REVIEW: Methods of Genome Engineering: a New Era of Molecular Biology
A. A. Chugunova1,2, O. A. Dontsova1,2, and P. V. Sergiev1*
1Lomonosov Moscow State University, Faculty of Chemistry, 119991 Moscow, Russia; E-mail: petya@genebee.msu.ru2Skolkovo Institute for Science and Technology, 143025 Skolkovo, Moscow Region, Russia
* To whom correspondence should be addressed.
Received February 15, 2016; Revision received March 17, 2016
Genome sequencing now progressing much faster than our understanding of the majority of gene functions. Studies of physiological functions of various genes would not be possible without the ability to manipulate the genome. Methods of genome engineering can now be used to inactivate a gene to study consequences, introduce heterologous genes into the genome for scientific and biotechnology applications, create genes coding for fusion proteins to study gene expression, protein localization, and molecular interactions, and to develop animal models of human diseases to find appropriate treatment. Finally, genome engineering might present the possibility to cure hereditary diseases. In this review, we discuss and compare the most important methods for gene inactivation and editing, as well as methods for incorporation of heterologous genes into the genome.
KEY WORDS: transgenosis, knockout, knockin, genome engineering, homologous recombination, integration, zinc finger nuclease (ZFN), TALEN, CRISPRDOI: 10.1134/S0006297916070038
Abbreviations: CRISPR, clustered regularly interspaced short palindromic repeat; crRNA, CRISPR RNA; HDR, homology directed repair; IR, inverted repeats; NHEJ, nonhomologous end joining; PAM, protospacer adjacent motif; RVD, repeat variable diresidue; TALE, transcription activator-like effector; TALEN, TALE-nuclease; tracrRNA, trans-activating CRISPR RNA; ZF, zinc-finger (protein); ZFN, zinc-finger nuclease.
It is well known that a genome contains all information about the
development and functioning of a living organism. Development of new
sequencing methods has allowed full genome sequencing of many different
organisms. It has become possible to look for individual genetic
differences that define predisposition to various diseases. At the same
time, new methods for genome engineering have been developed. Targeted
changes in the genome of living cells and organisms are powerful tools
and potential ways for therapy of genetic diseases. This field is
actively developing because of the large number of possible
applications. For example, gene function can be defined by inactivation
of a given gene and analysis of the consequences. Addition of an
affinity tag can be used for purification of a protein and its
partners, while addition of a fluorescent protein tag can facilitate
the analysis of tissue-specific expression and localization of a gene
product. By introducing a specific mutation in the genome of laboratory
animals, one can develop a model of human disease, which can be used
for optimization of treatment approaches. Finally, gene-modified
organisms are increasingly used in industry and agriculture.
In general, modifications of a genome comprise either inactivation of a gene (knockout), insertion of a foreign gene (knockin), or modification of a gene. Insertion of a foreign gene is mainly nonspecific; it can be inserted in any locus of a genome. Gene inactivation or modification is, on the contrary, more specific and should occur only in a certain locus of a genome. Nevertheless, this rule has some exceptions: sometimes, foreign genes are inserted into a chosen genome locus, where expression is efficient and does not have negative effects. Such methods as homology recombination [1-3], site-specific recombination [4], meganucleases [5], transposons [6, 7], or viral vectors [8, 9] have been widely used for genome engineering. Recently, methods using targeted cleavage of a chosen locus of a genome by new generation nucleases have become very popular.
METHODS OF NON-DIRECTED GENE INSERTION
Cell lines and transgenic animals constitutively expressing a foreign gene are usually generated using methods of random gene insertion, like direct injection of linear DNA in oocytes or viral vectors and transposons. Injection of linear DNA is the simplest, but not very efficient method, which often results in the insertion of multiple copies of a transgene that form concatemers [10]. Viral vectors are very useful for delivery of foreign DNA into cultured cells, but they have a number of drawbacks when working with transgenic animals as far as viruses are not able to get through the pellucid zone [11], while eggs without the pellucid zone dissociate easily to blastomeres [12]. Viral vectors can also have undesirable effects when used for gene therapy [13-16]. Generally, transposon-based vectors are used for insertion of various sequences into a genome (Fig. 1). Transposons are mobile elements capable of moving through a genome. DNA transposon consists of a gene coding for a transposase (an enzyme necessary for transposition) flanked by two inverted repeats (IR). Transposons move through the genome via the mechanism of cutting and insertion or copying and insertion. Integration into a genome occurs randomly. Some features of the transposition mechanism make transposons attractive as vectors for gene delivery. Only one protein – transposase – is necessary for integration both in vitro and in vivo. Transposase interacts with specific sequences of inverted repeats and assures excision and integration of DNA flanked by these repeats. Any gene can be inserted into a genome using a vector bearing a gene of interest flanked by two inverted repeats and a gene coding for transposase. The two components can be delivered into the cell either on separate vectors or within one.
Fig. 1. As soon as the transposase is expressed (oval), it binds to the inverted repeats of a transposon, inducing excision and further integration of a transgene.
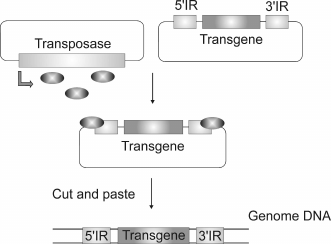
The only active transposons found in vertebrates – Tol2 transposons from the hAT family – were isolated from fish [17]. Two Tcl-like transposons – Sleeping Beauty and Frog Prince – were reconstructed from inactive transposons from fish [6] or frog [18] genomes, respectively. Transposon PiggyBac was identified when it was transferred spontaneously from the insect genome to the genome of a baculovirus [19]. Until now, Sleeping Beauty was the most frequently used system for transgenosis and random mutagenesis. Efficient integration and expression of a transgene using this system has been shown in fish, mouse, human, sheep, dog, cow, monkey, and rabbit cell cultures [20]. This transposon was also shown to be efficient in mouse [21] and human [22] embryonic stem cells.
Although viral vectors and transposons are quite efficient, in both cases insertion occurs randomly and not in a pre-chosen DNA locus. Such random genome changes can lead to undesirable consequences. An insertion can cause inactivation of a gene located near the insertion locus, or, to the contrary, can result in overexpression of a gene located nearby. This might trigger the development of cancer cells [23]. Moreover, these methods cannot be reproduced, since the probability that an insertion will occur in the same place of a genome is very low.
Directed modification of a genome fragment is possible using homologous recombination. For a long time, this method was efficiently applied only in several model organisms. Methods for modification of the genome of the yeast Saccharomyces cerevisiae were developed several decades ago [24, 25]. The success of yeast as a model organism resulted from a combination of convenient methods of DNA delivery into yeast cells, high probability of homologous recombination, and availability of selective markers for sorting of cells with insertion of foreign DNA.
Modification of the mouse genome became possible with the development of methods for manipulations with embryonic stem cells in culture, as well as methods for selection of clones with efficient integration [26]. Donor DNA for such manipulations should contain long flanking sequences homologous to the locus of insertion [27]. Markers for negative and positive selection increase the efficiency of the selection process [28]. Increasing the amounts of donor DNA has no positive effect; to the contrary, in mammalian cells it can considerably increase the nonhomologous integration [29].
Thus, directed mutagenesis of a chosen gene is not as easy. The main problem is that the undamaged target sequence is inert. The recombination level increases only after the target gene is damaged. Early experiments showed that DNA damaging agents stimulate homology recombination between sister chromatids [30]. It has been shown that natural recombination, including meiotic crossover [31] and the mating type switch in yeast [32], is initiated by double-stranded breaks in DNA. It was shown in 1990s that the frequency of homologous recombination could be increased by double-stranded DNA breaks in a site where recombination should take place [33, 34].
CONSEQUENCES OF DOUBLE-STRANDED DNA BREAKS
DNA breaks are regarded by a cell as potentially lethal damage, so one way to repair double-strand breaks is to use recombination with a homologous DNA sequence (Fig. 2).
Fig. 2. Double-stranded break (DSB) can be recovered via two pathways: error-prone NHEJ or homologous recombination (HR). NHEJ recovers double-stranded DNA breaks with random insertions and deletions. Homologous recombination occurs in the presence of a homologous template, resulting in precise gene correction or a precise insertion or gene replacement in case of artificial DNA template.
