REVIEW: Role of a Structurally Equivalent Phenylalanine Residue in Catalysis and Thermal Stability of Formate Dehydrogenases from Different Sources
V. I. Tishkov1,2,3*, K. V. Goncharenko1,3, A. A. Alekseeva2,3, S. Yu. Kleymenov2,4, and S. S. Savin1,2
1Lomonosov Moscow State University, Faculty of Chemistry, 119991 Moscow, Russia; E-mail: vitishkov@gmail.com2Bach Institute of Biochemistry, Federal Research Centre “Fundamentals of Biotechnology”, Russian Academy of Sciences, 119071 Moscow, Russia
3Innovations and High Technologies MSU Ltd., 109451 Moscow, Russia
4Koltzov Institute of Developmental Biology, Russian Academy of Sciences, 119334 Moscow, Russia
* To whom correspondence should be addressed.
Received August 3, 2015; Revision received September 4, 2015
Comparison of amino acid sequences of NAD+-dependent formate dehydrogenases (FDH, EC 1.2.1.2) from different sources and analysis of structures of holo-forms of FDH from bacterium Pseudomonas sp. 101 (PseFDH) and soya Glycine max (SoyFDH) as well as of structure of apo-form of FDH from yeast Candida boidinii (CboFDH) revealed the presence on the surface of protein globule of hydrophobic Phe residue in structurally equivalent position (SEP). The residue is placed in the coenzyme-binding domain and protects bound NAD+ from solvent. The effects of amino acid changes of the SEP on catalytic properties and thermal stability of PseFDH, SoyFDH, and CboFDH were compared. The strongest effect was observed for SoyFDH. All eight amino acid replacements resulted in increase in thermal stability, and in seven cases, increase in catalytic constant was achieved. Thermal stability of SoyFDH after mutations Phe290Asp and Phe290Glu increased 66- and 55-fold, respectively. KM values of the enzyme for substrate and coenzyme in different cases slightly increased or decreased. In case of one CboFDH, the mutein catalytic constant increased and thermal stability did not changed. In case of the second CboFDH mutant, results were the opposite. In one PseFDH mutant, amino acid change did not influence the catalytic constant, but in three others, the parameter was reduced. Two PseFDH mutants had higher and two mutants lower thermal stability in comparison with initial enzyme. Analysis of results of SEP mutagenesis in FDHs from bacterium, yeast, and plant shows that protein structure plays a key role for effect of the same amino acid changes in equivalent position in protein globule of formate dehydrogenases from different sources.
KEY WORDS: formate dehydrogenase, rational design, thermal stability, catalytic propertiesDOI: 10.1134/S0006297915130052
Abbreviations: FDH, formate dehydrogenase; PseFDH, CboFDH, and SoyFDH, recombinant formate dehydrogenases from bacterium Pseudomonas sp. 101, yeast Candida boidinii, and soya Glycine max; SEP, structurally equivalent Phe residue.
NAD+-dependent formate dehydrogenase (FDH, EC 1.2.1.2)
catalyzes the reaction of oxidation of a formate ion to carbon dioxide
coupled with reduction of NAD+ to NADH. Formate
dehydrogenase is a model enzyme for study of the catalytic mechanism of
hydride-ion transfer from the substrate to the C4-atom of the nicotine
amide moiety of NAD+ in the active site. FDH is also used in
practice for cofactor regeneration. The main advantages of FDH as a
biocatalyst for coenzyme regeneration compared to other enzymes are
irreversibility of the catalyzed reaction, wide pH-optimum of activity,
high specificity to both substrates, low price of formate, and the
simplicity of the removal of the second reaction product CO2
(bicarbonate). Genes encoding FDH have been found in bacteria, yeasts,
fungi, and plants [1-3].
Our laboratory engages in systematic studies of the structure and catalytic mechanism of FDHs from different sources. Formate dehydrogenase from the bacterium Pseudomonas sp. 101 (PseFDH) is the most thoroughly studied enzyme. In the PDB database, there are several structures for apo- (2NAC and 2GO1, resolutions 1.8 and 2.1 Å, respectively) as well as for holo- (2NAD, ternary complex with NAD+ and azide ion, resolution 2.05 Å) and for complex of FDH with formate (2GUG, resolution 2.28 Å). Structural data for PseFDH have been used for choosing amino acid residues for experiments using site-directed mutagenesis aimed to improving the catalytic properties and increasing the thermal and operational stability of the enzyme [2, 4-7]. It was shown that a combination of several single-point mutations into multiple mutants resulted in additive effects [5]. For example, the combination of seven amino acid substitutions in PseFDH produced the mutant PseFDH T7, which had 50-fold lower inactivation rate constant compared to the wild-type enzyme [2]. Three amino acid replacements responsible for increase in operational and thermal stability were combined in the mutant form PseFDH GAV [2, 8]. As a result, the affinity to NAD+ increased more than 2-fold, thermal stability 2.5-fold, and operational stability more than 1000-fold compared to wt-PseFDH [2, 8]. Mutant PseFDH GAV was successfully used for NADH regeneration in chiral syntheses [2, 9, 10]. Introduction of the additional substitution Cys145Ser into PseFDH GAV led to the new mutant PseFDH SM4 with improved parameters compared to PseFDH GAV: the Michaelis constant for formate decreased 1.9-fold, operational stability increased 2-fold [4], and thermal stability increased 1.6-fold. The increase in thermal stability allowed us to improve the purification of mutant PseFDHs significantly by introducing a heat-treatment step at 60°C for 20-30 min. As a result, the content of the target protein increased from 50% in cell-free extract to 80-90% without any loss of activity [2, 11].
