Regulation of cyclooxygenase 2 expression by agonists of PPAR nuclear receptors in the model of endotoxin tolerance in astrocytes
A. A. Astakhova1*, D. V. Chistyakov1, E. V. Pankevich2, and M. G. Sergeeva1
1Belozersky Research Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: +7 (495) 939-0338; E-mail: alina_astakhova@yahoo.com2Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: +7 (495) 939-4195
* To whom correspondence should be addressed.
Received April 8, 2015; Revision received May 20, 2015
Endotoxin tolerance (ET) represents a state of an altered immune response induced by multiple stimulations of a cell, a tissue, or an organism with lipopolysaccharide. Characteristics of ET include downregulation of induction of proinflammatory genes (TNFα, IL6, and others) and enhancement of induction of antiinflammatory genes (IL10, TGFβ). ET generally has protective functions; nevertheless, it might result in a state of innate immune deficiency and cause negative outcomes. A current issue is the search for the mechanisms controlling the level of inflammation in the course of endotoxin tolerance. In this work, we investigated the change in cyclooxygenase 2 (Cox2) expression in the model of endotoxin tolerance in astrocytes and analyzed the possibility of regulating this process applying nuclear receptor PPAR agonists. Our results indicate that: 1) endotoxin tolerance can be induced in astrocytes and results in TNFα and Cox2 mRNA induction decrease upon secondary stimulation; 2) tolerance is revealed on the level of TNFα release and Cox2 protein expression; 3) PPAR agonists GW7647, L-165041, and rosiglitazone control Cox2 mRNA expression levels under conditions of endotoxin tolerance. In particular, rosiglitazone (a PPARγ agonist) induces Cox2 mRNA expression, while GW7647 (a PPARα agonist) and L-165041 (a PPARβ agonist) suppress the expression. Our results demonstrate that Cox2 can be up- and downregulated during endotoxin tolerance in astrocytes, and PPAR agonists might be effective for controlling this target under conditions of multiple proinflammatory stimulations of brain tissues with endotoxin.
KEY WORDS: cyclooxygenase 2, nuclear receptors, PPAR agonists, rosiglitazone, endotoxin tolerance, innate immunity, astrocytesDOI: 10.1134/S0006297915100065
Abbreviations: CNS, central nervous tissue; Cox2, cyclooxygenase 2; ET, endotoxin tolerance; IL6, interleukin 6; LDH, lactate dehydrogenase; LPS, lipopolysaccharide (endotoxin); PPAR, peroxisome proliferator-activated receptors; TNFα, tumor necrosis factor alpha.
Endotoxin tolerance (ET) is a change in innate immune response to an
acute proinflammatory stimulation under conditions when an organism, a
tissue, or cells have been previously exposed to endotoxin [1, 2]. The change is characterized
by decreased induction of proinflammatory mediators (TNFα, IL6,
IL1β, and others) and enhanced upregulation of antiinflammatory
markers (IL10, TGFβ) in response to a second lipopolysaccharide
(LPS) stimulation [1, 3]. In
terms of outcomes, ET can be beneficial as well as detrimental for an
organism. Early studies demonstrated that a preceding endotoxin
challenge increased survival after subsequent injection of LPS at
lethal doses in animals [1]. Furthermore, there has
also been reported that LPS stimulation rendered nervous tissue
significantly resistant to a following stroke, reduced the degree of
damage, and shortened the functional recovery period [4, 5]. On the other hand, it has
been demonstrated that the later stages of sepsis involve mechanisms
that are similar to ET, and this condition is acknowledged among the
factors that determine sepsis-related lethality [6]. Thus, the mechanisms of cellular inflammatory
response regulation under conditions of endotoxin tolerance merit
special attention.
Astrocytes are auxiliary cells of the central nervous system and are recognized to take vitally important part in the processes of metabolism, nervous signal transduction, and immune responses [7, 8]. Astroglia expresses sensors of innate immunity (receptors of the TLR, NLR, and PAR groups and others) that induce synthesis of immune mediators (IL6, TNFα, IL10, and others) in response to a proinflammatory challenge (LPS, thrombin, or other substances) [9-12] and participate in immune processes within the central nervous system (CNS) tissues under conditions of proinflammatory reactions, including those that accompany strokes [13]. It is noteworthy that ET-like change in immune responses in astrocytes stimulated with LPS multiple times has been reported in two studies [14, 15]. It was demonstrated that ET in astroglial cultures is characterized by incomplete suppression of IL6 induction as well as differential expression of a number of potent immune mediators (TNFR, TGFβ, and others). Nevertheless, the question of regulation of inflammatory response in astrocytes in ET has remained overall uninvestigated.
Cyclooxygenase 2 (Cox2) is responsible for synthesis of prostaglandins and is induced upon proinflammatory stimulations in brain tissues [16, 17]. The enzyme has a dual role in immune response regulation [17, 18]. At early stages, Cox2 promotes the development of proinflammatory reactions, and Cox2 inhibition underlies the actions of aspirin, ibuprofen, and other nonsteroidal antiinflammatory drugs. Nevertheless, at later stages of an immune response Cox2 switches its role and produces antiinflammatory mediators [17, 18]. Inactivation of the enzyme may thus provide dysregulation of resolution of inflammation [19, 20]. Thus, it is important to investigate the mechanisms and outcomes of Cox2 inhibition under conditions of various models of inflammation dysregulation, including the model of endotoxin tolerance. Conditions of endotoxin tolerance have previously been reported to suppress Cox2 expression upon secondary proinflammatory challenges in immune cells [21, 22]. Data on Cox2 regulation in astrocytes in ET remain missing.
