceptor in PS I remain controversial [4, 5].
As mentioned above, out of 96 Chl molecules, six are electron transfer cofactors located near the contact surfaces of the PsaA and PsaB subunits. The chlorophyll dimer P700 consists of two Chl a molecules (Chl1A/Chl1B) whose porphyrin planes are parallel to each other (at a distance of 3.6 Å) and perpendicular to the plane of the membrane. The spatial localization of the other two molecules of Chl (Chl2A/Chl2B) roughly corresponds to the localization of two molecules of monomeric bacteriochlorophyll in the RC of purple bacteria, and two additional molecules of Chl (Chl3A and Chl3B) are arranged in a manner similar to the two molecules of bacteriopheophytin in the RC of purple bacteria [6, 7]. The planes of the porphyrin rings of Chl2A (Chl2B) and Chl3A (Chl3B) molecules are parallel (with distance of 3.9 Å), but they are slightly more shifted relative to each other than Chl1A and Chl1B. The mutual arrangement and relatively close distance between central Mg2+ atoms of Chl2A and Chl3A, and Chl2B and Chl3B (8.7 and 8.2 Å, respectively) imply the possibility of significant interaction between these pairs of Chl molecules. This fact allows us to consider A0A and A0B as a Chl2A/Chl3A (Chl2B/Chl3B) dimer.
As indicated above, PS I also includes two molecules of phylloquinone (A1A and A1B). Branch A includes Chl molecules labeled Chl1A, Chl2A, Chl3A and phylloquinone A1A, while branch B includes Chl1B, Chl2B, Chl3B and phylloquinone A1B. The two cofactor branches of the electron transfer chain merge onto the inter-polypeptide iron–sulfur cluster FX. Terminal electron acceptors – [4Fe-4S] clusters FA and FB – are located at the stromal subunit PsaC.
EXCITATION ENERGY AND ELECTRON TRANSFER IN PS I RC
Most of the previous studies implied that the primary charge separation step from P700* to A0 occur in PS I RC in the 0.8-4 ps time range, and the subsequent electron transfer from primary electron acceptor A0 to the secondary electron acceptor A1 was suggested to occur in the 10-50 ps range [8-10]. Recently, we have shown that after preferential excitation of P700 and A0, formation of the primary radical pair P700+A0– occurs within 100 fs, followed by electron transfer leading to formation of P700+A1– with characteristic time of ~25 ps [5]. Subsequent electron transfer reactions from A1A and A1B to FX take place with characteristic times of ~200 and ~20 ns, respectively [1, 3, 11, 12]. This is followed by electron transfer from FX to the terminal iron–sulfur clusters, FA and FB, and further to ferredoxin or flavodoxin. Photooxidized P700+ is reduced by plastocyanin or cytochrome c6, returning all the cofactors of PS I back to their initial state [1].
The main difference between PS I and bacterial RC is the impossibility of mechanically separating Chl molecules of the light-harvesting antenna from Chl molecules of the RC without affecting the structural and functional integrity of the enzyme complex. The difficulty is that protein subunits PsaA/PsaB bind not only six Chl molecules, which take part in primary steps of electron transfer, but also 79 antenna Chl molecules. All the 96 Chl molecules in PS I cannot be clearly separated from each other by their spectral features. According to [13], the major part of the antenna Chl pool has absorption maximum at 670-680 nm, whereas only some Chl molecules (possibly dimers or trimers) are characterized by an absorption band at 690-710 nm. It is generally accepted that a special pair of Chl molecules (P700) has absorption maximum at 700-705 nm, and the maximum absorption of the chlorophyll acceptor A0 is at 685-690 nm [14-16], but their spectra partially overlap with the spectra of several molecules of the antenna chlorophyll in the same spectral region.
Excitation energy transfer in the antenna and the electron transfer in RC of PS I have been studied by different approaches, in particular, using sub-picosecond pulse spectroscopy [5, 8-10]. However, primary reactions of electron transfer cannot be distinctly separated from reactions of the excitation energy transfer in the antenna because these reactions occur within the same time range. To separate the electron transfer and the excitation energy transfer and to clearly detect kinetics of the primary stages of electron transfer and the spectra of the corresponding intermediates, various approaches have been used. For example, Kumazaki et al. [17] extracted the major part of the antenna Chl and, on the resulting complex, recorded two kinetic components with lifetimes of ~0.8 and ~9 ps. These phases were assigned to the primary separation of charges. Note that a decrease in the total Chl content to 12-14 molecules is very likely to significantly affect both the structure and function of PS I complex.
