Molecular Dynamics Investigation of a Mechanism of Allosteric Signal Transmission in Ribosomes
G. I. Makarov1, A. V. Golovin2, N. V. Sumbatyan1, and A. A. Bogdanov1,3*
1Lomonosov Moscow State University, Faculty of Chemistry, 119991 Moscow, Russia2Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119991 Moscow, Russia
3Belozersky Institute of Physical-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: bogdanov@belozersky.msu.ru
* To whom correspondence should be addressed.
Received March 2, 2015; Revision received April 13, 2015
The ribosome is a molecular machine that synthesizes all cellular proteins via translation of genetic information encoded in polynucleotide chain of messenger RNA. Transition between different stages of the ribosome working cycle is strictly coordinated by changes in structure and mutual position both of subunits of the ribosome and its ligands. Therein, information regarding structural transformations is transmitted between functional centers of the ribosome through specific signals. Usually, functional centers of ribosomes are located at a distance reaching up to several tens of angstroms, and it is believed that such signals are transduced allosterically. In our study, we attempted to answer the question of how allosteric signal can be transmitted from one of the so-called sensory elements of ribosomal tunnel (RT) to the peptidyl transferase center (PTC). A segment of RT wall from the E. coli ribosome composed of nucleotide residues A2058, A2059, m2A2503, G2061, A2062, and C2063 of its 23S rRNA was examined by molecular dynamics simulations. It was found that a potential signal transduction pathway A2058-C2063 acted as a dynamic ensemble of interdependent conformational states, wherein cascade-like changes can occur. It was assumed that structural rearrangement in the A2058-C2063 RT segment results in reversible inactivation of PTC due to a strong stacking contact between functionally important U2585 residue of the PTC and nucleotide residue C2063. A potential role for the observed conformational transition in the A2058-C2063 segment for regulating ribosome activity is discussed.
KEY WORDS: ribosome, ribosomal tunnel, allostery, molecular dynamics simulationsDOI: 10.1134/S0006297915080106
The ribosome is a molecular machine that synthesizes all cellular proteins via translation of genetic information encoded in a polynucleotide chain of messenger RNA (mRNA). It is known that transition between different stages of the work cycle in ribosomes is modulated by strictly coordinated changes of structure and mutual position both of subunits of the ribosome and its ligands such as tRNA and protein translation factors [1-5]. Therein, information regarding such structural changes is transmitted between functional centers of the ribosome through specific signals [6]. Due to the fact that functional centers of the ribosome are usually separated by a long distance, sometimes reaching several tens of angstroms, it was assumed that transmission of such signals occurs allosterically [7]. Thus, strong evidence has been obtained indicating that ribosomal RNAs (rRNA) play a key role in this process [8, 9]. Quite likely, some antibiotics inhibiting the translation process can also influence ribosomal centers without directly contacting them, i.e. via allosteric interaction [10-13].
Consequences of the single mutations, insertions, and deletions of nucleotide residues in the rRNA located in the macromolecular structure of the ribosome at long distances away from its functional centers may be an example of allosteric impact on effective action of the ribosome. Such targeted local distortions in the primary structure of the rRNA usually result in alteration of mutual orientation of its multiple nucleotide residues, as was documented by methods of chemical modification (so-called “probing”) used while routinely analyzing secondary structure of RNAs. Importantly, these residues are located inside intra-ribosomal tertiary structure of rRNA within a relatively compact channel-like region, along which allosteric signals seem to be transduced [14-17].
Allosteric effects observed upon functioning of proteins, especially protein enzymes have been extensively studied and discussed for almost half a century [18], and important success has been achieved in understanding their nature (see review [19]). At the same time, mechanisms responsible for transmission of allosteric signals in macromolecular RNA and RNA–protein complexes began to be investigated only within recent years [20, 21], and today they are still basically unknown.
In our study, we tried to answer the question of how allosteric signal might be transmitted from the so-called sensory center of the ribosomal tunnel (RT) to the peptidyl transferase center (PTC) of the E. coli ribosome, where synthesis of polypeptide protein chain takes place. The RT is located inside the large subunit of the ribosome. It connects the PTC with a region on the surface of the ribosome, where the first stages of posttranslational modification and folding of polypeptide chain of newly synthesized proteins occur. Moreover, numerous antibiotics that inhibit protein synthesis by ribosomes bind to the RT [22]. Walls of the RT are formed by residues of rRNA and r-proteins, whereas its part adjacent to the PTC studied in our work is solely built up from nucleotide residues of 23S rRNA akin to the PTC [23]. The vast majority of polypeptide chains synthesized by ribosomes quite easily move along the RT. However, this rule has some exclusions: there are several cases (few so far) when synthesis polypeptide chain inside the PTC and its transport along the RT may stop. This results from the fact that while being inside the RT, specific amino acid residues at strictly determined positions in the nascent peptide chain contact to special (sensory) nucleotide residues located on the walls of the RT. Interaction between them signals to the PTC, so that it inactivates the latter and results in losing its ability to catalyze formation of new peptide bonds. When this happens, the work of the ribosome can be stopped either by the nascent polypeptide or by low molecular weight cofactors that should additionally be present in the RT [24]. In the latter, numerous examples have been documented showing that macrolide and ketolide antibiotics serve a cofactor in this process [25]. Due to the fact that the residues of 23S rRNA involved in these events are located outside the PTC, it is believed that transmission of a signal from sensor towards this center occurs allosterically.
Arrest of the work of ribosomes due to the above-mentioned events is an important way to regulate biosynthesis of proteins at the translation level, and it is not surprising that it has been extensively investigated (see review [24]). In particular, it is assumed that at least two chains of nucleotides from 23S rRNA creating the paths along which a signal may be transmitted from RT sensors to PTC exist in E. coli ribosome [26]. For instance, one potential path starting at nucleotide residue A752 was recently examined by combining cryoelectron microscopy, molecular dynamics simulations, and biochemical analysis [27]. It was demonstrated that the antibiotic erythromycin shifts the position of the C-terminus of the so-called leader peptide of the erythromycin-dependent methyltransferase ErmB so that it makes contact with nucleotide residues building up the PTC and transfers it into the inactive state that results in translation arrest.