NAD+-dependent formate dehydrogenase is a highly conserved enzyme. The homology between amino acid sequences of evolutionarily distant FDHs from bacteria and plants is more than 50% [2, 3]. The level of structure homology is even higher. The majority of amino acid residues localized in coenzyme-binding and catalytic domains of formate dehydrogenase have equivalent configuration. The Phe290 residue was revealed previously by structure analysis of the ternary complex of SoyFDH with the coenzyme NAD+ and the strong competitive inhibitor azide ion. This residue is localized on the surface of the protein globule in the coenzyme-binding domain, and it shields the active site-bound coenzyme from the solvent. Site-directed mutagenesis of this residue has been used to obtain eight mutant enzymes [12, 13]. The majority of the mutants have higher catalytic parameters and are more thermally stable compared to the wild-type enzyme [12, 13]. Analysis of 2NAD structure (ternary complex of PseFDH with NAD+ and azide ion) and apo-form of mutant FDH from yeast Candida boidinii (CboFDH, structures 2FSS and 2J6I) showed that Phe290 in SoyFDH corresponds to Phe311 in PseFDH and Phe285 in CboFDH. In the case of CboFDH, two mutants with replacements to Ser and Tyr were found after directed evolution experiments [14]. In the case of PseFDH, four mutants were obtained, but PseFDH SM4 was used as template instead of the wild-type enzyme. In this work, we compared results of the site-directed mutagenesis of the structurally equivalent Phe residue in formate dehydrogenase from three different sources – bacteria, yeasts, and plants. Special focus was made on the bacterial enzyme.
ANALYSIS OF POSITION OF STRUCTURALLY EQUIVALENT Phe RESIDUE IN
FORMATE DEHYDROGENASES
Now more than 100 amino acid sequences of NAD(P)+-dependent formate dehydrogenases from bacteria, yeasts, and plants can be found in databases. Figure 1 presents the fragment of alignment of amino acid sequences of FDHs from different sources in the region of Phe311 (numeration according to that in PseFDH sequence). Analysis of the multiple alignment of all reported FDH sequences showed that the frequency of occurrence of the Phe residue in this position depends on the type of the source. The highest conservatism is in bacterial enzymes. The Phe residue is located in this position in more than 80% of cases, and in other cases, a Tyr residue is found. Other amino acid residues have not been found in all studied amino acid sequences of bacterial FDHs. In the case of plant enzymes, in the position structurally equivalent to Phe290 in SoyFDH and Phe311 in PseFDH residues, Phe and Tyr have practically the same frequency – totally 70%. Other residues in this position are Asn, Asp, Ser, and His (arranged in decreasing frequency). For evolutionarily similar FDHs from yeasts and fungi, Phe residue can be found in more than 60% of cases. The second in frequency of occurrence is Asp (including FDH from baker’s yeast) rather than a Tyr residue.
Fig. 1. Fragment of multiple alignment of amino acid sequences in the region of 311 Phe residue. Bacterial FDHs are marked in blue: PseFDH – from Pseudomonas sp. 101, MorFDH – from Moraxella sp., ParFDH – from Paracoccus sp. 12-A, HypFDH – from Hyphomicrobium strain JT-17, SmeFDH – from Sinorhizobium meliloti, SauFDH – from Staphylococcus aureus. Plant FDHs are marked in green: SoyFDH2 – from soybean Glycine max, LesFDH1 – from tomato Lycopersicon esculentum, StuFDH1 – from potato Solanum tuberosum, CcaFDH1 – from coffee Coffea canephora, RcoFDH1 – from castor-oil plant Ricinus communis, BnaFDH2 – from raps Brassica napus, BolFDH1 – from cabbage Brassica oleracea, MdoFDH – from apple Malus domestica, AthFDH – from Arabidopsis thaliana, PtaFDH2 – from pine Pinus taeda. FDHs from yeasts are marked in rose: SceFDH – from bakery yeasts Saccharomyces cerevisiae, DhaFDH – from Debaryomyces hansenii, CmeFDH – from Candida methylica, CboFDH – from Candida boidinii, YliFDH – from Yarrowia lipolytica. FDHs from microfungi are marked in orange: NefFDH – from Neosartorya fischeri, AjcFDH1 – from Ajellomyces capsulatus, GzeFDH – from Gibberella zeae, CneFDH – from Cryptococcus neoformans.

The Phe290 position in the structure of the ternary complex (SoyFDH–NAD+–azide) was analyzed in [12, 13]. Molecular modeling of the mutant enzymes showed that in case of replacement of the Phe residue with Ala, the changes in structure are minimal, and no additional H-bonds occur. In the case of replacements to seven other residues – Asp, Asn, Glu, Gln, Tyr, Ser, and Thr, from two to four new additional H-bonds can appear. Moreover, in all mutants new H-bond(s) can appear between a mutated residue and Gln292 (Gln312 in PseFDH), which is in close contact with catalytically important His309 (His332 in PseFDH). It was shown for PseFDH and FDH from Moraxella sp. C1 that His332 is involved in formate ion binding [15, 16].
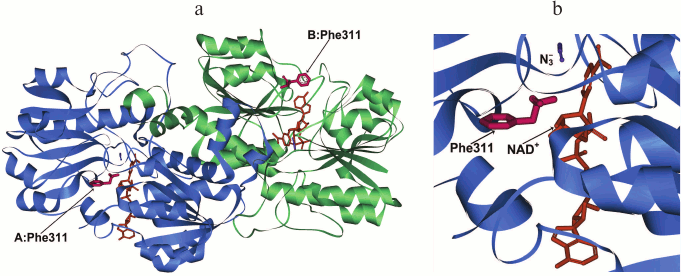
The structure of the dimer holo-form of PseFDH and the position of Phe311 in it are represented in Fig. 2. This residue is located on the surface in the coenzyme-binding domain of the active site. Its most probable function is shielding of NAD+ from the solvent, as it is in the plant SoyFDH. From the thermodynamic point of view, the location of a hydrophobic residue on the surface of the protein is not favorable for stability. Consequently, it is rather strange that Phe is located in this position in wild-type enzymes.