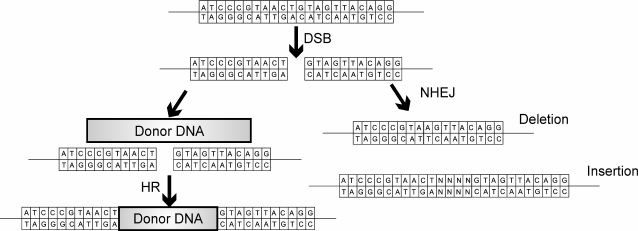
Homologous recombination. HDR (homology directed repair) is a way to recover from double-stranded DNA breaks using a homologous DNA template. The crucial step of HDR is the formation of a duplex composed of a damaged DNA and a complementary donor DNA. This interaction is catalyzed by RecA protein in bacteria and Rad51 protein in eukaryotes [35]. If compared to NHEJ (nonhomologous end joining), homologous recombination occurs generally in the late S/G2 phases of the cell cycle.
If a homologous sequence is absent, the double-stranded break can be repaired by NHEJ (Fig. 2), which can change the gene sequence, since this mechanism is error-prone [36, 37].
NHEJ. Nonhomologous end joining is a natural way of double-strand DNA break recovery through the joining of DNA ends. NHEJ is error-prone and can result in small deletions or insertions in the site of the damage. Such alterations can lead to a shift of an open reading frame, premature translation termination, and degradation of a transcript through the mechanism of recognition of premature stop codons (nonsense-mediated decay). NHEJ can occur during any phase of the cell cycle. In higher eukaryotes, NHEJ is a major mechanism for reparation of double-stranded DNA breaks [38, 39].
NHEJ can be Ku-dependent or Ku-independent. In the Ku-dependent process, the ends of DNA are protected by Ku70 and Ku80 proteins, which interact with DNA ends and recruit ligase IV and its cofactor. During NHEJ, single-stranded DNA ends hybridize with a short complementary sequence. In most cases, NHEJ occurs through hybridization of very short (1-4 nucleotides) overhangs [38].
The alternative Ku-independent pathway allows the restoration of DNA breaks without Ku proteins. The main Ku-independent NHEJ pathway is called microhomology-mediated end joining (MMEJ). MMEJ is based on homology sequences of 5-25 nucleotides. After hybridization of these sequences, extra nucleotides are removed and the gaps are filled in. Thus, this pathway allows longer deletions and insertions in the site of a break than the Ku-dependent NHEJ pathway does [40].
Both homologous recombination and nonhomologous end joining, when used with programmed nucleases, allow introducing changes in a given site of a genome, as, for example, a gene inactivation (knockout), insertion of a DNA fragment into a genome (knockin), or gene editing. Mutagenesis or gene replacement is carried out locally through a double-stranded DNA break, with subsequent repair via one of two pathways.
Low molecular weight substances showed rather low efficiency of double-stranded DNA break induction [41-43]. Nucleases with flexible specificity turned out to be the most efficient.
NUCLEASES USED FOR GENOME EDITING
Double-stranded DNA breaks can now be induced in a given DNA fragment by programmable DNA-binding zinc-finger proteins (ZF) [44] and TALE (transcription activator-like effector) [45], as well as the CRISPR (clustered regularly interspaced short palindromic repeat) system/CRISPR-associated protein 9 nuclease (Cas9) [46]. The latter system consists of the prokaryotic protein Cas9 and small guide RNA, which helps the nuclease to distinguish and cut the desired DNA sequence.
Zinc finger nucleases. Zinc fingers are small natural DNA-binding domains that are quite frequent in transcription factors. Relatively simple rules of DNA recognition by zinc fingers and the possibility of using several consecutive protein domains that recognize a continuous DNA sequence allow creation of artificial DNA-binding modules based on zinc fingers. Proteins composed of “programmable” DNA binding and “constant” endonuclease parts were the first representatives of a new generation of tools for directed genome modification. Type IIS restriction nucleases, for example FokI, are known to recognize short DNA sequences and to introduce a double-stranded break at some distance from the recognition site. This particularity is due to the fact that DNA-binding and nuclease domains of these proteins are separated and can function independently [47]. The nuclease domain has no obvious sequence-specificity. The site of the break can be changed by changing the specificity of the DNA-binding domain [48].
The most convenient zinc fingers used for generation of site-directed nucleases were those of the Cys2His2 family, so-called after four amino acids that coordinate a zinc atom. Each zinc finger is rather small, about 30 amino acids (a.a.); its secondary structure consists of one α-helix and two short β-sheets. The crystal structure of three zinc finger domains bound to DNA has shown that each domain recognizes specifically a three-nucleotide sequence [49].
Zinc fingers are widespread among eukaryotic transcription factors, including those of humans, and the specificity of some have been identified [50]. Many genetically engineered zinc fingers have been prepared [51, 52]. A number of new proteins were made that interact with all 5′-GNN triplets, where N is any base [53], or with several 5′-ANN and 5′-CNN triplets [54, 55]. Companies such as Gendaq, Ltd. and Sangamo Biosciences have a large database of efficient zinc fingers [56].
One of the special features of site-directed zinc finger endonucleases is that the FokI nuclease domain must dimerize for DNA cleavage to occur [57, 58]. Therefore, to induce one double-stranded DNA break, two proteins have to be designed, both containing DNA-binding and nuclease domains. Sites recognized by DNA-binding domains have to be located in the opposite DNA strands, and have to be inverted and be separated by a short fragment (Fig. 3). Upon interaction of DNA-binding domains with their targets, two nuclease domains become located rather close to each other to dimerize and induce DNA cleavage. The optimal distance between the zinc finger inverted binding sites is 5-7 nucleotides (n.) [59, 60].
Fig. 3. ZFN pair bound to DNA. Zinc finger nucleases consist of zinc finger binding domains (ZF) (shown as ovals) and nuclease domain FokI (shown as lozenge). Each ZF interacts with three nucleotides. Contacts of binding domains with DNA are shown as vertical lines. On average, 3-4 ZF are needed to recognize 9-12-n. sequence. Two nucleases are needed to induce a double-stranded DNA break because the nuclease domain has to form a dimer to be active.
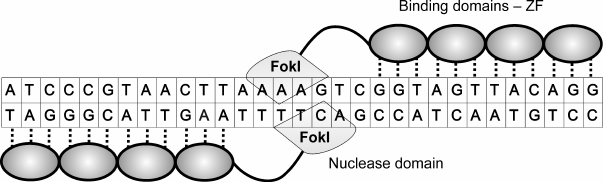
The need for dimerization of nuclease domains increases the accuracy of recognition as it doubles the length of the recognized sequence. In fact, two DNA-binding domains, each composed of three zinc fingers, recognize a sequence of 18 n., which is sufficient for cutting of one target even in a large genome.
The first genetically engineered enzyme composed of a set of zinc fingers connected to a nuclease domain was shown to be active in vitro in 1996 [61]. High efficiency of digestion and recombination was achieved upon injection of two nucleases and a DNA template in Xenopus oocytes [59]. The ZFN (zinc finger nuclease) pair designed de novo was used successfully to modify the drosophila genome [37]. Starting from that point, many ZFN pairs were designed and successfully used for modifications of individual genes in various organisms and cell lines (Table 1). Though modification efficiency varies, the average value is about 10%.
Table 1. Using of ZFN for editing of various
genomes
Despite all advantages, genome modification using zinc finger nucleases is a time-consuming process. Actually, most such proteins are not working [62, 63]. Having access to the database of zinc fingers, one can chose a set of proteins that will specifically interact with a given DNA sequence [63-65]. At the same time, it is possible that a protein that binds its target efficiently in certain conditions will not bind with the same efficiency under other conditions [62]. At least three binding domains are needed to provide sufficient affinity, but not all proteins bind DNA with the same efficiency. Additional domains can be added to increase the affinity, although it can decrease the activity [66]. Additional binding domains allow increasing not only the affinity, but also the specificity, which also plays an important role in overall performance of this system. Shortening of the distance between binding domains also improved specificity [67].
Finally, any gene in any organism can be modified using a correctly chosen pair of zinc finger nucleases. Everything depends on the interaction of a DNA-binding domain with a target sequence and on the DNA repair mechanism. Protein pairs that function well together have been selected and registered in the database [68, 69] and can be used in the future.
TALEN. Transcription activator-like effector nucleases (TALENs) are a new generation of “programmable” modular DNA-binding proteins. Like ZFN, TALEN proteins are composed of DNA-binding domains derived from TALE proteins and a nonspecific FokI endonuclease domain [70].
