Phosphorylation Regulates Interaction of 210-kDa Myosin Light Chain Kinase N-terminal Domain with Actin Cytoskeleton
E. L. Vilitkevich1#, A. Y. Khapchaev1#, D. S. Kudryashov2, A. V. Nikashin1, J. P. Schavocky3, T. J. Lukas3, D. M. Watterson3, and V. P. Shirinsky1*
1Russian Cardiology Research and Production Center, 121552 Moscow, Russia; fax: 7 (495) 414-6719; E-mail: shirinsky@cardio.ru2Ohio State University, Columbus, OH 43210 USA
3Northwestern University, Chicago, IL 60611 USA
# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received May 26, 2015; Revision received June 20, 2015
High molecular weight myosin light chain kinase (MLCK210) is a multifunctional protein involved in myosin II activation and integration of cytoskeletal components in cells. MLCK210 possesses actin-binding regions both in the central part of the molecule and in its N-terminal tail domain. In HeLa cells, mitotic protein kinase Aurora B was suggested to phosphorylate MLCK210 N-terminal tail at serine residues (Dulyaninova, N. G., and Bresnick, A. R. (2004) Exp. Cell Res., 299, 303-314), but the functional significance of the phosphorylation was not established. We report here that in vitro, the N-terminal actin-binding domain of MLCK210 is located within residues 27-157 (N27-157, avian MLCK210 sequence) and is phosphorylated by cAMP-dependent protein kinase (PKA) and Aurora B at serine residues 140/149 leading to a decrease in N27-157 binding to actin. The same residues are phosphorylated in a PKA-dependent manner in transfected HeLa cells. Further, in transfected cells, phosphomimetic mutants of N27-157 showed reduced association with the detergent-stable cytoskeleton, whereas in vitro, the single S149D mutation reduced N27-157 association with F-actin to a similar extent as that achieved by N27-157 phosphorylation. Altogether, our results indicate that phosphorylation of MLCK210 at distinct serine residues, mainly at S149, attenuates the interaction of MLCK210 N-terminus with the actin cytoskeleton and might serve to regulate MLCK210 microfilament cross-linking activity in cells.
KEY WORDS: 210-kDa myosin light chain kinase, phosphorylation-dependent actin binding, protein interactions, cAMP-dependent protein kinase, Aurora BDOI: 10.1134/S0006297915100090
Abbreviations: DFP, diisopropyl fluorophosphate; FBS, fetal bovine serum; IBMX, phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine; KRP, kinase-related protein; MLCK210, high molecular weight myosin light chain kinase (210 kDa); N27-157, genetic construction of N-terminal domain of avian MLCK210 encoding a.a. 27-157 as well as corresponding protein product; PKA, cAMP-dependent protein kinase; TRITC, tetramethylrhodamine.
High molecular weight myosin light chain kinase (MLCK210) is a
serine/threonine (S/T) protein kinase and the largest protein product
(210 kDa) encoded by the vertebrate non-muscle/smooth muscle myosin
light chain kinase (MLCK) genetic locus [1]. This
locus also encodes a shorter MLCK with a molecular mass of
108-135 kDa (MLCK108) and a small (17 kDa) Kinase-Related
Protein (KRP/telokin) [2]. The MLCK210 amino acid
sequence includes that of MLCK108 plus an additional, unique
N-terminal region. MLCK210 is abundant in cells such as
leukocytes, cultured fibroblasts, as well as in epithelial and
endothelial cells. Depending on physiological context or culture
conditions, MLCK210 is either the predominant form of the kinase or
co-expresses with MLCK108 [2-6].
Outside the catalytic region, the C-terminal KRP domain of MLCK210 was shown to represent a myosin-binding entity [7], the central DF/VRxxL repeat-enriched sequence was found to be a high-affinity microfilament-binding site, and the unique N-terminal tail was shown to possess an additional microfilament- and microtubule-binding region [8-10]. More recently, the N-terminal actin-binding region was confined to residues 1-201 [11]. In addition, sites of interaction with a number of other proteins, such as macrophage migration inhibitory factor (MIF), cortactin, supervillin, hARD1 acetylase, Pyk2, and p60src tyrosine protein kinases were mapped along the MLCK210 sequence [6, 12-16].
Relevant to the cytoskeleton interaction activities of MLCK210, a prevailing model suggests that in non-muscle cells, MLCK210 functions as a cytoskeleton integrator and motility organizer [9]. In this model, MLCK210 resides on microfilament bundles via its central high-affinity actin-binding site, whereas both the N- and C-terminal tails of the molecule are engaged in reversible interaction with other cytoskeletal and motile components of the cell. While MLCK210 resides on actin filaments, its KRP-domain binds to soluble monomeric non-muscle myosin II and facilitates the presentation of myosin II to the catalytic core of the kinase. Phosphorylation of myosin II allows self-assembly of myosin filaments, thereby supporting localized actomyosin motility. Similarly, the N-terminal microfilament and microtubule binding regions are viewed as important for integrating MLCK210 with other cytoskeletal structures to provide localized control of cytoplasm stiffness and motility. It has been demonstrated that the N-terminal domain of MLCK210 can be phosphorylated during interphase and mitosis of HeLa cells [17], but the phosphorylated residues have not been identified. On the other hand, a phosphopeptide derived from the N-terminal domain of MLCK210 and containing phosphoserine, homologous to S149 in the chicken protein, has been detected in mouse brain tissue [18, 19]. However, in both these cases, the functional significance of phosphorylations of MLCK210 N-terminal domain and the linkage of site-specific phosphorylation to candidate protein kinases remain to be clarified.
To address the gaps in knowledge about the MLCK210 N-terminal domain phosphorylation and its potential functional significance, we refined the localization of actin-binding activity, identified the phosphorylation sites, and linked the site-specific phosphorylation to changes in MLCK210 actin-binding and subcellular localization. Our results are consistent with phosphorylation at specific N-terminal tail domain sites being a mechanism to modulate the interaction of MLCK210 with the cytoskeleton, thereby providing for localized biological responses mediated by MLCK210.