Fig. 2. a) Structure of the ternary complex [PseFDH–NAD+–N3–] with residue Phe311 shown in red (PDB structure 2NAD). Enzyme domains are shown in blue and green, NAD+ is given in orange, and azide-ion is given in violet. b) Detailed picture of the position of Phe311 in relation to the location of NAD+ and azide ion.
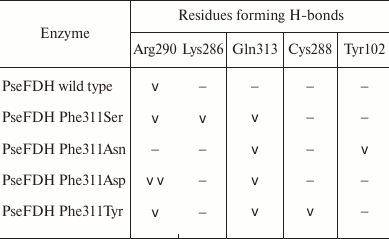
As mentioned earlier, in such positions of bacterial enzymes, Tyr can also be found. In formate dehydrogenases from other sources, after Phe and Tyr residues Asp, Asn, and Ser are most frequently occur. These residues were introduced in mutant PseFDH SM4. Computer modeling and structure alignment before and after mutation were made for all the mutant forms. The results of the computer modeling of mutant PseFDH with replacements of Phe311 to Ser, Asp, Asn, and Tyr are represented in Fig. 3, a-d. Positions of the amino acid residues before and after the mutation are shown in red and blue, respectively. The modeling showed that introduction of such mutations might cause formation of additional H-bonds (Table 1). In the case of wt-PseFDH, Phe311 is involved in only one H-bond – between the carbonyl oxygen of the main chain and the nitrogen of the guanidine group of Arg290. This bond is also present in the case of replacement Phe311 by Ser or Tyr and disappears in mutant PseFDH Phe311Asn. In the case of replacement to Asp, there are two additional H-bonds between residue 311 and Arg290 (Fig. 3a and Table 1). All four mutations lead to formation of an H-bond between the residue in position 311 and Gln313. Generally, we can observe one more (replacement to Asn) and two more (replacements to Ser, Asp, and Tyr) additional H-bonds compared to the structure of the initial bacterial enzyme. However, in comparison to the replacements of the structurally equivalent Phe290 residue in SoyFDH [13], the number of newly formed bonds in mutant PseFDH SM4 due to Phe311 substitution is less. For this reason, we expect that quantitative change in the properties of the bacterial mutant enzymes can be less than in the case of similar substitutions in SoyFDH.
Fig. 3. Molecular modeling of structure of mutant PseFDH SM4 with replacements of the Phe311 residue to Asn (a), Asp (b), Ser (c), and Tyr (d).
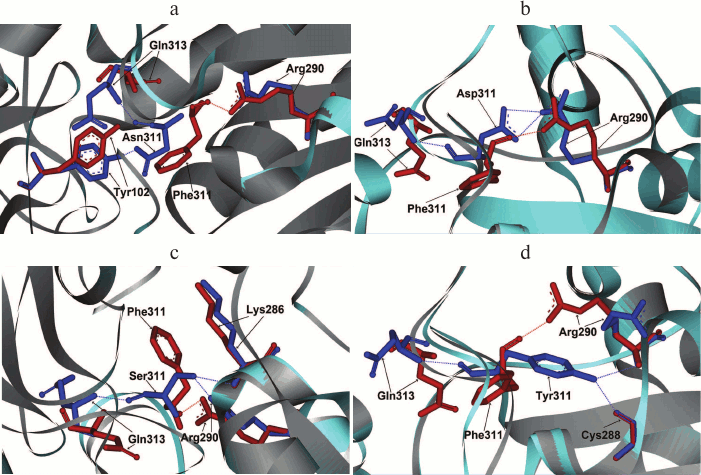
Table 1. Formation of H-bonds with the
residue in position 311 of PseFDH
In the case of FDH from the yeast C. boidinii in the PDB database of 3D structures of enzymes, there are only structures for apo-forms of two mutant enzyme (2FSS and 2J6I). Consequently, it is not possible to compare positions of the Phe residue in the holo-forms. However, the location of Phe285 in the apo-form of CboFDH (structure 2J6I; Fig. 4) suggests that the position of this residue is equivalent to Phe311 in the apo-form of PseFDH (not shown in the figure). Therefore, we suggest that the position of Phe285 while forming ternary complex of CboFDH with substrates corresponds to positions of Phe290 and Phe311 in holo-forms of SoyFDH and PseFDH, respectively.
Fig. 4. Position of structurally equivalent Phe285 residue in apo-form of formate dehydrogenase from the yeast C. boidinii (structure 2J6I).
INFLUENCE OF AMINO ACID CHANGES IN THE STRUCTURALLY EQUIVALENT
Phe RESIDUE ON CATALYTIC PROPERTIES
The influence of the replacement of Phe290 on the properties of SoyFDH was studied in works [12, 13], and data of mutagenesis of structurally equivalent Phe285 in CboFDH are presented in [14]. The results of replacement of Phe311 in PseFDH are shown in the present work. Kinetic parameters of mutant PseFDH SM4, CboFDH, and SoyFDH with replacements of structurally equivalent Phe residues are presented in Table 2. Note that the substitutions of Phe290 in SoyFDH showed the maximum effect. In practically all cases (except for the replacement Phe290Asn), the value of catalytic constant increased. Moreover, the value of kcat increased up to 1.76-fold. In the case of CboFDH, the catalytic constant increased 1.65-fold with the replacement Phe285Ser, but in the case of the Phe285Thr substitution, this parameter remained constant [14]. In bacterial PseFDH SM4, three replacements led to decrease in catalytic constant, and only PseFDH SM4 Phe311Tyr had practically the same kcat value as the initial enzyme.