In our work, we applied molecular dynamics (MD) simulation to investigate another segment of the RT wall in the E. coli ribosome, consisting of residues A2058, A2059, m2A2503, G2061, A2062 and C2063, which were convincingly shown to participate in transmitting signal from the A2058-A2059 sensory site using site-directed mutagenesis [25]. The beginning of this potential signaling pathway (residue A2058) is located 20 Å away from the PTC. Importantly, residues A2058 and A2059 located at the beginning of this pathway take part in formation of a binding site of all antibiotics such as macrolides and ketolides [28]. Moreover, it has long been known that N6-dimethylation of A2058 results in resistance of bacteria to antibiotics of this and other classes [29].
Despite the outstanding complexity of the ribosome, after deciphering its atomic structure, MD has been used repeatedly to describe dynamic features of the structure and functioning of both the entire ribosome and its separate parts (see a review [30]). Also, MD is used to study structure and functions of the RT [31]. In particular, it revealed some potential contacts of the key amino acid residues within peptides that arrest translation with sensory nucleotide residues of the RT [32]. Recently, using this method, we found possible reasons for abnormal biological activity of peptide derivatives of macrolides, which inhibit protein synthesis, and described a novel potential site in the walls of the RT that interacts with nascent polypeptide chains [33]. By using MD simulation, we observed that the potential signal-transmitting pathway A2058-C2063 behaved as a dynamic ensemble of interdependent conformational states, wherein cascade-like changes can originate. Hence, we assume that structural rearrangement in the A2058-C2063 RT segment results in reversible inactivation of the PTC. A potential role of the observed conformational transition in the A2058-C2063 segment in regulating ribosome activity is discussed.
MATERIALS AND METHODS
Simulation system. A starting system was made using the crystal structure of the 50S subunit from the E. coli ribosome complexed with erythromycin obtained using X-ray analysis with 3.1 Å resolution (PDB code: 3OFR) [34] supplemented with modified bases in accordance with the database [35]. The positions of modified bases were optimized by applying molecular mechanics energy minimization with the L-BFGS algorithm and limited memory usage [36] followed by further computation of 2-nsec molecular dynamics controlled by a velocity-rescaling thermostat with stochastic correction [37]. Hence, the positions of all unmodified bases were fixed, whereas modified bases, ions, and water moved, and erythromycin contained in the starting X-ray structure was removed. After optimization, regions having at least one atom within a cubic area with 7-nm edge that included the entire RT and PTC in a way that the center of this region was located at the ribosomal tunnel aligned along an imaginary z-axis were separated. The final separation was further used in all calculations.
Control systems were built using the starting system described above, wherein, in the first case, the position of nucleotide base G2057, A2058, and A2059 were selectively fixed; in the second case, 2-methyladenine in m2A2503 was replaced by guanine; in the third case, adenine in A2062 was replaced by uracil.
Simulation environment. Computation of molecular dynamics and analysis of the obtained trajectories were done using GROMACS [38, 39] software version 4.5.4 and 4.6.5 and parm99sb force field. All simulations were run at T = 300K with 0.1 psec coupling time controlled by a velocity-rescaling thermostat with additional stochastic correction [37] and isotropic constant-pressure boundary conditions controlled by a Berendsen barostat [40] with 5 psec coupling time. Electrostatic interactions were computed using the particle-mesh Ewald method for electrostatic interactions (PME) [41] with 0.125 nm grid and fourth-order interpolation. A simulation system was centered in the cubic box with 8.8-nm edge filled with 13,067 TIP4P water molecules [42] so that system edges were covered by a 0.9-nm-thick solvent layer. Negative charge of the system was neutralized by adding 95 magnesium and 381 sodium ions. The latter were used instead of potassium ions due to their incorrect parameterization in the parm99sb force field based on the data for crystals of potassium chloride. Upon simulation in water, potassium and chloride ions form crystals of potassium chloride at concentrations insufficient for this process [43]. Interactions between potassium ions and any negatively charged ions were overestimated, so that use of potassium ions as counterions can result in larger perturbations than for sodium ions in the simulated system. Magnesium ions were added so that they might form “magnesium bridges” between neighboring phosphate groups, whereas sodium ions were positioned close to negatively charged groups to compensate residual negative charge [44]. Residues having at least one atom located within 0.1 nm from the edge of simulated regions of the ribosome were positionally restrained, whereas the rest of the atoms were able to move freely. In all simulations, integration time step was 2 fsec, and coordinates were recorded into the trajectory file every 15 psec. To limit lengths of covalent bonds with hydrogen atoms, the LINCS algorithm was applied. Controlled dynamics and meta-dynamics were calculated using the PLUMED software [45] version 1.3 that works together with the GROMACS software suite. Position and frequency of hydrogen bonds were analyzed as described earlier [33].
RESULTS AND DISCUSSION
Our study was aimed at detailed analysis of conformational mobility for a segment of the E. coli ribosomal tunnel wall comprised by the 2058-2063 part of the polynucleotide 23S rRNA chain as well as a modified nucleotide residue m2A2503 that adjoins it within a tertiary ribosome structure. As mentioned above, this segment of the 23S rRNA macromolecule has been repeatedly considered as one of the most probable participants in the transmission of functional signals from the internal regions of the RT to the ribosomal PTC [24-26]. Quite recently, this assumption has been convincingly confirmed in experiments [13, 27, 46]. At the same time, a detailed picture of potential conformational transformations that might underlie transmission of functional signals through this rRNA segment has not yet been obtained. In our study, we tried to fill this gap by paying special attention to the search for an answer to the question of how changes in mutual position of nucleotide residues in the examined RT affect conformation of the ribosomal PTC. Our study was performed by the method of molecular dynamics simulations known for its ability to evaluate time-dependent changes of torsion angles in nucleotide residues of rRNA, changes in position and occurrence of hydrogen bonds among them, as well as changes in energy of association of heterocyclic nucleotides [47].