MATERIALS AND METHODS
Reagents were of analytical grade and purchased from Sigma and Fisher Scientific (USA).
Molecular biology. The structural relationship of MLCK210 N-terminal tail constructs used in this study and full-length MLCK210 is shown in Fig. 1. Amino acid residue numbers are given for chicken MLCK210 [1]. The N201 construct was described earlier [9, 11]. DNA restriction and modifying enzymes were from New England Biolabs (USA), Fermentas (Lithuania), and Sileks (Russia). Qiagen (Germany) kits were used for plasmid DNA purification and agarose gel extraction. Primers were ordered from Syntol (Russia); sequences of all the primers used in the present study are available on request. The N27-157 construct was produced by PCR using a pETN452 clone [9] as a template and subcloned in pAsk-IBA3 vector (IBA GmbH, Germany) at EcoRI and NcoI sites so that resulting N27-157 construct acquired the Strep-tag encoding sequence at its 3′-end. Next, N27-157-Strep-tag coding sequence was PCR-amplified and subcloned in pET21(d+) (Novagen, USA) at EcoRI and HindIII sites for protein production in bacteria. The final insert encoded for N27-157-Strep-tag followed by a stop codon (N27-157).
Fig. 1. MLCK210 N-terminal tail constructs used in the study. Relationships of various recombinant constructs to the full-length MLCK210 are depicted. White boxes, IgG-C2 domains. CaM-Reg, calmodulin binding and regulatory domain. KRP, Kinase-Related Protein domain.
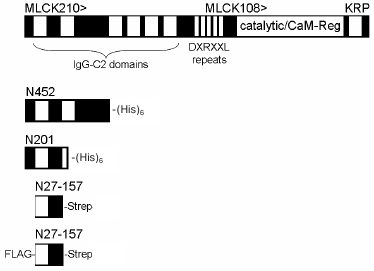
For expression in eukaryotic cells, the N27-157-Strep-tag sequence was PCR-amplified and subcloned in pFlag-CMV2 vector (Sigma) at EcoRI and BamHI sites to yield N27-157 with both 5′-Flag- and 3′-Strep-tags. For mutation of S140 and S149 into either aspartate or alanine residues, a site-directed mutagenesis approach (Stratagene, USA) was applied in compliance with the manufacturer’s directions. All the constructs were verified by DNA sequencing.
Protein preparation. Actin was isolated from rabbit skeletal muscle acetone powder according to the standard method of Spudich and Watt [20]. His-tagged N201 [11] and Strep-tagged N27-157 were isolated, respectively, on TALON-Resin (Clontech, USA) or Strep-Tactin Agarose (IBA GmbH) according to the manufacturers’ directions, except all buffers additionally contained 0.1 mM diisopropyl fluorophosphate (DFP; Sigma) to inhibit proteases. N27-157 was dialyzed in buffer containing 10 mM MOPS, pH 7.0, 100 mM NaCl, 1 mM MgCl2, 0.1 mM EGTA, and 1 mM dithiothreitol (DTT) and drop-frozen in liquid nitrogen. The proteins were stored at –70°C until use.
Protein concentration was measured by spectrophotometry using the following extinction coefficients and molecular weights: G-actin, OD290nm1mg/ml = 0.62 (43 kDa); N201, OD280nm1mg/ml = 1.08 (22 kDa); N27-157, OD280nm1mg/ml = 1.42 (16 kDa).
Co-sedimentation assay. Actin was polymerized by dialysis in a sedimentation buffer (10 mM MOPS, pH 7.0, 100 mM NaCl, 1 mM MgCl2, 0.1 mM EGTA, 1 mM DTT). To analyze the interaction of N27-157 and phospho-N27-157 (2.5 to 30 µM) with F-actin (5 to 15 µM), the proteins were mixed in the abovementioned buffer, incubated for 20 min at room temperature, and centrifuged at 100,000g for 30 min. Supernatant and pellet fractions were analyzed by 12% SDS-PAGE and Coomassie Brilliant Blue R-250 staining.
Electrophoresis and Western blotting. SDS-PAGE was conducted using the system of Laemmli [21]; gels were stained with Coomassie Brilliant Blue R-250. Western blotting was done according to the method of Towbin et al. [22] using a semi-dry electroblotter unit (Bio-Rad, USA). The proteins were transferred either to Immobilon P polyvinylidene difluoride (PVDF) or to nitrocellulose filters (Millipore, USA). Immunoblots were probed with either monoclonal antibody M2 to Flag epitope (Sigma), antibody to phosphoserine (GenTex, USA), anti-Strep-tag II antibody (IBA GmbH), or antibody to GAPDH (Sigma), and secondary antibodies conjugated with horseradish peroxidase (Pierce, USA) followed by treatment with ECL reagent (GE Healthcare, UK) and exposed to X-ray films (Fotochemische Werke GmbH, Germany). Gels and developed X-ray films were digitized using a Hewlett-Packard (USA) scanner and subjected to densitometric analysis using the NIH Image 1.59 software (NIH, USA).
In vitro phosphorylation and determination of phosphorylation sites. Phosphorylation of recombinant MLCK210 fragments in vitro (the final concentration varied from 10 to 18.3 µM) was carried out in a buffer containing 10 mM MOPS, pH 7.4, 100 mM NaCl, 1 mM MgCl2, 0.1 mM EGTA, 1 mM DTT in the presence of 0.5 mM γ-[32P]ATP (GE Healthcare). The reaction was started by addition of either the catalytic subunit of cAMP-dependent protein kinase (PKA; New England Biolabs, USA) or Aurora B protein kinase (Cell Signaling, USA) to a final concentration of 0.22 or 0.12 µM, respectively. Aliquots of the reaction mixture were spotted on Whatman (USA) filters in triplicates. Filters were washed with a phosphate buffer containing 10% trichloroacetic acid and subjected to liquid scintillation counting. Additionally, radioactive samples were separated by SDS-PAGE; the gels were stained with Coomassie Brilliant Blue R-250 and exposed to X-ray films. Alternatively, signal intensities of radioactive protein bands were quantified using a Storm Phosphorimager (Molecular Dynamics, USA).