Table 2. Kinetic parameters and relative
thermal stability of initial and mutant formate dehydrogenases with
substitutions of the structurally equivalent Phe residue (0.1 M
sodium-phosphate buffer, 0.01 M EDTA, pH 7.0)
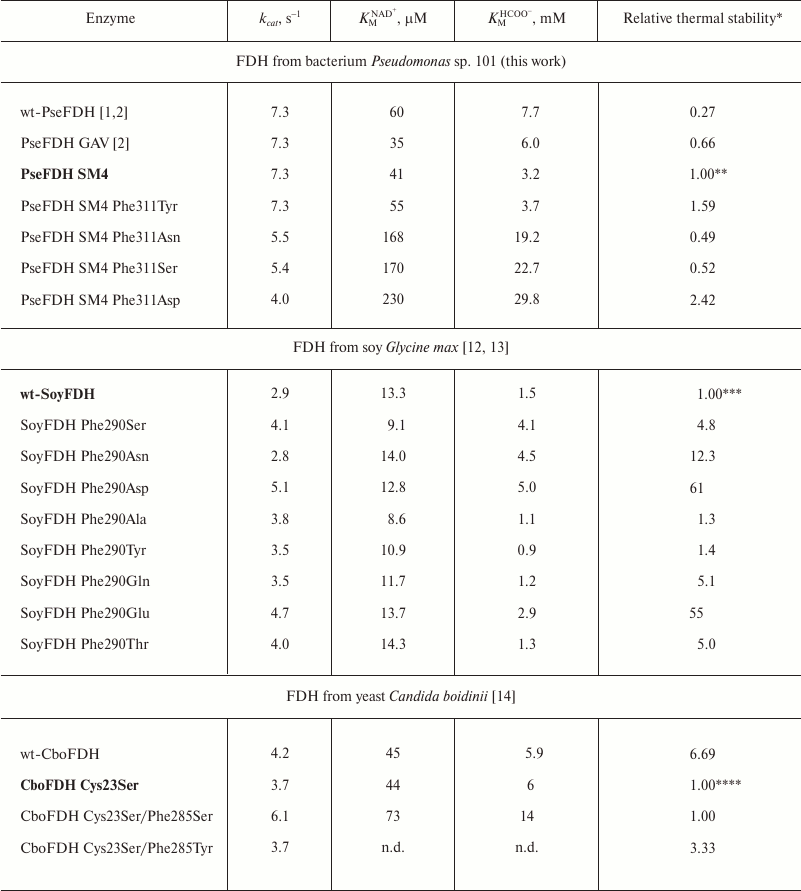
* Relative thermal stability is the ratio of inactivation rate constants
for initial enzyme and its mutant
(kinini/kinmut).Values
that are higher and lower than 1 mean stabilization and destabilization
of the enzyme, respectively. The initial enzyme form that was used for
thermal stability calculation is given in bold font.
** Measured at 64°C.
*** Measured at 54°C.
**** For the first three enzymes measured at 50°C. There are no kinetic
data (n.d.) for the mutant CboFDH Cys23Ser/Phe285Tyr. Stabilization
effect was evaluated based on indirect results. More details are given
in [2].
For the Michaelis constants of bacterial PseFDH, replacement of the Phe311 residue by Ser, Asn, and Asp led to notable increase in KM for NAD+ and formate. For the replacement of Phe311 by a tyrosine residue, KM for formate did not change within the limits of experimental error, and KMNAD+ increased slightly. For the Phe285 replacement in CboFDH [14], increase in catalytic constant was accompanied by a large increase in KM for the coenzyme and formate. Mutant SoyFDHs with substitutions in position 290 showed increase as well as decrease in Michaelis constants for both NAD+ and formate. However, such changes were not as large as for the bacterial and yeast enzymes.
THERMAL STABILITY STUDIED THROUGH INACTIVATION KINETICS
Thermal stability of mutant SoyFDHs with replacements of Phe290 have been studied over a wide temperature range [12, 13]. All mutations provided positive stabilization, and for the mutations Phe290Asp and Phe290Glu the stabilization effects were 55- and 61-fold at 54°C, respectively [12, 13]. Unfortunately, there are practically no data about the influence of substitutions in position 285 in CboFDH. In fact, only comparison of residual activity after 30 min incubation at one temperature was made [14]. However, monomolecular mechanism of CboFDH thermal inactivation [17] allowed us to calculate inactivation rate constants and stabilization effect. This method for stability comparison for mutant CboFDH is described in more details in work [2]. As mentioned above, the Phe285Ser substitution resulted in increase in catalytic constant but did not stabilize the enzyme. However, the Phe285Tyr substitution did not affect the catalytic constant, but it increased stability 3.3-fold [2, 14].
The thermal stability of mutant PseFDHs was studied in the temperature range 62-72°C. In this interval, the inactivation of PseFDH and the mutant PseFDH GAV is irreversible and proceeds through a monomolecular mechanism [2]. Figure 5 shows the dependence of residual activity on time for the initial enzyme PseFDH SM4 and its mutant forms at 64°C in semi-logarithmic coordinates. The data for widely practically used PseFDH GAV are also presented. As follows from Fig. 5, the time-dependences of residual activity for all mutant PseFDHs are exponential and linear in coordinates ln(A/A0) – t. The first order inactivation rate constants kin were calculated from the slopes of the dependences. It was shown additionally that change in enzyme concentration over a broad range (0.1-2.5 mg/ml) did not influence inactivation rate constant. Therefore, the inactivation of PseFDH SM4 and its mutants at high temperatures is considered a monomolecular process. Consequently, introducing such amino acid substitution does not influence the mechanism of enzyme inactivation according to these data.
Fig. 5. Dependence of residual activity on time for mutant PseFDHs in coordinates ln(A/A0) – t (64°C, 0.1 M sodium-phosphate buffer, pH 7.0).
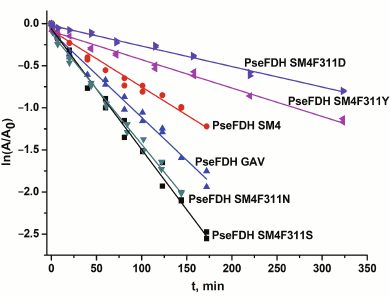
Table 2 shows that introducing Ser or Asn residues into position 311 destabilized the enzyme about 2-fold. However, substitution of Phe311 with Tyr or Asp increased thermal stability 1.6- and 2.4-fold, respectively. Therefore, two mutant forms of PseFDH with improved thermal stability were obtained.