There are two basic models describing charge separation in PS I. According to the first model, upon light absorption, primary donor P700 is excited to P700*, that followed by charge separation between P700* and the primary electron acceptor A0 with formation of ion-radical pair P700+A0–. Data obtained by picosecond absorption spectroscopy on the PS I particles from spinach suggested that immediate (less than 1.5 ps) P700 bleaching and 810 nm band formation are due to P700 excitation and P700* state formation [18]. In the near-infrared region of the spectrum, the 810 nm band induced by excitation of samples diminished to about 60% of its original intensity with the same ~14 ps time constant as the formation of the 690 nm band. These spectral changes were interpreted to be due to the formation of the state P700+A0–.
In one of the earliest papers, it was demonstrated that selective excitation of PS I by picosecond laser pulses, at 710 nm, results in the formation of P+A0–, which is characterized by two bleaching bands at 700 and 689 nm. These bands correspond to the formation of P700+ and to the reduction of primary acceptor A0, respectively [14].
Another approach to reveal the nature of the primary reactions in PS I complexes is based on subtracting the transient spectrum of the oxidized (closed) RC from the spectrum of the reduced (open) RC [19]. This approach allowed the authors to remove spectral changes caused by excitation energy transfer in the antenna and to reveal spectral features reflecting electron transfer in the RC. Hastings et al. [15] used this approach for PS I particles from cyanobacteria and observed two kinetic phases with lifetimes of 4 and 21 ps. These components were assigned to the generation and disappearance of the ion-radical pair P700+A0–. In that paper, a non-selective excitation at 590 nm was used, and therefore the phase with lifetime of 4 ps reflected not only the oxidation of P700, but also the energy transfer in the antenna. Thus, the time constant of ~4 ps could represent the upper limit of the rate constant in kinetics of the primary radical pair formation. Moreover, to obtain a good signal/noise ratio, Hastings et al. [15] used laser flashes with rather high energy that induced excitation annihilation and shortened the total time of reaching the state of one-exciton excitation in the antenna.
Savikhin et al. [16] used a similar approach and obtained 10 ps as the time of formation of P700+A0–. They excited PS I with 100-fs flashes with 660-nm maximum. Later, using a simple model of electron transfer and approximating the kinetics by two free parameters, same authors obtained a characteristic time of ~1.3 ps for the formation of P700+A0– and of ~13 ps for the subsequent reaction A0 → A1 [20]. Similar results were obtained earlier by White et al. [21] for PS I complexes isolated from spinach.
Melkozernov et al. [22] investigated fast spectral changes in PS I from the cyanobacterium Synechocystis sp. PCC 6803 using 150-fs laser flashes with maxima at 660, 693, and 710 nm. They suggested that the change in the absorption at 700 nm was caused by primary separation of charges and occurred in about 20 ps. In so doing, P700*A0 → P700+A0– should be the rate-limiting reaction, and the quasi-equilibrium in the antenna should be established in about 3 ps. Other authors consider that the excitation energy transfer in the antenna is the slower step with characteristic times about 20-25 ps [16, 23].
Muller et al. [24] proposed an alternative model for primary charge separation in PS I. They investigated PS I complex from Chlamydomonas reinhardtii, and based on modeling the kinetics and analysis of the differential absorption spectra they assumed that the primary charge separation should occur in 6-9 ps. They also suggested that the primary radical pair is not P700+A0–, but P700+Chl2– or Chl2+Chl3–, where Chl2 and Chl3 are, respectively, the distal and proximal molecules of a Chl monomer in the RC relative to P700. Data of Muller et al. [24] and Holzwarth et al. [25, 26] revealed kinetic components with characteristic times of 6, 20, and 40 ps. Holzwarth and coworkers [25, 26] proposed a scheme for the primary charge separation in PS I according to which the fastest component corresponds to the formation of the primary radical pair Chl2+Chl3– and the two slower components – to the radical pairs P700+Chl3– and P700+A1–. However, according to electrostatic calculations, the functioning of monomeric Chls 2A/2B as primary donors and monomeric Chls 3A/3B as primary acceptors seems unlikely, since in this case the electron transfer would be thermodynamically unfavorable [27].
PRIMARY STEPS OF CHARGE SEPARATION WITH PREFERENTIAL EXCITATION
OF REACTION CENTER CHLOROPHYLLS BY 20-FEMTOSECOND PULSES
In the majority of studies on the fast kinetics of spectral changes in PS I, flashes with duration >100 fs were used, as well as powerful flashes resulting in two quanta arriving into the same RC and annihilation of the exciton-induced excitation and distortion of the spectral change kinetics.