For determination of PKA phosphorylation sites in N201 using mass spectroscopy, an aliquot of N201 protein was phosphorylated by PKA in vitro for 2 h at 37°C. The reaction mixture was separated by 12% SDS-PAGE; the Coomassie-stained N201 protein band was excised and subjected to trypsinolysis. Peptides were extracted from the gel and separated by HPLC using a nanoscale C18 reverse-phase column (Zorbax C18, 0.075 × 50 mm). Peptides were eluted using a 0-60% acetonitrile gradient in aqueous 0.1% formic acid solution for 60 min at a flow rate of 0.3 µl/min. Spectral data were collected in an Agilent 1100 XCT ion trap LC-mass spectrometer run in data-dependent positive ion MS/MS mode. The instrument collected five MS/MS spectra per cycle and previously collected parent ions were excluded for 2 min. Phosphopeptide identifications were established by analysis of spectra using the Agilent Spectrum Mill (Agilent Technologies, USA) and Mascot (Matrix Science, USA) protein database search engines (restricted to avian species). Assessment of spectral quality and validation of peptides are described in [23].
Analysis of N27-157 actin binding in cells. Cell culture plastic ware was from Corning (Netherlands). HeLa cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (HyClone, USA) at 37°C in a 5% CO2 incubator. In 35-mm dishes, cells at a confluency of 50-70% were transfected with either pFlag-N27-157 wild-type or phospho-mutant constructs using a Maxifectin-21 kit (Pepline, Russia) according to the manufacturer’s protocol. Twenty-four hours post-transfection, the soluble fraction was extracted from transfected HeLa cells with a buffer containing 2 mM KH2PO4, 10 mM Na2HPO4, pH 7.4, 150 mM NaCl, 2.4 mM KCl, 1 mM EGTA, 0.5% Triton X-100, 0.1 mM DFP. Cells were incubated with 50 µl of the extraction buffer for 10 min on ice, and all liquid was recovered and mixed with 25 µl of 3× SDS-PAGE sample buffer. Then, 75 µl of 1× SDS-PAGE sample buffer was added to recover the detergent-insoluble cytoskeleton fraction from the dish with a rubber policeman. Samples of the soluble and cytoskeletal fractions of HeLa cells were boiled for 3 min and analyzed by 10% SDS-PAGE and immunoblotting.
Analysis of N27-157 phosphorylation in cells. HeLa cells were cultured in 35-mm dishes as above to reach 50-70% confluency and transfected with 3 µg of either pFlag-N27-157 or pFlag-N27-157(S140A, S149A) using a standard Ca2+-phosphate transfection method. Three days post-transfection, the culture medium was changed for 1.8 ml DMEM/10% FBS additionally containing [32P]orthophosphate (Leypunsky Institute for Physics and Power Engineering, Obninsk, Russia; 8 MBq per 60-mm plate) and incubated overnight. Next day, the medium was replaced with 1.8 ml of DMEM without FBS but containing [32P]orthophosphate (8 MBq). In 1 h, 1 ml of the medium was discarded and the cells were treated with a mixture containing 10 µM forskolin (Alexis Biochemicals, USA), 1 µM okadaic acid (LC Laboratories, USA), and 100 µM phosphodiesterase inhibitor IBMX (Loba Feinchemie GmbH, Austria) with or without 1 µM PKA inhibitor KT5720 (Calbiochem, USA) for 40 min at 37°C in a 5% CO2 atmosphere. Simultaneously, the corresponding volume of the vehicle (dimethyl sulfoxide, DMSO; Sigma) was added to control plates. The cells were rinsed with ice-cold PBS and lysed with 300 µl of a buffer containing 100 mM Tris-HCl, pH 8.0, 1 M NaCl, 1 mM EDTA, 0.1% Triton X-100, and both protease (Roche, Switzerland) and phosphatase inhibitor cocktails (Thermo Scientific, USA). The cellular DNA was needle-sheared followed by spinning at 13,000 rpm at 4°C in a tabletop centrifuge to remove cellular debris. The supernatant was incubated with 50 µl of Strep-Tactin Sepharose for 1 h at 4°C. Following incubation, the beads were washed with 100 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 1 mM EDTA. Next, 50 µl of 1× SDS-PAGE sample buffer was added to the Strep-Tactin beads, boiled for 3 min, and the soluble fraction was subjected to 10% SDS-PAGE and electrotransferred onto a PVDF filter. The filter was dried, stained with Ponceau S, and exposed to a Storage Phosphor Screen for 3 weeks. Phosphate content in N27-157 protein bands was analyzed using a Storm Phosphorimager. The resulting images were processed with the ImageJ software (NIH, USA). The same PVDF filter was immunostained using a monoclonal antibody to Flag epitope to normalize radioactivity signal by N27-157 protein loading, i.e. the relative radioactive phosphate content in N27-157 constructs was calculated as a ratio of the phosphorimager signal intensity to the intensity of anti-Flag ECL signal in the same samples. In selected experiments, PVDF filter was then stripped of anti-Flag and secondary antibodies [24] and re-probed with an anti-phosphoserine monoclonal antibody.
