REVIEW: Phylogeny of Aging and Related Phenoptotic Phenomena
G. Libertini
Independent Researcher, Via Cavour 13, Caivano, 80023 Naples, Italy; E-mail: giacinto.libertini@tin.it
Received July 31, 2015; Revision received August 28, 2015
The interpretation of aging as adaptive, i.e. as a phenomenon genetically determined and modulated, and with an evolutionary advantage, implies that aging, as any physiologic mechanism, must have phylogenetic connections with similar phenomena. This review tries to find the phylogenetic connections between vertebrate aging and some related phenomena in other species, especially within those phenomena defined as phenoptotic, i.e. involving the death of one or more individuals for the benefit of other individuals. In particular, the aim of the work is to highlight and analyze similarities and connections, in the mechanisms and in the evolutionary causes, between: (i) proapoptosis in prokaryotes and apoptosis in unicellular eukaryotes; (ii) apoptosis in unicellular and multicellular eukaryotes; (iii) aging in yeast and in vertebrates; and (iv) the critical importance of the DNA subtelomeric segment in unicellular and multicellular eukaryotes. In short, there is strong evidence that vertebrate aging has clear similarities and connections with phenomena present in organisms with simpler organization. These phylogenetic connections are a necessary element for the sustainability of the thesis of aging explained as an adaptive phenomenon, and, on the contrary, are incompatible with the opposite view of aging as being due to the accumulation of random damages of various kinds.
KEY WORDS: IMICAW, aging, phenoptosis, telomere, telomerase, apoptosis, proapoptosisDOI: 10.1134/S0006297915120019
Aging, here precisely defined as “increasing mortality with increasing chronological age in populations in the wild” (IMICAW [1]), is observed in many species under natural conditions [2-9], our species included [10]. The definitions “actuarial senescence in the wild” [7] and “progressive loss of function accompanied by decreasing fertility and increasing mortality with advancing age” [11] are synonyms of the above-said descriptive definition of aging. On the contrary, statements as “ageing, or senescence, results from a waning of the force of natural selection with respect to the age of gene effects” [12] or “aging is caused... by evolved limitations in somatic maintenance, resulting in a build-up of damage” [13] do not describe aging in a neutral way but are only short expressions of not proven hypotheses about aging (see below).
In fact, aging is interpreted in two very different ways [14], which, for their opposite numberless implications and for the importance of the subject, deserve to be considered incompatible paradigms in the meaning of the term “paradigm” proposed by Kuhn [15]. The first (“old paradigm”) includes a vast patchwork of disparate hypotheses that try to explain aging as the inevitable consequence of damaging factors that progressively jeopardize fitness [16-43]. In the older theories, damage accumulation is conceived without considering any evolutionary mechanism, with the implicit incorrect assumption that aging and natural selection act in two different contexts. The newer theories, or at least some of them, try to take into account the mechanisms of natural selection, which, according to them, would be able to counteract damaging factors only partially and to an age-related decreasing extent, also because of pleiotropic effects and contrasting physiological necessities. The second paradigm (“new paradigm”) includes hypotheses that define and interpret aging as a physiological phenomenon, i.e. a phenomenon that, in spite of its undoubted disadvantages for the aging individual, is determined and modulated by natural selection as being evolutionarily advantageous in terms of supra-individual selection [1, 44-65], although for some hypotheses only in particular conditions [1, 65].
The new paradigm sees aging as a particular type of phenoptosis (see definition below), a term coined in 1997 by Skulachev [48]. Subsequently, the same author, to emphasize the peculiarity of the phenomenon within the phenoptotic universe, coined for aging the definition of “slow phenoptosis” [66]. The concept of phenoptosis can be shortly defined as “the programmed death of an individual” [49] or described more extensively: “phenoptosis is the death of an individual caused by its own actions or by actions of close relatives (siblicide; in particular, the parent-caused death of an offspring or filial infanticide) and not caused primarily by accidents or diseases or external factors, which is determined, regulated or influenced by genes favored by natural selection” [63]. It includes a large and very heterogeneous category of phenomena, most well known and documented for a long time [6], but not appreciated, before Skulachev’s definition, in their entirety and in terms of their important implications [63].
For the new paradigm, aging is a physiological phenomenon and must necessarily have, like every other phenomenon of this nature: (i) a normal function (or physiology); (ii) pathological alterations in specific cases; (iii) evolutionary causes; (iv) a phylogeny. In this review, I will not discuss or reiterate the arguments or the evidence in support of the new paradigm and against the old paradigm, which have already been debated elsewhere [14, 67], nor a description of aging physiology and pathology, already expounded in general in other works [60, 68, 69]. The aim of this work is only to indicate, or at least to hypothesize, phylogenetic aspects of aging in relation to other similar or related phenoptotic phenomena.
It is opportune to stress that the definition of phenoptosis includes a wide and heterogeneous set of phenomena that do not necessarily involve the same evolutionary advantages or a single monophyletic origin. In fact, by choosing among many phenoptotic phenomena, as examples: (i) endotokic matricide, a particular phenomenon shown by some invertebrates, where maternal death is obligatory in reproduction: “the young kill their mother by boring through her body wall” or cannibalizing her body [6]; (ii) cryptic female choice [70], i.e. non-pathologic miscarriages to eliminate before birth offspring with lesser antigen variability and possible reduced resistance to infections [71]; (iii) semelparity and sudden death after reproduction shown by many species of Anguilliformes, Salmoniformes, dasyurid marsupials, rodents, and plants [6]; (iv) aphagy in adult insects: “aphagy from defective mouthparts or digestive organs is very common during the adult phases of insects… and is the limiting factor in the adult lifespan of many short-lived species. This phenomenon is, inarguably, programmed senescence...” [6]; (v) bacterial suicide activated by phage infection [72]; (vi) filial infanticide [73]; it is quite unlikely that these phenomena can be explained based on common evolutionary advantages and/or with a single phylogenetic origin. Thus, the phylogenetic investigation will be restricted to the relationships and similarities between aging and certain types of phenoptotic phenomena that appear to have a probable common phylogenetic origin, with or without a common evolutionary advantage.
PROKARYOTE WORLD
Phenoptosis is well documented among prokaryotes, e.g. (i) bacterial suicide activated by phage infection “thereby curtailing viral multiplication and protecting nearby E. coli from infection” [72]; “In E. coli, three suicide mechanisms that are activated by the appearance of a phage in the cell interior have been described” [74]; (ii) mass suicide of bacterial phytoplankton as defense against viruses [75]; (iii) in E. coli, the “built-in suicide module” that is activated by antibiotics [76].
The mechanisms that activate phenoptosis in bacteria have been defined as “proapoptosis” and proposed as phylogenetic precursors of apoptosis in eukaryotes [77], because they share various features with apoptosis: “Several key enzymes of the apoptotic machinery, including the paracaspase and metacaspase families of the caspase-like protease superfamily, apoptotic ATPases and NACHT family NTPases, and mitochondrial HtrA-like proteases, have diverse homologs in bacteria, but not in archaea. Phylogenetic analysis strongly suggests a mitochondrial origin for metacaspases and the HtrA-like proteases, whereas acquisition from Actinomycetes appears to be the most likely scenario for AP-ATPases. The homologs of apoptotic proteins are particularly abundant and diverse in bacteria that undergo complex development, such as Actinomycetes, Cyanobacteria and α-proteobacteria, the latter being progenitors of the mitochondria” [78].
In the prokaryote world, phenoptosis is not at all a curiosity limited to a few rare cases but appears to be a very common occurrence that determines spectacular mass suicide [75]. Intrinsic to the definition of phenoptosis and therefore necessary to explain a “programmed death in bacteria” [74, 79] is that these phenomena are favored by natural selection. In prokaryotes, the main causes of selective pressure favoring phenoptosis appear to be: (i) defense against infections by phages [72, 75]; (ii) elimination of somehow impaired individuals that take away resources from other individuals: “Most bacterial species actually do not live as planktonic suspensions in vivo but form complex biofilms, tightly knit communities of cells. From this perspective, programmed death of damaged cells may be beneficial to a multicellular bacterial community” [79]. In both cases, it is essential to envisage mechanisms of kin or group selection (as already proposed by others: “As most plankton in a bloom are near identical genetically, from the perspective of their genes, a die-off that creates enough scorched earth to stop the viral advance can make sense” [75]), despite the old theoretical anathema against group selection by Maynard Smith [80, 81]. Regarding the type of selection that would favor these phenomena, it is important to stress that for kin selection, which is well-known and accepted [82-85], if a species is divided into demes, each consisting of closely related individuals or even with monoclonal origin, the distinction between kin selection and group selection thins or disappears.
In fact, kin selection calculates the inclusive fitness of a C gene that acts in the individual defined as 1, and that has also some consequences for the fitness of other individuals (2, 3, …n) in which the probability of the existence of C is equal to the coefficient of kinship (r). In each generation, C is favored by natural selection if the variation of the inclusive fitness is positive, i.e. if:
where Sx is advantage/disadvantage for the individual x (–1 ≥ Sx ≤ +1); Px is reproductive value of individual x (0 ≥ Px ≤ 1); rx is coefficient of kinship between individual x and individual 1 (0 ≥ rx ≤ 1).
In cases where the gene acts only on individual 1, since by definition r1 = 1, formula (1) becomes:
which is the classic formula for individual selection.
Now, let us consider a species divided into monoclonal demes and subjected to a catastrophic event. In such cases, for each of them, if there is no sacrifice of any individual, there is a disadvantage for every individual equal to S. On the contrary, if, by action of the C gene, among n individuals having the C gene, some (nd) sacrifice themselves and die (Sd = –1), while the survivors (ns) have an advantage Ss, as in bacteria a constant reproductive value at any age can be assumed (Px = 1) and as in a monoclonal deme rx is always equal to 1, the C gene will be favored by natural selection if:
that is:
a formula that is a development of Eq. (1).
In cases where the deme is composed of several clones (1, 2, ... z), if C exists in all the individuals of clone 1, it will exist in clone x with a probability equal to the coefficient of kinship of the individuals of clone x with those of clone 1 (rx), and gene C is favored by selection if:
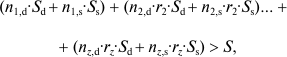 (5)
(5)where, in a clone x, nx,d are the individuals that sacrifice themselves and nx,s the survivors.
It should be noted that kin selection formula (1) has been transformed into a formula that describes a type of group selection; thus, there is no insurmountable distinction between individual selection, kin selection, and group selection.