In previous works [5, 28, 29], we have used a complex approach including short (~20 fs) laser flashes with relatively low energy (20 nJ) with maximum at 720 nm. Under these conditions, time-dependent changes of differential spectra of PS I from Synechocystis sp. PCC 6803 were measured. Since the femtosecond pulse with 20 fs duration has a bandwidth of 40 nm, antenna pigments (chlorophylls and carotenoids), which absorb in a short wavelength region of the Qy-band, are not excited. Under these conditions, about half of the RC chlorophyll molecules (i.e. molecules of P700 and A0) were excited directly, whereas the other half received excitation energy from the nearest molecules of the antenna chlorophyll. Each PS I complex absorbed about 0.10-0.15 quanta due to relatively low pumping pulse energy, which was less than 20 nJ. These conditions allowed us to reduce the contribution of bi- and multiexcitonic excitation of PS I into spectral-kinetic dependencies, so it became negligible.
These conditions were chosen to maximally increase the relative contribution of the direct excitation of the RC in order to separate the kinetics of primary stages of the charge separation in the RC from the kinetics of the excitation energy transfer in the antenna. Excitation in antenna is transferred to P700 in a non-radiative way. According to spectral-kinetic measurements, this process was observed as a separate kinetic component, which clearly differs from charge transfer component(s). Based on these findings, it was possible to reveal the difference spectra of the intermediates (P700A0)*, P700+A0– and P700+A1–, and to describe the kinetics of transitions between these intermediates.
Figure 2a shows these difference spectra of PS I obtained upon excitation of sample (720 nm) by a 22-fs pulse at different time delays. During the time delay shorter than 100 fs (Fig. 2, a and b) spectra undergo significant evolution: primary peak of the bleaching at 690 nm at time delay t = 0 transformed to bleaching band with two peaks at 690 and 705 nm until t = 100 fs. The bleaching peak at 690 and 705 nm can be attributed to A0 (690 nm) and P700 (705 nm), which indicates formation of the primary ion-radical pair P700+A0–A1 with characteristic time less than 100 fs.
Fig. 2. a) Differential absorption spectra evolution within 0-100-fs time region at time delays (fs): 1) 0; 2) 20; 3) 50; 4) 60; 5) 100. Excitation energy 20 nJ, wavelength 720 nm, pulse duration 22 fs. The dotted line represents the inverted overlap between absorption spectrum of PS I ε(λ) and the spectrum of the excitation pulse I(λ) (right axis). Inset: absorption spectrum of PS I RCs at the Qy band (left axis) and normalized laser pulse spectrum (right axis). b) Kinetics of the ratio of differential absorption ΔA705/ΔA690 (left axis, experimental points are shown by filled squares, solid line is a theoretical curve simulated by the quantum-mechanical mixing of two excited energy levels) and kinetics of differential absorption of ΔA660 after 60 fs (right axis, hourglass symbol).
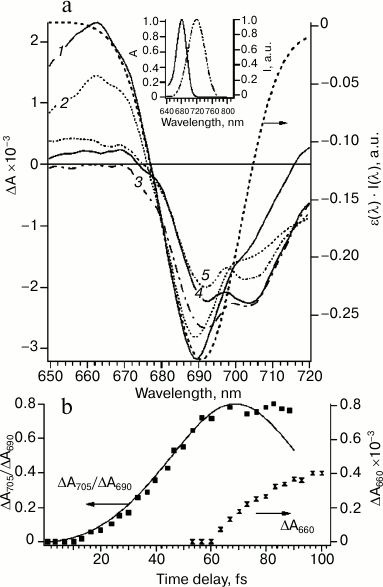
At zero time delay, the bleaching peak is at 690 nm, which corresponds to maximum overlapping of absorption bands of Chl molecules in the Qy band and the spectrum of the excitation pulse (Fig. 2a). At time delay more than 10 fs, bleaching at 690 nm decreased and an additional absorption band at 705 nm appeared. This process occurred between first 10 and 60 fs (Fig. 2b) and, probably, corresponded to the energy transfer. The kinetic curve of the bleaching peaks at 705 and 690 nm ratio (ΔA705/ΔA690) can be modeled within the framework of the quantum beat formalism [30, 31]. The quantum beat theory for two interacting states A0* and P700* has been described in detail [5]. According to this model, excitation oscillates between A0 and P700 chromophores (Fig. 2b). These oscillations will be damped if an additional process would not lead to coherence relaxation. In RC of PS I, such a process of coherence dissipation evidently corresponds to electron transfer from P700 to A0 (Fig. 2b). The formation of the primary ion-radical pair P700+A0– explains the deviation from theoretical curve of the quantum beat model at time delays more than 80 fs and almost constant ratio ΔA705/ΔA690 in the time range between 80 fs and ~10 ps.