Nuclear receptors of the PPAR group – PPARα, PPARβ, and PPARγ – are recognized to be among the important regulators of immune responses within the CNS. These transcription factors control the expression of multiple inflammation related genes including Cox2. Agonists having antiinflammatory functions are known for each PPAR isoform. A number of agonists, including rosiglitazone and fibrates, are already used in clinics as hypolipidemic substances and drugs for treatment of diabetes type 2. Many substances are subject to trials of neurodegenerative condition treatment, including Alzheimer’s disease, Parkinson’s disease, and other pathologies [23-26]. Previously, our group has demonstrated involvement of PPARs in the onset of inflammation in astrocytes. Particularly, PPAR agonists were shown to mediate regulation of phospholipase A2, the enzyme that provides Cox2 with arachidonic acid (its major substrate [27]), as well as to modulate Cox2 expression [9, 11]. Importantly, not only Cox2 suppression, but also Cox2 upregulation mediated by PPAR ligands was possible in astrocytes. Specifically, PPARγ stimulation resulted in Cox2 expression upregulation in PPARβ-dependent manner, and cell treatment with PPARα and PPARβ agonists suppressed LPS-induced inflammation within cells [9]. However, these effects have been demonstrated within the model of acute inflammatory stimulation, and it remains unknown whether similar effects can be observed for cells under ET conditions.
In the present work, we investigated the development of ET in astrocytes stimulated with LPS for a prolonged period, traced the change in Cox2 expression, and analyzed the possibility of regulating Cox2 expression at the mRNA level by applying GW7647 (a PPARα agonist), L-165041 (a PPARβ agonist), and rosiglitazone (a PPARγ agonist). Our results demonstrate for the first time that induction of Cox2 expression is downregulated in ET in astrocytes and can be modulated by agonists of PPARα, PPARβ, and PPARγ.
MATERIALS AND METHODS
Materials and reagents. Culture medium DMEM, fetal bovine serum, streptomycin-penicillin, trypsin, and EDTA were from PanEco (Russia); LPS from Sigma-Aldrich (USA); GW7647, L-165041, and rosiglitazone from Cayman; mAb against Cox2 (E1412) and against β-tubulin (L0512) and HRP-conjugated antibodies (L1510) from Santa Cruz (USA); substrate SuperSignal™ West Pico from Pierce (USA); TNFα release kit (Rat TNF alpha ELISA Kit), First Strand cDNA Synthesis Kit, and Maxima SYBR Green/Fluorescein qPCR Master Mix (2x) from Thermo Scientific (USA); PowerLyzer RNA Isolation and Cytotoxicity Detection Kit (LDH) from Roche (Switzerland).
Cell culture and stimulations. Primary cultures of rat astrocytes were obtained from neonatal Wistar rats according to a protocol used previously [11]. In brief, the animals were aseptically decapitated, and the brains were isolated, washed in ice-cold Puck’s buffer (137 mM NaCl, 5.4 mM KCl, 0.2 mM KH2PO4, 0.17 mM Na2HPO4, 5 mM glucose, 58.4 mM sucrose, pH 7.4), and minced against meshes of 250 and 136 µm size. Then the tissue fragments were placed into culture flasks. Material was supplied with DMEM (1 g/liter D-glucose, 10% bovine fetal serum (FBS), 50 units/ml streptomycin, 50 µg/ml penicillin) and incubated at 37°C, 5% CO2, 10% humidity. Five days later, cultures were shaken to detach microglial cells and given fresh portions of media with the same composition. The cells were cultured for an additional 6 days with media being changed every 2 days. After monolayer formation, the cells were washed with warm PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) and treated with trypsin plus EDTA solution. Trypsin action was stopped with DMEM with FBS addition, and the cells were washed free from the enzyme solution. The cells were plated into 6-well culture plates at 750,000 cells/well and allowed to attach for 24 h. Then the media were changed. To model the conditions of endotoxin tolerance, cells were incubated with LPS at 10 ng/ml concentration. The cells were incubated for the following 46 h. Then, the media in all wells were changed to DMEM with 1% FBS. Two hours later, the cells were treated as described for particular experiments (Fig. 1).
Fig. 1. Scheme of stimulations used in the modeled conditions for the research. Cells were placed into 6-well plates and allowed to attach for 24 h (37°C, 5% CO2, 10% humidity). The media were then substituted by fresh media of the same composition (DMEM, 10% FBS) with 10 ng/ml LPS (model 10/100 and models GW7647/L-165041/Ros) or without LPS (models 0/100 and 0/0). After 46 h, media were changed for fresh potions of DMEM with 1% FBS (in all cultures), and 2 h later cells were challenged with LPS (100 ng/ml) or PPAR agonists plus LPS (agonists (Ag.) were applied 15 min prior to LPS).