TALE protein is a virulence factor from bacteria of the Xanthomonas family that infects plants. TALE proteins consist of several identical domains that have some structural particularities: a signal of nuclear localization, N-terminal translocation signal, acid active domain, and central repeating DNA-binding domain. TALE proteins differ only by the number and sequence of their DNA-binding domains [71]. Each repeat consists of 34-35 a.a. Amino acids in positions 12 and 13 represent a variable site (repeat variable diresidue, RVD) that defines the specificity of the DNA binding domain to one nucleotide [71, 72]. Bioinformatic analysis and practical experiments have shown the mutual correspondence between RVD of TALE proteins and nucleotides in a target DNA sequence. Various combinations of amino acids in the RVD can play a role in recognition of one or more nucleotides. For example, the asparagine–isoleucine pair (NI) corresponds to A; histidine–aspartic acid (HD) corresponds to C; asparagine–glycine (NG) – to T; asparagine–asparagine (NN) – to A and G; asparagine–serine (NS) – to A, C, G, and T; asparagine–lysine (NK) – to G [73].
Many laboratories have shown that TALENs are as efficient as ZFNs in cutting of the same genome DNA sequences [74, 75]. One advantage of TALENs compared to ZFNs is their lower cytotoxicity [74, 75]. Some data shows that TALENs have higher specificity, which means that they induce fewer off-target breaks and stimulate higher frequency of homologous recombination compared to other nucleases, including CRISPR-Cas9 [76, 77].
Similar to ZFNs, TALENs can be designed to act on any target DNA (Fig. 4). TALEN genes are easier to manipulate than zinc finger nucleases. Generally, TALEN proteins that can recognize a sequence of 18-20 n. are used. Increasing the number of DNA-binding domains in TALENs can result in lower specificity [78]. Generation of nucleases with a large number of repeating units is difficult because of recombination, which can be stimulated by high sequence similarities of individual TALE domains [79]. Several methods of TALE assembly are used [80-83]. It should be noted that the gene coding for a TALEN is three times larger than one coding for a ZFN. Since a TALE protein is almost as large as a ZF but recognizes only one base instead of three, one can assume that the final construct will be rather large. This raises problems with the delivery of such constructs into cells. Because of the large number of TALE protein repeats, viral delivery to a mammalian genome is also problematic [79].
Fig. 4. Pair of TALENs bound to DNA. TALENs consist of TALE binding domains (shown as vertical ovals) and FokI nuclease domain (shown as lozenges). Specificity of a binding domain is defined by a hypervariable RVD motif. Each TALE domain interacts with one nucleotide; contacts of binding domains with DNA are shown as vertical lines. Two nucleases are needed to induce a double-stranded DNA break because the nuclease domain has to form a dimer to be active.
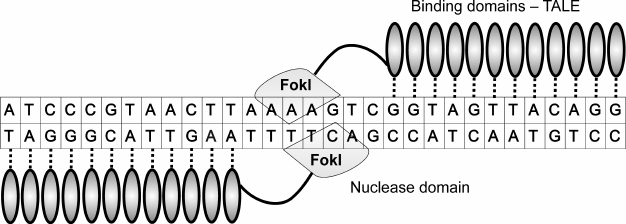
Like other nucleases, a TALEN introduces off-target mutations through breaks in DNA sequences partially similar to the target sequence [84]. This problem can be overcome by choosing a unique site that differs from all other sites in the genome, at least in 7 n. [85]. Such a site can be chosen using the website www.talenlibrary.net.
TALEN technology has been successfully used for modification of various organisms and cell cultures (Table 2).
Table 2. Use of TALENs for editing of
various genomes

Before TALENs started to be widely used as good alternatives to zinc finger nucleases, the CRISPR-Cas9 system came to the stage.
CRISPR-Cas9. Generation of genetic constructs coding for ZFNs and TALENs is rather expensive and time-consuming. Discovery of the CRISPR-Cas9 system significantly simplified genome modification. It is now the most popular system for genome editing because it is efficient and simple in use.
The CRISPR-Cas9 system is an adaptive immune system of bacteria and archaea against phages or plasmids [86, 87]. A cell “memorizes” a genome sequence of a phage that has infected it. A fragment of heterologous DNA about 20 n. (called a spacer) is taken and inserted into the genome of the bacterium or archaean to elongate the CRISPR cassette. The type II CRISPR system is used for artificial genome editing. In this system, DNA digestion occurs through interaction of CRISPR RNA (crRNA), which is generated from the spacer; trans-activating CRISPR RNA (tracrRNA) necessary for the formation of mature CRISPR RNA [88], and endonuclease CRISPR-associated Cas9 protein. Two RNAs with Cas9 form active endonuclease, which cuts the DNA protospacer corresponding to the spacer with a small three-nucleotide fragment called PAM (protospacer adjacent motif). Currently, one chimeric guide RNA composed of crRNA and tracrRNA is used for genome editing [89], which simplifies even more the use of the CRISPR system.
The presence of a PAM motif is a prerequisite for choosing a site of cleavage. In the Streptococcus pyogenes system (Fig. 5), the source of the first genetically engineered CRISPR-Cas9 system, the endonuclease recognizes a target sequence of 23 n. composed of 20 bases of a guide sequence, corresponding to crRNA (protospacer), and 5′-NGG-3′ (or 5′-NAG-3′ to a lesser extent) [90-92]. The PAM sequence is recognized by Cas9 endonuclease [93] and helps to distinguish between the native sequence of DNA spacer and the heterologous protospacer sequence. Cas9 proteins from other organisms recognize other PAM motifs [94-97]. The PAM site imposes some limitations when choosing a site for a DNA break.
Fig. 5. The CRISPR-Cas9 system induces double-strand breaks in genomic DNA. Cas9 that is bound to guide RNA (composed of crRNA and tracrRNA) interacts with a 20-n. sequence.
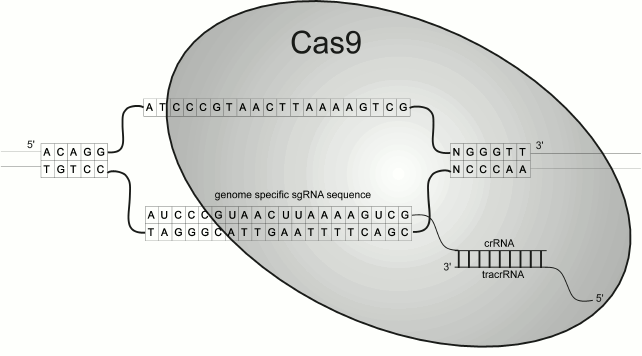
Specificity of this system is defined by the small guide RNA and not by the protein as in case of ZFNs and TALENs. One advantage of this system compared to ZFNs and TALENs is the ability to digest methylated DNA [90].
Another important advantage of this system relative to ZFNs and TALENs is simplicity. It does not require generation of complicated genetically engineered constructs coding for modular DNA-binding proteins. Since Cas9 is an invariable component, the new construct can be obtained by simple cloning a short fragment corresponding to the guide RNA [94, 98]. Such simplicity allows generating a huge set of vectors for a large number of targets, including whole genome libraries [99-101].
It is relatively simple to introduce multiple changes in a genome using the CRISPR-Cas9 system, one just needs to clone several guide RNAs in one vector. This method was used to create animals [102, 103] and plants [104] (Table 3) with knockout of several genes simultaneously. In the case of ZFN and TALEN systems, two constructs are needed to generate one double-stranded DNA break [105]. Not only DNA coding for the components of the CRISPR-Cas9 system can be used, but also recombinant Cas9 protein or Cas9 mRNA and in vitro transcribed guide RNA. This approach is safer for therapeutic purposes [106, 107] and is more convenient in case of microinjections in fertilized oocytes.
Table 3. Use of CRISPR-Cas9 for editing of
various genomes

The biggest disadvantage of the CRISPR-Cas9 system is probably the rather high frequency of undesirable DNA breaks in sites partially complementary to the guide RNA. Several approaches have been developed to overcome this problem. One consists of decreasing the Cas9 protein activity [90, 108, 109]. Another possibility is to use Cas9 nickase, which cuts only one DNA strand. In this case two guide RNAs are needed to direct cleavage of two complementary strands [92, 110]. Thus, the actual sequence that defines where to cut DNA is two times longer, which increases considerably the specificity of the cleavage. Another way to improve specificity of the system is to create a hybrid Cas9 protein bearing a FokI nuclease domain. This approach combines the simplicity of generating the addressing part, based on guide RNA, and the need to use two addressing blocks specific for FokI nuclease [78, 111]. In this system, the FokI nuclease domain is fused with catalytically inactive Cas9 protein (dCas9). Two guide RNAs are needed to induce a double-stranded break, since the FokI domain has to form a dimer to cut the DNA as in the case of ZFNs and TALENs. Thus, the specificity of digestion increases, leading to a decreased frequency of undesired mutations.