Twenty-one trajectories lasting from 200 to 360 nsec were calculated for the examined system. While analyzing them, various conformational transitions were documented that affected both nucleotide residues of the examined RT segment and some nucleotide residues that participate in functioning of the ribosomal PTC. The following main conformational transitions were found: 1) adenine bases A2058 and A2059 come closer; 2) the C2063 base loses contact with guanine base G2061, which results in emerging of the opportunity for the A2062 base to intercalate between them and create an element of the structure wherein all three bases are tightened through stacking interactions; 3) the conformation of residue U2585, a ribosomal PTC component, changes in a way that its base makes a stacking contact with cytosine base C2063.
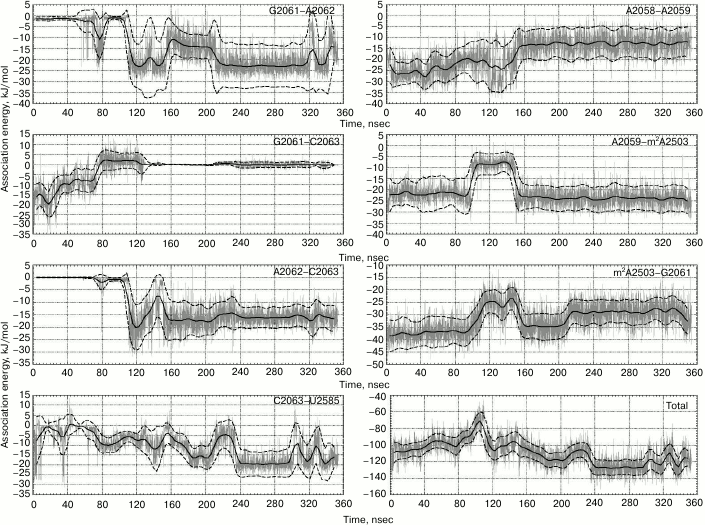
Sometimes, such events occur separately, and, importantly, in one of the trajectories they sometimes occur together by making an interconnected conformational transition wherein bases A2058 and A2059 come closer for approximately 60 nsec followed by all events mentioned above taking place (Fig. 1). As a result, A2058-A2059-m2A2503-G2061 (existing in the original structure) and A2062-C2063-U2585 (newly generated) blocks being stabilized via stacking interactions unite into a single stack-like structure. Such coordinated movement is clearly exhibited in the first main component obtained while analyzing the magnitude of the main components of β, γ, δ, and χ torsion angles for thirty-four nucleotide residues in the nearest vicinity of the examined RT segment and the PTC (in part, results of the analysis are shown in Fig. 2). Also, it was well illustrated by time-dependent changes for energy of noncovalent (electrostatic and van der Waals) interactions responsible for association of heterocyclic bases of neighbor nucleotide residues (Fig. 3). Thus, it must be emphasized that changes in magnitude of angles and association energy occur sequentially rather than synchronously, being split into three main groups described above. If conformational changes stand alone, then they do not complete and do not result in formation of stable structures; however, when all are involved in a united conformational transition, the process ends up with generation of a stable structure. It is important to note that the observed changes in mutual position of nucleotide residues in the A2058-C2063 segment as well as nucleotide residues m2A2503 and U2585 contacting to them turned out to be reversible, as found using the meta-dynamics approach described by Laio and Parrinello [48].
Fig. 1. Main phases of conformational transition in the A2058-C2063 region of the 23S rRNA E. coli ribosome observed using molecular dynamics (MD) simulation: a) baseline (4 nsec); b, c) intermediate (71 and 117 nsec); d) final (353 nsec) stage. Nucleotide A2062 is highlighted in solid dark gray color.
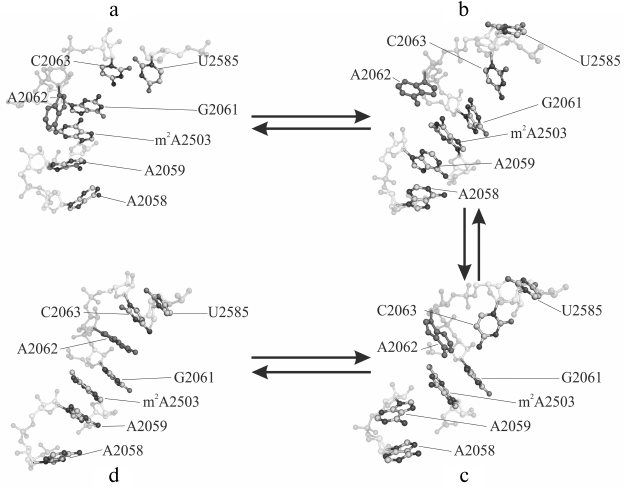
Fig. 2. Values of torsion angles for some nucleotide residues in the A2058-C2063 region and U2585 undergoing the maximum changes during the examined conformational transitions.
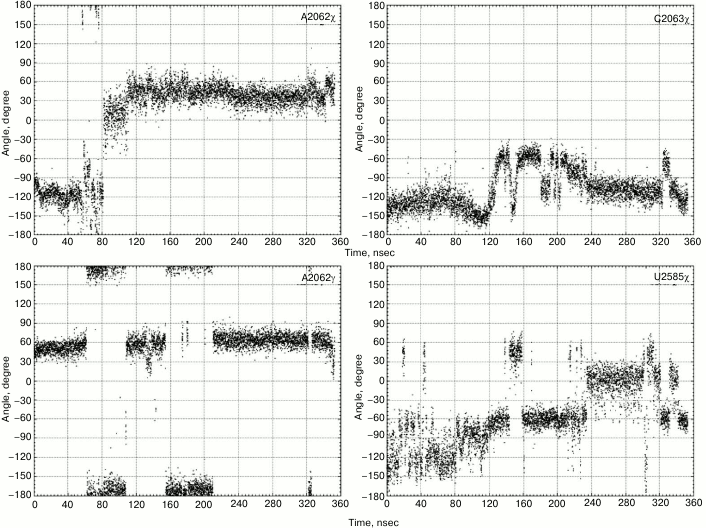
Fig. 3. Energy of noncovalent interactions for nitrogen base pairs involved in stacking interactions during the examined conformational transition. Magnitude of energy of noncovalent interactions is depicted in gray color, value of energy smoothed by sliding average with 9-nsec window – solid black line, moving standard deviation – dashed black line. Lower right plots depict changes in total energy of noncovalent interactions between bases in the examined region of 23S rRNA.