Western blot. After the end of the stimulation period, media were withdrawn from the wells, and the cells were washed with ice-cold PBS and lysed in RIPA buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1% Nonidet P-40, 1% C24H39NaO4) for 10 min. The lysates were then centrifuged (10,000 rpm, 10 min). The total protein content was determined by a standard procedure using Bradford reagent. The lysed cells were mixed with Laemmli buffer for SDS-PAGE. The SDS-PAGE procedure and protein transfer to nitrocellulose membranes (0.2 µm) were performed in accordance with the conventional protocol. Transfer quality was determined by membrane staining with Ponceau S (Helicon, Russia). Membranes were blocked in TBS (40 mM NaCl, 3 mM KCl, 25 mM Tris, pH 7.4) with 5% fat-free milk and 0.05% Tween. The membranes were then treated with TBST with primary antibodies against Cox2 diluted at 1 : 500 and incubated for 8 h in the cold on a shaker. After procedures of washing and treatment with secondary antibodies, proteins were visualized using chemiluminescent substrate from Thermo Scientific (USA). After the analysis of Cox2 expression, the membranes were treated with Restore Buffer (Thermo Scientific) and used for β-tubulin expression analysis. Images were obtained using Gel Doc™ XR+ System camera from BioRad (USA). Quantitative analysis was performed using the application QuantityOne (BioRad 4.6.9).
RNA isolation, reverse transcription, and real-time PCR. Total mRNA was isolated using a PowerLyzer RNA Isolation kit. Nucleic acid concentration was determined spectrophotometrically. To obtain the first strand, a First Strand cDNA Synthesis Kit was used. The reaction was performed in accordance with the manufacturer’s instructions using oligo(dT) primers. The relative mRNA Cox2 levels were determined by real-time PCR using a DTlite 4 thermocycler (DNK-Tekhnologiya, Russia) and a Maxima SYBR Green/Fluorescein qPCR Master Mix kit. One reaction mixture had volume of 25 µl and contained 70 ng of cDNA. Levels of β-actin were used as the constitutive for normalization. The following pairs of primers were used:
| COX-2: | FW 5′-TGTACAAGCAGTGGCAAAGG-3′; |
| RV 5′-TAGCATCTGGACGAGGCTTT-3′; | |
| TNFα: | FW 5′-ACGTCGTAGCAAACCACCAA-3′; |
| RV 5′-AAATGGCAAATCGGCTGAC-3′; | |
| β-actin: | FW 5′-TCATCACTATCGGCAATGAGCGGT-3′; |
| RV 5′-ACAGCACTGTGTTGGCATAGAGGT-3′. |
Analysis of lactate dehydrogenase (LDH) release. The media were taken from the wells and saved for the further analysis of LDH release. The levels of released LDH were determined by a Cytotoxicity Detection (LDH) kit (Roche) following the manufacturer’s instructions.
Analysis of TNFα release. The media were taken from the wells and saved for the further analysis of tumor necrosis factor alpha (TNFα) release. The levels of released TNFα were determined by a Rat TNF alpha ELISA kit (Thermo Scientific) following the manufacturer’s instructions.
Statistics. All experiments were reproduced three times. Values are given as mean ± S.D. Data were analyzed using ANOVA. The level of significance was taken as 0.05 (* relative to control, p < 0.05).
RESULTS
Conditions of endotoxin tolerance are nontoxic for cells. The general scheme of cell stimulations in the experiments is given in Fig. 1.
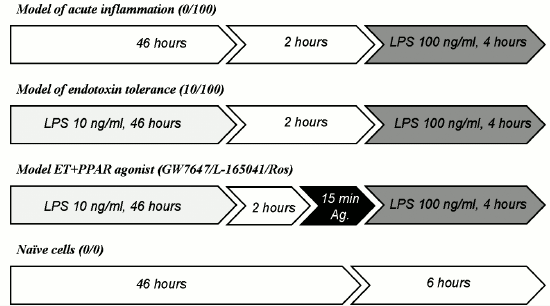
To determine if the short-term and the long-term stimulation of astrocytes with endotoxin have cytotoxic effects, we analyzed LDH release in experimental and control conditions. In all investigated conditions, the level of released LDH comprised 8-10% of the total cell LDH content (Fig. 2). The values obtained for the ET model and the model of acute inflammation were not significantly different from the values obtained for the control cells (0/0). The data indicate that acute stimulation of astrocytes with LPS and ET conditions are not toxic for cells.
Fig. 2. Release of lactate dehydrogenase (LDH) in astrocytic cultures under conditions of the model of acute inflammation and the model of endotoxin tolerance. Cultures of astrocytes were treated in accordance with the scheme of stimulations given in Fig. 1. Culture media from experimental and control samples were collected for LDH release analysis; 100% represents the LDH content from total cell lysates.