UNICELLULAR EUKARYOTE WORLD
For the sake of brevity and to propose evidence and arguments based on sound grounds, in this section the discussion will be limited to a single but well-studied eukaryotic unicellular species, i.e. the yeast Saccharomyces cerevisiae. In this species, a phenomenon that closely resembles apoptosis of multicellular eukaryotes is a relatively recent finding [86]. In particular, the phenomenon was shown to be elicited by the overexpression of a factor that triggered apoptosis (mammalian BAX) [87], while the overexpression of another factor that inhibited apoptosis (human Bcl-2) appeared to delay the processes leading to it [88]. The similarity between this phenomenon and apoptosis in multicellular eukaryotes is corroborated by a growing body of evidence. This implies that the two types of phenomena deserve the same name and, moreover, suggests a common phylogenetic origin [56, 89, 90]: “... since the first description of apoptosis in a yeast Saccharomyces cerevisiae strain carrying a CDC48 mutation…, several yeast orthologues of crucial mammalian apoptotic proteins have been discovered…, and conserved proteasomal, mitochondrial, and histone-regulated apoptotic pathways have been delineated...” [91].
In yeast, apoptosis is triggered or favored by: (i) harmful chemical alterations to the habitat [90]; (ii) a decrease in nutrients [92]; (iii) unsuccessful mating [91]; (iv) killer toxins that are secreted by competing yeast tribes [91]. A crucial fact, analogous to what happens in multicellular eukaryotes, is that when a yeast cell dies by apoptosis, its parts are not harmful to other individuals and, on the contrary, are usefully phagocytosed or absorbed by other cells, which, consequently, “are able to survive longer with substances released by dying cells” [93].
Yeast apoptotic patterns have been explained as adaptive because they appear to be useful for the survival of the deme [56, 57, 74, 93-96]. The adaptive interpretation of mass suicide by apoptosis appears plausible in many cases if a yeast species is divided into small demes, each consisting of one or a few clones. A different case is suicide by apoptosis triggered by toxins that are secreted by enemy yeast tribes, where the abovementioned adaptive mechanism is clearly exploited by competitors [91].
So far, it is possible to notice evident analogies between prokaryotic mass phenoptosis through proapoptotic mechanisms and yeast mass phenoptosis through apoptosis, and in both cases group selection is a likely evolutionary cause of these phenomena. However, yeast show more sophisticated mechanisms and something other that, as we shall see, is connected with phenomena shown by multicellular organisms.
Yeast reproduction occurs by asymmetric division into two cells, one defined as “mother” and the other as “daughter”. Cells of the daughter lineage show no limit to reproduction, while those of mother lineage can reproduce only a limited number of times; Jazwinski found a limit of 25-35 duplications in about three days [97]. As the number of duplications increases, there is a growing vulnerability to apoptosis and replicative senescence [91, 93, 98, 99], and this explains how in particular stress conditions part of the population dies and the other survives. However, this is more than a sophisticated mechanism to select a list of the individuals that must sacrifice themselves if necessary (with a priority for sacrifice proportional to the number of previous duplications in mother lineage).
In yeast, mother lineage cells show, proportionally to the number of duplications, besides increasing susceptibility to replicative senescence and apoptosis, increasing metabolic alterations [91, 93, 98-100]. The consequent age-related death rate increase follows an exponential dynamic [101], similar to that shown by individuals of many multicellular species [8, 9]. Wild yeast cells of the mother lineage show a decline in fitness and an increase in mortality that is proportional to the number of duplications, so this phenomenon is somehow within the concept of aging (“increasing mortality with increasing chronological age in populations in the wild” [1]).
It should be noted that while it is possible to say that yeast ages, this is inappropriate for bacteria. Now, it is essential to consider how this happens in yeast. Eukaryotic cells, both of unicellular and of multicellular species, unlike prokaryotes that have circular DNA, have linear DNA. It is well known that, at each replication, DNA polymerase leaves out part of the terminal section of linear DNA (the telomere) and the molecule becomes shorter [102, 103]. The progressive shortening of DNA leads to duplication impairment and, so, it was predicted that an indispensable but hypothetical enzyme restored the lost part of the telomere [104]. The enzyme (telomerase) was subsequently discovered [105]. In yeast, telomerase is always active and at any duplication faithfully restores the length of the DNA molecule. Therefore, yeast cells of both mother and daughter lineage show no telomere length decrease at each replication [106-108]. This indicates that the metabolic alterations and the vulnerability to apoptosis and replicative senescence shown, proportionally to the number of divisions, by the individuals of the mother lineage are due to another mechanism that has been identified.
In yeast mother cells of the wild type, particular molecules, i.e. extrachromosomal ribosomal DNA circles (ERCs), accumulate proportionally to the number of duplications [109] and “several lines of evidence suggest that accumulation of ERCs is one determinant of life span” [100]. In this regard, two yeast mutant types show interesting data. The mutants of dna2-1 type suffer from anomalous DNA replication and so manifest increased rates of ERC accumulation, which causes precocious alterations in gene expression. In short, in mother lineages, young individuals of these mutants have a transcriptome that is similar to those of older individuals of normal yeast [100].
Another type of mutant yeast, tlc1Δ mutants, which is telomerase deficient, show telomere shortening in both mother and daughter cells. Moreover, older individuals belonging to daughter cell lineages, which – as normal strains – have no ERC accumulation, manifest an overall expression of genes, i.e. a transcriptome, similar to that of mother lineage older individuals of normal strains and of mother lineage young individuals of dna2-1 mutants [100].
As we shall see below for multicellular eukaryotes, it is possible that in yeast mutants that are telomerase deficient, as in cells of multicellular eukaryotes with inactive telomerase, the shortening of the telomere causes the sliding of a heterochromatin hood over the telomere, and this interferes with critical parts of subtelomeric DNA. From evidence largely based on experiments in yeast: “One model of telomere–gene expression linkage is an altered chromosomal structure..., such as a heterochromatin “hood” that covers the telomere and a variable length of the subtelomeric chromosome... As the telomere shortens, the hood slides further down the chromosome (the heterochromatin hood remains invariant in size and simply moves with the shortening terminus) or the hood shortens (as the telomere is less capable of retaining heterochromatin). In either case, the result is an alteration of transcription from portions of the chromosome immediately adjacent to the telomeric complex, usually causing transcriptional silencing, although the control is doubtless more complex than merely telomere effect through propinquity… These silenced genes may in turn modulate other, more distant genes (or set of genes). There is some direct evidence for such modulation in the subtelomere…” [110].
While in tlc1Δ yeast mutant, the silencing of subtelomeric DNA could be a consequence of telomere shortening, in non-mutant yeast cells of the mother lineage, silencing could be caused by progressive ERC accumulation that covers and inhibits the subtelomeric region. (For further considerations about gradual subtelomeric silencing and the related metabolic alterations, see the next section, i.e. the subsection “Gradual cell senescence”.)
Regarding the increasing susceptibility to apoptosis and replicative senescence, in proportion to duplication number, a mechanism analogous to that found in multicellular eukaryotes could be proposed (see the subsection “On/off cell senescence”). However, these data raise an important question. A mechanism that causes a differential resistance to apoptosis and therefore establishes a kind of priority list for individual sacrifice in case of need is perfectly consistent with the logic of group selection that appears to favor mass phenoptosis if this is useful for the survival of the deme. On the contrary, the fact that the same mechanism (or something related) progressively impairs cellular metabolism and the vulnerability to replicative senescence, and thus fitness, does not appear to be necessary for the purposes of possible sacrifice and, therefore, is not explained by the abovementioned group selection. We must therefore assume: (i) an obligatory link between increasing vulnerability to apoptosis and progressive metabolic impairment (and likewise the critical importance of the DNA subtelomeric region), (ii) or that there is an evolutionary alternative explanation.
In fact, Büttner et al. already suggested “apoptosis coupled to chronological and replicative aging limits longevity that would maintain ancient genetic variants within the population and, therefore, favor genetic conservatism” [91]. This is not at all a new hypothesis. Well before the acquisition of the above-reported data from yeast, it was proposed that, for species subject to K-selection (see definition below) [111], an age-related fitness decline, i.e. aging, would be adaptive [1, 58]. The original papers should be read, but here a short exposition may be useful. Within any species, the speed of diffusion of any gene G depends both on its advantage (S) over an allele that is assumed neutral and on the generation time, i.e. the reciprocal of the “mean duration of life” (= 1/ML; see Fig. 1). Now, a hypothetical gene C that brings about the untimely death of the individual I, in which C is present, and therefore reduces its ML and determines a disadvantage S′, quickens the diffusion of any advantageous gene G in the individual I′ that replaces I. If I′ is relative to I, C will be increased in its frequency by natural selection, if:
where MLC indicates the ML of individuals having gene C, and MLC′ is the ML of those having a neutral allele C′; Σ(Sx) is the summation of the advantages of n favorable genes G spreading within the species; S′ is the disadvantage of a shorter ML; r indicates the mean coefficient of kinship between I and I′.
Fig. 1. A) Diffusion of a gene G in relation to the value of S; B) diffusion of a gene G in relation to ML variation. An increase/decrease in S or of the inverse in the ML value has the same results on the spreading speed of a gene G within a species (figure redrawn from [1]).
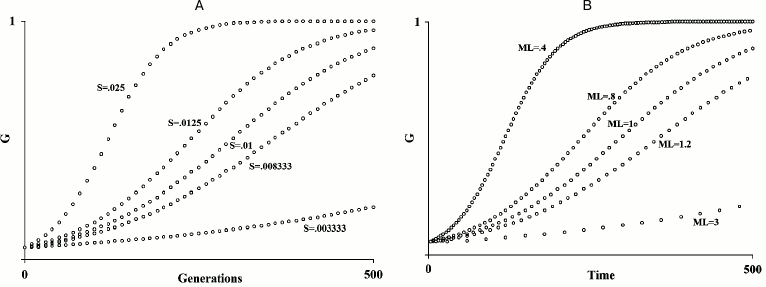
Three brief annotations: (i) formula (6) is another development of the general formula (1) for kin selection; (ii) if G is a harmful gene, gene C accelerates its elimination; (iii) kin selection as an explanation for aging should not be confused with its use to explain the survival of post-reproductive individuals, e.g. as suggested elsewhere [112].