Researchers also managed to improve the Cas9 protein specificity by means of mutagenesis [112]. The crystal structure of S. pyogenes Cas9 protein (SpCas9) in complex with guide RNA showed that a positively charged groove located between HNH, RuvC, and PAM-interacting domains is involved in interaction of Cas9 protein with DNA. It was assumed that neutralization of a positive charge in this locus would increase the Cas9 protein specificity, which was shown to be true in a triple mutant [112].
Comparison of nucleases used for genome editing. Each nuclease has advantages and disadvantages as described below and summarized in Table 4. The majority of ZF and TALE nucleases are not functional or have very low specificity. Despite continuous progress in this field [63, 68, 69], commercially available proteins work better in general. The efficiency of mutagenesis using TALENs varies from 1 to 60% [82, 85], while the efficiency when using a CRISPR-Cas9 system varies from 2.3 to 79% [94, 113, 114]. It is not possible to predict in advance how functional and efficient each system will be. Efficiency and accuracy of each system depends on several factors including the cell type and delivery method.
Table 4. Comparative analysis of nucleases
used for genome editing
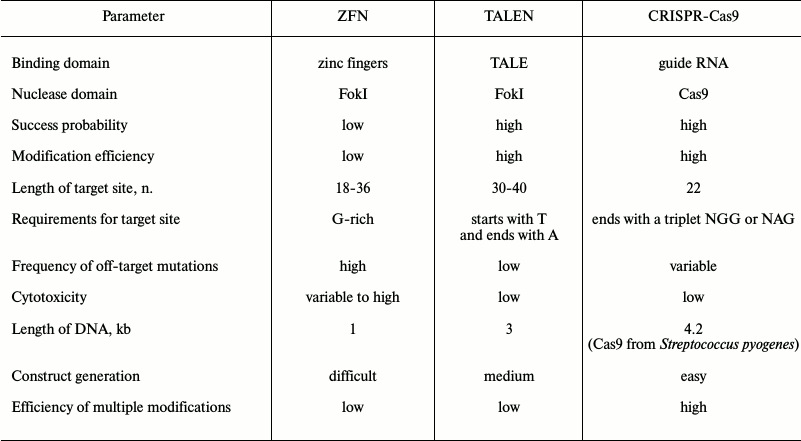
Specificity of nucleases can be improved by changing the number of binding domains. Proteins containing more binding domains recognize longer DNA sequences, which results in higher efficiency and specificity. Nevertheless, higher affinity to a target DNA can also lead to an increase in the number of potential partially corresponding off-targets. Nucleases forming dimers, such as nucleases containing a FokI nuclease domain or Cas9 nickase, have higher specificity. Theoretically, recognition sites longer than 16 n. (416 = 4.3·109) should appear, on average, once in a genome such as the human genome (3.2·109). Nevertheless, all three nucleases being used cause off-target mutations [84, 90, 115, 116], which explains their cytotoxicity [117].
TALEN and CRISPR-Cas9 systems have lower cytotoxicity compared to ZFNs. Among all nucleases, CRISPR-Cas9 is the simplest and most convenient in use. However, it was shown to induce many off-target mutations [90, 92, 118]. Cas9 protein can cut sequences that differ in 5 n. from a target sequence, which means that in the human genome there are thousands of potential off-target sequences. Moreover, such off-target interactions can lead to various chromosome reorganizations [119]. The CRISPR-Cas9 system is extremely insensitive to noncomplementary nucleotides in the 5′-terminus of the guide RNA, located far from the PAM site. This particularity of the CRISPR-Cas9 system can be explained by the fact that bacteria and archaea need to protect themselves from hypervariable DNA sequences of infecting agents.
Although zinc finger nucleases and TALENs have the same nuclease domain, they introduce different off-target mutations. It turned out that TALENs cause deletions more often than insertions (89 against 1.6% in mammalian cells), while ZFNs cause deletions and insertions with the same frequency [120]. According to some data, the CRISPR-Cas9 system causes mainly one- or two-nucleotide insertions [114].
Limitations for target site choice. Although each ZFN DNA-binding domain recognizes 3 n., and an open database of nucleases able to recognize all possible triplet combinations is available [53, 121], the choice of a target site for a DNA break is limited. Zinc finger nucleases recognize efficiently a target sequence if the latter is guanine-rich and consists of 5′-GNN-3′ triplets. Thus, one potential site efficiently recognized by zinc fingers occurs on average each 100 n. [50]. Various programs helping to search for potential recognition sites for a ZFN in a given sequence are available.
A target site for TALENs requires only the presence of a thymine on the 5′-terminus, which is recognized by two N-terminal repeats [122]. However, new TALE variants able to recognize other bases on the 5′-terminus of a target sequence were recently developed [123, 124]. Ordinary TALENs are not able to cut sequences containing methylated cytosine [85, 125]. For recognition of methylated cytosine, one can use a TALE domain recognizing thymine and containing the RVD Asn-Gly [126, 127].
The CRISPR-Cas9 system has two limitations for the target cleavage site. First, the PAM sequence recognized by Cas9 is either 5′-NGG-3′ or 5′-NAG-3′. Second, there should be a guanine on the 5′-end, since the guide RNA is transcribed by RNA polymerase III from the U6 promoter. RNA transcripts without 5′-terminal guanine will be poorly synthesized in the cell. Addition of one or several guanines to the 5′-end of the guide RNA can solve this problem [114, 119], since this fragment of the guide RNA is not crucial for recognition specificity. Generally, a PAM sequence occurs every 8 n. Unlike zinc finger nucleases and TALENs, the CRISPR-Cas9 system can cut methylated DNA [90]. Efficiency of cleavage varies for different DNA target sites; it is not always possible to predict what potential site will be efficiently cleaved [128].
Multiple genome modifications. It is possible to modify several genes simultaneously using all three nucleases. In case of ZFNs and TALENs, it is necessary to keep in mind that wrong pairs of dimers can be formed if more than two nucleases are introduced into the cell. This increases the frequency of off-target mutations [105]. The CRISPR-Cas9 system does not have such problems, as Cas9 is constant and works as a monomer. Simultaneous inactivation of up to four targets was described in literature [129].
DELIVERY OF PROGRAMMABLE NUCLEASES
One of the steps crucial for overall modification efficiency is the delivery of nucleases into cells. Nucleases can be introduced into a cell as a vector coding for a protein, as in vitro transcribed mRNA, or as a protein. Generally, DNA is delivered into cells in culture by electroporation or using a liposome transfecting agent [130, 131]. In vitro transcribed mRNA coding for nucleases ZFN, TALEN, or Cas9, and guide RNA, are usually injected in oocytes to obtain knockout or knockin animals [102, 132, 133]. Direct injection of mRNA results in faster expression compared to transfection with DNA and helps to avoid undesirable integration of vector DNA into the genome.
Non-integrating viral vectors are also used for delivery. Integrase deficient lentivirus vectors (IDLVs), adenoviruses, and adeno-associated viruses (AAV) can deliver genes coding nucleases, both in vitro and in vivo. ZFNs are the most compact representatives of this class of modifying agents. DNA coding for nucleases can be packed into a small AAV vector [134], which is widely used for gene therapy. IDLVs were successfully used for simultaneous delivery of a ZFN with a homologous template in hemopoietic and embryonic stem cells [135]. Unfortunately, when using IDLVs for delivery of a TALEN into cells, undesirable recombination can occur because of high similarity of DNA-binding domain sequences [79].
For stable expression of a Cas9 protein and a guide RNA in mammalian cells, an integrating viral vector has been used [101, 136]. A high level of nuclease Cas9 expression leads to increase in the number of off-target mutations. When a Cas9 protein is used instead of Cas9 protein-coding DNA, gene coding for the nuclease is not integrated into the genome. Protein degradation also limits the time when a protein is functional, therefore reducing the level of off-target mutations [137, 138].