Immunocytochemistry. For fluorescent microscopy, cells were fixed in 3.7% formaldehyde in PBS, permeabilized with 1% Triton X-100 in PBS, and processed for immunofluorescent staining (secondary antibodies labeled with Alexa Fluor 488 and TRITC-labeled phalloidin were from Molecular Probes, USA). Immunofluorescent images were obtained using a Zeiss Axiovert 200 microscope (Zeiss, Germany) equipped with an AxioCam cooled CCD camera and Axiovision v.3.1 software and assembled in Adobe Photoshop v.6.0 (Adobe Systems, USA).
RESULTS
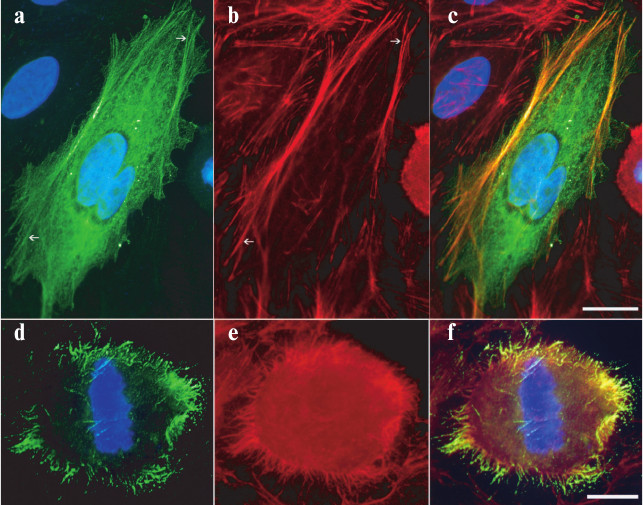
N-terminal actin-binding region of MLCK210. The intracellular localization of Flag-tagged N27-157 was assessed in transiently transfected HeLa cells. Figure 2 (a-c) demonstrates that in spread interphase cells, N27-157 colocalizes with microfilament bundles as visualized by TRITC-phalloidin staining (arrows). In rounded mitotic (metaphase) cells, anti-Flag immunostaining was associated with F-actin positive hair-like projections on cell membrane, whereas no Flag-positive structures are detected in the inner cytoplasm (Fig. 2, d-f).
Fig. 2. Colocalization of N27-157-Flag with actin cytoskeleton in HeLa cells. Cell were transfected with N27-157 containing Flag epitope at the N-terminus and processed for immunostaining 24 h later. a, d) Anti-Flag immunostaining; b, e) filamentous actin staining with TRITC-phalloidin; c, f) merged images. Arrows point to microfilament bundles. Nuclei are stained with 4′,6-diamidino-2-phenylindole (DAPI). Bar, 10 µm (a-c), 5 µm (d-f).
PKA phosphorylation sites in the N-terminal actin-binding region of MLCK210. The N-terminal actin-binding region of MLCK210 possesses several consensus sites recognized by PKA such as R/KxS, where x is any amino acid. We determined sites affected by PKA in chicken MLCK210 N-terminus using mass spectroscopy and a longer version of the actin-binding region, N201 [11]. PKA incorporated 1.5-2.0 moles of phosphate per mole of N201 in vitro (data not shown). Analysis of mass spectra revealed S140 and S149 as phosphorylated residues in a single phosphopeptide 134TPGGRLSVPPVEHRPSIWGESPPK157 derived from N201. The m/z of the peptide was 914.5; it corresponds to a triple-charged peptide with a parent mass of 2740 Da. Phosphorylation site assignments were based upon neutral loss ions corresponding to the underlined serine residues in the peptide.
PKA and Aurora B kinase phosphorylate common serine residues in N-terminal actin-binding region of MLCK210. Given that the shorter version of MLCK210 N-terminal actin-binding region N27-157 includes PKA phosphorylation sites determined in N201, we continued analysis of these sites using the N27-157 construct. To confirm the phosphorylation sites within N27-157, S140 and S149 were mutated to alanine residues. Recombinant wild-type N27-157 and its double alanine mutant were treated in vitro by two protein kinases, PKA and Aurora B. As judged by radioactive phosphate incorporation, both PKA and Aurora B kinase more readily phosphorylated wild-type N27-157 (Fig. 3a). PKA (Fig. 3a, left) incorporated 1.730 ± 0.116 moles of phosphate per mole of N27-157 and 0.545 ± 0.037 moles of phosphate per mole of N27-157(S140A, S149A). Aurora B kinase (Fig. 3a, right) incorporated 0.287 ± 0.030 mole of phosphate per mole of N27-157 and only 0.025 ± 0.004 mole of phosphate per mole of N27-157(S140A, S149A). Thus, the substitution of S140 and S149 for alanine residues led to a marked decrease in phosphate incorporation by PKA and Aurora B and confirmed these sites as available for posttranslational modification in N27-157. Noteworthy, N27-157(S140A, S149A) still incorporates a significant amount of radioactivity in the presence of PKA, raising the possibility that cryptic phosphorylation site(s) are present in this fragment.
Fig. 3. Phosphorylation of N27-157 and N27-157(S140A, S149A) in vitro and in transfected HeLa cells. a) Recombinant wild-type N27-157 and N27-157(S140A, S149A) mutant were phosphorylated in vitro by either PKA (left) or Aurora B kinase (right) in the presence of γ-[32P]ATP as described under “Materials and Methods”. Representative images of Coomassie R-250 stained gels (upper panels) and autoradiograms (lower panels) are shown. b) HeLa cells expressing at comparable expression levels either N27-157 (1-3) or N27-157(S140A, S149A) (4-6) were loaded with [32P]orthophosphate and treated either with vehicle (1, 4), or with a mixture containing 10 µM forskolin, 1 µM okadaic acid, and 100 µM IBMX (2, 5), or with the same mixture supplemented with PKA inhibitor KT5720 to a final concentration of 1 µM (3, 6). Upper panel, top row, [32P]phosphate incorporation in N27-157 and N27-157(S140A, S149A) recovered from HeLa cells and assessed using a Storm Phosphorimager. Upper panel, bottom row, total protein content of N27-157 and N27-157(S140A, S149A) mutant assessed in the same samples using Western blot with anti-Flag antibody. Lower panel, bar graph showing normalized 32P radioactivity content of N27-157 (1-3) and N27-157(S140A, S149A) mutant (4-6) under cell treatment conditions specified above. Each bar corresponds to a representative radioactivity/immunoblot data shown above it. Mean ± S.D., n = 3; * p < 0.05 vs. basal phosphorylation level.