This hypothesis was proposed for multicellular eukaryotes, but for its application to unicellular eukaryotes, there is no opposing theoretical argument against it. Büttner et al. do not propose alternative evolutionary explanations besides the above-mentioned suggestion [91], which may be considered a short enunciation of the hypothesis described [1], in particular if, for yeast, we suppose ecological life conditions of the K-selection type. On the other hand, the above-described adaptive interpretation of aging applied to yeast may be considered within Büttner’s et al. suggestion formulated in terms of kin selection.
Lewis, in his criticism of this hypothesis, argues against the “suggestion that yeast cells provide a precedent for programmed death” [79] proposed by others [113], by the following remark: if a mother lineage yeast cell dies after the 25-35 duplications reported in laboratory conditions [97], the presence of a single individual with the greatest possible number of duplications among 225-235 (= 3.36·107-3.44·1010) descendants appears unlikely and so its death would be insignificant for any adaptive theory of programmed death. However, Lewis’ argument misses the pivotal point: the death at the last possible duplication of a single individual among countless others is a very rare or impossible event, while the increasing and progressive probability of apoptosis – proportionally to the number of duplications – plus the differences in fitness (i.e. mortality rates) and in the capability of having offspring in the comparison between “younger” and “older” individuals, is real: (“in a population of [yeast] cells the lifespan distribution follows the Gompertz law” [101], i.e. an age-related progressive increase of mortality; “The probability that an individual yeast cell will produce daughters declines exponentially as a function of its age in cell divisions or generations (Jazwinski et al., 1998)” [100]). If, in the wild, the death of “older” individuals significantly reduces the ML of yeast and thus causes a quicker generation turnover, Lewis’ argument is trifling for the hypothesis that yeast duplication-related increasing mortality has a favorable selective value. However, Lewis’ argument is very interesting as it echoes an analogous objection against programmed aging hypotheses for multicellular organisms, which will be debated in the next section.
Another possible criticism is that, in yeast, generations follow one another within a few days, a very fast rate when compared with that of species such as ours, and therefore the reduction of a few days of yeast ML might appear irrelevant. However, this objection does not consider that the adaptive aging theory proposed in 1988 [1] (illustrated in Fig. 1), is based on the relative acceleration of the evolution rate and not on the absolute value of it. For example, if the ML of a species passes from a value t to a value t/2, the spreading rate of a gene within a species doubles, both if t = 30 years and if t = 5 days (and if t has any other value).
TRANSITION FROM UNICELLULAR TO MULTICELLULAR EUKARYOTES
The transition from a unicellular and monoclonal eukaryotic deme to a multicellular eukaryotic organism with undifferentiated or minimally differentiated cells likely means a gradual transition and not a drastic break. In a monoclonal deme composed of unicellular individuals, the sacrifice of an individual by apoptosis is clearly a phenoptotic phenomenon explainable in terms of group selection. In a multicellular individual with undifferentiated cells, it is possible to define the sacrifice of some cells, e.g. by apoptosis, as a phenomenon distinct from phenoptosis, but the difference of this sacrifice in comparison with the apoptosis–phenoptosis of an individual in a monoclonal deme of single-cell individuals is small and difficult to define.
As, in multicellular species, the cells of an individual increase their differentiation and, in particular, as a fundamental divide, when reproductive function is entrusted solely to some differentiated cells, apoptosis differs markedly in its meaning from apoptosis–phenoptosis of unicellular organisms and acquires its distinct functions in the context of the more complex organization of a multicellular organism with differentiated cells and organs.
MULTICELLULAR EUKARYOTIC WORLD WITH DIFFERENTIATED CELLS
Multicellular eukaryotes show a series of phenomena that are important for our discussion.
Programmed cell death. In addition to apoptosis, multicellular organisms show various kinds of programmed cell death (PCD), e.g. keratinization of epidermis or hair cells, osteocyte phagocytosis by osteoclasts, detachment of cells from the internal walls of body cavities, and erythrocytes, which are specialized cells that lose their nucleus and are subsequently removed by macrophages.
For the first time apoptosis was described and clearly differentiated from necrosis in multicellular eukaryotic organisms in a study of the normal liver [114]. While necrosis is cell death determined by acute cellular damage, apoptosis can be defined as an ordered form of cell self-destruction. It is ubiquitous in eukaryotic species [56] and, similarly to what occurs in the yeast [93], a cell that dies by apoptosis does not harm other cells; cell fragments are removed by phagocytes in an orderly manner and do not elicit an inflammatory response [115].
PCD by apoptosis, which is selectively triggered at specific times and for specific cells, is indispensable for a series of functions that necessarily represent a development and an adaptation of the original functions in single-celled organisms: morphogenetic mechanisms (neural development in embryo [116], wound healing [117], etc.), lymphocyte selection [118, 119], removal of infected or damaged cells [120, 121], etc.
In vertebrates, apoptosis occurs in many tissues and organs [122-134] and is an essential fact for cell turnover in healthy organs [135-138]. Short telomeres and inactivated telomerase increase the probability of apoptosis [110, 139-142].
Limits to cell duplication capacity and cell turnover. In healthy organ and tissues, to ensure normal cell turnover, continuous cell death by apoptosis and other PCD types must be balanced by substitution with an equal number of cells from the duplication of specific stem cells. Before the 1960s, Nobel laureate Alexis Carrel’s old thesis of unlimited duplication capacity in non-germline cells of multicellular organisms was undisputed [143], but, at the same time, the so-called Hayflick limit of cell duplication capacity had been demonstrated in vitro [144, 145] and then in vivo [146]. This limit is documented for many cell types [147-149], is in inverse relation with the age of the individual [150] and, approximately, in direct relation with the longevity of the species [151]. As mentioned in a previous section, this limit is the consequence of the incomplete duplication of the telomere by polymerase. Telomeres, highly conserved repetitive sequences of DNA [152-154], shorten at each cell duplication [155], but telomerase, if active, elongates telomere with each replication. This explains why germline cells have unlimited duplication capacity [105]. If telomerase is inactive, duplications cause telomere shortening and a cell culture or a tissue shows a reduction in its duplication capacity [156], while a cell with activated telomerase becomes capable of unlimited duplications [157-161].
Telomerase activity is regulated by specific proteins [162] and is active without restrictions in immortal human cell lines [163]. On the contrary, for most cell types, telomerase activity is limited, likely in inverse proportion to the cell turnover rate. In fact, it is well known that the cell turnover rate is quite variable according to cell, tissue, and organ type: “bone has a turnover time of about 10 years in humans” [164] and “the heart is replaced roughly every 4.5 years” [165], but “cells [of the intestinal epithelium] are replaced every three to six days” [164] (for other data about cell turnover rhythms, see [166]). This implies that the various types of stem cells must allow the regulation of telomerase activity that varies enormously according to cell type. Therefore, any limitation in the activity of telomerase must be genetically determined and finely modulated and cannot be the result of insurmountable biochemical restrictions.
In multicellular eukaryotes, in short, the limits of cell replication are determined and modulated by restrictions in telomerase activity. This is different from what happens in yeast, where, as already emphasized, telomerase is always active and the progressive limitation in the ability to duplicate is determined by the accumulation – which is proportional to the number of duplications and happens only in the mother lineage – of particular molecules (ERCs) over the subtelomeric segment.
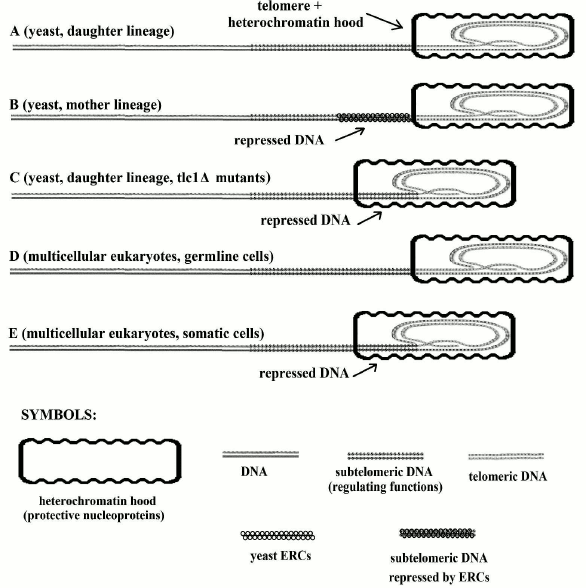
However, it has been also pointed out that for yeast tlc1Δ mutants, which suffer from telomerase inactivation, cells of the daughter lineage, which have no accumulation of ERCs, show telomere shortening and limitations in their ability to duplicate and other alterations such as those caused by ERC accumulation in mother lineage cells (Fig. 2). The similarities between yeast tlc1Δ mutants and the cells of multicellular organisms are impressive and have allowed us to envisage a common phylogenetic relationship.
Fig. 2. In A and D, telomeres are not shortened and the subtelomere (subtelomeric section of DNA molecule) is not repressed. In C and E, telomeres are shortened and the subtelomere is repressed due to protein hood sliding. In B, the telomere is not shortened and the subtelomere is repressed due to binding to ERC molecules (the “symbols” in the first line are used in the subsequent figures). Modified and redrawn from Fig. 6 of [67].
“On/off” cell senescence. In a cell culture, replicative senescence, i.e. the final incapability to cell duplication, was shown to be a progressive reduction in the growth potential of a cell culture, related to telomere length reduction, and not a sudden contemporaneous event for all cells [167, 168].
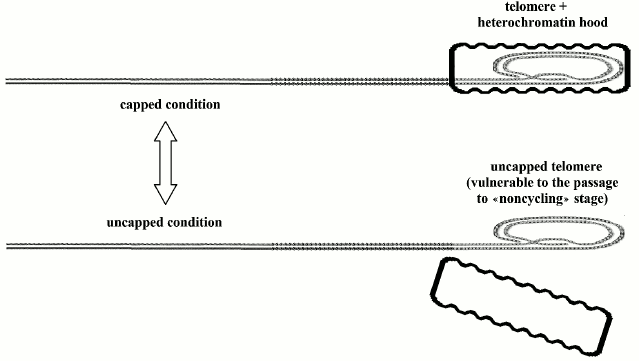
According to Blackburn’s model [169], a protein hood caps the telomere, which oscillates between “uncapped” and “capped” conditions: the first state is susceptible to passage to replicative senescence, i.e. non-cycling conditions, while the duration of the other state is directly related to telomere length (Fig. 3). Even if cells have activated telomerase and maintain telomeres at the maximum length, with each division, a small percentage of them should pass into the non-cycling state [169].