Directed genome editing technologies have become simpler and more available to researchers. Double-stranded breaks generated by specially designed nucleases facilitate the process of genome editing. Zinc finger nucleases – the first representatives of this technology – have been developed and improved for 20 years. The next generation TALEN and CRISPR-Cas9 systems were also improved. Nevertheless, some aspects of these technologies, including efficiency, decrease of off-target mutations, constructs generation, and delivery can be improved. All modifications are aimed to increase the overall system efficiency and safety for therapeutic approaches for genetic diseases. More precise genome editing without off-target effects will allow manipulating a genome of a living organism without serious consequences. Programmable nucleases with improved efficiency and specificity will open a new era in biological research, medicine, and biotechnology.
This work was done with financial support from the Russian Science Foundation (project No. 14-14-00072) and the Russian Foundation for Basic Research (projects Nos. 16-04-01100, 14-04-01061).
REFERENCES
1.Hinnen, A., Hicks, J. B., and Fink, G. R. (1978)
Transformation of yeast, Proc. Natl. Acad. Sci. USA, 75,
1929-1933.
2.Szostak, J. W., Orr-Weaver, T. L., Rothstein, R.
J., and Stahl, F. W. (1983) The double-strand-break repair model for
recombination, Cell, 33, 25-35.
3.Thomas, K. R., Folger, K. R., and Capecchi, M. R.
(1986) High frequency targeting of genes to specific sites in the
mammalian genome, Cell, 44, 419-428.
4.Akagi, K., Sandig, V., Vooijs, M., Van der Valk,
M., Giovannini, M., Strauss, M., and Berns, A. (1997) Cre-mediated
somatic site-specific recombination in mice, Nucleic Acids Res.,
25, 1766-1773.
5.Epinat, J. C., Arnould, S., Chames, P., Rochaix,
P., Desfontaines, D., Puzin, C., Patin, A., Zanghellini, A., Paques,
F., and Lacroix, E. (2003) A novel engineered meganuclease induces
homologous recombination in yeast and mammalian cells, Nucleic Acids
Res., 31, 2952-2962.
6.Ivics, Z., Hackett, P. B., Plasterk, R. H., and
Izsvak, Z. (1997) Molecular reconstruction of Sleeping Beauty, a
Tc1-like transposon from fish, and its transposition in human cells,
Cell, 91, 501-510.
7.Kawakami, K., Shima, A., and Kawakami, N. (2000)
Identification of a functional transposase of the Tol2 element, an
Ac-like element from the Japanese medaka fish, and its transposition in
the zebrafish germ lineage, Proc. Natl. Acad. Sci. USA,
97, 11403-11408.
8.Lois, C., Hong, E. J., Pease, S., Brown, E. J., and
Baltimore, D. (2002) Germline transmission and tissue-specific
expression of transgenes delivered by lentiviral vectors,
Science, 295, 868-872.
9.Khan, I. F., Hirata, R. K., and Russell, D. W.
(2011) AAV-mediated gene targeting methods for human cells, Nat.
Protoc., 6, 482-501.
10.Palmiter, R. D., and Brinster, R. L. (1986)
Germ-line transformation of mice, Annu. Rev. Genet., 20,
465-499.
11.Soriano, P., Cone, R. D., Mulligan, R. C., and
Jaenisch, R. (1986) Tissue-specific and ectopic expression of genes
introduced into transgenic mice by retroviruses, Science,
234, 1409-1413.
12.Brown, G. A., and Corbin, T. J. (2002) Methods
in Molecular Biology (Clarke, A. R., ed.) Humana Press Inc, Totowa,
New Jersey, pp. 39-70.
13.Aiuti, A., and Roncarolo, M. G. (2009) Ten years
of gene therapy for primary immune deficiencies, Hematol. Am. Soc.
Hematol. Educ. Program, 682-689.
14.Baum, C., Von Kalle, C., Staal, F. J., Li, Z.,
Fehse, B., Schmidt, M., Weerkamp, F., Karlsson, S., Wagemaker, G., and
Williams, D. A. (2004) Chance or necessity? Insertional mutagenesis in
gene therapy and its consequences, Mol. Ther., 9,
5-13.
15.Raper, S. E., Chirmule, N., Lee, F. S., Wivel, N.
A., Bagg, A., Gao, G. P., Wilson, J. M., and Batshaw, M. L. (2003)
Fatal systemic inflammatory response syndrome in a ornithine
transcarbamylase deficient patient following adenoviral gene transfer,
Mol. Genet. Metab., 80, 148-158.
16.Mingozzi, F., and High, K. A. (2011) Immune
responses to AAV in clinical trials, Curr. Gene Ther.,
11, 321-330.
17.Koga, A., Suzuki, M., Inagaki, H., Bessho, Y.,
and Hori, H. (1996) Transposable element in fish, Nature,
383, 30.
18.Miskey, C., Izsvak, Z., Plasterk, R. H., and
Ivics, Z. (2003) The Frog Prince: a reconstructed transposon from
Rana pipiens with high transpositional activity in vertebrate
cells, Nucleic Acids Res., 31, 6873-6881.
19.Fraser, M. J., Brusca, J. S., Smith, G. E., and
Summers, M. D. (1985) Transposon-mediated mutagenesis of a baculovirus,
Virology, 145, 356-361.
20.Izsvak, Z., Ivics, Z., and Plasterk, R. H. (2000)
Sleeping Beauty, a wide host-range transposon vector for genetic
transformation in vertebrates, J. Mol. Biol., 302,
93-102.
21.Luo, G., Ivics, Z., Izsvak, Z., and Bradley, A.
(1998) Chromosomal transposition of a Tc1/mariner-like element in mouse
embryonic stem cells, Proc. Natl. Acad. Sci. USA, 95,
10769-10773.
22.Orban, T. I., Apati, A., Nemeth, A., Varga, N.,
Krizsik, V., Schamberger, A., Szebenyi, K., Erdei, Z., Varady, G.,
Karaszi, E., Homolya, L., Nemet, K., Gocza, E., Miskey, C., Mates, L.,
Ivics, Z., Izsvak, Z., and Sarkadi, B. (2009) Applying a
“double-feature” promoter to identify cardiomyocytes
differentiated from human embryonic stem cells following
transposon-based gene delivery, Stem Cells, 27,
1077-1087.
23.Hacein-Bey-Abina, S., Von Kalle, C., Schmidt, M.,
Le Deist, F., Wulffraat, N., McIntyre, E., Radford, I., Villeval, J.
L., Fraser, C. C., Cavazzana-Calvo, M., and Fischer, A. (2003) A
serious adverse event after successful gene therapy for X-linked severe
combined immunodeficiency, N. Engl. J. Med., 348,
255-256.
24.Rothstein, R. J. (1983) One-step gene disruption
in yeast, Methods Enzymol., 101, 202-211.
25.Scherer, S., and Davis, R. W. (1979) Replacement
of chromosome segments with altered DNA sequences constructed in
vitro, Proc. Natl. Acad. Sci. USA, 76, 4951-4955.
26.Capecchi, M. R. (2005) Gene targeting in mice:
functional analysis of the mammalian genome for the twenty-first
century, Nat. Rev. Genet., 6, 507-512.
27.Capecchi, M. R. (1989) Altering the genome by
homologous recombination, Science, 244, 1288-1292.
28.Mansour, S. L., Thomas, K. R., and Capecchi, M.
R. (1988) Disruption of the proto-oncogene int-2 in mouse
embryo-derived stem cells: a general strategy for targeting mutations
to non-selectable genes, Nature, 336, 348-352.
29.Vasquez, K. M., Marburger, K., Intody, Z., and
Wilson, J. H. (2001) Manipulating the mammalian genome by homologous
recombination, Proc. Natl. Acad. Sci. USA, 98,
8403-8410.
30.Latt, S. A. (1981) Sister chromatid exchange
formation, Annu. Rev. Genet., 15, 11-55.
31.Youds, J. L., and Boulton, S. J. (2011) The
choice in meiosis – defining the factors that influence
crossover or non-crossover formation, J. Cell Sci., 124,
501-513.
32.Haber, J. E. (2012) Mating-type genes and MAT
switching in Saccharomyces cerevisiae, Genetics,
191, 33-64.
33.Rouet, P., Smih, F., and Jasin, M. (1994)
Expression of a site-specific endonuclease stimulates homologous
recombination in mammalian cells, Proc. Natl. Acad. Sci. USA,
91, 6064-6068.
34.Smih, F., Rouet, P., Romanienko, P. J., and
Jasin, M. (1995) Double-strand breaks at the target locus stimulate
gene targeting in embryonic stem cells, Nucleic Acids Res.,
23, 5012-5019.