It should be noted that Gumbart et al. previously investigated potential changes in conformation of the A2062-G2061-C2063 region of the examined 23S rRNA segment under the influence of SecM stalling-peptide using MD simulation [32]. Their data indicated that a functional signal was transmitted to the PTC without rearranging the order of bases relative to the position of nucleotide residues within this element. However, in this case MD simulations were terminated at 20 nsec, i.e. long before such transition might occur according to our data.
A hydrophobic effect and stacking interactions might be considered as a driving force in the observed coordinated conformational changes in the A2028-C2063 segment. During such structural transition, the hydrophobic area of the system wanes, as the bases of A2062 and U2585 nucleotides located in the original structure inside the tunnel lumen began to make stacking interactions with their new neighbors. Indeed, energy gain is achieved due to insertion of the A2062 base between C2063 and G2061 and formation of contact between C2063 and U2585 (Fig. 3); the change in energy of noncovalent interactions was –19 ± 10 kJ/mol. Thus, between 80 and 160 nsec the system overcomes an energy barrier corresponding to conformations wherein bases G2061 and C2063 had already diverged, but A2062 and U2585 did not contact them yet, being stabilized by stacking interactions.
Now let us consider properties of participants in the examined conformational transition in the light of published data. It was already noted that it starts when nucleotide residues A2058 and A2059 come closer to each other. Their adenine bases form a hydrophobic “pocket” exposed on the RT wall – one of the main sites for binding macrolides and ketolides in the large subunit of the ribosome [49, 50]. A convergence effect was anticipated, as the macrolide antibiotic erythromycin in the starting system was removed from this “pocket”, whereas in a ribosome bound to erythromycin vs. free ribosome bases A2058 and A2059 diverged (e.g. compare structures with PDB 2AW4 and 3OFR codes, respectively). It is important that in the control system, where the position of these nucleotides was “frozen” at the baseline, a conformational transition in the examined segment was not observed.
However, the strongest conformational rearrangements in the examined system are related to altered position of A2062 (Fig. 1), a fully conservative nucleotide residue in rRNA of the large ribosomal subunit that possesses extremely high mobility. Such extraordinary mobility of A2062 has also been evolutionarily conserved, suggesting that this capacity is necessary for normal functioning of the ribosome. In many currently known crystal structures of the ribosome, A2062 exhibits a conformation that we call here as “open”. Thus, its heterocyclic base is directed into the free lumen of the RT (e.g. see structures in PDB with codes 1VQL, 2WDN, and 3OFR). However, in some crystal structures of the 50S subunit of the bacterial ribosome the adenine residue in A2062 is rotated with respect to its carbohydrate moiety, so that its plane is rather parallel to the RT walls (e.g. see structures in PDB with codes 1KQS, 3OHK, and 3OFQ). Such conformation will be called as “closed”. Our calculations of molecular dynamics for A2062 show that it indeed steadily transits from “open” to “closed” conformation, wherein the former is preferred (Fig. 4). While doing MD simulation of the A2058-A2063 segment, we use the structure of the 50S subunit of E. coli ribosome (code 3OFR in PDB), where A2062 is present in the “open” conformation. However, after the simulation was started, its conformation rapidly rearranged into “closed” conformation (see structure in Fig. 1 that corresponds to 4 nsec), which is also clearly observed in a scheme illustrating dynamic changes in position of hydrogen bonds between A2062 and its neighbors (Fig. 5a). It is known that in crystal structures of the 50S subunit of the ribosome from Haloarcula marismortui and Escherichia coli heterocyclic base of A2062, being both in “open” and “closed” conformation, is connected through hydrogen bonds with the residue of the modified adenine in m2A2503 [51], which was noted above to be inserted into the A2058-C2063 segment in the 3D-structure of the ribosome. Initially, A2062 makes an unstable trans-Watson–Crick/Hoogsteen A–A pair with m2A2503 (Fig. 5a) (according to the classification by Westhof et al. [52]). This pair is destroyed at early stages of the examined conformational transition, later allowing A2062 to intercalate between nucleotide bases G2061 and C2063. It was already noted that this event is preceded by altered position of C2063 and loss of its contact with G2061 (Figs. 1 and 2). Regarding nucleotide base m2A2503, we noted that during simulation it largely has a stacking contact with neighbor A2059 and G2061, except for the time between 90 and 150 nsec when this contact is markedly weakened (Fig. 2).
Fig. 4. Distribution of magnitude for χ torsion angles of nucleotide residues A2062 and U2585. For distribution of magnitude of χ torsion angle for base A2062 peak I corresponds to phases (a) and (b) (Fig. 1), peak II – phases (c) and (d) (Fig. 1). For distribution of magnitude of χ torsion angle for base U2585, peak I corresponds to phase (a), peak II – phases (b) and (c), peak III – phase (d) (Fig. 1).
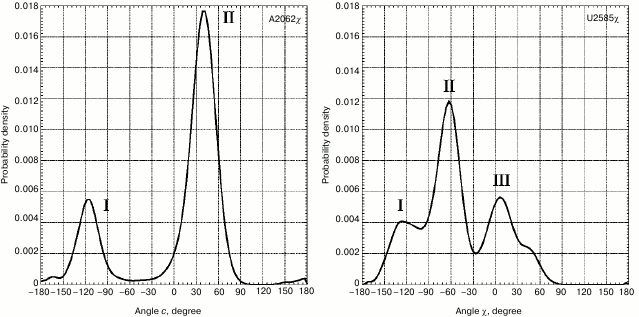
Fig. 5. a) Scheme depicting formation of the main hydrogen bonds of nucleotide A2062 during molecular dynamics simulations. The gray vertical line depicts the occurrence of the noted hydrogen bond at a particular time point. b) Map of hydrogen bonds of G2503 obtained in silico from trajectory with A2503G mutation.