Fig. 3. Telomeres oscillate between the “capped” and “uncapped” conditions. The probability of uncapped telomeres increases in proportion to telomere shortening. A non-protected telomere is a free end of the DNA molecule and is susceptible to end-to-end joining that blocks cell replication.
For a population of cells with inactive telomerase and telomeres at their maximum length, a progressive decline of the replication capacity, proportional to the number of duplications, has been demonstrated. Moreover, stem cells, unlike germ cells, should show levels of telomerase activity only partially able to preserve telomere length [170] and, therefore, they cannot indefinitely replace the elements eliminated by PCD for cell populations in renewal [110].
The absolute length of telomeres is not constantly or strictly related to the life span of a species, e.g. (i) the hamster and the mouse have long telomeres [171], but they age more precociously than humans who have shorter telomeres; (ii) in rodents, there is no relationship between telomerase activity and maximum lifespan [172].
In connection with the mean number of cell duplications in a tissue or a cell culture, there is a growing probability of cell senescence, which has been indicated as a “fundamental cellular program” [173] and is characterized by the modified expression of many genes, in a way that compromises cell functions, and by replicative senescence (i.e. Blackburn’s “noncycling state” [169]). A senescent cell has harmful consequences both on the extracellular matrix and on other cells that are physiologically interdependent or physically nearby. Cell senescence (replicative senescence, its main characteristic, included) certainly derives somehow from relative telomere shortening (Fossel’s “cell senescence limited model”) [110].
“Gradual” cell senescence. Telomere shortening influences the expression of subtelomeric DNA. This phenomenon has been known for some time and has been called the “telomere position effect” [174], but I prefer the definition of “gradual” cell senescence to this quite prudent expression. Apart from the references reported in the section dedicated to unicellular eukaryotes, a recent paper [175] confirms this phenomenon – “Our results demonstrate that the expression of a subset of subtelomeric genes is dependent on the length of telomeres and that widespread changes in gene expression are induced by telomere shortening long before telomeres become rate-limiting for division or before short telomeres initiate DNA damage signaling. These changes include up-regulation and down-regulation of gene expression levels” – and highlights that telomere shortening, by repressing subtelomeric DNA, modifies gene expression even for distant non-subtelomeric parts of DNA. Additionally, the likelihood of a mechanism of this type for multicellular eukaryotes as well is discussed at length by Fossel (see pp. 45-56 in [110]; a scheme of the phenomenon is illustrated in Fig. 4).
Fig. 4. Telomeres shorten at each replication and repress an increasing portion of subtelomeric DNA. This allows for alterations in gene expression in different and distant parts of the DNA molecule.

The capping nucleoproteins of “on/off” senescence [169] and the heterochromatin “hood” of “gradual” cell senescence [110] are very probably the same thing as: (i) they cover necessarily the same end part of the DNA molecule; and (ii) the activation of telomerase and the consequent lengthening of the telomere determine the reversal of all the characteristics of cell senescence [157-161].
Relationship between aging and relative telomere shortening and not with absolute telomere length. For germline cells and for the somatic cells of a donor from which a cloned animal is originated, resetting the telomere clock is indispensable before the first cell duplication [110]. The initial telomere length must be established in the reset phase because, with each following telomere shortening, the probability of cell senescence will increase. In the “reset” phase, the absolute value of the “telomere length is irrelevant” [110]. For example, two murine (Mus) strains, with a telomere length of 10 and 20 kb, respectively, have equal life spans and patterns in the timing of cell senescence; the same has been demonstrated for cloned animals derived from somatic cells with shortened telomeres and their donor animals [110]. In the “reset” phase, an appropriate modeling of the heterochromatin hood based on telomere length could justify the related timing of “gradual” and “on/off” senescence in spite of the different lengths of their telomeres (Fig. 5).
Fig. 5. When the telomere clock is reset, the heterochromatin hood is likely shaped in proportion to the telomere length and should have the same size during all cell life. Replication related telomere shortening determines the sliding of the above-said heterochromatin hood over the subtelomeric DNA with the above-mentioned negative consequences on cell functions and on the equilibrium between the capped/uncapped telomere states. This hypothetical model may explain the irrelevancy of initial telomere length as regards the consequences of its subsequent shortening [110].
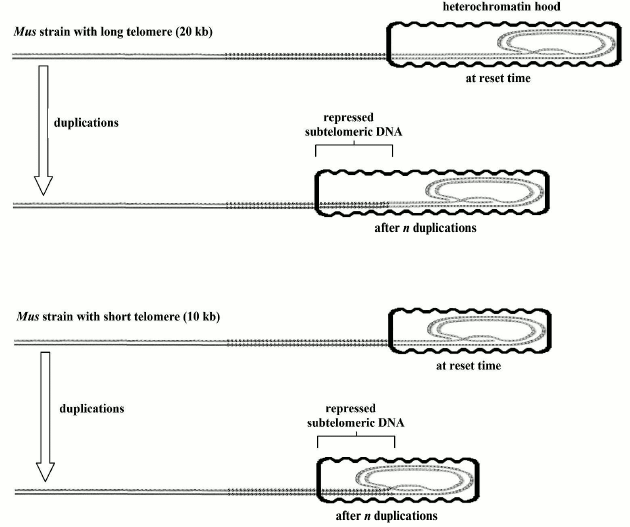
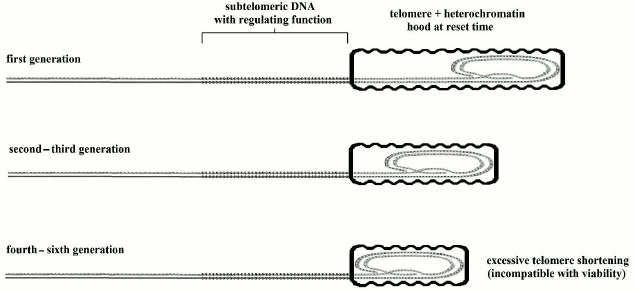
Mice and other animals show a shorter life span, despite much longer telomeres in comparison with our species [171] and a baseline activity of telomerase in most somatic cells [176]. (However, in mice microglia cells, it has been observed that telomeres shorten with age and “the low levels of telomerase activity present may be preferentially recruited to maintain the shortest telomeres while allowing the longer ones to shorten more rapidly” [177].) Moreover, in telomerase knockout (mTR–/–) mice, characterized by genetically inactivated telomerase, we see that only after four [178] to six [179] generations, when telomeres become very shortened, fertility and viability are jeopardized, but in organs with high cell turnover, the dysfunctions appear in early generations [178, 180] (in any case, the fitness reduction caused by this alterations should be considered in relation to wild conditions and not in the artificial protected conditions of the laboratory). The model of Fig. 5, as developed in Fig. 6, could easily explain this apparently paradoxical phenomenon.
Fig. 6. In knockout mice, heterochromatin hood length, which is defined in the reset phase, must be proportional to telomere length. Afterwards, the heterochromatin hood slides over subtelomeric DNA and progressively represses it, but this is not influenced by the length of the hood. If the telomere, in the reset phase, is exceedingly shortened, the mechanism is jeopardized and the viability of the cell is lost.
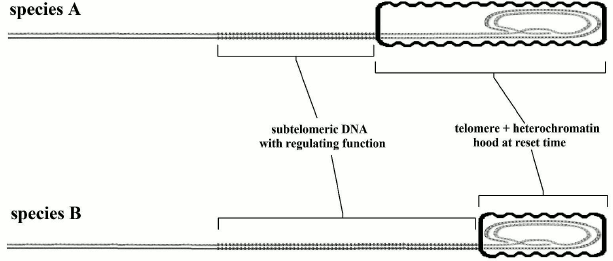
The evidence highlighted before (with its falsely misleading apparent contradictions) leads to an immediate crucial consideration. If (i) the telomere length in the reset phase (if it is not below a critical level) does not affect longevity; (ii) aging is proportional to telomere shortening; (iii) telomere shortening is directly related to the gradual repression of subtelomeric DNA; the trivial conclusion is that for the greater or lesser longevity we should investigate the relationship not with telomere length but with the length and other properties of the subtelomeric segment. This hypothesis, restricted to the length of the subtelomeric region, is illustrated in Fig. 7 and is compatible with the evidence and the arguments expounded above and illustrated in Figs. 2-6. Moreover, we should recall that in yeast, which has a fixed telomere length, aging is caused solely by ERC accumulation over the subtelomeric segment. An implication of this hypothesis is that no necessary correlation between telomere length and longevity is predicted; in fact, a correlation is contradicted by the evidence [171, 172].
Fig. 7. Species A has longer telomeres and shorter subtelomeric segments than species B. For species A, telomere shortening causes a greater relative impairment of the subtelomeric area and this should result in earlier aging.
In short, subtelomeric DNA has been shown to have both essential importance for cell functions and a position vulnerable to repression by telomere shortening itself. If we exclude inexplicable evolutionary contradictions, this coincidence may be justified only as something favored by selection to determine “gradual” and “on/off” senescence. A likely interpretation of the evolution of the system that includes telomeres, telomerase, subtelomeric DNA, and cell senescence is proposed below.
The suggested and very probable cause for age-related fitness decline is the progressive slowing of cell turnover, which means a progressive prevalence of PCD on cell substitution by stem cell duplication (Fossel’s “cell senescence general model of aging” [58, 110]) coupled with an increasing fraction of cells more or less altered by “gradual” and “on/off” cell senescence [69, 110]. In support of this hypothesis, in some species (rockfish and lobsters), both mortality rate and telomere length do not vary with the age [181, 182].