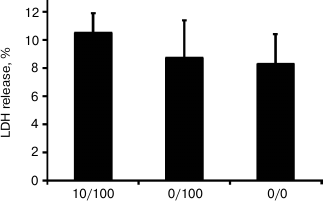
Repeated stimulation with LPS results in endotoxin tolerance in astrocytes. The conventional characteristic of ET is a decrease in TNFα synthesis in response to a second proinflammatory challenge [1-3]. To verify that the stimulations applied in the present study induce ET, we analyzed TNFα expression levels induced by LPS stimulation (100 ng/ml) under conditions when cells were previously incubated with LPS at lower concentrations (10 ng/ml, 10/100 model). As a positive control, we took the level of TNFα in cells stimulated with LPS in acute manner without preceding treatment with endotoxin (0/100 model). Our results indicated significant decrease in TNFα levels in the cultures with repeated LPS stimulation. However, under ET conditions, the TNFα level was 25 times higher than in the control cells (0/0 model) (Fig. 3). A similar effect was observed at the level of TNFα release into culture media: TNFα concentration was equal to 750 pg/ml (754 ± 13) in the samples from acutely stimulated cells (0/100 model), while the concentrations for naïve cultures and cultures treated within the ET model comprised 280 ± 12 pg/ml (0/0) and 333 ± 9 pg/ml (10/100), respectively. The data indicate ET establishment.
Fig. 3. Change in mRNA expression (a) and release to culture media (b) of TNFα in astrocytes stimulated with LPS under conditions of acute inflammation (0/100) and endotoxin tolerance (10/100). Cultures of astrocytes were treated in accordance with the scheme of stimulations given in Fig. 1. a) RNA was isolated from cell lysates. Reverse transcription and real-time PCR were applied to determine TNFα and actin expression levels, and the values for TNFα were normalized to actin levels. b) Culture media from experimental and control samples were used to determine TNFα release. The levels of TNFα in cultures of astrocytes stimulated with LPS under conditions of endotoxin tolerance (10/100) were taken for control.
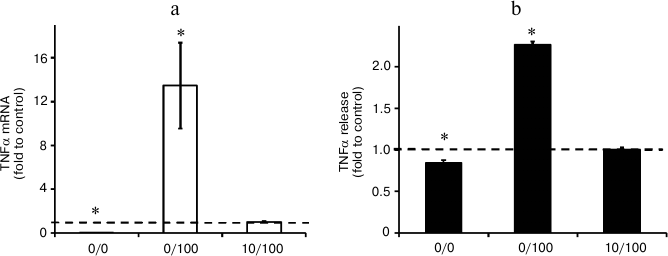
Endotoxin tolerance partially blocks expression of Cox2. In the next step, we analyzed Cox2 expression changes under the conditions of our model. Our results indicated that acute stimulation of astrocytes with LPS (100 ng/ml, 0/100 model) induced approximately 30-fold upregulation of Cox2 mRNA expression comparing to control (naïve cells). LPS treatment (10 ng/ml, 48 h) decreased LPS (100 ng/ml)-induced levels of Cox2 mRNA expression approximately 6-fold. Cox2 induction inhibition in response to LPS stimulation under conditions of ET was also detected at the protein level (Fig. 4). The data indicate that ET partially but not completely blocks Cox2 expression in astrocytes.
Fig. 4. Change in mRNA (a) and protein expression (b, c) of Cox2 in astrocytes stimulated with LPS under conditions of acute inflammation (0/100) and endotoxin tolerance (10/100). Cultures of astrocytes were treated in accordance with the scheme of stimulations given in Fig. 1. a) RNA was isolated from cell lysates. Reverse transcription and real-time PCR were applied to determine Cox2 and actin expression levels, and the values for Cox2 were normalized to actin levels. b, c) Cell lysates were used to obtain protein. The levels of Cox2 and β-tubulin expression were measured by conventional methods of immunoblotting. The Cox2 levels were normalized to β-tubulin values. The levels of Cox2 in cultures of astrocytes stimulated with LPS under ET conditions (10/100) were taken as the control.
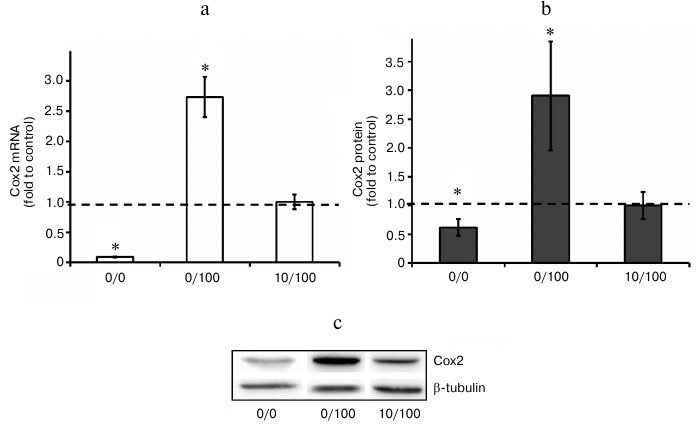
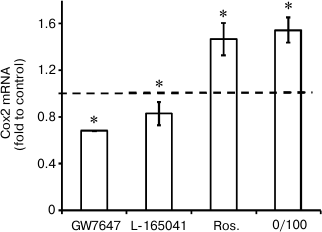
Agonists of PPARs regulate expression of Cox2 mRNA under conditions of endotoxin tolerance. It was demonstrated previously that LPS-induced Cox2 expression in astrocytes could be modulated by nuclear receptor PPAR agonists. Specifically, PPARγ activation by rosiglitazone induces Cox2 expression, while agonists of PPARβ and PPARα decrease cyclooxygenase expression [9-11, 28]. That sets the question of whether or not PPAR agonists can regulate Cox2 expression in astrocytes under conditions of endotoxin tolerance. To answer this question, we treated cells with endotoxin, incubated them with PPAR agonists GW7647 (PPARα agonist, 1 µM), L-165041 (PPARβ agonist, 1 µM), and rosiglitazone (PPARγ agonist, 20 µM), and stimulated them with LPS (100 ng/ml, 4 h). Further real-time PCR analysis revealed Cox2 mRNA increase in cells stimulated with LPS in combination with rosiglitazone and Cox2 expression decrease in cells stimulated with LPS in combination with GW7647 (the PPARα agonist) and to lesser extent in combination with L-165041 (the PPARβ agonist) under ET conditions (Fig. 5). The resulting data indicate that under conditions of endotoxin tolerance astrocyte Cox2 expression can be regulated by modulation of nuclear receptor PPAR activity.