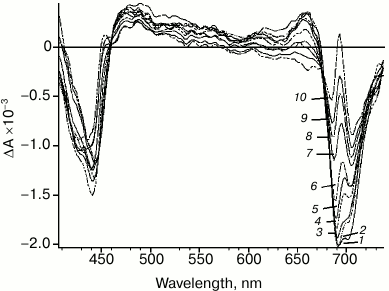
Significant changes in differential spectra emerge in the time scale from picoseconds to hundreds of picoseconds (Fig. 3). These changes are caused by two processes: energy transfer to primary electron donor P700 followed by rapid charge separation with sequential formation of both primary P700+A0–A1 and secondary ion-radical pairs P700+A0A1–. Specific spectral features of the differential spectrum of P700+A0A1– are shown in Fig. 3. The features include: bleaching band at 705 nm, which is related to P700+ formation; bleaching band at about 685 nm and a specific peak at 692 nm, which are caused by Kerr shift of the A0 absorption band; bleaching band between 658 and 578 nm, which is probably related to Kerr shift of antenna carotenoid absorption band and, finally, absorption dip at 458 nm. Besides, the bleaching in the Soret band (about 440 nm) and wide absorption band between 449 and 578 nm can be attributed to P700+ absorption. In the short-wavelength region of the spectrum (<413 nm), the edge of the secondary acceptor phylloquinone absorption band appears.
Fig. 3. PS I differential absorption spectra after excitation by 22-fs pulse at 720 nm. Time delay: 1) 85 fs; 2) 150 fs; 3) 500 fs; 4) 2 ps; 5) 4 ps; 6) 8 ps; 7) 20 ps; 8) 30 ps; 9) 50 ps; 10) 90 ps.
These results can be summarized by the following scheme.
| (P700A0)* | → | (P700+A0–) | → | (P700+A1–) |
| <100 fs | ~25 ps | |||
In our recent work on cyanobacterial PS I complexes, time-resolved differential spectra were obtained upon excitation of PS I complexes by femtosecond pulses with maxima at different wavelengths (670, 700, and 720 nm). It was found that the ratio of number of excited antenna Chls to the number of excited Chls in RC depended on spectral characteristics of the exciting pulse [28]. Upon excitation by light pulses with maxima at 670 and 700 nm, the bleaching, caused by excitation of antenna chlorophylls, hinders registration of the processes in the RC. These data allowed us to explain possible causes of mismatching of the results of primary steps of electron transfer kinetic investigation in different laboratories. In the case of significant contribution of antenna excitation, it is difficult to reveal the kinetics of the primary stages of electron transfer between redox-cofactors P700, A0, and A1 because of energy transfer to the primary electron donor.
Chl2A/Chl2B and Chl3A/Chl3B molecules, which form the primary acceptor A0, are characterized by unusual axial ligands: water molecules in case of Chl2A/Chl2B and methionine residues in case of Chl3A/Chl3B. The methionine residues (M688PsaA and M668PsaB) are present in all known species of plants and cyanobacteria. These methionine residues have been mutated to leucine, histidine, and asparagine [29, 32, 33].
Ultrafast optical measurements showed that branch A mutants (M688LPsaA, M688NPsaA and M688HPsaA) demonstrate A0 → A1 electron transfer deceleration, whereas branch B mutants’ (M668LPsaB, M668NPsaB and M668HPsaB) kinetics do not reveal any difference from the kinetics observed in the case of wild-type PS I [29, 33]. These data indicate asymmetrical contribution of the A and B cofactor branches to the secondary ion-radical pair P700+A1– formation. Time-resolved EPR-spectra of PS I from M688NPsaA and M668NPsaB mutants at a frequency of 95 GHz (in W-band) at 100 K showed that charge recombination kinetics of wild-type PS I and B-branch mutant were similar and significantly differed from the kinetics in the case of PS I from A-branch mutant [34]. Electron spin echo envelope modulation (ESEEM) analysis revealed that distances between the centers of ion-radical pairs P700+A1– for wild-type PS I and B-branch mutant correspond to electron transfer only through branch A [34]. Ultrafast optical measurements at room temperature showed that electron transfer in PS I from Synechocystis sp. PCC 6803 is asymmetrical with the contribution of branch A being up to 80% [29].