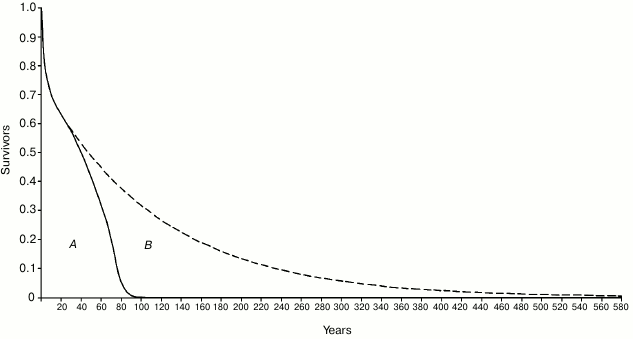
There is evidence for an evolutionary advantage of the age-related mortality increase phenomenon [14, 67], which in its more advanced manifestations, common in artificially protected conditions, is usually defined as “aging”, a term that is inaccurate in this context [58]. A theory, identical to that mentioned above to explain “on/off” and “gradual” cell senescence in yeast, justifies this fitness decline, or mortality increase, as evolutionarily advantageous, in terms of supra-individual selection, by a mechanism based on kin selection that, due to a faster generation turnover, allows a quicker diffusion of any advantageous mutations. In accordance with the theory, this advantage exists only when there is K-selection (i.e. species divided into demes that are composed of kin individuals and living in saturated habitats where only the disappearance of an individual allows the existence of a new individual) [1, 58]. An objection against this hypothesis is that “As a rule, wild animals simply do not live long enough to grow old. Therefore, natural selection has limited opportunity to exert a direct influence over the process of senescence” [11]. This criticism, analogous to Lewis’ argument for yeast and as mentioned above, misses an essential point: the absence in wild conditions of “old” individuals (e.g. for lions (Panthera leo), individuals that are older than 15 years) is irrelevant. Individuals of P. leo with an age below 15 years are “not old” individuals according to Kirkwood and Austad’s concept [11], yet they show increasing mortality at ages existing under natural conditions: this significantly reduces the ML with a consequent quicker generation turnover and the proposed selective advantage. “Senescence reduces average life span... by almost 80% when m0 = 0.01 year–1” [8]. For the individuals that survived the high mortality of the first life phases, in eight mammal species studied in the wild, the ratio between ML with age-related increasing mortality (wild condition) and ML without the mortality increase (hypothetical condition) has been calculated to be in the range of 2.5-5. If we do not disregard the individuals that died in the first life phases, the ratio has been shown to be in the range of 1.55-3.21 [1]. Moreover, the study of a human population under wild conditions [10, 64] has shown that: (i) survival for 60- and 70-year-old individuals was about 30 and nearly 20%, respectively, and (ii) the proportion of ML reduction was considerable and, so, undoubtedly subjected to natural selection (Fig. 8).
Fig. 8. Area A (delimited by the continuous line): life table of Ache in the wild, data from Hill and Hurtado [10] (the figure was modified and reprinted with permission from [64]). Area B (delimited by the dashed line): hypothetical life table without age-related increasing mortality. The proportion of senescent death (Ps, as defined by Ricklefs [8]) is given by the ratio between B and A + B.
In short, in the wild, ML reduction determined by age-related fitness decline is not at all negligible, although the equivalents of centenarians or older individuals for animals are most likely non-existent under wild conditions.
CONCLUSION
The main points of the above-mentioned evidence and arguments can be summarized as follows.
1. Prokaryote mass phenoptosis by proapoptosis and unicellular eukaryote mass phenoptosis by apoptosis, in particular cases, are favored by similar supra-individual selective mechanisms and likely have a common phylogenetic origin [77].
2. For both types of phenomena, a mechanism that activates the suicide pattern is indispensable only in a fraction of the population and should be proportional to necessity (e.g. the scarcity of a critical resource). Yeast have evolved an effective mechanism depending on the number of preceding duplications and on ERC accumulation over subtelomeric DNA that creates a kind of ranking for individuals destined to sacrifice themselves [91, 93, 98, 101].
3. Apoptosis in multicellular eukaryotic species has an evident phylogenetic relationship with unicellular eukaryotic apoptosis [183] but the evolved clock is based not on the accumulation of ERCs but on telomere shortening due to restrictions in telomerase activity analogously to the case of yeast tlc1Δ mutants in which telomerase is inactive [110]. In both cases, the damaging effects on the cell are caused by the progressive repression of subtelomeric DNA, which is clearly a pivotal aspect of aging mechanisms.
4. In multicellular eukaryote organisms with undifferentiated cell functions, if we consider each individual as a clone that has all cells with the same genes, apoptosis of a cell may be regarded as caused by mechanisms analogous to kin selection. In multicellular eukaryote organisms, as cells assume different functions and, in particular, with the limitations of reproductive function to some specific cells, this interpretation is inappropriate and the multicellular individual is subjected to natural selection as a single entity.
5. In multicellular eukaryote organisms, apoptosis assumes a long series of functions (morphogenetic mechanisms, lymphocyte selection, removal of damaged or infected cells, etc.), which are unquestionably a derived function, as they are impossible in unicellular organisms.
6. In yeast, apoptosis is essential for the phenomenon summarized in point (1), but, moreover, the phenomena defined as “on/off” and “gradual” senescence cause a duplication-related progressive fitness decline, which may be defined “aging”. This appears to contrast “genetic conservatism” [91], and might be explained in the same way as aging in multicellular organisms [1, 58] (see point (7)).
7. In eukaryotic multicellular organisms, PCD (by apoptosis and other types of PCD), “on/off” senescence, “gradual” senescence, and limits to cell duplication capacity appear to be elements of a highly sophisticated system that is essential both for cell turnover (i.e. continuous renewal of the organism) and for age-related progressive fitness decline [58, 110]. This decline, i.e. aging, could be explained as a powerful system to accelerate evolution by means of supra-individual selection, which is possible only under conditions of K-selection [1, 58].
8. Aging in yeast has been proposed as adaptive [91]. For multicellular eukaryotes, aging as an adaptive phenomenon is excluded by the prevailing gerontological paradigm [11], but this is contrasted by both empirical evidence and theoretical arguments [1, 14, 56, 58, 67, 95, 184, 185].
9. The limits to cell duplication capacity determined by the telomere–telomerase system are currently justified as a general defensive mechanism against cancer [186-189], but evidence and strong arguments contradict this hypothesis [14, 64, 110, 190, 191], e.g. the secretion by senescent cells of substances that increase both mutation rates and oncogenic risk [192, 193]. The obstinate affection by the advocates of non-adaptive aging theory to the hypothetical anti-cancer role of telomerase restrictions is likely caused by the absence of any explanation compatible with the non-adaptive hypotheses and because of philosophical bias [192]. “The hypothesis that telomerase is restricted to achieve a net increase in lifespan via cancer prevention is certainly false. Were it not for the unthinkability of the alternative – programmed death – the theory would be dead in the water” [190].
In short, aging shows clear phylogenetic connections regarding both underlying physiological mechanisms, and evolutionary causes. This represents a further set of arguments and evidence in support of the paradigm that interprets aging as a phenoptotic phenomenon, i.e. a type of death that is genetically determined and modulated with specific evolutionary causes.
REFERENCES
1.Libertini, G. (1988) An adaptive theory of the
increasing mortality with increasing chronological age in populations
in the wild, J. Theor. Biol., 132, 145-162.
2.Deevey, E. S., Jr. (1947) Life tables for natural
populations of animals, Quart. Rev. Biol., 22,
283-314.
3.Laws, R. M., and Parker, I. S. (1968) Recent
studies on elephant populations in East Africa, Symp. Zool. Soc.
Lond., 21, 319-359.
4.Spinage, C. A. (1970) Population dynamics of the
Uganda Defassa Waterbuck (Kobus defassa Ugandae Neumann) in the
Queen Elizabeth park, Uganda, J. Anim. Ecol., 39,
51-78.
5.Spinage, C. A. (1972) African ungulate life tables,
Ecology, 53, 645-652.
6.Finch, C. E. (1990) Longevity, Senescence, and
the Genome, The University of Chicago Press, Chicago.
7.Holmes, D. J., and Austad, S. N. (1995) Birds as
animal models for the comparative biology of aging: a prospectus, J.
Gerontol. A Biol. Sci., 50, 59-66.
8.Ricklefs, R. E. (1998) Evolutionary theories of
aging: confirmation of a fundamental prediction, with implications for
the genetic basis and evolution of life span, Am. Nat.,
152, 24-44.
9.Nussey, D. H., Froy, H., Lemaitre, J. F., Gaillard,
J. M., and Austad, S. N. (2013) Senescence in natural populations of
animals: widespread evidence and its implications for bio-gerontology,
Ageing Res. Rev., 12, 214-225.
10.Hill, K., and Hurtado, A. M. (1966) Ache Life
History, Aldine De Gruyter, New York.
11.Kirkwood, T. B., and Austad, S. N. (2000) Why do
we age? Nature, 408, 233-238.
12.Martin, G. M., and Oshima, J. (2000) Lessons from
human progeroid syndromes, Nature, 408, 263-266.
13.Kirkwood, T. B. (2005) Understanding the odd
science of aging, Cell, 120, 437-447.
14.Libertini, G. (2008) Empirical evidence for
various evolutionary hypotheses on species demonstrating increasing
mortality with increasing chronological age in the wild, Sci. World
J., 8, 182-193.
15.Kuhn, T. S. (1962) The Structure of Scientific
Revolutions, The University of Chicago Press, Chicago.
16.Minot, C. S. (1907) The Problem of Age,
Growth, and Death; A Study of Cytomorphosis, Based on lectures at
the Lowell Institute, London.
17.Carrel, A., and Ebeling, A. H. (1921)
Antagonistic growth principles of serum and their relation to old age,
J. Exp. Med., 38, 419-425.
18.Brody, S. (1924) The kinetics of senescence,
J. Gen. Physiol., 6, 245-257.
19.Bidder, G. P. (1932) Senescence, Br. Med.
J., 115, 5831-5850.
20.Lansing, A. I. (1948) Evidence for aging as a
consequence of growth cessation, Proc. Natl. Acad. Sci. USA,
34, 304-310.
21.Lansing, A. I. (1951) Some physiological aspects
of ageing, Physiol. Rev., 31, 274-284.
22.Medawar, P. B. (1952) An Unsolved Problem in
Biology, H. K. Lewis, London; reprinted as Medawar, P. B. (1957)
The Uniqueness of the Individual, Methuen, London.
23.Williams, G. C. (1957) Pleiotropy, natural
selection, and the evolution of senescence, Evolution,
11, 398-411.
24.Hamilton, W. D. (1966) The moulding of senescence
by natural selection, J. Theor. Biol., 12, 12-45.
25.Edney, E. B., and Gill, R. W. (1968) Evolution of
senescence and specific longevity, Nature, 220,
281-282.
26.Harman, D. (1972) The biologic clock: the
mitochondria? J. Am. Geriatr. Soc., 20, 145-147.
27.Kirkwood, T. B. (1977) Evolution of ageing,
Nature, 270, 301-304.
28.Comfort, A. (1979) The Biology of
Senescence, Elsevier North Holland, New York.
29.Kirkwood, T. B., and Holliday, R. (1979) The
evolution of ageing and longevity, Proc. R. Soc. Lond. B Biol.