Fig. 5. Effect of GW7647 (agonist of PPARα), L-165041 (agonist of PPARβ), and rosiglitazone (agonist of PPARγ) on LPS-induced Cox2 mRNA expression in astrocytes under ET conditions. Cultures of astrocytes were treated in accordance with the scheme of stimulations given in Fig. 1. RNA was isolated from cell lysates. Reverse transcription and real-time PCR were applied to determine Cox2 and actin expression levels, and the values for Cox2 were normalized to actin levels. The levels of Cox2 in cultures of astrocytes stimulated with LPS under ET conditions (10/100) were taken as the control.
DISCUSSION
Activation of innate immunity has nonspecific cytotoxic effect on the host tissues and is extremely dangerous for components of the CNS due to their limited regenerative capacity [29, 30]. Endotoxin tolerance is assumed to have protective functions preventing excessive activation of immune response in peripheral structures as well as the CNS [4, 31-33]. Treatment of cells with endotoxin at concentrations from 1 to 1000 ng/ml for several hours results in desensitization of proinflammatory mediator induction in response to a subsequent proinflammatory stimulation that in turn prevents tissues from being damaged [3, 34]. Although ET development in brain tissues has been known for a long time, little data is available on the impact of various cellular types into the process [4, 5]. One research team demonstrated that endotoxin tolerance is accompanied by suppression of IL6 upregulation in astrocytic cultures [14, 15]. However, they used a model where cultures were challenged with LPS at concentration 100 ng/ml for 24 h, and then were stimulated with endotoxin at a lower concentration – 10 ng/ml for 1 or 24 h [14, 15]. Suppression of IL6 expression might thus be explained by insufficient second challenge due to a decrease in sensitivity of the cells. Indeed, usual models of ET implement a scheme where prolonged stimulations with moderate concentrations of endotoxin are followed by stimulations with higher concentrations of proinflammatory substances [1-3]. Thus, within our model of ET in astroglial cells we used a scheme where lower concentration of LPS (10 ng/ml) was used for adaptation (48 h), and a higher concentration (100 ng/ml) was used for acute stimulation (4 h). Analysis of TNFα, the classic ET marker, and Cox2 expression within our ET model (10/100) revealed the suppression of expression induction in response to the second challenge in comparison to the response observed in naïve cells stimulated with LPS in acute manner (0/100). However, similarly to IL6 [14, 15], the second stimulation upregulated the expression of the markers comparing to naïve cells (0/0). These effects were observed at the mRNA level as well as at the level of protein synthesis for TNFα and Cox2. Together the data indicate the establishment of endotoxin tolerance in astrocytes.
Cytotoxic effect of LPS under conditions of ET was previously revealed for macrophages [35]. Thus, we investigated whether the conditions of our model are toxic for cells. The results indicate the absence of cytotoxicity during short-term (0/100) and combined treatment (10/100) of cells with endotoxin. Consequently, the observed phenomena reflect the general reorganization of the inflammatory response and do not represent the results of cell damage.
At present, the general strategy for inflammatory condition control includes immune response suppression. However, long-term outcomes for such approach are often detrimental. In a number of reports including those focused on brain investigations, negative consequences of switch-off of proinflammatory mediators including TNFα, IL6, and transcription factor NFκB have been reported [36-38]. Indeed, instead of expected improved prognosis (due to decreased tissue damage induced by cytokines), proinflammatory gene switch-off exacerbated healing processes, delayed the onset of resolution, impaired damaged site regeneration, and switched the pattern of inflammation to a chronic type [36-38]. Similar results were obtained for prolonged Cox2 inhibition in peripheral tissues of an organism [19, 20]. Dysregulation of inflammation due to Cox2 inactivation has also been demonstrated for brain tissues [39, 40]. Palliative rather than curative effect of inflammatory response in CNS tissue demonstrates interconnections between the processes of immune response onset and termination and sets questions of long-term outcomes of ET and the mechanisms that could regulate inflammation in the CNS under conditions of ET.