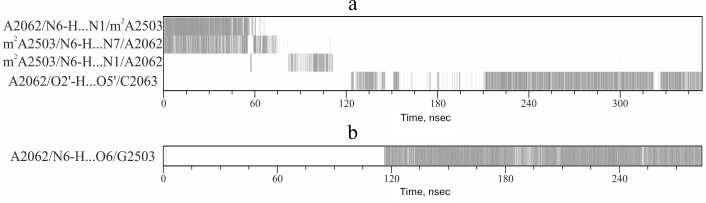
An assumption that nucleotide base A2062 plays an important role in functioning of the ribosome was stated soon after deciphering its atomic structure [53]. Later, it was convincingly shown by Mankin et al. that mutation in this position within 23S rRNA entirely excluded arrest of the ribosome in systems with stalling peptides [54]. Because a mere substitution of adenine in A2062 by other nucleotide bases did not notably affect efficacy of ribosomal PTC, it was assumed that mutation of this nucleotide residue interrupts transmission of allosteric signal in the PTC from deeper regions of the RT. Similar results were obtained by the same authors for m2A2503 as well [51]. Following these data, we substituted in the starting system for MD simulation a base in A2062 for uracil, whereas N2-methyladenine in m2A2503 was substituted with guanine. These in silico mutations resulted in hindering of the above-mentioned conformational transition. Thus, U2062 intercalated for a short time between G2061 and C2063 that, however, was unable to make with them a stable triad due to smaller size of the heterocyclic ring and weakened stacking interactions. Regarding G2503, it can be noted that its base tended to make a quite strong non-Watson–Crick G–A pair with the base of A2062, thus letting the latter be held in its initial state (Fig. 5b).
Although the order of conservative nucleotide residues in the G2061-A2062-C2063 triad we described corresponds to the secondary structures of all known rRNAs of the large ribosomal subunit, it has not been observed in any of its 3D structures obtained with X-ray analysis or cryoelectron microscopy with near-atomic resolution. Therein, nucleotide residue G2061 directly contacts C2063, whereas A2062 is either taken outside (“open” conformation) or located nearby G2061, but not C2063 (“closed” conformation). Nucleotide residues G2061 and C2063 belong to the so-called “outer shell” of the PTC [55] and participate in structuring of A2451, one of the key functional nucleotide residues of the ribosomal PTC [56, 57], whereas their site-directed mutagenesis dramatically affected activity of the ribosome [58, 59]. Thus, this suggests that gradual shifting of C2063 relative to its position in the starting structure as well as complete loss of its contacts with G2061 by 80 nsec can also affect efficacy of the peptidyl transferase (PT) reaction.
Formation of a stable association between C2063 and U2585 at 240 nsec represents another important consequence observed after the A2062-G2061-C2063 segment was reorganized into G2061-A2062-C2063 triad (Fig. 2). Although investigation of the mechanism of the PT reaction is not yet complete (e.g. see recent paper by Polikanov et al. [57]), by now it has been firmly established that at least four nucleotide bases of rRNA such as C2063, A2451, U2585, and U2586 play a key role in catalysis in generating peptide bonds in the polypeptide chain of proteins synthesized in the ribosome [60]. It was shown that at each step of the PT reaction one of substrates of PTC –aminoacyl-tRNA – induces rearrangement of conformation in an ensemble composed of these nucleotides, so that it fits the structure of the 3′-terminal region of tRNA and the amino acid residue used for its acylation [61]. In other words, this ensemble should be easily rearranged. Thus, it turned out that the mobility of U2585 in it was particularly prominent. Hence, it is clear that the observed deviation of U2585 from the position optimal for effective course of the PT reaction as well as its arrest should result in inactivation of the ribosome. Moreover, we found that U2585, being associated with C2063, imposes difficulties for binding of 3′-terminal adenosine in peptidyl-tRNA to the P-site of the PTC.
It is important to note that the conformational transition of A2058-C2063 segment we observed, first, is reversible, and second, it proceeds at a rate several orders exceeding the rate of the PT reaction. Therefore, the conformational changes under discussion could affect the activity of the ribosome (resulting, for example, in long-lasting arrest of translation), if the conformational transition of A2058-C2063 segment would be fixed somehow, e.g. by a stalling peptide. Very recently, owing to significant progress in increasing resolving power of cryoelectron microscopy, various stalling peptides and their surroundings inside the RT were observed [27, 62-65]. However, only Arenz et al. mentioned above [27] were able to observe fixation of the position of U2585, which occurred in the case when the ErmBL leader peptide and the antibiotic erythromycin were simultaneously present inside the E. coli ribosome. Moreover, in this case mobility of U2585 was restrained due to its stacking interaction with U2586 rather than C2063. Furthermore, no pronounced change in conformation of the RT region examined in our study was noted by Arenz et al. (see structure with code PDB 3J5L).
So, how might “freezing” of the unusual conformation in the A2058-C2063 segment that we observe happen? It was already noted that a conformational transition in this RT segment is initiated by alteration of mutual position, namely convergence of adenine bases within dinucleotide fragment A2058-A2059 of 23S rRNA. One can imagine that while a newly synthesized polypeptide moves along the RT, one of its hydrophobic amino acid residues (X) enters the hydrophobic cavity comprised by A2058 and 2059 so that it moves the adenine bases apart. Most probably, binding of X to this site would be easily reversible (in contrast to macrolides and ketolides, which bind to this region of RT by a number of firm hydrogen bonds along with hydrophobic contact). Then, before the next step of synthesis of the polypeptide chain occurs, residue X may periodically leave the hydrophobic cavity of the A2058-A2059 segment and bind it again, thus resulting in periodic changes in conformation of the A2058-C2063 segment as described above. Afterwards, further events could develop by one of two scenarios depending on the primary structure of the C-terminal fragment of the nascent polypeptide chain. Indeed, it was recently found that peptides with specific amino acid sequence only 3-amino-acid-long bound to tRNA in the PTC could arrest translation under certain circumstances [66]. This occurs due to their interaction with nucleotide residues of the RT directly adjacent to the PTC. In case the structure of the C-terminal fragment of the synthesized polypeptide allows it to fix the G2061-A2062-C2063 fragment in stacking conformation, then protein synthesis in the ribosome would be slowed or even arrested due to stabilizing contact of U2585 with C2063. If the C-terminal fragment of the synthesized polypeptide had a distinct amino acid sequence, then arrest or significant slowing of translation might not occur. A process regulating translation by any factors reversibly binding to the A2058-A2059 sensory element of the RT might be accomplished according to the same mechanism, which certainly must be experimentally tested.