The true monomolecular mechanism of thermal inactivation of PseFDH SM4 and its mutant forms over the whole temperature range allowed the use of transition state theory for the analysis of the process. The equation of transition state theory for the dependence of inactivation rate constant on temperature can be presented in linear form:
where 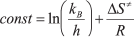 ,
kB and h are the Boltzmann and Plank
constants, respectively, R is the universal gas constant, and
ΔH≠ and ΔS≠ are
activation parameters.
,
kB and h are the Boltzmann and Plank
constants, respectively, R is the universal gas constant, and
ΔH≠ and ΔS≠ are
activation parameters.
Figure 6 shows the dependences of inactivation rate constant on temperature for mutant PseFDHs in coordinates ln(kin/T) – 1/T. From this picture, we see that the dependences are linear, so the equation of transition state theory can be applied to them. The activation parameters – enthalpy and entropy – can be calculated from the slope and the intercept, respectively (Table 3). Similar parameters for other formate dehydrogenases are also provided in the table.
Fig. 6. Dependence of inactivation rate constant on temperature for mutant forms of PseFDH in coordinates ln(kin/T) – 1/T, K–1 (0.1 M sodium-phosphate buffer, pH 7.0).
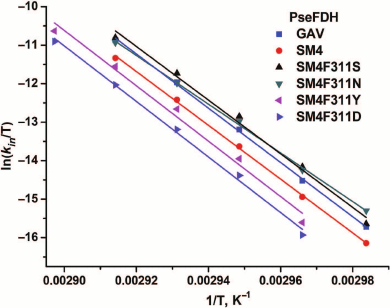
Table 3. Inactivation parameters for mutant
and wild-type FDHs from different sources*
* 0.1 M sodium-phosphate buffer, pH 7.0.
** wt-PseFDH, wt-MorFDH, wt-SoyFDH, wt-AthFDH, and wt-CboFDH –
recombinant wild type formate dehydrogenases from bacteria
Pseudomonas sp. 101 and Moraxella sp. C-1, plants soya
Glycine max and A. thaliana, and yeast C.
boidinii, respectively.
*** n.d., no data.
STUDY OF THERMAL STABILITY BY DIFFERENTIAL SCANNING
CALORIMETRY
Thermal stabilities of mutant forms of SoyFDH and PseFDH SM4 with substitution of the structurally equivalent Phe residue were also studied by differential scanning calorimetry (DSC). Results for SoyFDH are discussed in details in [12, 13]. Melting curves for the initial enzyme, the mutant with replacement Phe311Tyr, and for some other formate dehydrogenases are presented in Fig. 6. Numerical values of melting temperature are presented in Table 3. The data are in good agreement with data of the thermal inactivation kinetics. For replacements Phe311Asn and Phe311Ser, the thermal stability decreased, and for mutations Phe311Tyr and Phe311Asp, it increased compared to the initial PseFDH SM4. According to both methods, the proteins can be arranged by thermal stability in the same order: PseFDH SM4 F311N < PseFDH SM4 F311S < PseFDH GAV < PseFDH SM4 < PseFDH SM4 F311Y < PseFDH SM4 F311D.
ANALYSIS OF INFLUENCE OF AMINO ACID REPLACEMENTS OF THE
STRUCTURALLY EQUIVALENT PHENYLALANINE ON PROPERTIES OF FORMATE
DEHYDROGENASE
The data show that structurally equivalent Phe plays a very important role in stability and catalysis of formate dehydrogenases from the bacterium Pseudomonas sp. 101 (Phe311), soybean G. max (Phe290), and methylotrophic yeast C. boidinii (Phe285). However, the results for mutants of PseFDH SM4, SoyFDH [12, 13], and CboFDH [14] with replacements of residues Phe311, Phe290, and Phe285 differ greatly in direction (increase or decrease) and are strictly characteristic for each enzyme.
Catalytic constant. The increase in the catalytic constant caused by mutagenesis depends on the initial specific activity of the enzyme, as shown in Table 2. In the case of bacterial PseFDH SM4, which has higher activity compared to enzymes from yeasts and plants, there were no replacements that increased kcat (Table 2). Amino acid substitutions Phe290Asn in SoyFDH and Phe285Tyr in CboFDH Cys23Ser do not influence the value of kcat, but for mutant SoyFDHs with replacements Phe290Ser, Phe290Asp, Phe290Ala, Phe290Tyr, Phe290Gln, Phe290Glu, and Phe290Thr, kcat increases from 20 to 76% [12, 13], and for mutant CboFDH Cys23Ser/Phe285Ser, kcat increases by 65% compared to the initial enzyme CboFDH Cys23Ser. However, if we compare this result with wt-CboFDH, the effect will be less, 42%, and it is rather similar to SoyFDH – the Phe290Ser mutation increased kcat by 43%. In addition, it should be noted that specific activity of the best mutant of SoyFDH, Phe290Asp, is less than the same value for the mutant CboFDH Cys23Ser/Phe285Ser.
Michaelis constants. The replacement Phe311Tyr in PseFDH SM4 has practically no influence on KMNAD+ and KMHCOO– (Table 1). Unfortunately, there are no data in the literature for mutant CboFDH Cys23Ser/Phe285Tyr. Replacement Phe285Ser in the yeast enzyme and replacements of Phe311 to Ser, Asn, and Asp in PseFDH SM4 increased Michaelis constants for both substrates several-fold. This effect is rather high in bacterial enzymes. Different data were obtained for SoyFDH (Table 2). The value of KMHCOO– increased from 2- to 3.3-fold (for four of eight replacements – Phe290Asp, Phe290Ser, Phe290Asn, and Phe290Glu) and decreased 1.4-fold (for replacement Phe290Tyr). For NAD+, the value of KM becomes better (Phe290Ser and Phe290Ala) or remains constant within the experimental error (Phe290Glu, Phe290Asn, Phe290Gln, Phe290Glu, Phe290Thr, and Phe290Asp).