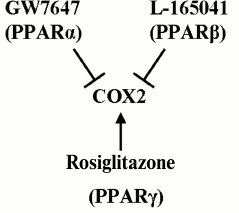
Cox2 might be promising with respect to ET due to the (i) key role that the enzyme plays in regulation of inflammation onset and termination [19, 20]; (ii) involvement of the protein in inflammatory processes within nervous tissue [41-43]; and (iii) availability of multiple drugs, including ones that have already been approved for clinical application and can regulate the enzyme activity [26]. Previously, our group and other research teams demonstrated that various agonists of PPARs modulate LPS-induced upregulation of Cox2 expression in astroglial cells [9-11, 28, 44-46]. Nuclear PPARs regulate the expression of multiple genes involved into inflammatory response and are themselves targets for many antiinflammatory drugs [23-25, 47]. Three PPAR isoforms have been recognized so far – PPARα, PPARβ, and PPARγ – and all three are involved in immune response control in astrocytes and can differentially regulate Cox2 activity [9, 28, 44, 46, 48]. It has been demonstrated that stimulation of cells with PPARγ agonists induced Cox2. Feedback mechanisms that co-regulated three PPARs have been revealed, with PPARβ playing the central role in the regulation [9, 11, 28]. In the present work, we demonstrated that under the conditions of ET astrocytes retain the ability to induce Cox2 expression upon stimulation with rosiglitazone (PPARγ agonist) and are able to suppress it upon treatment with GW7647 (PPARα agonist) and L-165041 (PPARβ agonist). The general scheme of Cox2 regulation in astrocytes under conditions of ET is given in Fig. 6. Similar effects of PPARγ activation have been described for astrocytes challenged with LPS within the model of acute inflammation [28]. However, treatment of astroglial cultures with PPARβ and PPARα agonists under conditions of ET resulted in a decrease in Cox2 expression, while these effects were not observed in naïve cells [9, 11, 28]. The mentioned change in PPAR agonist effects might indicate their antiinflammatory activity that is present under ET but is absent under conditions of acute inflammation and point the involvement of additional regulatory mediators connected to PPARβ. This possibility is in accordance with previously reported activity of PPARβ in the processes of resolution within spinal cord tissues and supports the hypothesis of a dual pro- and antiinflammatory role of PPARβ in astrocytes in inflammation [10, 11, 49]. The antiinflammatory role of PPARs in ET requires further study.
Fig. 6. Scheme of Cox2 regulation by PPAR agonists: GW7647 (PPARα), L-165041 (PPARβ), and rosiglitazone (PPARγ). Designations: ↑, stimulatory effect; ┴, inhibitory effect.
In total, our results demonstrate for the first time that Cox2 can be differentially regulated in astroglial cells by PPAR agonists under conditions of endotoxin tolerance. This might have potential for investigation of ET phenomenon as well as for therapeutic applications.
The authors thank Dr. L. R. Gorbacheva from Lomonosov Moscow State University for assistance in obtaining cell material.
This study was partially supported by the Russian Foundation for Basic Research (grant 13-04-00833).
REFERENCES
1.Biswas, S. K., and Lopez-Collazo, E. (2009)
Endotoxin tolerance: new mechanisms, molecules and clinical
significance, Trends Immunol., 30, 475-487.
2.West, M. A., and Heagy, W. (2002) Endotoxin
tolerance: a review, Crit. Care Med., 30, S64-S73.
3.Nahid, M. A., Satoh, M., and Chan, E. K. L. (2011)
MicroRNA in TLR signaling and endotoxin tolerance, Cell Mol.
Immunol., 8, 388-403.
4.Rosenzweig, H. L., Lessov, N. S., Henshall, D. C.,
Minami, M., Simon, R. P., and Stenzel-Poore, M. P. (2004) Endotoxin
preconditioning prevents cellular inflammatory response during ischemic
neuroprotection in mice, Stroke, 35, 2576-2581.
5.Vartanian, K. B., Stevens, S. L., Marsh, B. J.,
Williams-Karnesky, R., Lessov, N. S., and Stenzel-Poore, M. P. (2011)
LPS preconditioning redirects TLR signaling following stroke: TRIF-IRF3
plays a seminal role in mediating tolerance to ischemic injury, J.
Neuroinflamm., 8, 160.
6.Cavaillon, J. M., and Adib-Conquy, M. (2006)
Bench-to-bedside review: endotoxin tolerance as a model of leukocyte
reprogramming in sepsis, Crit. Care, 10, 233.
7.Farina, C., Aloisi, F., and Meinl, E. (2007)
Astrocytes are active players in cerebral innate immunity, Trends
Immunol., 28, 138-145.
8.Gorina, R., Font-Nieves, M., Marquez-Kisinousky,
L., Santalucia, T., and Planas, A. M. (2011) Astrocyte TLR4 activation
induces a proinflammatory environment through the interplay between
MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways,
Glia, 59, 242-255.
9.Aleshin, S., Grabeklis, S., Hanck, T., Sergeeva,
M., and Reiser, G. (2009) Activated receptor PPAR-γ positively
controls and PPARα negatively controls cyclooxygenase-2
expression in rat brain astrocytes through a convergence on
PPARβ/δ via mutual control of PPAR expression levels,
Mol. Pharmacol., 76, 414-424.
10.Chistyakov, D. V., Aleshin, S. E., Astakhova, A.