The authors are thankful to the Research Computing Center of Lomonosov Moscow State University for the opportunity to compute molecular dynamics by using “Lomonosov” supercomputer.
This work was done with financial support of the Russian Scientific Foundation (grant 14-24-00061).
REFERENCES
1.Polacek, N., Patzke, S., Nierhaus, K. H., and
Barta, A. (2000) Periodic conformational changes in rRNA: monitoring
the dynamics of translating ribosomes, Mol. Cell, 6,
159-171.
2.Zhou, J., Lancaster, L., Donohue, J. P., and
Noller, H. F. (2013) Crystal structures of EF-G–ribosome
complexes trapped in intermediate states of translocation,
Science, 340, 1236086.
3.Pulk, A., and Cate, J. H. D. (2013) Control of
ribosomal subunit rotation by elongation factor G, Science,
340, 1235970.
4.Ogle, J. M., Brodersen, D. E., Clemons, W. M.,
Tarry, M. J., Carter, A. P., and Ramakrishnan, V. (2001) Recognition of
cognate transfer RNA by the 30S ribosomal subunit, Science,
292, 897-902.
5.Munro, J. B., Sanbonmatsu, K. Y., Spahn, C. M., and
Blanchard, S. C. (2009) Navigating the ribosome’s metastable
energy landscape, Trends Biochem. Sci., 34, 390-400.
6.Steitz, T. A. (2008) A structural understanding of
the dynamic ribosome machine, Nat. Rev. Mol. Cell Biol.,
9, 242-253.
7.Rheinberger, H.-J., and Nierhaus, K. H. (1986)
Allosteric interactions between the ribosomal transfer RNA-binding
sites A and E, J. Biol. Chem., 261, 9133-9139.
8.Bogdanov, A. A., Dontsova, O. A., Dokudovskaya, S.
S., and Lavrik, I. N. (1995) Structure and function of 5S rRNA in the
ribosome, Biochem. Cell Biol., 73, 869-876.
9.Chan, Y.-L., Dresios, J., and Wool, I. G. (2006) A
pathway for the transmission of allosteric signals in the ribosome
through a network of RNA tertiary interactions, J. Mol.
Biol., 355, 1014-1025.
10.Blaha, G., Gurel, G., Schroeder, S. J., Moore, P.
B., and Steitz, T. A. (2008) Mutations outside the anisomycin-binding
site can make ribosomes drug-resistant, J. Mol. Biol.,
379, 505-519.
11.Davidovich, C., Bashan, A., Auerbach-Nevo, T.,
Yaggie, R. D., Gontarek, R., and Yonath, A. (2007) Induced-fit tightens
pleuromutilins binding to ribosomes and remote interactions enable
their selectivity, Proc. Natl. Acad. Sci. USA, 104,
4291-4296.
12.Wang, L., Pulk, A., Wasserman, M. R., Feldman, M.
B., Altman, R. B., Cate J. H., and Blanchard, S. C. (2012) Allosteric
control of the ribosome by small-molecule antibiotics, Nat. Struct.
Mol. Biol., 19, 957-963.
13.Sothiselvam, S., Liu, B., Han, W., Ramu, H.,
Klepacki, D., Atkinson, G. C., Brauer, A., Remm, M., Tenson, T.,
Schulten, K., Vazquez-Laslop, N., and Mankin, A. S. (2014) Macrolide
antibiotics allosterically predispose the ribosome for translation
arrest, Proc. Natl. Acad. Sci. USA, 111, 9804-9809.
14.Sergiev, P. V., Bogdanov, A. A., Dahlberg, A. E.,
and Dontsova, O. (2000) Mutations at position A960 of E. coli
23S ribosomal RNA influence the structure of 5S ribosomal RNA and the
peptidyltransferase region of 23S ribosomal RNA, J. Mol. Biol.,
299, 379-389.
15.Sergiev, P. V., Lesnyak, D. V., Burakovsky, D.
E., Kiparisov, S. V., Leonov, A. A., Bogdanov, A. A., Brimacombe, R.,
and Dontsova, O. A. (2005) Alteration in location of a conserved
GTPase-associated center of the ribosome induced by mutagenesis
influences the structure of peptidyltransferase center and activity of
elongation factor G, J. Biol. Chem., 280,
31882-31889.
16.Sergiev, P. V., Kiparisov, S. V., Burakovsky, D.
E., Lesnyak, D. V., Leonov, A. A., Bogdanov, A. A., and Dontsova, O. A.
(2005) The conserved A-site finger of the 23S rRNA: just one of the
intersubunit bridges or a part of the allosteric communication pathway?
J. Mol. Biol., 353, 116-123.
17.Burakovsky, D. E., Sergiev, P. V., Steblyanko, M.
A., Konevega, A. L., Bogdanov, A. A., and Dontsova, O. A. (2011) The
structure of helix 89 of 23S rRNA is important for peptidyl transferase
function of Escherichia coli ribosome, FEBS Lett.,
585, 3073-3078.
18.Monod, J., Wyman, J., and Changeux, J.-P. (1965)
On the nature of allosteric transitions: a plausible model, J. Mol.
Biol., 12, 88-118.
19.Goodey, N. M., and Benkovic, S. J. (2008)
Allosteric regulation and catalysis emerge via a common route, Nat.