Structural aspects of kinetic parameters change. Different effects on kinetic parameters of bacterial and plant formate dehydrogenases are in good agreement with results of structure modeling of mutant enzymes. As mentioned earlier, a new H-bond is formed between the introduced residue and Gln313 (Table 1). The latter residue is not involved in catalysis, but it is in tight contact with residue His332, which takes part in formate ion binding in the enzyme active site [15, 16]. Formation of the new H-bond with Gln313 leads to change in its conformation (Fig. 3, a-d). Such change primarily should influence formate binding in the active site, and then change in catalytic properties. At the same time, the results of structure modeling of mutant SoyFDHs based on the crystal structure of the ternary complex [SoyFDH–NAD+–N3–] [12, 13] showed that the location of Gln292 (equivalent of Gln313 in PseFDH) remains practically constant. All substitutions in position 290 except for Phe290Ala lead to formation of from two to four new H-bonds, and one of them is always with Gln292. Consequently, increase in KMHCOO– in the plant enzyme is not related to conformation change in the formate-binding domain of the active site. This effect relates to change in microdielectric permittivity in the active site by means of the replacement of hydrophobic Phe by more polar residues. The same should be valid for bacterial and yeast formate dehydrogenases. For PseFDH SM4 and SoyFDH, the greatest increase in KMHCOO– occurs on substitution to negatively charged Asp. The substitution to the least polar of the studied residues, Tyr, has the smallest effect on KMHCOO–. The residues with intermediate polarity (Ser and Ala) produce intermediate effects on the KM values.
Apparently, increase in catalytic constant in the case of plant and yeast formate dehydrogenase and the lack of such effect for the bacterial enzyme relates to the difference of catalytic and kinetics mechanisms of the enzymes. PseFDH has a random quasi-equilibrium kinetic mechanism [19, 20]. The data of pre-steady-state kinetics and experiments for determination of primarily deuterium kinetic isotope effect (HVmax/DVmax 3.0 and H(Vmax/KM)/D(Vmax/KM) 2.5) at different pH values show that hydride-ion transfer in the ternary complex is the rate-limiting step of the PseFDH kinetic mechanism [20]. However, ordered substrate binding is typical for yeast and plant enzymes [21, 22]. The primary deuterium kinetic isotope effects for the reaction maximum velocity HVmax/DVmax are 2.13 and 2.10 for yeast FDH from C. boidinii [22] and C. methylica [23], respectively, and definitely lower than for PseFDH (3.0) [20]. For FDH from C. boidinii, the ratio H(Vmax/KM)/D(Vmax/KM) 2.27 corresponds to the ratio HVmax/DVmax within experimental error [22], but for PseFDH such value is reliably lower (2.50) [20]. This means that the effective catalytic constant may depends on the step of formate binding (or on the rate of CO2 dissociation). A somewhat different way of formate ion binding in the active site of bacterial and yeast formate dehydrogenases leads to the fact that PseFDH cannot catalyze the oxidation of the nearest structural analog – thioformate. On the contrary, the enzyme from the yeast Hansenula polymorpha catalyzes this reaction [2, 24]. Probably slight decrease in formate ion binding and the product of its oxidation, carbon dioxide, in the active site of mutants with replacement of hydrophobic Phe to polar Ser and Asp residues is connected with increase in their dissociation rate from the active site, and, as a result, the maximum rate of the reaction increases. Nevertheless, bacterial formate dehydrogenases are considered more efficient as biocatalysts than their analogs from yeasts and plants because even under the best of circumstances (replacement Phe285Ser in CboFDH), the value of the catalytic constant 6.1 s–1 [17] is still less than kcat for the bacterial wild-type enzyme (7.3 s–1) [2].
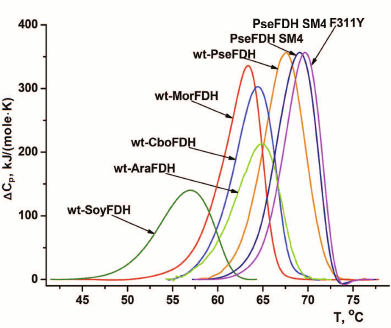
The data on thermal stability of formate dehydrogenases from different sources follow the rule “the highest effect is observed for the least perfect enzyme”. SoyFDH is one of the less stable enzymes among studied formate dehydrogenases (Fig. 6). Only the enzyme from baker’s yeasts is less stable than SoyFDH [18, 25]. Therefore, it is not surprise that the highest stabilization effect, 61-fold, was observed for the mutant form SoyFDH Phe290Asp (Table 2). This single-point mutation resulted in thermal stability level comparable with that of FDH from other plant, A. thaliana, which is inferior only to that of PseFDH (Fig. 7). For the same replacement in PseFDH SM4, the effect is much lower – only 2.4-fold. However, if we compare this mutein with wt-PseFDH, the stabilization effect is 9-fold. Taking into account that PseFDH is the most stable among the studied formate dehydrogenases, this result seems to be very successful.
Fig. 7. DSC melting curves for wild-type formate dehydrogenases from plants soya (wt-SoyFDH) and A. thaliana (wt-AthFDH), yeast C. boidinii (wt-CboFDH), bacteria Moraxella sp. C1 (wt-MorFDH) and Pseudomonas sp. 101 (wt-PseFDH), and mutants PseFDH SM4 and PseFDH SM4 F311Y (0.1 M sodium phosphate buffer, pH 7.0). Heating rate, 1°C/min.