Sci., 205, 531-546.
30.Miquel, J., Economos, A. C., Fleming, J., and
Johnson, J. E., Jr. (1980) Mitochondrial role in cell aging, Exp.
Gerontol., 15, 575-591.
31.Mueller, L. D. (1987) Evolution of accelerated
senescence in laboratory populations of drosophila, Proc. Natl.
Acad. Sci. USA, 84, 1974-1977.
32.Rose, M. R. (1991) Evolutionary Biology of
Aging, Oxford University Press, New York.
33.Partridge, L., and Barton, N. H. (1993)
Optimality, mutation, and the evolution of ageing, Nature,
362, 305-311.
34.Bohr, V. A., and Anson, R. M. (1995) DNA damage,
mutation, and fine structure DNA repair in aging, Mutat. Res.,
338, 25-34.
35.Croteau, D. L., and Bohr, V. A. (1997) Repair of
oxidative damage to nuclear and mitochondrial DNA in mammalian cells,
J. Biol. Chem., 272, 25409-25412.
36.Beckman, K. B., and Ames, B. N. (1998) The free
radical theory of aging matures, Physiol. Rev., 78,
547-581.
37.Weinert, B. T., and Timiras, P. S. (2003) Invited
review: theories of aging, J. Appl. Physiol., 95,
1706-1716.
38.Trifunovic, A., Wredenberg, A., Falkenberg, M.,
Spelbrink, J. N., Rovio, A. T., Bruder, C. E., Bohlooly, Y, M., Gidlof,
S., Oldfors, A., Wibom, R., Tornell, J., Jacobs, H. T., and Larsson, N.
G. (2004) Premature ageing in mice expressing defective mitochondrial
DNA polymerase, Nature, 429, 417-423.
39.Balaban, R. S., Nemoto, S., and Finkel, T. (2005)
Mitochondria, oxidants, and aging, Cell, 120,
483-495.
40.Blagosklonny, M. V. (2006) Aging and immortality:
quasi-programmed senescence and its pharmacologic inhibition, Cell
Cycle, 5, 2087-2102.
41.Sanz, A., and Stefanatos, R. K. (2008) The
mitochondrial free radical theory of aging: a critical view, Curr.
Aging Sci., 1, 10-21.
42.Oliveira, B. F., Nogueira-Machado, J.-A., and
Chaves, M. M. (2010) The role of oxidative stress in the aging process,
Sci. World J., 10, 1121-1128.
43.Blagosklonny, M. V. (2013) MTOR-driven
quasi-programmed aging as a disposable soma theory: blind watchmaker
vs. intelligent designer, Cell Cycle, 12, 1842-1847.
44.Weismann, A. (1889) Essays upon Heredity and
Kindred Biological Problems, Vol. I, Clarendon Press, Oxford.
45.Weismann, A. (1892) Essays upon Heredity and
Kindred Biological Problems, Vol. II, Clarendon Press, Oxford.
46.Kirkwood, T. B. L., and Cremer, T. (1982)
Cytogerontology since 1881: a reappraisal of August Weissmann and a
review of modern progress, Hum. Genet., 60, 101-121.
47.Libertini, G. (2011) Evolutionary Arguments on
Aging, Disease, and Other Topics, Azinet Press, Crownsville.
48.Skulachev, V. P. (1997) Aging is a specific
biological function rather than the result of a disorder in complex
living systems: biochemical evidence in support of Weismann’s
hypothesis, Biochemistry (Moscow), 62, 1191-1195.
49.Skulachev, V. P. (1999) Phenoptosis: programmed
death of an organism, Biochemistry (Moscow), 64,
1418-1426.
50.Skulachev, V. P. (1999) Mitochondrial physiology
and pathology; concepts of programmed death of organelles, cells, and
organisms, Mol. Aspects Med., 20, 139-184.
51.Skulachev, V. P. (2001) The programmed death
phenomena, aging, and the Samurai law of biology, Exp.
Gerontol., 36, 995-1024.
52.Bredesen, D. E. (2004) The non-existent aging
program: how does it work? Aging Cell, 3, 255-259.
53.Goldsmith, T. C. (2004) Aging as an evolved
characteristic – Weismann’s theory reconsidered, Med.
Hypotheses, 62, 304-308.
54.Mitteldorf, J. (2004) Aging selected for its own
sake, Evol. Ecol. Res., 6, 1-17.
55.Travis, J. M. (2004) The evolution of programmed
death in a spatially structured population, J. Gerontol. A Biol.
Sci. Med. Sci., 59, 301-305.
56.Longo, V. D., Mitteldorf, J., and Skulachev, V.
P. (2005) Programmed and altruistic ageing, Nat. Rev. Genet.,
6, 866-872.
57.Skulachev, V. P., and Longo, V. D. (2005) Aging
as a mitochondria-mediated atavistic program: can aging be switched
off? Ann. N. Y. Acad. Sci., 1057, 145-164.
58.Libertini, G. (2006) Evolutionary explanations of
the “actuarial senescence in the wild” and of the
“state of senility”, Sci. World J., 6,
1086-1108.
59.Goldsmith, T. C. (2008) Aging, evolvability, and
the individual benefit requirement; medical implications of aging
theory controversies, J. Theor. Biol., 252, 764-768.
60.Libertini, G. (2009) The role of
telomere-telomerase system in age-related fitness decline, a tameable
process, in Telomeres: Function, Shortening, and Lengthening
(Mancini, L., ed.) Nova Science Publishers, New York, pp. 77-132.
61.Mitteldorf, J., and Pepper, J. (2009) Senescence
as an adaptation to limit the spread of disease, J. Theor.
Biol., 260, 186-195.
62.Martins, A. C. (2011) Change and aging senescence
as an adaptation, PLoS One, 6, e24328.
63.Libertini, G. (2012) Classification of
phenoptotic phenomena, Biochemistry (Moscow), 77,
707-715.
64.Libertini, G. (2013) Evidence for aging theories
from the study of a hunter-gatherer people (Ache of Paraguay),
Biochemistry (Moscow), 78, 1023-1032.
65.Mitteldorf, J., and Martins, A. C. (2014)
Programmed life span in the context of evolvability, Am. Nat.,
184, 289-302.
66.Skulachev, V. P. (2002) Programmed death
phenomena: from organelle to organism, Ann. N. Y. Acad. Sci.,
959, 214-237.
67.Libertini, G. (2015) Non-programmed versus
programmed aging paradigm, Curr. Aging Sci., 8, in
press.
68.Libertini, G. (2009) Prospects of a longer life
span beyond the beneficial effects of a healthy lifestyle, in:
Handbook on Longevity: Genetics, Diet, and Disease (Bentely, J.
V., and Keller, M., eds.) Nova Science Publishers Inc., New York, pp.
35-96.
69.Libertini, G. (2014) Programmed aging paradigm:
how we get old, Biochemisry (Moscow), 79, 1004-1016.
70.Loisel, D. A., Alberts, S. C., and Ober, C.
(2008) Functional significance of MHC variation in mate choice,
reproductive outcome, and disease risk, in Evolution in Health and
Disease (Stearns, S. C., and Koella, J. C., eds.) 2nd Edn., Oxford
University Press, Oxford.
71.Apanius, V., Penn, D., Slev, P. R., Ruff, L. R.,
and Potts, W. K. (1997) Crit. Rev. Immunol., 17,
179-224.
72.Raff, M. C. (1998) Cell suicide for beginners,
Nature, 396, 119-122.
73.Hausfater, G., and Hrdy, S. B. (1984)
Infanticide: Comparative and Evolutionary Perspectives, Aldine,
New York.
74.Skulachev, V. P. (2003) Aging and the programmed
death phenomena, in Topics in Current Genetics (Nystrom, T., and
Osiewacz, H. D., eds.) Vol. 3, Model Systems in Aging,
Springer-Verlag, Berlin, Heidelberg.
75.Lane, N. (2008) Marine microbiology: origins of
death, Nature, 453, 583-585.
76.Engelberg-Kulka, H., Sat, B., Reches, M., Amitai,
S., and Hazan, R. (2004) Bacterial programmed cell death systems as
targets for antibiotics, Trends Microbiol., 12,
66-71.
77.Hochman, A. (1997) Programmed cell death in
prokaryotes, Crit. Rev. Microbiol., 23, 207-214.
78.Koonin, E. V., and Aravind, L. (2002) Origin and
evolution of eukaryotic apoptosis: the bacterial connection, Cell
Death Differ., 9, 394-404.
79.Lewis, K. (2000) Programmed death in bacteria,
Microbiol. Mol. Biol. Rev., 64, 503-514.
80.Maynard Smith, J. (1964) Group selection and kin
selection, Nature, 201, 1145-1147.
81.Maynard Smit, J. (1976) Group selection,
Quart. Rev. Biol., 51, 277-283.
82.Hamilton, W. D. (1964) The genetical evolution of
social behaviour, I, II, J. Theor. Biol., 7, 1-52.
83.Hamilton, W. D. (1970) Selfish and spiteful
behaviour in an evolutionary model, Nature, 228,
1218-1220.
84.Trivers, R. L. (1971) The evolution of reciprocal
altruism, Quart. Rev. Biol., 46, 35-57.
85.Trivers, R. L., and Hare, H. (1976)
Haploidiploidy and the evolution of the social insect, Science,
191, 249-263.
86.Madeo, F., Frohlich, E., and Frohlich, K. U.
(1997) A yeast mutant showing diagnostic markers of early and late
apoptosis, J. Cell Biol., 139, 729-734.
87.Ligr, M., Madeo, F., Frohlich, E., Hilt, W.,
Frohlich, K. U., and Wolf, D. H. (1998) Mammalian Bax triggers
apoptotic changes in yeast, FEBS Lett., 438, 61-65.
88.Longo, V. D., Ellerby, L. M., Bredesen, D. E.,
Valentine, J. S., and Gralla, E. B. (1997) Human Bcl-2 reverses
survival defects in yeast lacking superoxide dismutase and delays death
of wild-type yeast, J. Cell Biol., 137, 1581-1588.
89.Kaeberlein, M., Burtner, C. R., and Kennedy, B.
K. (2007) Recent developments in yeast aging, PLoS Genet.,
3, e84.
90.Madeo, F., Frohlich, E., Ligr, M., Grey, M.,
Sigrist, S. J., Wolf, D. H., and Frohlich, K. U. (1999) Oxygen stress:
a regulator of apoptosis in yeast, J. Cell Biol., 145,
757-767.