REACTIONS OF LIGHT-DEPENDENT QUINONE OXIDATION IN THE
A1-SITE
PS I RC contains two molecules of phylloquinone (PhQ) that are characterized by extremely low midpoint redox potential (from –700 to –820 mV) [27, 35, 36] in comparison with ubiquinones and plastoquinones in the QA site of PS II and purple bacteria RCs (from –100 to 0 mV) [37, 38]. One of the promising approaches for investigation of electron transfer involving quinone cofactors in PS I is substitution of the PhQ molecule in the A1 site by quinones with different redox potentials (such as benzoquinones, naphthoquinones, and anthraquinones). This can be accomplished by using cyanobacterial mutants, such as menB deletion strain, which carry a mutation in the PhQ biosynthetic pathway [39]. At the same time, biosynthesis of other quinones in this mutant is not affected. The empty A1 site in this case is occupied by plastoquinone-9 (PQ), which can be easily replaced by other quinones [40].
Since the PQ redox potential in A1 is ~95 mV more positive than the potential of native PhQ, the forward electron transfer from A1 to FX in the PS I from the MenB mutant becomes ~1000 times slower, while the charge recombination – ~25 times faster in comparison with wild-type PS I [41].
One of the most important advantages of menB PS I as a model system for investigation of the electron transfer mechanism involving quinone is that it requires neither the use of organic solvents nor the removal of the PsaC subunit to replace the PQ in the A1-site. This minimizes possible changes in electrostatic properties and conformation of the protein. Thus, the menB mutant provides new ways for the study of kinetic and thermodynamic aspects of the electron transfer.
An interesting approach to investigation of electron transfer involving quinone is the prevention of electron transfer from the quinone to the iron–sulfur clusters. This can be achieved by substitution of PQ by the naphthoquinone derivative 2,3-dichloro-1,4-naphthoquinone (Cl2NQ), which has much more positive midpoint potential. The redox potential of Cl2NQ in dimethylformamide is about –50 mV, which is 400 mV more positive than the corresponding value for PhQ. In this case, the forward electron transfer to FX is thermodynamically unfavorable, and the electron recombines from the quinone cofactor in the A1-site to P700+ (Fig. 4).
Fig. 4. Forward and backward reactions in PS I from wild type (PhQ, solid line) and menB mutant, which contains PQ (dashed line) or Cl2NQ (dash-dot line) in the A1-site. The diagram also shows the sites of oxygen reduction in PS I and the pathway of electron transfer in the presence of high concentrations of ascorbate (dotted line) under steady state illumination.
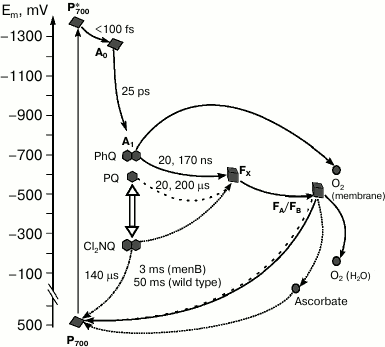
Charge recombination kinetics of PS I with Cl2NQ in the A1 site obtained by time-resolved spectrometry and EPR spectroscopy demonstrate significant decrease of characteristic time of the main kinetic component in comparison with intact complexes from menB containing PQ [42]. In the case of Cl2NQ, at room temperature charge recombination is ~30 times faster than in PS I from menB and is ~900 times faster in comparison with PS I from the wild type (Fig. 4). Furthermore, analysis of the low-temperature EPR spectra of the iron–sulfur centers FA/FB showed that in contrast to native complexes isolated from the wild-type cyanobacteria and menB mutant, in PS I containing Cl2NQ the photoreduction of iron–sulfur clusters does not occur. These data indicate that in the latter case electron transfer from quinone cofactor to the iron–sulfur clusters is completely prevented. Moreover, it was shown that at low temperature, only the quinone molecule in the A-branch participates in electron transfer.
X-band EPR-spectroscopy data obtained under steady-state illumination revealed that upon substitution of PQ by Cl2NQ, almost twofold decrease of the P700+ signal was observed [43].
In the presence of a high concentration of ascorbate (5 mM) under steady-state illumination, even in the case of PS I with Cl2NQ in the A1-site, electron transfer from A1 acceptor through iron–sulfur clusters to ascorbate becomes possible (Fig. 4). This is proved by the observed increase of the P700+ EPR signal amplitude in the presence of ascorbate [43]. This can be explained considering ascorbate as a cyclic electron flow mediator: in the reduced state, it can donate electrons to the photooxidized P700+, and in the oxidized state, it can accept electrons from the terminal iron–sulfur clusters (FA/FB)–.