The data indicate that replacements of Phe311 in PseFDH and Phe290 in SoyFDH result in different temperature dependences of the inactivation rate constant. For PseFDH SM4, the dependences of inactivation rate constant in coordinates ln(kin/T) – T–1 have the same slope. This means that the activation enthalpy remains constant for the initial PseFDH SM4 and the mutants (Table 3). Consequently, change in thermal stability depends only on change in activation entropy. These results are in contrast to data obtained for SoyFDH (Table 3) [12, 13]. For SoyFDH mutants with substitutions in position 290, the values of activation enthalpy and activation entropy for thermal denaturation may be lower (replacements Phe290Tyr and Phe290Gln) or higher compared to those for the wild-type enzyme. Activation parameters for mutant PseFDH Phe290Ala with no additional hydrogen bonds are similar to those for wild-type SoyFDH. Nevertheless, all of the mutants are more stable than the initial enzyme, and this is not influenced by the activation parameters. However, some mutant forms of SoyFDH have better catalytic parameters than the initial enzyme. Differences in activation enthalpy of wt-SoyFDH and its mutants with substitutions in position 290 change the stabilization effect. When ΔH≠ increases, the stabilization effect becomes greater while the temperature decreases. For the values of ΔH≠ less than that of the wild-type enzyme, the difference in stability disappears, and at temperatures less than 45°C, mutant forms SoyFDH Phe290Tyr and SoyFDH Phe290Gln are less stable than the initial enzyme. Only for the mutant SoyFDH Phe290Ala, which has the same value of ΔH≠ as the native enzyme, the stabilization effect does not depend on temperature.
The same substitution differently influences the stability of formate dehydrogenases. The replacement Phe285Ser in FDH from the yeast C. boidinii makes the catalytic constant better, but the Michaelis constants for both substrates become worse, while the thermal stability remains constant. In the case of bacterial FDH, thermal stability decreases nearly two-fold as a result of the same substitution. Replacement Phe285Tyr in CboFDH does not influence the catalytic constant, but it increases the stability 3.3-fold (Table 2). The same mutation in PseFDH leads to less increase in stability (only 1.5-fold). For the enzyme from soybean, the catalytic constant and KM for the coenzyme become better and thermal stability increases 4.8-fold. It should be noted that the replacement Phe/Asp increases stability of both bacterial PseFDH and plant SoyFDH. However, this mutation causes the opposite effect on values of catalytic constant and KM for formate.
Analysis of results of mutagenesis of the structurally equivalent Phe residue in formate dehydrogenases from three sources types – bacteria, yeasts, and plants – shows that this residue plays a very important role in catalysis and stability. All eight amino acid replacements in SoyFDH resulted in mutants with improved thermal stability. Moreover, in two cases the stabilization effect was extremely high compared to common stabilization values caused by single-point mutations. Combination with two other positive mutations [26] led to preparation of triple mutants and the best mutant thermal stability very close that for wild-type PseFDH [27]. The catalytic constant increased for seven mutants of SoyFDH with replacements in position 290. Mutants of SoyFDH with substitutions Phe290Asp and Phe290Glu show very high catalytic efficiency and are considered the most perspective for biotechnology. For the yeast enzyme, only replacement Phe285Tyr that led to increased stability is interesting for practical application. Substitution Phe285Ser increased the catalytic constant 1.65-fold, but at the same time, the KM for the coenzyme increased twice. As a result, catalytic efficiency did not change, but in practice in the process of coenzyme regeneration, the concentration of NAD+ should be increased 2-fold. For PseFDH, two mutant forms of the enzyme with higher thermal stability compared with initial mutant PseFDH SM4 were obtained by single point mutation. Thus, the enzyme with replacement Phe311Tyr does not have significantly changed catalytic properties. Comparison of the influence of Phe residue replacement on properties of formate dehydrogenases from bacteria, yeasts, and plants proves that the structure of the enzyme plays a crucial role for the same replacements in structurally equivalent positions in different formate dehydrogenases.
This work was supported by the Russian Foundation for Basic Research (project No. 14-04-01625).
REFERENCES
1.Tishkov, V. I., and Popov, V. O. (2004) Catalytic
mechanism and application of formate dehydrogenase, Biochemistry
(Moscow), 69, 1252-1267.
2.Tishkov, V. I., and Popov, V. O. (2006) Protein
engineering of formate dehydrogenase, Biomol. Eng., 23,
89-110.
3.Alekseeva, A. A., Savin, S. S., and Tishkov, V. I.
(2011) NAD+-dependent formate dehydrogenase from plants,
Acta Naturae, 3, No. 4(11), 38-54.
4.Savin, S. S., and Tishkov, V. I. (2010) Assessment
of formate dehydrogenase stress stability in vivo using
inactivation by hydrogen peroxide, Acta Naturae, 2, No.
1(4), 78-82.
5.Rojkova, A. M., Galkin, A. G., Kulakova, L. B.,
Serov, A. E., Savitsky, P. A., Fedorchuk, V. V., and Tishkov, V. I.
(1999) Bacterial formate dehydrogenase. Increasing the enzyme thermal
stability by hydrophobization of alpha helices, FEBS Lett.,
445, 183-188.
6.Fedorchuk, V. V., Galkin, A. G., Yasny, I. E.,
Kulakova, L. B., Rojkova, A. M., Filippova, A. A., and Tishkov, V. I.
(2002) Influence of interactions between amino acid residues 43 and 61
on thermal stability of bacterial formate dehydrogenases,
Biochemistry (Moscow), 67, 1145-1151.
7.Serov, A. E., Odintzeva, E. R., Uporov, I. V., and
Tishkov, V. I. (2005) Use of Ramachandran map for the increasing
thermal stability of bacterial formate dehydrogenase, Biochemistry
(Moscow), 70, 804-808.
8.Alekseeva, A. A., Fedorchuk, V. V., Zarubina, S.
A., Sadykhov, E. G., Matorin, A. D., Savin, S. S., and Tishkov, V. I.
(2015) Role of Ala198 in stability and coenzyme specificity of
bacterial formate dehydrogenases, Acta Naturae, 7, No.