91.Büttner, S., Eisenberg, T., Herker, E.,
Carmona-Gutierrez, D., Kroemer, G., and Madeo, F. (2006) Why yeast
cells can undergo apoptosis: death in times of peace, love, and war,
J. Cell Biol., 175, 521-525.
92.Granot, D., Levine, A., and Dor-Hefetz, E. (2003)
Sugar-induced apoptosis in yeast cells, FEMS Yeast Res.,
4, 7-13.
93.Herker, E., Jungwirth, H., Lehmann, K. A.,
Maldener, C., Frohlich, K. U., Wissing, S., Büttner, S., Fehr, M.,
Sigrist, S., and Madeo, F. (2004) Chronological aging leads to
apoptosis in yeast, J. Cell Biol., 164, 501-507.
94.Fabrizio, P., Battistella, L., Vardavas, R.,
Gattazzo, C., Liou, L. L., Diaspro, A., Dossen, J. W., Gralla, E. B.,
and Longo, V. D. (2004) Superoxide is a mediator of an altruistic aging
program in Saccharomyces cerevisiae, J. Cell Biol.,
166, 1055-1067.
95.Mitteldorf, J. (2006) How evolutionary thinking
affects people’s ideas about aging interventions, Rejuvenation
Res., 9, 346-350.
96.Skulachev, V. P. (2002) Programmed death in yeast
as adaptation? FEBS Lett., 528, 23-26.
97.Jazwinski, S. M. (1993) The genetics of aging in
the yeast Saccharomyces cerevisiae, Genetica, 91,
35-51.
98.Fabrizio, P., and Longo, V. D. (2008)
Chronological aging-induced apoptosis in yeast, Biochim. Biophys.
Acta, 1783, 1280-1285.
99.Laun, P., Pichova, A., Madeo, F., Fuchs, J.,
Ellinger, A., Kohlwein, S., Dawes, I., Frohlich, K.-U., and
Breitenbach, M. (2001) Aged mother cells of Saccharomyces
cerevisiae show markers of oxidative stress and apoptosis, Mol.
Microbiol., 39, 1166-1173.
100.Lesur, I., and Campbell, J. L. (2004) The
transcriptome of prematurely aging yeast cells is similar to that of
telomerase-deficient cells, MBC Online, 15,
1297-1312.
101.Laun, P., Bruschi, C. V., Dickinson, J. R.,
Rinnerthaler, M., Heeren, G., Schwimbersky, R., Rid, R., and
Breitenbach, M. (2007) Yeast mother cell-specific ageing, genetic
(in)stability, and the somatic mutation theory of ageing, Nucleic
Acids Res., 35, 7514-7526.
102.Olovnikov, A. M. (1971) Principle of
marginotomy in template synthesis of polynucleotides, Dokl. Akad.
Nauk SSSR, 201, 1496-1499.
103.Watson, J. D. (1972) Origin of concatemeric T7
DNA, Nat. New Biol., 239, 197-201.
104.Olovnikov, A. M. (1973) A theory of
marginotomy: the incomplete copying of template margin in enzyme
synthesis of polynucleotides and biological significance of the
problem, J. Theor. Biol., 41, 181-190.
105.Greider, C. W., and Blackburn, E. H. (1985)
Identification of a specific telomere terminal transferase activity in
Tetrahymena extracts, Cell, 51, 405-413.
106.D’Mello, N. P., and Jazwinski, S. M.
(1991) Telomere length constancy during aging of Saccharomyces
cerevisiae, J. Bacteriol., 173, 6709-6713.
107.Smeal, T., Claus, J., Kennedy, B., Cole, F.,
and Guarente, L. (1996) Loss of transcriptional silencing causes
sterility in old mother cells of Saccharomyces cerevisiae,
Cell, 84, 633-642.
108.Maringele, L., and Lydall, D. (2004)
Telomerase- and recombination-independent immortalization of budding
yeast, Genes Dev., 18, 2663-2675.
109.Sinclair, D. A., and Guarente, L. (1997)
Extrachromosomal rDNA circles – cause of aging in yeast,
Cell, 91, 1033-1042.
110.Fossel, M. B. (2004) Cells, Aging and Human
Disease, Oxford University Press, New York.
111.Pianka, E. R. (1970) On r- and K-selection,
Am. Nat., 104, 592-597.
112.Lee, R. (2008) Sociality, selection, and
survival: simulated evolution of mortality with intergenerational
transfers and food sharing, Proc. Natl. Acad. Sci. USA,
105, 7124-7128.
113.Sinclair, D., Mills, K., and Guarente, L.
(1998) Aging in Saccharomyces cerevisiae, Annu. Rev.
Microbiol., 52, 533-560.
114.Kerr, J. F. R., Wyllie, A. H., and Currie, A.
R. (1972) Apoptosis: a basic biological phenomenon with wide-ranging
implications in tissue kinetics, Br. J. Cancer, 26,
239-257.
115.Erwig, L.-P., and Henson, P. M. (2008)
Clearance of apoptotic cells by phagocytes, Cell Death Differ.,
15, 243-250.
116.Nijhawan, D., Honarpour, N., and Wang, X.
(2000) Apoptosis in neural development and disease, Annu. Rev.
Neurosci., 23, 73-87.
117.Greenhalgh, D. G. (1998) The role of apoptosis
in wound healing, Int. J. Biochem. Cell Biol., 30,
1019-1030.
118.Cohen, J. J. (1993) Programmed cell death and
apoptosis in lymphocyte development and function, Chest,
103, 99-101.
119.Opferman, J. T. (2008) Apoptosis in the
development of the immune system, Cell Death Differ., 15,
234-242.
120.Tesfaigzi, Y. (2006) Roles of apoptosis in
airway epithelia, Am. J. Respir. Cell Mol. Biol., 34,
537-547.
121.White, E. (2006) Mechanisms of apoptosis
regulation by viral oncogenes in infection and tumorigenesis, Cell
Death Differ., 13, 1371-1377.
122.Pontèn, J., Stein, W. D., and Shall, S.
(1983) A quantitative analysis of the aging of human glial cells in
culture, Cell Phys., 117, 342-352.
123.Harada, K., Iwata, M., Kono, N., Koda, W.,
Shimonishi, T., and Nakanuma, Y. (2000) Distribution of apoptotic cells
and expression of apoptosis-related proteins along the intrahepatic
biliary tree in normal and non-biliary diseased liver,
Histopathology, 37, 347-354.
124.Cardani, R., and Zavanella, T. (2000)
Age-related cell proliferation and apoptosis in the kidney of male
Fischer 344 rats with observations on a spontaneous tubular cell
adenoma, Toxicol. Pathol., 28, 802-806.
125.Finegood, D. T., Scaglia, L., and Bonner-Weir,
S. (1995) Dynamics of β-cell mass in the growing rat pancreas.
Estimation with a simple mathematical model, Diabetes,
44, 249-256.
126.Benedetti, A., Jezequel, A. M., and Orlandi, F.
(1988) A quantitative evaluation of apoptotic bodies in rat liver,
Liver, 8, 172-177.
127.Dremier, S., Golstein, J., Mosselmans, R.,
Dumont, J. E., Galand, P., and Robaye, B. (1994) Apoptosis in dog
thyroid cells, Biochem. Biophys. Res. Commun., 200,
52-58.
128.Sutherland, L. M., Edwards, Y. S., and Murray,
A. W. (2001) Alveolar type II cell apoptosis, Comp. Biochem.
Physiol., 129, 267-285.
129.Heraud, F., Heraud, A., and Harmand, M. F.
(2000) Apoptosis in normal and osteoarthritic human articular
cartilage, Ann. Rheum. Dis., 59, 959-965.
130.Xia, S. J., Xu, C. X., Tang, X. D., Wang, W. Z,
and Du, D. L. (2001) Apoptosis and hormonal milieu in ductal system of
normal prostate and benign prostatic hyperplasia, Asian J.
Androl., 3, 131-134.
131.Prins, J. B., and O’Rahilly, S. (1997)
Regulation of adipose cell number in man, Clin. Sci. (London),
92, 3-11.
132.Spelsberg, T. C., Subramaniam, M., Riggs, B.
L., and Khosla, S. (1999) The actions and interactions of sex steroids
and growth factors/cytokines on the skeleton, Mol. Endocrinol.,
13, 819-828.
133.Migheli, A., Mongini, T., Doriguzzi, C.,
Chiado-Piat, L., Piva, R., Ugo, I., and Palmucci, L. (1997) Muscle
apoptosis in humans occurs in normal and denervated muscle, but not in
myotonic dystrophy, dystrophinopathies or inflammatory disease,
Neurogenetics, 1, 81-87.
134.Pollack, M., and Leeuwenburgh, C. (2001)
Apoptosis and aging: role of the mitochondria, J. Gerontol. A Biol.
Sci. Med. Sci., 56, 475-482.
135.Israels, L. G., and Israels, E. D. (1999)
Apoptosis, Stem Cells, 17, 306-313.
136.Lynch, M. P., Nawaz, S., and Gerschenson, L. E.
(1986) Evidence for soluble factors regulating cell death and cell
proliferation in primary cultures of rabbit endometrial cells grown on
collagen, Proc. Natl. Acad. Sci. USA, 83, 4784-4788.
137.Medh, R. D., and Thompson, E. B. (2000)
Hormonal regulation of physiological cell turnover and apoptosis,
Cell Tissue Res., 301, 101-124.
138.Wyllie, A. H., Kerr, J. F. R., and Currie, A.
R. (1980) Cell death: the significance of apoptosis, Int. Rev.
Cytol., 68, 251-306.
139.Ozen, M., Imam, S. A., Datar, R. H., Multani,
A. S., Narayanan, R., Chung, L. W., Von Eschenbach, A. C., and Pathak,
S. (1998) Telomeric DNA: marker for human prostate cancer development?
Prostate, 36, 264-271.
140.Holt, S. E., Glinsky, V. V., Ivanova, A. B.,
and Glinsky, G. V. (1999) Resistance to apoptosis in human cells
conferred by telomerase function and telomere stability, Mol.
Carcinog., 25, 241-248.
141.Seimiya, H., Tanji, M., Oh-hara, T., Tomida,
A., Naasani, I., and Tsuruo, T. (1999) Hypoxia up-regulates telomerase
activity via mitogen-activated protein kinase signaling in human solid
tumor cells, Biochem. Biophys. Res. Commun., 260,
365-370.