35.Sarbajna, S., and West, S. C. (2014) Holliday
junction processing enzymes as guardians of genome stability, Trends
Biochem. Sci., 39, 409-419.
36.Rouet, P., Smih, F., and Jasin, M. (1994)
Introduction of double-strand breaks into the genome of mouse cells by
expression of a rare-cutting endonuclease, Mol. Cell. Biol.,
14, 8096-8106.
37.Bibikova, M., Golic, M., Golic, K. G., and
Carroll, D. (2002) Targeted chromosomal cleavage and mutagenesis in
Drosophila using zinc-finger nucleases, Genetics,
161, 1169-1175.
38.Lieber, M. R. (2010) The mechanism of
double-strand DNA break repair by the nonhomologous DNA end-joining
pathway, Annu. Rev. Biochem., 79, 181-211.
39.Puchta, H. (2005) The repair of double-strand
breaks in plants: mechanisms and consequences for genome evolution,
J. Exp. Bot., 56, 1-14.
40.McVey, M., and Lee, S. E. (2008) MMEJ repair of
double-strand breaks (director’s cut): deleted sequences and
alternative endings, Trends Genet., 24, 529-538.
41.Chin, J. Y., and Glazer, P. M. (2009) Repair of
DNA lesions associated with triplex-forming oligonucleotides, Mol.
Carcinog., 48, 389-399.
42.Doss, R. M., Marques, M. A., Foister, S.,
Chenoweth, D. M., and Dervan, P. B. (2006) Programmable oligomers for
minor groove DNA recognition, J. Am. Chem. Soc., 128,
9074-9079.
43.Kim, K. H., Nielsen, P. E., and Glazer, P. M.
(2006) Site-specific gene modification by PNAs conjugated to psoralen,
Biochemistry, 45, 314-323.
44.Carroll, D. (2011) Genome engineering with
zinc-finger nucleases, Genetics, 188, 773-782.
45.Chandrasegaran, S., and Carroll, D. (2015)
Origins of programmable nucleases for genome engineering, J. Mol.
Biol., 428, 963-989.
46.Doudna, J. A., and Charpentier, E. (2014) Genome
editing. The new frontier of genome engineering with CRISPR-Cas9,
Science, 346, 1258096.
47.Li, L., Wu, L. P., and Chandrasegaran, S. (1992)
Functional domains in Fok I restriction endonuclease, Proc. Natl.
Acad. Sci. USA, 89, 4275-4279.
48.Kim, Y. G., and Chandrasegaran, S. (1994)
Chimeric restriction endonuclease, Proc. Natl. Acad. Sci. USA,
91, 883-887.
49.Pavletich, N. P., and Pabo, C. O. (1991) Zinc
finger-DNA recognition: crystal structure of a Zif268-DNA complex at
2.1 Å, Science, 252, 809-817.
50.Kim, H. J., Lee, H. J., Kim, H., Cho, S. W., and
Kim, J. S. (2009) Targeted genome editing in human cells with zinc
finger nucleases constructed via modular assembly, Genome Res.,
19, 1279-1288.
51.Desjarlais, J. R., and Berg, J. M. (1992) Toward
rules relating zinc finger protein sequences and DNA binding site
preferences, Proc. Natl. Acad. Sci. USA, 89,
7345-7349.
52.Desjarlais, J. R., and Berg, J. M. (1993) Use of
a zinc-finger consensus sequence framework and specificity rules to
design specific DNA binding proteins, Proc. Natl. Acad. Sci.
USA, 90, 2256-2260.
53.Segal, D. J., Dreier, B., Beerli, R. R., and
Barbas, C. F., 3rd. (1999) Toward controlling gene expression at will:
selection and design of zinc finger domains recognizing each of the
5′-GNN-3′ DNA target sequences, Proc. Natl. Acad. Sci.
USA, 96, 2758-2763.
54.Dreier, B., Beerli, R. R., Segal, D. J., Flippin,
J. D., and Barbas, C. F., 3rd. (2001) Development of zinc finger
domains for recognition of the 5′-ANN-3′ family of DNA
sequences and their use in the construction of artificial transcription
factors, J. Biol. Chem., 276, 29466-29478.
55.Dreier, B., Fuller, R. P., Segal, D. J., Lund, C.
V., Blancafort, P., Huber, A., Koksch, B., and Barbas, C. F., 3rd.
(2005) Development of zinc finger domains for recognition of the
5′-CNN-3′ family DNA sequences and their use in the
construction of artificial transcription factors, J. Biol.
Chem., 280, 35588-35597.
56.Carroll, D. (2014) Genome engineering with
targetable nucleases, Annu. Rev. Biochem., 83,
409-439.
57.Bitinaite, J., Wah, D. A., Aggarwal, A. K., and
Schildkraut, I. (1998) FokI dimerization is required for DNA cleavage,
Proc. Natl. Acad. Sci. USA, 95, 10570-10575.
58.Smith, J., Bibikova, M., Whitby, F. G., Reddy, A.
R., Chandrasegaran, S., and Carroll, D. (2000) Requirements for
double-strand cleavage by chimeric restriction enzymes with zinc finger
DNA-recognition domains, Nucleic Acids Res., 28,
3361-3369.
59.Bibikova, M., Carroll, D., Segal, D. J.,
Trautman, J. K., Smith, J., Kim, Y. G., and Chandrasegaran, S. (2001)
Stimulation of homologous recombination through targeted cleavage by
chimeric nucleases, Mol. Cell. Biol., 21, 289-297.
60.Shimizu, Y., Bhakta, M. S., and Segal, D. J.
(2009) Restricted spacer tolerance of a zinc finger nuclease with a six
amino acid linker, Bioorg. Med. Chem. Lett., 19,
3970-3972.
61.Kim, Y. G., Cha, J., and Chandrasegaran, S.
(1996) Hybrid restriction enzymes: zinc finger fusions to FokI cleavage
domain, Proc. Natl. Acad. Sci. USA, 93, 1156-1160.
62.Ramirez, C. L., Foley, J. E., Wright, D. A.,
Muller-Lerch, F., Rahman, S. H., Cornu, T. I., Winfrey, R. J., Sander,
J. D., Fu, F., Townsend, J. A., Cathomen, T., Voytas, D. F., and Joung,
J. K. (2008) Unexpected failure rates for modular assembly of
engineered zinc fingers, Nat. Methods, 5, 374-375.
63.Kim, J. S., Lee, H. J., and Carroll, D. (2010)
Genome editing with modularly assembled zinc-finger nucleases, Nat.
Methods, 7, 91; author reply 91-92.
64.Carroll, D., Morton, J. J., Beumer, K. J., and
Segal, D. J. (2006) Design, construction and in vitro testing of
zinc finger nucleases, Nat. Protoc., 1, 1329-1341.
65.Gonzalez, B., Schwimmer, L. J., Fuller, R. P.,
Ye, Y., Asawapornmongkol, L., and Barbas, C. F., 3rd. (2010) Modular
system for the construction of zinc-finger libraries and proteins,
Nat. Protoc., 5, 791-810.
66.Shimizu, Y., Sollu, C., Meckler, J. F.,
Adriaenssens, A., Zykovich, A., Cathomen, T., and Segal, D. J. (2011)
Adding fingers to an engineered zinc finger nuclease can reduce
activity, Biochemistry, 50, 5033-5041.
67.Moore, M., Klug, A., and Choo, Y. (2001) Improved
DNA binding specificity from polyzinc finger peptides by using strings
of two-finger units, Proc. Natl. Acad. Sci. USA, 98,
1437-1441.
68.Gupta, A., Christensen, R. G., Rayla, A. L.,
Lakshmanan, A., Stormo, G. D., and Wolfe, S. A. (2012) An optimized
two-finger archive for ZFN-mediated gene targeting, Nat.
Methods, 9, 588-590.
69.Sander, J. D., Dahlborg, E. J., Goodwin, M. J.,
Cade, L., Zhang, F., Cifuentes, D., Curtin, S. J., Blackburn, J. S.,
Thibodeau-Beganny, S., Qi, Y., Pierick, C. J., Hoffman, E., Maeder, M.
L., Khayter, C., Reyon, D., Dobbs, D., Langenau, D. M., Stupar, R. M.,
Giraldez, A. J., Voytas, D. F., Peterson, R. T., Yeh, J. R., and Joung,
J. K. (2011) Selection-free zinc-finger-nuclease engineering by
context-dependent assembly (CoDA), Nat. Methods, 8,
67-69.