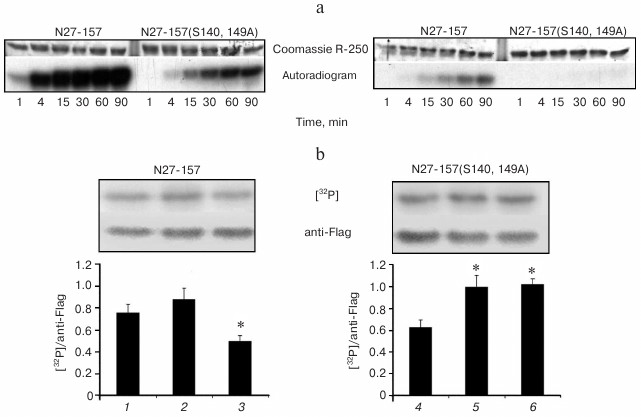
PKA is involved in phosphorylation of N-terminal actin-binding region of MLCK210 in HeLa cells. For evaluation of N27-157 phosphorylation in living cells, we transiently expressed N27-157 and N27-157(S140A, S149A) in HeLa cells, loaded them with [32P]orthophosphate, and challenged to activate or inhibit PKA activity. Then, N27-157 was recovered from cell lysates using an affinity resin; the incorporated radioactivity was quantified and normalized to N27-157 protein content.
As shown in Fig. 3b (left), in HeLa cells radioactive phosphate was incorporated in N25-157 under basal cell growth conditions. Simultaneous treatment of HeLa cells with adenylate cyclase activator forskolin and phosphodiesterase inhibitor IBMX (aimed at endogenous PKA activation) as well as with protein serine/threonine phosphatase inhibitor, okadaic acid, resulted in a modest increase in [32P]phosphate incorporation in N27-157. When PKA inhibitor KT5720 was added to the challenging agent mixture, radioactive phosphate content of N27-157 decreased 1.5-fold below the level observed under basal cell growth conditions and about 1.8-fold below the level in cells treated with forskolin, IBMX, and okadaic acid.
In HeLa cells, there was a lower basal level of radioactive phosphate incorporation in N27-157(S140A, S149A) mutant as compared to N27-157 (Fig. 3b, right). However, upon simultaneous treatment with forskolin, IBMX, and okadaic acid, the normalized radioactivity of N27-157(S140A, S149A) increased above that of N27-157 and remained elevated even in the presence of the PKA inhibitor. Similar results were obtained when we used anti-phosphoserine monoclonal antibody to determine the phosphoserine content in both wild-type N27-157 and its double alanine mutant (data not shown).
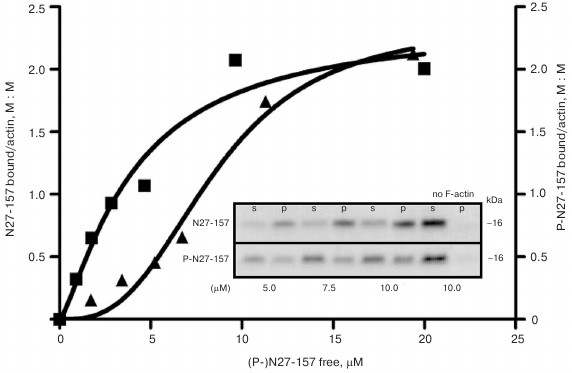
Phosphorylation by PKA attenuates actin-binding activity of N27-157 in vitro. Interaction of N27-157 with F-actin was confirmed in vitro by a co-sedimentation assay using recombinant purified N27-157. Figure 4 demonstrates that, under the conditions used, N27-157 was found in the pellet fraction only if F-actin was included in the probes. Phosphorylation by PKA in vitro affects the mode of interaction of N27-157 with purified F-actin leading to attenuation of protein–protein binding, as judged by less phospho-N27-157 protein being distributed to the pellet fraction with F-actin.
Fig. 4. Binding curves of N27-157 and phospho-N27-157 to F-actin in vitro. GraphPad Prizm 4 nonlinear fitting of binding data for N27-157 (boxes) and phospho-N27-157 (triangles) interaction with F-actin. Inset, a representative Coomassie R-250 stained gel demonstrating the distribution of N27-157 and its PKA phosphorylated version P-N27-157 between supernatant (s) and F-actin pellet (p) in a co-sedimentation assay at various concentrations of the MLCK210 construct (µM). No F-actin, self-sedimentation of N27-157 and P-N27-157.
Phosphomimetic mutants of N27-157 demonstrate reduced association with the cytoskeleton in living cells and in vitro. To elucidate the role of S140 and S149 phosphorylation in living cells, we developed phosphomimetic mutants of N27-157 by replacing S140 and S149 with aspartate residues. N27-157(S149D) and N27-157(S140D, S149D) were expressed in HeLa cells, and association of these phospho-mutants with the cytoskeleton was studied using a differential extraction approach. As shown in Fig. 5a, wild-type N27-157 distributed equally between cytoskeletal and soluble fractions of HeLa cells extracted with a detergent-containing buffer. In contrast, single and double phosphomimetic mutants of N27-157 were predominantly found in the soluble fraction, indicating their decreased association with the cytoskeletal components. For both N27-157(S149D) and N27-157(S140D, S149D), a similar extent of interaction with cellular cytoskeleton was observed, suggesting the leading role of the S149D mutation in attenuation of N27-157 binding to the cytoskeletal structures. The N27-157 to GAPDH ratio did not significantly differ between wild-type N27-157, N27-157(S149D), and N27-157(S140D, S149D) transfectants (data not shown).