On the other hand, it is possible that the exogenous electron acceptors directly interact with the quinone cofactor in the A1-site. Measurements of oxygen uptake under steady-state illumination suggested the involvement of the quinone cofactor in oxygen reduction in isolated PS I complexes [44]. It was shown that under low and medium light intensities, oxygen uptake rates in PS I from the wild type and menB strain were similar and increased with increasing light intensity. At the same time, under high light intensity, oxygen uptake rate by PS I continues to increase in the case of the wild type and reaches a steady state in case of menB. The only significant difference between the samples was the difference in midpoint redox potentials of the quinones in the A1 site, which is more auspicious for interaction with oxygen in the case of PS I from the wild type (PhQ). The authors suggested that under high light intensity, two different ways of electron transfer to oxygen coexist: reaction of oxygen with iron–sulfur clusters and its reduction by quinone cofactor in the A1 site (Fig. 4). It was proposed that the first process occurs under moderate illumination, whereas the second occurs at high light intensities.
Thus, the role of the quinone cofactor in electron transfer in PS I may not be restricted only to participation in the forward transfer from the primary acceptor A0 to the iron–sulfur clusters. There are reasons to believe that phylloquinone molecules A1A and A1B in symmetrical branches of cofactors A and B are significant for electron transfer asymmetry and may participate in the interaction with oxygen, preventing overreduction of the cofactors of the PS I electron transfer chain under high light intensity.
This work was supported by the Russian Foundation for Basic Research (grants 15-04-04252, 13-04-40298, 14-04-00519 and 13-04-40299) and the Russian Science Foundation (grant 14-14-00789).
REFERENCES
1.Golbeck, J. H. (ed.) (2006) Photosystem I: The
Light-Driven Plastocyanin:Ferredoxin Oxidoreductase, Springer,
Dordrecht.
2.Jordan, P., Fromme, P., Witt, H. T., Klukas, O.,
Saenger, W., and Krauß, N. (2001) Three-dimensional structure of
photosystem I at 2.5 Å resolution, Nature, 411,
909-917.
3.Guergova-Kuras, M., Boudreaux, B., Joliot, A.,
Joliot, P., and Redding, K. (2001) Evidence for two active branches for
electron transfer in photosystem I, Proc. Natl. Acad. Sci. USA,
98, 4437-4442.
4.Srinivasan, N., and Golbeck, J. H. (2009)
Protein–cofactor interactions in bioenergetics complexes: the
role of the A1A and A1B phylloquinones in
photosystem I, Biochim. Biophys. Acta, 1787,
1057-1088.
5.Shelaev, I. V., Gostev, F. E.,
Mamedov, M. D., Sarkisov, O. M., Nadtochenko, V. A.,
Shuvalov, V. A., and Semenov, A. Y. (2010) Femtosecond primary
charge separation in Synechocystis sp. PCC 6803 photosystem I,
Biochim. Biophys. Acta, 1797, 1410-1420.
6.Deisenhofer, J., Epp, O., Miki, K., Huber, R., and
Michel, H. (1985) Structure of the protein subunits in the
photosynthetic reaction centre of Rhodopseudomonas viridis at 3
Å resolution, Nature, 318, 618-624.
7.Komiya, H., Yeates, T. O., Rees, D. C., Allen, J.
P., and Feher, G. (1988) Structure of reaction center from
Rhodobacter sphaeroides R-26 and 2.4.1: symmetry reactions and
sequence comparison between different species, Proc. Natl. Acad.
Sci. USA, 85, 9012-9016.
8.Brettel, K., and Leibl, W. (2001) Electron transfer
in photosystem I, Biochim. Biophys. Acta, 1507,
100-114.
9.Savikhin, S. (2006) Ultrafast optical spectroscopy
of photosystem I, in Photosystem I. The Light-Driven
Plastocyanin:Ferredoxin Oxidoreductase (Golbeck, J., ed.)
Advances in Photosynthesis and Respiration, Vol. 24, Springer,
Dordrecht, pp. 155-175.
10.Savikhin, S., and Jankowiak, R. (2014) Mechanism
of primary charge separation in photosynthetic reaction centers, in
The Biophysics of Photosynthesis (Golbeck, J., and van der Est,
A., eds.) Springer, pp. 193-240.
11.Brettel, K. (1988) Electron transfer in
photosystem I, FEBS Lett., 239, 93-98.
12.Setif, P., and Bottin, H. (1989) Identification
of electron-transfer reactions involving the acceptor AI of
photosystem I at room temperature, Biochemistry, 28,
2689-2697.
13.Karapetyan, N. V., Schlodder, E., van
Grondelle, R., and Dekker, P. (2006) in Photosystem I. The
Light-Driven Plastocyanin:Ferredoxin Oxidoreductase (Golbeck, J.,
ed.) Springer, Dordrecht, pp. 177-192.
14.Shuvalov, V. A., Nuijs, A. M., van
Gorkom, H. J., Smit, H. W. J., and Duysens, L. N. M. (1986)
Picosecond absorbance changes upon selective excitation of the primary
electron donor P700 in photosystem I, Biochim. Biophys. Acta,
850, 319-323.