Chem. Biol., 4, 474-482.
20.Sethi, A., Eargle, J., Black, A. A., and
Luthey-Schulten, Z. (2009) Dynamical networks in tRNA:protein
complexes, Proc. Natl. Acad. Sci. USA, 106,
6620-6625.
21.Williams, S. G., and Hall, K. B. (2014) Linkage
and allostery in snRNP protein/RNA complexes, Biochemistry,
53, 3529-3539.
22.Wilson, D. N. (2009) The A-Z of bacterial
translation inhibitors, Crit. Rev. Biochem. Mol. Biol.,
44, 393-433.
23.Ban, N., Nissen, P., Hansen, J., Moore, P. B.,
and Steitz, T. A. (2000) The complete atomic structure of the large
ribosomal subunit at 2.4 Å resolution, Science,
289, 905-920.
24.Ito, K., and Chiba, S. (2013) Arrest peptides:
cis-acting modulators of translation, Annu. Rev.
Biochem., 82, 171-202.
25.Vazquez-Laslop, N., Ramu, H., and Mankin, A. S.
(2011) Nascent peptide mediated ribosome stalling promoted by
antibiotics, in Ribosomes Structure, Function and Dynamics
(Rodnina, M. V., Wintermeyer, W., and Green, R., eds.) Springer,
Vienna, pp. 377-392.
26.Seidelt, B., Innis, C. A., Wilson, D. N.,
Gartmann, M., Armache, J.-P., Villa, E., Trabuco, L. G., Becker, T.,
Mielke, T., Schulten, K., Steitz, T. A., and Beckmann, R. (2009)
Structural insight into nascent polypeptide chain-mediated
translational stalling, Science, 326, 1412-1415.
27.Arenz, S., Ramu, H., Gupta, P., Berninghausen,
O., Beckmann, R., Vazquez-Laslop, N., Mankin, A. S., and Wilson, D. N.
(2014) Molecular basis for erythromycin-dependent ribosome stalling
during translation of the ErmBL leader peptide, Nat. Commun.,
5, 3501.
28.Kannan, K., and Mankin, A. S. (2011) Macrolide
antibiotics in the ribosomal tunnel: species-specific binding and
action, Ann. N.Y. Acad. Sci., 1241, 33-47.
29.Weisblum, B. (1995) Erythromycin resistance by
ribosome modification, Antimicrob. Agents Chemother., 39,
577-585.
30.Sanbonmatsu, K. Y. (2012) Computational studies
of molecular machines: the ribosome, Curr. Opin. Struct. Biol.,
22, 168-174.
31.Trabuco, L. G., Harrison, C. B., Schreiner, E.,
and Schulten, K. (2010) Recognition of the regulatory nascent chain
TnaC by the ribosome, Structure, 18, 627-637.
32.Gumbart, J., Schreiner, E., Wilson, D., Beckmann,
R., and Schulten, K. (2012) Mechanism of SecM-mediated stalling in the
ribosome, Biophys. J., 103, 331-341.
33.Shishkina, A., Makarov, G., Tereshchenkov, A.,
Korshunova, G., Sumbatyan, N., Golovin, A., Svetlov, M., and Bogdanov,
A. (2013) Conjugates of amino acids and peptides with
5-O-mycaminosyltylonolide and their interaction with the ribosomal exit
tunnel, Bioconj. Chem., 24, 1861-1869.
34.Jack, A., Dunkle, J. A., Xiong, L., Mankin A. S.,
and Cate, J. H. (2010) Structures of the Escherichia coli
ribosome with antibiotics bound near the peptidyl transferase center
explain spectra of drug action, Proc. Natl. Acad. Sci. USA,
107, 17152-17157.
35.Cannone, J. J., Subramanian, S., Schnare, M. N.,
Collett, J. R., DʼSouza, L. M., Du, Y., Feng, B., Lin, N.,
Madabusi, L. V., Muller, K. M., Pande, N., Shang, Z., Yu, N., and
Gutell, R. R. (2002) The comparative RNA web (CRW) site: an online
database of comparative sequence and structure information for
ribosomal, intron, and other RNAs, BMC Bioinformatics, 3,
2.
36.Byrd, R. H., Lu, P., and Nocedal, J. (1995) A
limited memory algorithm for bound constrained optimization, SIAM J.
Sci. Comput., 16, 1190-1208.
37.Bussi, G., Donadio, D., and Parrinello, M. (2007)
Canonical sampling through velocity rescaling, J. Chem. Phys.,
126, 41.
38.Van der Spoel, D., Lindahl, E., Hess, B.,
Groenhof, G., Mark, A. E., and Berendsen, H. J. C. (2005) GROMACS:
fast, flexible, free, J. Comput. Chem., 26,
1701-1718.
39.Hess, B., Kutzner, C., van der Spoel, D., and
Lindahl, E. (2008) GROMACS 4: algorithms for highly efficient,
load-balanced, and scalable molecular simulation, J. Chem. Theory
Comput., 4, 435-447.
40.Berendsen, H. J. C., Postma, J. P. M., van
Gunsteren, W. F., DiNola, A., and Haak, J. R. (1984) Molecular dynamics
with coupling to an external bath, J. Chem. Phys., 81,
3684-3690.
41.Darden, T., York, D., and Pedersen, L. (1993)
Particle mesh Ewald: an N log(N) method for Ewald sums in large
systems, J. Chem. Phys., 98, 10089-10092.
42.Jorgensen, W. L., Chandrasekhar, J., and Madura,
J. D. (1983) Comparison of simple potential functions for simulating
liquid water, J. Chem. Phys., 79, 926-935.
43.Reshetnikov, R. V., Sponer, J., Rassokhina, O.
I., Kopylov, A. M., Tsvetkov, P. O., Makarov, A. A., and Golovin, A. V.
(2011) Cation binding to 15-TBA quadruplex DNA is a multiple-pathway
cation-dependent process, Nucleic Acids Res., 39,
9789-9802.