Fig. 5. Effect of wild-type N27-157 phosphorylation or phosphomimetic mutations on actin binding in cells and in vitro. a) Relative distribution of N27-157 and its phosphomimetic mutants between detergent-soluble (open bars) and cytoskeletal fractions (filled bars) of transfected HeLa cells was determined by immunoblotting with anti-Flag antibody. Data are presented as mean ± S.D., n = 6, * p < 0.05. Typical immunoblot results are shown above the corresponding bars. b) Binding of N27-157, PKA phosphorylated N27-157, and N27-157(S149D) to F-actin in vitro. Distribution of N27-157 between the supernatant (open bars) and F-actin pellet (filled bars) in a co-sedimentation assay was calculated using densitometry of Coomassie-stained gels. Representative gel fragments are shown above the corresponding bars. Data are presented as mean ± S.D., n = 3.
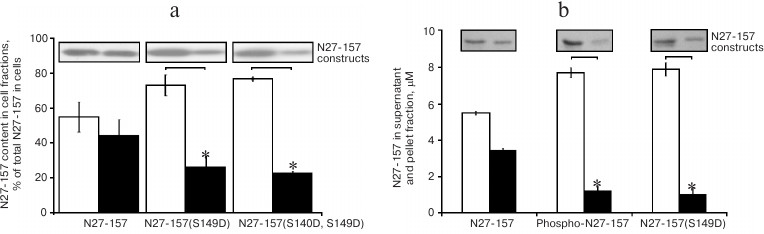
The effect of the S149D mutation was confirmed using an in vitro co-sedimentation assay. Figure 5b demonstrates that phosphorylation of the wild-type N27-157 and incorporation of a single S149D mutation attenuated the binding of N27-157 to actin filaments to similar extent.
DISCUSSION
In this study, we localized the N-terminal actin-binding region of MLCK210 to the first IgG-like domain and its C-terminal flanking sequence (a.a. 27-157 in the avian sequence used in these studies, which corresponds to a.a. 33-162 in the human sequence) and demonstrated that actin binding is regulated by site-specific phosphorylation at a nearby S/T protein kinase substrate segment. The phosphorylation-dependent change in actin-binding activity was observed in both in vitro reconstitution and cell-based assays. N27-157, the protein construct based on the segment of MLCK210, co-localized with microfilament bundles in spread transfected HeLa cells, which normally express MLCK210 at low levels. In addition, in rounded metaphase HeLa cells, N27-157 is concentrated in peripheral projections where filamentous actin is preserved (Fig. 2). These structures most likely represent retraction/anchoring fibers that are developed by substrate-dependent cultured cells during mitosis [25].
Previously, we showed that longer MLCK210 N-terminal fragments bind actin [8, 11]. In the present study, we show that the N27-157 MLCK210 fragment retains actin-binding activity in cells and in vitro (Figs. 2 and 4). These findings indicate that the N27-157 fragment contains an actin-binding site sufficient to target N27-157 to actin-containing filaments in cells. The N-terminal MLCK210 fragment containing two N-terminal IgG-like domains was able to bundle actin in vitro [26], indicating that the N-terminal MLCK210 actin-binding site might have a composite structure as it is the case for the actin-binding site located in the central part of the MLCK210 molecule and consisting of five D(F/V)RxxL motifs. Our results indicate that an N-terminal actin-binding site, or at least an independent part of the putative composite N-terminal actin-binding site, is located within the N27-157 MLCK210 fragment. Motif analysis of N27-157 identified a six-residue segment, EHRPSI, with amino acid composition similar to the DF/VRxxL actin-binding motifs that are located in the central part of MLCK210 and provide for high affinity binding to actin filaments [27, 28]. Noteworthy, the EHRPSI motif is phylogenetically conserved across avian, rodent, and human. A single DF/VRxxL-like motif in the N-terminal actin-binding site versus five DF/VRxxL motifs in the central actin-binding region of MLCK210 is consistent with markedly different affinities [8-11] of these sites to actin filaments. Therefore, a lower-affinity N-terminal actin-binding region is likely to be involved in transient interactions with adjacent actin filaments, whereas the central actin-binding region is more likely to keep MLCK210 permanently attached to a particular actin filament. Based on the model of MLCK210 N-terminal tail as a flexible chain of six IgG-like domains connected by unstructured spacers, it is suggestive that MLCK210 has two actin-binding sites positioned quite distantly from each other. As such, MLCK210 can be regarded as a long-range microfilament cross-linker capable of forming loose mesh-works of actin filaments. This model is supported by previous electron microscopy data demonstrating the full-length N-terminal tail of MLCK210 to form networks and loose bundles of actin filaments [8]. Hence, in the cellular context, MLCK210 may complement the function of another high molecular weight actin-binding protein, filamin [29], and cooperate with the latter in regulation of the local sol–gel conversion of the cytoplasm.
In the N-terminal actin-binding region of MLCK210, bioinformatics analyses reveal several consensus PKA phosphorylation sites, but only the sites containing S140 and S149 were identified experimentally in the studies reported here. Both S140 and S149 map to the linker sequence between the first and second IgG-like domains of MLCK210. Moreover, S149 is located within the putative actin-binding motif 145EHRPSI150. The potential significance to cellular function and integration with other signal transduction pathways (“cross-talk”) is indicated by the reduction of actin binding brought about by phosphorylation at S140/149. Further support is provided by the finding that phosphomimetic mutants of N27-157 (S149D and S140D, S149D) show less cytoskeleton association in transfected cells (Fig. 5a). Additional in vitro experiments using N27-157(S149D) mutant and wild-type N27-157 phosphorylated by PKA (Fig. 5b) provided evidence that S149 phosphorylation in N27-157 has a more pronounced effect in the regulation of N27-157 interaction with actin filaments, whereas S140 and other yet unidentified phospho-S/T residues within the N27-157 sequence exert minor if any effect.