15.Hastings, G., Kleinherenbrink, F. A., Lin, S.,
McHugh, T. J., and Blankenship, R. E. (1994) Observation of the
reduction and reoxidation of the primary electron acceptor in
photosystem I, Biochemistry, 33, 3193-3200.
16.Savikhin, S., Xu, W., Martinsson, P., Chitnis, P.
R., and Struve, W. S. (2001) Kinetics of charge separation and
A0 → A1 electron transfer in photosystem I
reaction centers, Biochemistry, 40, 9282-9290.
17.Kumazaki, S., Ikegami, I., Furusawa, H., Yasuda,
S., and Yoshihara, K. (2001) Energy equilibration among the
chlorophylls in the electron-transfer system of photosystem I reaction
center from spinach, J. Phys. Chem. B, 105,
1093-1099.
18.Wasielewski, M. R., Fenton, J. M., and
Govindjee (1987) The rate of formation of P700(+)-A0(–) in
photosystem I particles from spinach as measured by picosecond
transient absorption spectroscopy, Photosynth. Res., 12,
181-189.
19.Nuijs, A. M., Shuvalov, V. A., van
Gorkom, H. J., Plijter, J. J., and Duysens, L. N. M. (1986)
Picosecond absorbance difference spectroscopy on the primary reactions
and the antenna-excited states in photosystem I particles, Biochim.
Biophys. Acta, 850, 310-318.
20.Savikhin, S., Xu, W., Chitnis, P. R., and Struve,
W. S. (2000) Ultrafast primary processes in PS I from
Synechocystis sp. PCC 6803: roles of P700 and A(0), Biophys.
J., 79, 1573-1586.
21.White, N. T. H., Beddard, G. S., Thorne, J. R.
G., Tim, M., Feehan, T. M., Keyes, T. E., and Heathcote, P.
(1996) Primary charge separation and energy transfer in the photosystem
I reaction center of higher plants, J. Phys. Chem., 100,
12086-12099.
22.Melkozernov, A. N., Lin, S., and Blankenship, R.
E. (2000) Excitation dynamics and heterogeneity of energy equilibration
in the core antenna of photosystem I from the cyanobacterium
Synechocystis sp. PCC 6803, Biochemistry, 39,
1489-1498.
23.Gobets, B., and van Grondelle, R. (2001) Energy
transfer and trapping in photosystem I, Biochim. Biophys. Acta,
1507, 80-99.
24.Muller, M. G., Niklas, J., Lubitz, W., and
Holzwarth, A. R. (2003) Ultrafast transient absorption studies on
photosystem I reaction centers from Chlamydomonas reinhardtii.
1. A new interpretation of the energy trapping and early electron
transfer steps in photosystem I, Biophys. J., 85,
3899-3922.
25.Holzwarth, A. R., Muller, M. G.,
Reus, M., Nowaczyk, M., Sander, J., and Rogner, M.
(2006) Kinetics and mechanism of electron transfer in intact
photosystem II and in the isolated reaction center: pheophytin is the
primary electron acceptor, Proc. Natl. Acad. Sci. USA,
103, 6895-6900.
26.Holzwarth, A. R., Muller, M. G., Niklas, J.,
and Lubitz, W. (2006) Ultrafast transient absorption studies on
photosystem I reaction centers from Chlamydomonas reinhardtii.
2. Mutations near the P700 reaction center chlorophylls provide new
insight into the nature of the primary electron donor, Biophys.
J., 90, 552-565.
27.Ptushenko, V. V., Cherepanov, D. A.,
Krishtalik, L. I., and Semenov, A. Y. (2008) Semicontinuum
electrostatic calculations of redox potentials in photosystem I,
Photosynth. Res., 97, 55-74.
28.Semenov, A. Y., Shelaev, I. V., Gostev, F.
E., Mamedov, M. D., Shuvalov, V. A., Sarkisov, O. M.,
and Nadtochenko, V. A. (2012) Primary steps of electron and energy
transfer in photosystem I: effect of excitation pulse wavelength,
Biochemistry (Moscow), 77, 1011-1020.
29.Sun, J., Hao, S., Raddle, M., Xu, W.,
Shelaev, I., Nadtochenko, V., Shuvalov, V., Semenov, A.,
Gordon, H., van der Est, A., and Golbeck, J. H. (2014) Evidence that
histidine forms a coordination bond to the A0A and A0B chlorophylls and
a second H-bond to the A1A and A1B phylloquinones in M688HPsaA and
M668HPsaB variants of Synechocystis sp. PCC 6803, Biochim.
Biophys. Acta, 1837, 1362-1375.