44.Athavale, S. S., Petrov, A. S., Hsiao, C.,
Watkins, D., Prickett, C. D., Gossett, J. J., Lie, L., Bowman, J. C.,
O’Neill, E., Bernier, C. R., Hud, N. V., Wartell, R. M., Harvey,
S. C., and Williams, L. D. (2012) RNA folding and catalysis mediated by
iron(II), PloS One, 7, e38024.
45.Bonomi, M., Branduardi, D., Bussi, G., Camilloni,
C., Provasi, D., Raiteri, P., Donadio, D., Marinelli, F., Pietrucci,
F., Broglia, R. A., and Parrinello, M. (2009) PLUMED: a portable plugin
for free-energy calculations with molecular dynamics, Comput. Phys.
Commun., 180, 1961-1972.
46.Arenz, S., Meydan, S., Starosta, A. L.,
Berninghausen, O., Beckmann, R., Vazquez-Laslop, N., and Wilson, D. N.
(2014) Drug sensing by the ribosome induces translational arrest via
active site perturbation, Mol. Cell, 56, 446-452.
47.Hashem, Y., and Auffinger, P. (2009) A short
guide for molecular dynamic simulation of RNA systems, Methods,
47, 187-197.
48.Laio, A., and Parrinello, M. (2002) Escaping free
energy minima, Proc. Natl. Acad. Sci. USA, 99,
12562-12566.
49.Hansen, J., Ippolito, J., Ban, N., Nissen, P.,
Moore, P., and Steitz, T. (2002) The structures of four macrolide
antibiotics bound to the large ribosomal subunit, Mol. Cell,
10, 117-128.
50.Hansen, J. L., Moore, P. B., and Steitz, T. A.
(2003) Structures of five antibiotics bound at the peptidyl transferase
center of the large ribosomal subunit, J. Mol. Biol.,
330, 1061-1075.
51.Vazquez-Laslop, N., Ramu, H., Klepacki, D.,
Kannan, K., and Mankin, A. S. (2010) The key function of a conserved
and modified rRNA residue in the ribosomal response to the nascent
peptide, EMBO J., 29, 3108-3117.
52.Leontis, N. B., Stombaugh, J., and Westhof, E.
(2000) The non-Watson–Crick base pairs and their associated
isostericity matrices, Nucleic Acids Res., 30,
3497-3531.
53.Hansen, J. L., Schmeing, T. M., Moore, P. B., and
Steitz, T. A. (2002) Structural insights into peptide bond formation,
Proc. Natl. Acad. Sci. USA, 99, 11670-11765.
54.Vazquez-Laslop, N., Thum, C., and Mankin, A. S.
(2008) Molecular mechanism of drug-dependent ribosome stalling, Mol.
Cell, 30, 190-202.
55.Youngman, E. M., Brunelle, J. L., Kochaniak, A.
B., and Green, R. (2004) The active site of the ribosome is composed of
two layers of conserved nucleotides with distinct roles in peptide bond
formation and peptide release, Cell, 117, 589-599.
56.Nissen, P., Hansen, J., Ban, N., Moore, P. B.,
and Steitz, T. A. (2000) The structural basis of ribosome activity in
peptide bond synthesis, Science, 289, 920-930.
57.Polikanov, Y. S., Steitz, T. A., and Innis, C. A.
(2014) A proton wire to couple aminoacyl-tRNA accommodation and
peptide-bond formation on the ribosome, Nat. Struct. Mol. Biol.,
21, 787-793.
58.Sergiev, P. V., Lesnyak, D. V., Burakovsky, D.
E., Svetlov, M., Kolb, V. A., Serebryakova, M. V., Demina, I. A.,
Govorun, V. M., Dontsova, O. A., and Bogdanov, A. A. (2012)
Non-stressful death of 23S rRNA mutant G2061C defective in puromycin
reaction, J. Mol. Biol., 416, 656-667.
59.Chirkova, A., Erlacher, M. D., Clementi, N.,
Zywicki, M., Aigner, M., and Polacek, N. (2010) The role of the
universally conserved A2450–C2063 base pair in the ribosomal
peptidyl transferase center, Nucleic Acids Res., 38,
4844-4855.
60.Leung, E. K., Suslov, N., Tuttle, N., Sengupta,
R., and Piccirilli, J. A. (2011) The mechanism of peptidyl transfer
catalysis by the ribosome, Annu. Rev. Biochem., 80,
527-555.
61.Schmeing, T. M., Huang, K. S., Strobel, S. A.,
and Steitz, T. A. (2005) An induced-fit mechanism to promote peptide
bond formation and exclude hydrolysis of peptidyl-tRNA, Nature,
438, 520-524.
62.Bhushan, S., Hoffmann, T., Seidelt, B.,
Frauenfeld, J., Mielke, T., Berninghausen, O., Wilson, D. N., and
Beckmann, R. (2011) SecM-stalled ribosomes adopt an altered geometry at
the peptidyl transferase center, PLoS Biol., 18,
e1000581.
63.Tsai, A., Kornberg, G., Johansson, M., Chen, J.,
and Puglisi, J. D. (2014) The dynamics of SecM-induced translational
stalling, Cell Rep., 7, 1521-1533.
64.Sothiselvam, S., Liu, B., Han, W., Ramu, H.,
Klepacki, D., Atkinson, G. C., Brauer, A., Remm, M., Tenson, T.,
Schulten, K., Vazquez-Laslop, N., and Mankin, A. S. (2014) Macrolide
antibiotics allosterically predispose the ribosome for translation
arrest, Proc. Natl. Acad. Sci. USA, 111, 9804-9809.
65.Bischoff, L., Berninghausen, O., and Beckmann, R.
(2014) Molecular basis for the ribosome functioning as an L-tryptophan
sensor, Cell Rep., 9, 469-475.
66.Kannan, K., Kanabar, P., Schryerm, D., Florin,
T., Oh, E., Bahroos, N., Tenson, T., Weissman, J. S., and Mankin, A. S.
(2014) The general mode of translation inhibition by macrolide
antibiotics, Proc. Natl. Acad. Sci. USA, 111,
15958-15963.