Although the molecular mechanisms of the effect of phosphorylation on actin binding remain to be established, the results reported here are consistent with a parsimonious model in which the phosphorylatable serine residue is within, or in close proximity to, sequences intimately involved in actin–MLCK210 interaction. Finally, identification of MLCK210 phosphorylation at S149 in mouse brain during high-throughput proteomic screen [18, 19] (see also http://www.phosphosite.org/proteinAction.do?id=15552&showAllSites=true) provides additional support for the in vivo significance of this posttranslational modification that has a measurable and functionally relevant outcome in various experimental models.
Clearly, the independent localization of phosphorylation sites and actin-binding regions to this common N-terminal domain, combined with their direct linkage through activity, provide a strong argument in favor of the N-terminal domain being essential to selective MLCK210 roles in cellular responses. However, there is a diverse set of possible mechanisms that could be exploited by the cell to add further complexity and potential biological selectivity to responses. For example, a PKA-mediated response antecedent to a cellular response using another signal transduction pathway might result in PKA phosphorylation of the two serine residues, preventing subsequent phosphorylations by other kinases at the consensus but unused phosphorylation sites present in the N27-157 sequence. The technical details on our phosphorylation studies with the double serine-to-alanine mutant proteins are consistent with the possibility of such cryptic phosphorylation sites. Alternatively, the timing of cellular stimuli could be critical for different kinases to exploit the same phosphorylation sites depending on which cellular pathways are dominant under a given set of circumstances. An example in this regard is the protein kinase Aurora B, which is capable of phosphorylating the N-terminal domain of MLCK210 [17]. We showed that Aurora B can phosphorylate N27-157 and, further, that this capability is lost when the double phosphorylation site mutant N27-157(S140A, S149A) is used as the substrate. Although both kinases might be capable of using the same phosphorylation sites within this domain, the physiological context could dictate which one would be engaged in a given time window or in response to cellular stimuli. Aurora B is most active in mitosis, while PKA does not appear to have such a restriction, so it could transduce cAMP-mediated responses in interphase cells. In this context, PKA and Aurora B might comprise a minimum set of protein kinases needed to regulate MLCK210 phosphorylation throughout the cell cycle.
In summary, we have mapped the actin-binding activity of the MLCK210 unique N-terminal tail domain to a more restricted region and mapped a functional set of phosphorylation sites to the same region. Aurora B and cAMP-dependent protein kinase are two examples of S/T protein kinases to specifically phosphorylate these sites. Further, we have linked phosphorylation of this proximal site to attenuation of the same domain’s actin-binding activity and cellular cytoskeleton localization function. The results add to an increasing body of knowledge about the functions of MLCK210 domains outside of the catalytic domain and provide a foundation for future research into the molecular basis of phosphorylation-dependent modulation of this key regulatory protein.
Assistance of Dr. Olga V. Stepanova with initial immunofluorescence studies is acknowledged.
This work was supported by the Russian Foundation for Basic Research grants 08-04-01158 (VPS), 11-04-01343 (VPS) and 12-04-32265 (ELV).
REFERENCES
1.Watterson, D. M., Collinge, M., Lukas, T. J., Van
Eldik, L. J., Birukov, K. G., Stepanova, O. V., and Shirinsky, V. P.
(1995) Multiple gene products are produced from a novel protein kinase
transcription region, FEBS Lett., 373, 217-220.
2.Birukov, K. G., Schavocky, J. P., Shirinsky, V. P.,
Chibalina, M. V., Van Eldik, L. J., and Watterson, D. M. (1998)
Organization of the genetic locus for chicken myosin light chain kinase
is complex: multiple proteins are encoded and exhibit differential
expression and localization, J. Cell. Biochem., 70,
402-413.
3.Clayburgh, D. R., Rosen, S., Witkowski, E. D.,
Wang, F., Blair, S., Dudek, S., Garcia, J. G., Alverdy, J. C., and
Turner, J. R. (2004) A differentiation-dependent splice variant of
myosin light chain kinase, MLCK1, regulates epithelial tight junction
permeability, J. Biol. Chem., 279, 55506-55513.
4.Gallagher, P. J., Garcia, J. G., and Herring, B. P.
(1995) Expression of a novel myosin light chain kinase in embryonic
tissues and cultured cells, J. Biol. Chem., 270,
29090-29095.
5.Garcia, J. G., Lazar, V., Gilbert-McClain, L. I.,
Gallagher, P. J., and Verin, A. D. (1997) Myosin light chain kinase in
endothelium: molecular cloning and regulation, Am. J. Respir. Cell
Mol. Biol., 16, 489-494.
6.Xu, J., Gao, X. P., Ramchandran, R., Zhao, Y. Y.,
Vogel, S. M., and Malik, A. B. (2008) Nonmuscle myosin light-chain
kinase mediates neutrophil transmigration in sepsis-induced lung
inflammation by activating beta2 integrins, Nat. Immunol.,
9, 880-886.
7.Shirinsky, V. P., Vorotnikov, A. V., Birukov, K.
G., Nanaev, A. K., Collinge, M., Lukas, T. J., Sellers, J. R., and
Watterson, D. M. (1993) A kinase-related protein stabilizes
unphosphorylated smooth muscle myosin minifilaments in the presence of
ATP, J. Biol. Chem., 268, 16578-16583.
8.Kudryashov, D. S., Chibalina, M. V., Birukov, K.
G., Lukas, T. J., Sellers, J. R., Van Eldik, L. J., Watterson, D. M.,
and Shirinsky, V. P. (1999) Unique sequence of a high molecular weight
myosin light chain kinase is involved in interaction with actin
cytoskeleton, FEBS Lett., 463, 67-71.