A., Sergeeva, M. G., and Reiser, G. (2015) Regulation of peroxisome
proliferator-activated receptors (PPAR) α and γ of rat
brain astrocytes in the course of activation by toll-like receptor
agonists, J. Neurochem., March 25, DOI: 10.1111/jnc.13101 [E-pub
ahead of print].
11.Chistyakov, D. V., Aleshin, S., Sergeeva, M. G.,
and Reiser, G. (2014) Regulation of peroxisome proliferator-activated
receptor β/δ expression and activity levels by toll-like
receptor agonists and MAP kinase inhibitors in rat astrocytes, J.
Neurochem., 130, 563-574.
12.Donovan, F. M., Pike, C. J., Cotman, C. W., and
Cunningham, D. D. (1997) Thrombin induces apoptosis in cultured neurons
and astrocytes via a pathway requiring tyrosine kinase and RhoA
activities, J. Neurosci., 17, 5316-5326.
13.Chen, Y. M., and Swanson, R. A. (2003) Astrocytes
and brain injury, J. Cereb. Blood Flow Metab., 23,
137-149.
14.Beurel, E., and Jope, R. S. (2010) Glycogen
synthase kinase-3 regulates inflammatory tolerance in astrocytes,
Neuroscience, 169, 1063-1070.
15.Beurel, E. (2011) HDAC6 regulates LPS-tolerance
in astrocytes, PLoS One, 6, e25804.
16.Kaufmann, W. E., Andreasson, K. I., Isakson, P.
C., and Worley, P. F. (1997) Cyclooxygenases and the and the central
nervous system, Prostaglandins, 54, 601-624.
17.Varfalomeeva, A. T., and Sergeeva, M. G. (2006)
Arachidonic Acid Cascade [in Russian], Narodnoe Obrazovanie,
Moscow.
18.Ricciotti, E., and FitzGerald, G. A. (2011)
Prostaglandins and inflammation, Arterioscler. Thromb. Vasc.
Biol., 31, 986-1000.
19.Chan, M. M.-Y., and Moore, A. R. (2010)
Resolution of inflammation in murine autoimmune arthritis is disrupted
by cyclooxygenase-2 inhibition and restored by prostaglandin
E2-mediated lipoxin A(4) production, J. Immunol., 184,
6418-6426.
20.Rajakariar, R., Yaqoob, M. M., and Gilroy, D. W.
(2006) COX-2 in inflammation and resolution, Mol. Interv.,
6, 199-207.
21.Learn, C. A., Mizel, S. B., and McCall, C. E.
(2000) mRNA and protein stability regulate the differential expression
of pro- and anti-inflammatory genes in endotoxin-tolerant THP-1 cells,
J. Biol. Chem., 275, 12185-12193.
22.Spitzer, J. A., Zheng, M. Q., Kolls, J. K.,
Stouwe, C. V., and Spitzer, J. J. (2002) Ethanol and LPS modulate
NF-κB activation, inducible NO synthase and COX-2 gene expression
in rat liver cells in vivo, Front. Biosci., 7,
A99-A108.
23.Bensinger, S. J., and Tontonoz, P. (2008)
Integration of metabolism and inflammation by lipid-activated nuclear
receptors, Nature, 454, 470-477.
24.Heneka, M. T., and Landreth, G. E. (2007)
Peroxisome proliferator-activated receptors (PPARs) in skin health,
repair and disease, PPARs in the brain, Biochim. Biophys. Acta,
1771, 1031-1045.
25.Michalik, L., and Wahli, W. (2007) Peroxisome
proliferator-activated receptors (PPARs) in skin health, repair and
disease, Biochim. Biophys. Acta, 1771, 991-998.
26.Rainsford, K. D. (2007) Anti-inflammatory drugs
in the 21st century, Subcell. Biochem., 42, 3-27.
27.Sergeeva, M. G., Aleshin, S. E., Grabeklis, S.,
and Reiser, G. (2010) PPAR activation has dichotomous control on the
expression levels of cytosolic and secretory phospholipase A2 in
astrocytes; inhibition in naive, untreated cells and enhancement in
LPS-stimulated cells, J. Neurochem., 115,
399-410.
28.Aleshin, S., Strokin, M., Sergeeva, M., and
Reiser, G. (2013) Peroxisome proliferator-activated receptor (PPAR)
β/δ, a possible nexus of PPARα- and
PPARγ-dependent molecular pathways in neurodegenerative diseases:
review and novel hypotheses, Neurochem. Int., 63,
322-330.
29.Fitch, M. T., and Silver, J. (2008) CNS injury,
glial scars, and inflammation: inhibitory extracellular matrices and
regeneration failure, Exp. Neurol., 209, 294-301.
30.Siniscalchi, A., Gallelli, L., Malferrari, G.,
Pirritano, D., Serra, R., Santangelo, E., and De, S. G. (2014) Cerebral
stroke injury: the role of cytokines and brain inflammation, J.
Basic Clin. Physiol. Pharmacol., 25, 131-137.
31.Dirnagl, U., Becker, K., and Meisel, A. (2009)
Preconditioning and tolerance against cerebral ischemia: from
experimental strategies to clinical use, Lancet Neurol.,
8, 398-412.
32.Hafenrichter, D. G., Roland, C. R., Mangino, M.