142.Ren, J. G., Xia, H. L., Tian, Y. M., Just, T.,
Cai, G. P., and Dai, Y. R. (2001) Expression of telomerase inhibits
hydroxyl radical-induced apoptosis in normal telomerase negative human
lung fibroblasts, FEBS Lett., 488, 133-138.
143.Carrel, A., and Ebeling, A. H. (1921) Age and
multiplication of fibroblasts, J. Exp. Med., 34,
599-623.
144.Hayflick, L., and Moorhead, P. S. (1961) The
serial cultivation of human diploid cell strains, Exp. Cell
Res., 25, 585-621.
145.Hayflick, L. (1965) The limited in vitro
lifetime of human diploid cell strains, Exp. Cell Res.,
37, 614-636.
146.Schneider, E. L., and Mitsui, Y. (1976) The
relationship between in vitro cellular aging and in vivo
human age, Proc. Natl. Acad. Sci. USA, 73, 3584-3588.
147.Rheinwald, J. G., and Green, H. (1975) Serial
cultivation of strains of human epidermal keratinocytes: the formation
of keratinizing colonies from single cells, Cell, 6,
331-344.
148.Bierman, E. L. (1978) The effect of donor age
on the in vitro life span of cultured human arterial
smooth-muscle cells, In vitro, 14, 951-955.
149.Tassin, J., Malaise, E., and Courtois, Y.
(1979) Human lens cells have an in vitro proliferative capacity
inversely proportional to the donor age, Exp. Cell Res.,
123, 388-392.
150.Martin, G. M., Sprague, C. A., and Epstein, C.
J. (1970) Replicative life span of cultivated human cells. Effects of
donor’s age, tissue, and genotype, Lab. Invest.,
23, 86-92.
151.Rohme, D. (1981) Evidence for a relationship
between longevity of mammalian species and life spans of normal
fibroblasts in vitro and erythrocytes in vivo, Proc.
Natl. Acad. Sci. USA, 78, 5009-5013.
152.Blackburn, E. H., and Gall, J. G. (1978) A
tandemly repeated sequence at the termini of the extrachromosomal
ribosomal RNA genes in Tetrahymena, J. Mol. Biol.,
120, 33-53.
153.Moyzis, R. K., Buckingham, J. M., Cram, L. S.,
Dani, M., Deaven, L. L., Jones, M. D., Meyne, J., Ratliff, R. L., and
Wu, J. R. (1988) A highly conserved repetitive DNA sequence, (TTAGGG)n,
present at the telomeres of human chromosomes, Proc. Natl. Acad.
Sci. USA, 85, 6622-6626.
154.Blackburn, E. H. (1991) Structure and function
of telomeres, Nature, 350, 569-573.
155.Harley, C. B., Futcher, A. B., and Greider, C.
W. (1990) Telomeres shorten during ageing of human fibroblasts,
Nature, 345, 458-460.
156.Yu, G. L., Bradley, J. D., Attardi, L. D., and
Blackburn, E. H. (1990) In vivo alteration of telomere sequences
and senescence caused by mutated Tetrahymena telomerase RNAs,
Nature, 344, 126-132.
157.Bodnar, A. G., Ouellette, M., Frolkis, M.,
Holt, S. E., Chiu, C., Morin, G. B., Harley, C. B., Shay, J. W.,
Lichsteiner, S., and Wright, W. E. (1998) Extension of life span by
introduction of telomerase into normal human cells, Science,
279, 349-352.
158.Counter, C. M., Hahn, W. C., Wei, W., Caddle,
S. D., Beijersbergen, R. L., Lansdorp, P. M., Sedivy, J. M., and
Weinberg, R. A. (1998) Dissociation among in vitro telomerase
activity, telomere maintenance, and cellular immortalization, Proc.
Natl. Acad. Sci. USA, 95, 14723-14728.
159.De Lange, T., and Jacks, T. (1999) For better
or worse? Telomerase inhibition and cancer, Cell,
98, 273-275.
160.Vaziri, H. (1998) Extension of life span in
normal human cells by telomerase activation: a revolution in cultural
senescence, J. Anti Aging Med., 1, 125-130.
161.Vaziri, H., and Benchimol, S. (1998)
Reconstitution of telomerase activity in normal cells leads to
elongation of telomeres and extended replicative life span, Curr.
Biol., 8, 279-282.
162.Van Steensel, B., and De Lange, T. (1997)
Control of telomere length by the human telomeric protein TRF1,
Nature, 385, 740-743.
163.Morin, G. B. (1989) The human telomere terminal
transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG
repeats, Cell, 59, 521-529.
164.Alberts, B., Bray, D., Hopkin, K., Johnson, A.,
Lewis, J., Raff, M., Roberts, K., and Walter, P. (2013) Essential
Cell Biology, 4th Edn., Garland Science, New York.
165.Anversa, P., Kajstura, J., Leri, A., and Bolli,
R. (2006) Life and death of cardiac stem cells, Circulation,
113, 1451-1463.
166.Richardson, B. R., Allan, D. S., and Le, Y.
(2014) Greater organ involution in highly proliferative tissues
associated with the early onset and acceleration of ageing in humans,
Exp. Gerontol., 55, 80-91.
167.Jones, R. B., Whitney, R. G., and Smith, J. R.
(1985) Intramitotic variation in proliferative potential: stochastic
events in cellular aging, Mech. Ageing Dev., 29,
143-149.
168.Ponten, J., Stein, W. D., and Shall, S. (1983)
A quantitative analysis of the aging of human glial cells in culture,
J. Cell Phys., 117, 342-352.
169.Blackburn, E. H. (2000) Telomere states and
cell fates, Nature, 408, 53-56.
170.Holt, S. E., Shay, J. W., and Wright, W. E.
(1996) Refining the telomere–telomerase hypothesis of aging and
cancer, Nat. Biotechnol., 14, 836-839.
171.Slijepcevic, P., and Hande, M. P. (1999)
Chinese hamster telomeres are comparable in size to mouse telomeres,
Cytogenet. Cell Genet., 85, 196-199.
172.Gorbunova, V., Bozzella, M. J., and Seluanov,
A. (2008) Rodents for comparative aging studies: from mice to beavers,
Age, 30, 111-119.
173.Ben-Porath, I., and Weinberg, R. (2005) The
signals and pathways activating cellular senescence, Int. J.
Biochem. Cell Biol., 37, 961-976.
174.Gottschling, D. E., Aparicio, O. M.,
Billington, B. L., and Zakian, V. A. (1990) Position effect at S.
cerevisiae telomeres: reversible repression of Pol II
transcription, Cell, 63, 751-762.
175.Robin, J. D., Ludlow, A. T., Batten, K.,
Magdinier, F., Stadler, G., Wagner, K. R., Shay, J. W., and Wright, W.
E. (2014) Telomere position effect: regulation of gene expression with
progressive telomere shortening over long distances, Genes Dev.,
28, 2464-2476.
176.Prowse, K. R., and Greider, C. W. (1995)
Developmental and tissue-specific regulation of mouse telomerase and
telomere length, Proc. Natl. Acad. Sci. USA, 92,
4818-4822.
177.Flanary, B. E. (2003) Telomeres shorten with
age in rat cerebellum and cortex in vivo, J. Anti Aging
Med., 6, 299-308.
178.Herrera, E., Samper, E., Martin-Caballero, J.,
Flores, J. M., Lee, H. W., and Blasco, M. A. (1999) Disease states
associated with telomerase deficiency appear earlier in mice with short
telomeres, EMBO J., 18, 2950-2960.
179.Blasco, M. A., Lee, H. W., Hande, M. P.,
Samper, E., Lansdorp, P. M., DePinho, R. A., and Greider, C. W. (1997)
Telomere shortening and tumor formation by mouse cells lacking
telomerase RNA, Cell, 91, 25-34.
180.Lee, H. W., Blasco, M. A., Gottlieb, G. J.,
Horner, J. W., 2nd, Greider, C. W., and DePinho, R. A. (1998) Essential
role of mouse telomerase in highly proliferative organs, Nature,
392, 569-574.
181.Klapper, W., Heidorn, H., Kuhne, K.,
Parwaresch, R., and Krupp, G. (1998) Telomerase activity in
“immortal” fish, FEBS Lett., 434,
409-412.
182.Klapper, W., Kuhne, K., Singh, K. K., Heidorn,
K., Parwaresch, R., and Krupp, G. (1998) Longevity of lobsters is
linked to ubiquitous telomerase expression, FEBS Lett.,
439, 143-146.
183.Longo, V. D., and Finch, C. E. (2003)
Evolutionary medicine: from dwarf model systems to healthy
centenarians? Science, 299, 1342-1346.
184.Goldsmith, T. C. (2003) The Evolution of
Aging: How Darwin’s Dilemma is Affecting Your Chance for a Longer
and Healthier Life, iUniverse, Lincoln, Nebraska.
185.Skulachev, V. P. (1997) Aging is a specific
biological function rather than the result of a disorder in complex
living systems: biochemical evidence in support of Weismann’s
hypothesis, Biochemistry (Moscow), 62, 1191-1195.
186.Campisi, J. (1997) The biology of replicative
senescence, Eur. J. Cancer, 33, 703-709.
187.Campisi, J. (2003) Cancer and ageing: rival
demons? Nat. Rev. Cancer, 3, 339-349.
188.Troen, B. (2003) The biology of aging, Mt.
Sinai J. Med., 30, 3-22.
189.Wright, W. E., and Shay, J. W. (2005) Telomere
biology in aging and cancer, J. Am. Geriatr. Soc., 53,
292-294.
190.Mitteldorf, J. (2013) Telomere biology: cancer
firewall or aging clock? Biochemistry (Moscow), 78,
1054-1060.
191.Milewski, L. A. K. (2010) The evolution of
ageing, Biosci. Horizons, 3, 77-84.
192.Parrinello, S., Coppe, J.-P., Krtolica, A., and
Campisi, J. (2005) Stromal-epithelial interactions in aging and cancer:
senescent fibroblasts alter epithelial cell differentiation, J. Cell
Biol., 118, 485-496.
193.Coppe, J.-P., Patil, C. K., Rodier, F., Sun,
Y., Munoz, D. P., Goldstein, J., Nelson, P. S., Desprez, P.-Y., and
Campisi, J. (2008) Senescence-associated secretory phenotypes reveal
cell-nonautonomous functions of oncogenic RAS and the p53 tumor
suppressor, PLoS Biol., 6, 2853-2568.