70.Miller, J. C., Tan, S., Qiao, G., Barlow, K. A.,
Wang, J., Xia, D. F., Meng, X., Paschon, D. E., Leung, E., Hinkley, S.
J., Dulay, G. P., Hua, K. L., Ankoudinova, I., Cost, G. J., Urnov, F.
D., Zhang, H. S., Holmes, M. C., Zhang, L., Gregory, P. D., and Rebar,
E. J. (2011) A TALE nuclease architecture for efficient genome editing,
Nat. Biotechnol., 29, 143-148.
71.Boch, J., Scholze, H., Schornack, S., Landgraf,
A., Hahn, S., Kay, S., Lahaye, T., Nickstadt, A., and Bonas, U. (2009)
Breaking the code of DNA binding specificity of TAL-type III effectors,
Science, 326, 1509-1512.
72.Moscou, M. J., and Bogdanove, A. J. (2009) A
simple cipher governs DNA recognition by TAL effectors, Science,
326, 1501.
73.Morbitzer, R., Romer, P., Boch, J., and Lahaye,
T. (2010) Regulation of selected genome loci using de
novo-engineered transcription activator-like effector (TALE)-type
transcription factors, Proc. Natl. Acad. Sci. USA, 107,
21617-21622.
74.Ramalingam, S., Annaluru, N., Kandavelou, K., and
Chandrasegaran, S. (2014) TALEN-mediated generation and genetic
correction of disease-specific human induced pluripotent stem cells,
Curr. Gene Ther., 14, 461-472.
75.Ramalingam, S., London, V., Kandavelou, K.,
Cebotaru, L., Guggino, W., Civin, C., and Chandrasegaran, S. (2013)
Generation and genetic engineering of human induced pluripotent stem
cells using designed zinc finger nucleases, Stem Cells Dev.,
22, 595-610.
76.Kim, H., and Kim, J. S. (2014) A guide to genome
engineering with programmable nucleases, Nat. Rev. Genet.,
15, 321-334.
77.Xu, P., Tong, Y., Liu, X. Z., Wang, T. T., Cheng,
L., Wang, B. Y., Lv, X., Huang, Y., and Liu, D. P. (2015) Both TALENs
and CRISPR/Cas9 directly target the HBB IVS2-654 (C > T) mutation in
beta-thalassemia-derived iPSCs, Sci. Rep., 5, 12065.
78.Guilinger, J. P., Thompson, D. B., and Liu, D. R.
(2014) Fusion of catalytically inactive Cas9 to FokI nuclease improves
the specificity of genome modification, Nat. Biotechnol.,
32, 577-582.
79.Holkers, M., Maggio, I., Liu, J., Janssen, J. M.,
Miselli, F., Mussolino, C., Recchia, A., Cathomen, T., and Goncalves,
M. A. (2013) Differential integrity of TALE nuclease genes following
adenoviral and lentiviral vector gene transfer into human cells,
Nucleic Acids Res., 41, e63.
80.Briggs, A. W., Rios, X., Chari, R., Yang, L.,
Zhang, F., Mali, P., and Church, G. M. (2012) Iterative capped
assembly: rapid and scalable synthesis of repeat-module DNA such as TAL
effectors from individual monomers, Nucleic Acids Res.,
40, e117.
81.Cermak, T., Doyle, E. L., Christian, M., Wang,
L., Zhang, Y., Schmidt, C., Baller, J. A., Somia, N. V., Bogdanove, A.
J., and Voytas, D. F. (2011) Efficient design and assembly of custom
TALEN and other TAL effector-based constructs for DNA targeting,
Nucleic Acids Res., 39, e82.
82.Reyon, D., Tsai, S. Q., Khayter, C., Foden, J.
A., Sander, J. D., and Joung, J. K. (2012) FLASH assembly of TALENs for
high-throughput genome editing, Nat. Biotechnol., 30,
460-465.
83.Schmid-Burgk, J. L., Schmidt, T., Kaiser, V.,
Honing, K., and Hornung, V. (2013) A ligation-independent cloning
technique for high-throughput assembly of transcription activator-like
effector genes, Nat. Biotechnol., 31, 76-81.
84.Mussolino, C., Morbitzer, R., Lutge, F.,
Dannemann, N., Lahaye, T., and Cathomen, T. (2011) A novel TALE
nuclease scaffold enables high genome editing activity in combination
with low toxicity, Nucleic Acids Res., 39, 9283-9293.
85.Kim, Y., Kweon, J., Kim, A., Chon, J. K., Yoo, J.
Y., Kim, H. J., Kim, S., Lee, C., Jeong, E., Chung, E., Kim, D., Lee,
M. S., Go, E. M., Song, H. J., Kim, H., Cho, N., Bang, D., Kim, S., and
Kim, J. S. (2013) A library of TAL effector nucleases spanning the
human genome, Nat. Biotechnol., 31, 251-258.
86.Barrangou, R., Fremaux, C., Deveau, H., Richards,
M., Boyaval, P., Moineau, S., Romero, D. A., and Horvath, P. (2007)
CRISPR provides acquired resistance against viruses in prokaryotes,
Science, 315, 1709-1712.
87.Makarova, K. S., Grishin, N. V., Shabalina, S.
A., Wolf, Y. I., and Koonin, E. V. (2006) A putative
RNA-interference-based immune system in prokaryotes: computational
analysis of the predicted enzymatic machinery, functional analogies
with eukaryotic RNAi, and hypothetical mechanisms of action, Biol.
Direct, 1, 7.
88.Deltcheva, E., Chylinski, K., Sharma, C. M.,
Gonzales, K., Chao, Y., Pirzada, Z. A., Eckert, M. R., Vogel, J., and
Charpentier, E. (2011) CRISPR RNA maturation by trans-encoded small RNA
and host factor RNase III, Nature, 471, 602-607.
89.Jinek, M., Chylinski, K., Fonfara, I., Hauer, M.,
Doudna, J. A., and Charpentier, E. (2012) A programmable
dual-RNA-guided DNA endonuclease in adaptive bacterial immunity,
Science, 337, 816-821.
90.Hsu, P. D., Scott, D. A., Weinstein, J. A., Ran,
F. A., Konermann, S., Agarwala, V., Li, Y., Fine, E. J., Wu, X.,
Shalem, O., Cradick, T. J., Marraffini, L. A., Bao, G., and Zhang, F.
(2013) DNA targeting specificity of RNA-guided Cas9 nucleases, Nat.
Biotechnol., 31, 827-832.
91.Jiang, W., Bikard, D., Cox, D., Zhang, F., and
Marraffini, L. A. (2013) RNA-guided editing of bacterial genomes using
CRISPR-Cas systems, Nat. Biotechnol., 31, 233-239.
92.Mali, P., Aach, J., Stranges, P. B., Esvelt, K.
M., Moosburner, M., Kosuri, S., Yang, L., and Church, G. M. (2013) CAS9
transcriptional activators for target specificity screening and paired
nickases for cooperative genome engineering, Nat. Biotechnol.,
31, 833-838.
93.Mojica, F. J., Diez-Villasenor, C.,
Garcia-Martinez, J., and Almendros, C. (2009) Short motif sequences
determine the targets of the prokaryotic CRISPR defense system,
Microbiology, 155, 733-740.
94.Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto,
R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A., and
Zhang, F. (2013) Multiplex genome engineering using CRISPR/Cas systems,
Science, 339, 819-823.
95.Fonfara, I., Le Rhun, A., Chylinski, K.,
Makarova, K. S., Lecrivain, A. L., Bzdrenga, J., Koonin, E. V., and
Charpentier, E. (2014) Phylogeny of Cas9 determines functional
exchangeability of dual-RNA and Cas9 among orthologous type II
CRISPR-Cas systems, Nucleic Acids Res., 42,
2577-2590.
96.Hou, Z., Zhang, Y., Propson, N. E., Howden, S.
E., Chu, L. F., Sontheimer, E. J., and Thomson, J. A. (2013) Efficient
genome engineering in human pluripotent stem cells using Cas9 from
Neisseria meningitidis, Proc. Natl. Acad. Sci. USA,
110, 15644-15649.
97.Shan, Q., Wang, Y., Li, J., Zhang, Y., Chen, K.,
Liang, Z., Zhang, K., Liu, J., Xi, J. J., Qiu, J. L., and Gao,
C. (2013) Targeted genome modification of crop plants using a
CRISPR-Cas system, Nat. Biotechnol., 31, 686-688.