1(24), 60-69.
9.Hofstetter, K., Lutz, J., Lang, I., Witholt, B.,
and Schmid, A. (2004) Coupling of biocatalytic asymmetric epoxidation
with NADH regeneration in organic–aqueous emulsions, Angew.
Chem. Int. Ed. Engl., 43, 2163-2166.
10.Maurer, S. C., Kuehnel, K., Kaysser, L. A.,
Eiben, S., Schmid, R. D., and Urlacher, V. B. (2005) Catalytic
hydroxylation in biphasic systems using CYP102A1 mutants, Adv.
Synth. Catal., 347, 1090-1098.
11.Tishkov, V. I., Galkin, A. G., Fedorchuk, V. V.,
Savitsky, P. A., Rojkova, A. M., Gieren, H., and Kula, M.-R. (1999)
Pilot scale production and isolation of recombinant NAD+-
and NADP+-specific formate dehydrogenase, Biotechnol.
Bioeng., 64, 187-193.
12.Alekseeva, A. A., Serenko, A. A., Kargov, I. S.,
Savin, S. S., Kleymenov, S. Yu., and Tishkov, V. I. (2012) Engineering
catalytic properties and thermal stability of plant formate
dehydrogenase by single-point mutations, Prot. Eng. Des.
Select., 25, 781-788.
13.Kargov, I. S., Kleymenov, S. Y., Savin, S. S.,
Tishkov, V. I., and Alekseeva, A. A. (2015) Improvement of the soy
formate dehydrogenase properties by rational design, Protein Eng.
Des. Sel., 28, 171-178.
14.Felber, S. (2001) Optimierung der
NAD+-Abhaengigen Formiatdehydrogenase aus Candida boidinii
fuer den Einsatz in der Biokatalyse: Ph. D. Thesis, Heinrich-Heine
University of Duesseldorf. URL: http://diss.ub.uni-duesseldorf.de/ebib/diss/file?dissid=78.
15.Tishkov, V. I., Matorin, A. D., Rojkova, A. M.,
Fedorchuk, V. V., Savitsky, A. P., Dementieva, L. A., Lamzin, V. S.,
Mezentzev, A. V., and Popov, V. O. (1996) Site-directed mutagenesis of
formate dehydrogenase active centre: role of the His332-Gln313 pair in
enzyme catalysis, FEBS Lett., 390, 104-108.
16.Shabalin, I. G., Polyakov, K. M., Tishkov, V. I.,
and Popov, V. O. (2009) Atomic resolution crystal structure of
NAD+-dependent formate dehydrogenase from bacterium
Moraxella sp. C-1, Acta Naturae, 1, No. 3,
89-93.
17.Tishkov, V. I., Uglanova, S. V., Fedorchuk, V.
V., and Savin, S. S. (2010) Influence of ion strength and pH on thermal
stability of yeast formate dehydrogenase, Acta Naturae,
2, No. 2, 82-87.
18.Sadykhov, E. G., Serov, A. E., Voinova, N. S.,
Uglanova, S. V., Petrov, A. S., Alexeeva, A. A., Kleimenov, S. Yu.,
Popov, V. O., and Tishkov, V. I. (2006) A comparable study of thermal
stability of formate dehydrogenases from microorganisms and plants,
Appl. Biochem. Microbiol., 42, 236-240.
19.Tishkov, V. I., Popov, V. O., and Egorov, A. M.
(1983) Bacterial formate dehydrogenase. Substrate specificity and
kinetic mechanism of oxidation of S-formyl glutathione, Biochemistry
(Moscow), 48, 1003-1010.
20.Tishkov, V. I., Galkin, A. G., and Egorov, A. M.
(1989) Kinetic isotope effect and pre-steady state kinetics of the
reaction catalyzed by bacterial formate dehydrogenase,
Biochimie, 71, 551-557.
21.Zaks, A. M., Avilova, T. V., Egorova, O. A.,
Popov, V. O., and Egorov, A. M. (1982) Kinetic mechanism of
NAD+-dependent formate dehydrogenase from methylotrophic
yeast Candida methylica, Biochemistry (Moscow),
47, 546-551.
22.Hermes, J. D., Morrical, S. W., O’Leary, M.
H., and Cleland, W. W. (1984) Variation of transition-state structure
as a function of the nucleotide in reactions catalyzed by
dehydrogenases. 2. Formate dehydrogenase, Biochemistry,
23, 5479-5488.
23.Tishkov, V. I., Galkin, A. G., and Egorov, A. M.
(1989) NAD-dependent formate dehydrogenase of methylotrophic yeast:
preparation and characterization, Biochemistry (Moscow),
54, 231-237.
24.Mezentzev, A. V., Ustinnikova, T. B., Tikhonova,
T. V., and Popov, V. O. (1996) Obtaining and kinetic mechanism of
NAD-dependent formate dehydrogenase from methylotrophic yeast
Hansenula polymorpha, Appl. Biochem. Microbiol.
(Moscow), 32, 529-534.
25.Serov, A. E., and Tishkov, V. I. (2006)
Baker’s yeast formate dehydrogenase: unusual mechanism of
inactivation and stabilization by ionic strength and cofactor,
Moscow Univ. Chem. Bull., 47, 1-5.
26.Alekseeva, A. A., Savin, S. S., Kleymenov, S.
Yu., Uporov, I. V., Pometun, E. V., and Tishkov, V. I. (2012)
Stabilization of plant formate dehydrogenase by rational design,
Biochemistry (Moscow), 77, 1199-1209.
27.Alekseeva, A. A., Kargov, I. S., Kleymenov, S.
Yu., Savin, S. S., and Tishkov, V. I. (2015) Additivity of the
stabilization effect of single amino acid substitutions in triple
mutants of recombinant formate dehydrogenase from the soybean
Glycine max, Acta Naturae, 7, No. 3(26),
55-64.