9.Kudryashov, D. S., Stepanova, O. V., Vilitkevich,
E. L., Nikonenko, T. A., Nadezhdina, E. S., Shanina, N. A., Lukas, T.
J., Van Eldik, L. J., Watterson, D. M., and Shirinsky, V. P. (2004)
Myosin light chain kinase (210 kDa) is a potential cytoskeleton
integrator through its unique N-terminal domain, Exp. Cell Res.,
298, 407-417.
10.Smith, L., Su, X., Lin, P., Zhi, G., and Stull,
J. T. (1999) Identification of a novel actin binding motif in smooth
muscle myosin light chain kinase, J. Biol. Chem., 274,
29433-29438.
11.Vilitkevich, E. L., Kudriashev, D. S., Stepanova,
O. V., and Shirinsky, V. P. (2004) A new actin-binding area of the
myosin light chains’ high-molecular kinase, Ross. Fiziol. Zh.
im. I. M. Sechenova, 90, 577-585.
12.Birukov, K. G., Csortos, C., Marzilli, L., Dudek,
S., Ma, S. F., Bresnick, A. R., Verin, A. D., Cotter, R. J., and
Garcia, J. G. (2001) Differential regulation of alternatively spliced
endothelial cell myosin light chain kinase isoforms by p60(Src), J.
Biol. Chem., 276, 8567-8573.
13.Dudek, S. M., Birukov, K. G., Zhan, X., and
Garcia, J. G. (2002) Novel interaction of cortactin with endothelial
cell myosin light chain kinase, Biochem. Biophys. Res. Commun.,
298, 511-519.
14.Shin, D. H., Chun, Y. S., Lee, K. H., Shin, H.
W., and Park, J. W. (2009) Arrest defective-1 controls tumor cell
behavior by acetylating myosin light chain kinase, PLoS One,
4, e7451.
15.Takizawa, N., Ikebe, R., Ikebe, M., and Luna, E.
J. (2007) Supervillin slows cell spreading by facilitating myosin II
activation at the cell periphery, J. Cell Sci., 120,
3792-3803.
16.Wadgaonkar, R., Dudek, S. M., Zaiman, A. L.,
Linz-McGillem, L., Verin, A. D., Nurmukhambetova, S., Romer, L. H., and
Garcia, J. G. (2005) Intracellular interaction of myosin light chain
kinase with macrophage migration inhibition factor (MIF) in
endothelium, J. Cell. Biochem., 95, 849-858.
17.Dulyaninova, N. G., and Bresnick, A. R. (2004)
The long myosin light chain kinase is differentially phosphorylated
during interphase and mitosis, Exp. Cell Res., 299,
303-314.
18.Huttlin, E. L., Jedrychowski, M. P., Elias, J.
E., Goswami, T., Rad, R., Beausoleil, S. A., Villen, J., Haas, W.,
Sowa, M. E., and Gygi, S. P. (2010) A tissue-specific atlas of mouse
protein phosphorylation and expression, Cell, 143,
1174-1189.
19.Goswami, T., Li, X., Smith, A. M., Luderowski, E.
M., Vincent, J. J., Rush, J., and Ballif, B. A. (2012) Comparative
phosphoproteomic analysis of neonatal and adult murine brain,
Proteomics, 12, 2185-2189.
20.Spudich, J. A., and Watt, S. (1971) The
regulation of rabbit skeletal muscle contraction. I. Biochemical
studies of the interaction of the tropomyosin troponin complex with
actin and the proteolytic fragments of myosin, J. Biol. Chem.,
246, 4866-4871.
21.Laemmli, U. K. (1970) Cleavage of structural
proteins during the assembly of the head of bacteriophage T4,
Nature, 227, 680-685.
22.Towbin, H., Staehelin, T., and Gordon, J. (1979)
Electrophoretic transfer of proteins from polyacrylamide gels to
nitrocellulose sheets: procedure and some applications, Proc. Natl.
Acad. Sci. USA, 76, 4350-4354.
23.Lukas, T. J., Wang, A. L., Yuan, M., and Neufeld,
A. H. (2009) Early cellular signaling responses to axonal injury,
Cell Commun. Signal., 7, 5.
24.Yeung, Y. G., and Stanley, E. R. (2009) A
solution for stripping antibodies from polyvinylidene fluoride
immunoblots for multiple reprobing, Anal. Biochem., 389,
89-91.
25.Mitchison, T. J. (1992) Actin based motility on
retraction fibers in mitotic PtK2 cells, Cell Motil.
Cytoskeleton, 22, 135-151.
26.Yang, C. X., Chen, H. Q., Chen, C., Yu, W. P.,
Zhang, W. C., Peng, Y. J., He, W. Q., Wei, D. M., Gao, X., and Zhu, M.
S. (2006) Microfilament-binding properties of N-terminal extension of
the isoform of smooth muscle long myosin light chain kinase, Cell
Res., 16, 367-376.
27.Smith, L., Parizi-Robinson, M., Zhu, M. S., Zhi,
G., Fukui, R., Kamm, K. E., and Stull, J. T. (2002) Properties of long
myosin light chain kinase binding to F-actin in vitro and in
vivo, J. Biol. Chem., 277, 35597-35604.
28.Poperechnaya, A., Varlamova, O., Lin, P. J.,
Stull, J. T., and Bresnick, A. R. (2000) Localization and activity of
myosin light chain kinase isoforms during the cell cycle, J. Cell
Biol., 151, 697-708.
29.Stossel, T. P., Condeelis, J., Cooley, L.,
Hartwig, J. H., Noegel, A., Schleicher, M., and Shapiro, S. S. (2001)
Filamins as integrators of cell mechanics and signaling, Nat. Rev.
Mol. Cell Biol., 2, 138-145.