30.Macomber, G. D. (1976) The Dynamics of
Spectroscopic Transition, John Wiley and Sons, New
York-London-Sydney-Toronto.
31.Shuvalov, A. (1984) Main features of the primary
charge separation in photosynthetic reaction centers, in Advances in
Photosynthesis Research (Sybesma, C., ed.) The
Hague-Boston-Lancaster, pp. 93-100.
32.Cohen, R. O., Shen, G., Golbeck, J. H., Xu, W.,
Chitnis, P. R., Valieva, A. I., van der Est, A., Pushkar, Y., and
Stehlik, D. (2004) Evidence for asymmetric electron transfer in
cyanobacterial photosystem I: analysis of a methionine-to-leucine
mutation of the ligand to the primary electron acceptor A0,
Biochemistry, 43, 4741-4754.
33.Dashdorj, N., Xu, W., Cohen, R. O., Golbeck, J.
H., and Savikhin, S. (2005) Asymmetric electron transfer in
cyanobacterial photosystem I: charge separation and secondary electron
transfer dynamics of mutations near the primary electron acceptor
A0, Biophys. J., 88, 1238-1249.
34.Savitsky, A., Gopta, O., Mamedov, M.,
Golbeck, J. H., Tikhonov, A., Moebius, K., and Semenov, A. (2010)
Alteration of the axial met ligand to electron acceptor A0
in photosystem I: effect on the generation of
P700+A1– radical
pairs as studied by W-band transient EPR, Appl. Magnet. Reson.,
37, 85-102.
35.Vos, M. H., and Van Gorkom, H. J. (1990)
Thermodynamic and structural information on photosynthetic systems
obtained from electroluminescence kinetics, Biophys. J.,
58, 1547-1555.
36.Ishikita, H., and Knapp, E. W. (2003) Redox
potential of quinones in both electron transfer branches of photosystem
I, J. Biol. Chem., 278, 52002-52011.
37.Krieger, A., Rutherford, A. W., and Johnson, G.
N. (1995) On the determination of redox potential of the primary
quinone electron acceptor, QA, in photosystem II,
Biochim. Biophys. Acta, 1229, 193-201.
38.Wraight, C. A. (1981) Oxidation-reduction
physical chemistry of the acceptor quinone complex in bacterial
photosynthetic reaction centers: evidence for a new model of herbicide
activity, Israel J. Chem., 21, 348-354.
39.Jhonson, T. W., Zybailov, B., Jhones, A. D.,
Bittl, R., Zech, S., Stehlik, D., Golbeck, J. H., and Chitnis, P. R.
(2001) Recruitment of a foreign quinone into A1 site of photosystem I:
in vivo replacement of plastoquinone-9 by media-supplemented
naphthoquinones in phylloquinone biosynthetic pathway mutants of
Synechocystis sp. PCC 6803, J. Biol. Chem., 276,
39512-39521.
40.Van der Est, A., Pushkar, Yu., Karyagina, I.,
Fonovic, B., Dudding, T., Niklas, J., Lubitz, W., and Golbeck, J. H.
(2010) Incorporation of 2,3-disubstituted-1,4-naphthoquinones into the
A1 binding site of photosystem I studied by EPR and ENDOR spectroscopy,
Appl. Magn. Reson., 37, 65-83.
41.Semenov, A. Yu., Vassiliev, I. R., van der Est,
A., Mamedov, M. D., Zybailov, B., Shen, G. Z., Stehlik, D., Diner,
B. A., Chitnis, P. R., and Golbeck, J. H. (2000) Recruitment of a
foreign quinone into the A1 site of photosystem I: altered kinetics of
electron transfer in phylloquinone biosynthetic pathway mutants studied
by time-resolved optical, EPR, and electrometric techniques, J.
Biol. Chem., 275, 23429-23438.
42.Mula, S., Savitsky, A., Mobius, K., Lubitz, W.,
Golbeck, J. H., Mamedov, M. D., Semenov, A. Yu., and van der Est,
A. (2012) Incorporation of a high potential quinone reveals that
electron transfer in photosystem I becomes highly asymmetric at low
temperature, Photochem. Photobiol. Sci., 11, 946-956.
43.Trubitsin, B. V., Mamedov, M. D.,
Semenov, A. Yu., and Tikhonov, A. N. (2014) Interaction of ascorbate
with photosystem I, Photosynth. Res., 122, 215-231.
44.Kozuleva, M. A., Petrova, A. A.,
Mamedov, M. D., Semenov, A. Yu., and Ivanov, B. N. (2014)
O2 reduction by photosystem I involves phylloquinone under
steady-state illumination, FEBS Lett., 588,
4364-4368.