J., and Flye, M. W. (1994) The Kupffer cell in endotoxin tolerance:
mechanisms of protection against lethal endotoxemia, Shock,
2, 251-256.
33.Pena, O. M., Pistolic, J., Raj, D., Fjell, C. D.,
and Hancock, R. E. (2011) Endotoxin tolerance represents a distinctive
state of alternative polarization (M2) in human mononuclear cells,
J. Immunol., 186, 7243-7254.
34.Lopez-Collazo, E., and del Fresno, C. (2013)
Pathophysiology of endotoxin tolerance: mechanisms and clinical
consequences, Crit. Care, 17, 242.
35.Karahashi, H., and Amano, F. (2003)
Endotoxin-tolerance to the cytotoxicity toward a macrophage-like cell
line, J774.1, induced by lipopolysaccharide and cycloheximide: role of
p38 MAPK in induction of the cytotoxicity, Biol. Pharm. Bull.,
26, 1249-1259.
36.Erta, M., Quintana, A., and Hidalgo, J. (2012)
Interleukin-6, a major cytokine in the central nervous system, Int.
J. Biol. Sci., 8, 1254-1266.
37.Lawrence, T., Gilroy, D. W., Colville-Nash, P.
R., and Willoughby, D. A. (2001) Possible new role for NF-κB in
the resolution of inflammation, Nat. Med., 7,
1291-1297.
38.Nadeau, S., Filali, M., Zhang, J., Kerr, B. J.,
Rivest, S., Soulet, D., Iwakura, Y., de Rivero Vaccari, J. P., Keane,
R. W., and Lacroix, S. (2011) Functional recovery after peripheral
nerve injury is dependent on the pro-inflammatory cytokines IL-1β
and TNF: implications for neuropathic pain, J. Neurosci.,
31, 12533-12542.
39.Aid, S., Langenbach, R., and Bosetti, F. (2008)
Neuroinflammatory response to lipopolysaccharide is exacerbated in mice
genetically deficient in cyclooxygenase-2, J. Neuroinflamm.,
5, 17.
40.Blais, V., Turrin, N. P., and Rivest, S. (2005)
Cyclooxygenase 2 (COX-2) inhibition increases the inflammatory response
in the brain during systemic immune stimuli, J. Neurochem.,
95, 1563-1574.
41.Gupta, A., Kumar, A., and Kulkarni, S. K. (2011)
Targeting oxidative stress, mitochondrial dysfunction and
neuroinflammatory signaling by selective cyclooxygenase (COX)-2
inhibitors mitigates MPTP-induced neurotoxicity in mice, Prog.
Neuropsychopharmacol. Biol. Psychiatry, 35,
974-981.
42.Phillis, J. W., Horrocks, L. A., and Farooqui, A.
A. (2006) Cyclooxygenases, lipoxygenases, and epoxygenases in CNS:
their role and involvement in neurological disorders, Brain Res.
Rev., 52, 201-243.
43.Trepanier, C. H., and Milgram, N. W. (2010)
Neuroinflammation in Alzheimer’s disease: are NSAIDs and
selective COX-2 inhibitors the next line of therapy? J.
Alzheimer’s Dis., 21, 1089-1099.
44.Luna-Medina, R., Cortes-Canteli, M., Alonso, M.,
Santos, A., Martinez, A., and Perez-Castillo, A. (2005) Regulation of
inflammatory response in neural cells in vitro by
thiadiazolidinones derivatives through peroxisome
proliferator-activated receptor gamma activation, J. Biol.
Chem., 280, 21453-21462.
45.Pahan, K., Jana, M., Liu, X., Taylor, B. S.,
Wood, C., and Fischer, S. M. (2002) Gemfibrozil, a lipid-lowering drug,
inhibits the induction of nitric oxide synthase in human astrocytes,
J. Biol. Chem., 277, 45984-45991.
46.Xu, J., Chavis, J. A., Racke, M. K., and Drew, P.
D. (2006) Peroxisome proliferator-activated receptor-alpha and retinoid
X receptor agonists inhibit inflammatory responses of astrocytes, J.
Neuroimmunol., 176, 95-105.
47.Michalik, L., Auwerx, J., Berger, J. P.,
Chatterjee, V. K., Glass, C. K., Gonzalez, F. J., Grimaldi, P. A.,
Kadowaki, T., Lazar, M. A., O’Rahilly, S., Palmer, C. N.,
Plutzky, J., Reddy, J. K., Spiegelman, B. M., Staels, B., and Wahli, W.
(2006) International Union of Pharmacology. LXI. Peroxisome
proliferator-activated receptors, Pharmacol. Rev., 58,
726-741.
48.Cristiano, L., Cimini, A., Moreno, S., Ragnelli,
A. M., and Paola, C. M. (2005) Peroxisome proliferator-activated
receptors (PPARs) and related transcription factors in differentiating
astrocyte cultures, Neuroscience, 131,
577-587.
49.Paterniti, I., Impellizzeri, D., Crupi, R.,
Morabito, R., Campolo, M., Esposito, E., and Cuzzocrea, S. (2013)
Molecular evidence for the involvement of PPAR-delta and PPAR-gamma in
anti-inflammatory and neuroprotective activities of
palmitoylethanolamide after spinal cord trauma, J.
Neuroinflamm., 10, 20.