98.Ding, Q., Regan, S. N., Xia, Y., Oostrom, L. A.,
Cowan, C. A., and Musunuru, K. (2013) Enhanced efficiency of human
pluripotent stem cell genome editing through replacing TALENs with
CRISPRs, Cell Stem Cell, 12, 393-394.
99.Findlay, G. M., Boyle, E. A., Hause, R. J.,
Klein, J. C., and Shendure, J. (2014) Saturation editing of genomic
regions by multiplex homology-directed repair, Nature,
513, 120-123.
100.Gilbert, L. A., Horlbeck, M. A., Adamson, B.,
Villalta, J. E., Chen, Y., Whitehead, E. H., Guimaraes, C., Panning,
B., Ploegh, H. L., Bassik, M. C., Qi, L. S., Kampmann, M., and
Weissman, J. S. (2014) Genome-scale CRISPR-mediated control of gene
repression and activation, Cell, 159, 647-661.
101.Shalem, O., Sanjana, N. E., Hartenian, E., Shi,
X., Scott, D. A., Mikkelsen, T. S., Heckl, D., Ebert, B. L., Root, D.
E., Doench, J. G., and Zhang, F. (2014) Genome-scale CRISPR-Cas9
knockout screening in human cells, Science, 343,
84-87.
102.Niu, Y., Shen, B., Cui, Y., Chen, Y., Wang, J.,
Wang, L., Kang, Y., Zhao, X., Si, W., Li, W., Xiang, A. P., Zhou, J.,
Guo, X., Bi, Y., Si, C., Hu, B., Dong, G., Wang, H., Zhou, Z., Li, T.,
Tan, T., Pu, X., Wang, F., Ji, S., Zhou, Q., Huang, X., Ji, W., and
Sha, J. (2014) Generation of gene-modified cynomolgus monkey via
Cas9/RNA-mediated gene targeting in one-cell embryos, Cell,
156, 836-843.
103.Wang, H., Yang, H., Shivalila, C. S., Dawlaty,
M. M., Cheng, A. W., Zhang, F., and Jaenisch, R. (2013) One-step
generation of mice carrying mutations in multiple genes by
CRISPR/Cas-mediated genome engineering, Cell, 153,
910-918.
104.Xie, K., Minkenberg, B., and Yang, Y. (2015)
Boosting CRISPR/Cas9 multiplex editing capability with the endogenous
tRNA-processing system, Proc. Natl. Acad. Sci. USA, 112,
3570-3575.
105.Sollu, C., Pars, K., Cornu, T. I.,
Thibodeau-Beganny, S., Maeder, M. L., Joung, J. K., Heilbronn, R., and
Cathomen, T. (2010) Autonomous zinc-finger nuclease pairs for targeted
chromosomal deletion, Nucleic Acids Res., 38,
8269-8276.
106.Ramakrishna, S., Kwaku Dad, A. B., Beloor, J.,
Gopalappa, R., Lee, S. K., and Kim, H. (2014) Gene disruption by
cell-penetrating peptide-mediated delivery of Cas9 protein and guide
RNA, Genome Res., 24, 1020-1027.
107.Zuris, J. A., Thompson, D. B., Shu, Y.,
Guilinger, J. P., Bessen, J. L., Hu, J. H., Maeder, M. L., Joung, J.
K., Chen, Z. Y., and Liu, D. R. (2015) Cationic lipid-mediated delivery
of proteins enables efficient protein-based genome editing in
vitro and in vivo, Nat. Biotechnol., 33,
73-80.
108.Zetsche, B., Volz, S. E., and Zhang, F. (2015)
A split-Cas9 architecture for inducible genome editing and
transcription modulation, Nat. Biotechnol., 33,
139-142.
109.Davis, K. M., Pattanayak, V., Thompson, D. B.,
Zuris, J. A., and Liu, D. R. (2015) Small molecule-triggered Cas9
protein with improved genome-editing specificity, Nat. Chem.
Biol., 11, 316-318.
110.Ran, F. A., Hsu, P. D., Lin, C. Y., Gootenberg,
J. S., Konermann, S., Trevino, A. E., Scott, D. A., Inoue, A., Matoba,
S., Zhang, Y., and Zhang, F. (2013) Double nicking by RNA-guided CRISPR
Cas9 for enhanced genome editing specificity, Cell, 154,
1380-1389.
111.Tsai, S. Q., Wyvekens, N., Khayter, C., Foden,
J. A., Thapar, V., Reyon, D., Goodwin, M. J., Aryee, M. J., and Joung,
J. K. (2014) Dimeric CRISPR RNA-guided FokI nucleases for highly
specific genome editing, Nat. Biotechnol., 32,
569-576.
112.Slaymaker, I. M., Gao, L., Zetsche, B., Scott,
D. A., Yan, W. X., and Zhang, F. (2016) Rationally engineered Cas9
nucleases with improved specificity, Science, 351,
84-88.
113.Jinek, M., East, A., Cheng, A., Lin, S., Ma,
E., and Doudna, J. (2013) RNA-programmed genome editing in human cells,
Elife, 2, e00471.
114.Cho, S. W., Kim, S., Kim, J. M., and Kim, J. S.
(2013) Targeted genome engineering in human cells with the Cas9
RNA-guided endonuclease, Nat. Biotechnol., 31,
230-232.
115.Pattanayak, V., Ramirez, C. L., Joung, J. K.,
and Liu, D. R. (2011) Revealing off-target cleavage specificities of
zinc-finger nucleases by in vitro selection, Nat.
Methods, 8, 765-770.
116.Fu, Y., Foden, J. A., Khayter, C., Maeder, M.
L., Reyon, D., Joung, J. K., and Sander, J. D. (2013) High-frequency
off-target mutagenesis induced by CRISPR-Cas nucleases in human cells,
Nat. Biotechnol., 31, 822-826.
117.Cornu, T. I., Thibodeau-Beganny, S., Guhl, E.,
Alwin, S., Eichtinger, M., Joung, J. K., and Cathomen, T. (2008)
DNA-binding specificity is a major determinant of the activity and
toxicity of zinc-finger nucleases, Mol. Ther., 16,
352-358.
118.Cradick, T. J., Fine, E. J., Antico, C. J., and
Bao, G. (2013) CRISPR/Cas9 systems targeting β-globin and
CCR5 genes have substantial off-target activity, Nucleic
Acids Res., 41, 9584-9592.
119.Cho, S. W., Kim, S., Kim, Y., Kweon, J., Kim,
H. S., Bae, S., and Kim, J. S. (2014) Analysis of off-target effects of
CRISPR/Cas-derived RNA-guided endonucleases and nickases, Genome
Res., 24, 132-141.
120.Kim, Y., Kweon, J., and Kim, J. S. (2013)
TALENs and ZFNs are associated with different mutation signatures,
Nat. Methods, 10, 185.
121.Bae, K. H., Kwon, Y. D., Shin, H. C., Hwang, M.
S., Ryu, E. H., Park, K. S., Yang, H. Y., Lee, D. K., Lee, Y., Park,
J., Kwon, H. S., Kim, H. W., Yeh, B. I., Lee, H. W., Sohn, S. H., Yoon,
J., Seol, W., and Kim, J. S. (2003) Human zinc fingers as building
blocks in the construction of artificial transcription factors, Nat.
Biotechnol., 21, 275-280.
122.Mak, A. N., Bradley, P., Cernadas, R. A.,
Bogdanove, A. J., and Stoddard, B. L. (2012) The crystal structure of
TAL effector PthXo1 bound to its DNA target, Science,
335, 716-719.
123.Doyle, E. L., Hummel, A. W., Demorest, Z. L.,
Starker, C. G., Voytas, D. F., Bradley, P., and Bogdanove, A. J. (2013)
TAL effector specificity for base 0 of the DNA target is altered in a
complex, effector- and assay-dependent manner by substitutions for the
tryptophan in cryptic repeat-1, PLoS One, 8, e82120.
124.Lamb, B. M., Mercer, A. C., and Barbas, C. F.,
3rd. (2013) Directed evolution of the TALE N-terminal domain for
recognition of all 5′ bases, Nucleic Acids Res.,
41, 9779-9785.
125.Bultmann, S., Morbitzer, R., Schmidt, C. S.,
Thanisch, K., Spada, F., Elsaesser, J., Lahaye, T., and Leonhardt, H.
(2012) Targeted transcriptional activation of silent oct4
pluripotency gene by combining designer TALEs and inhibition of
epigenetic modifiers, Nucleic Acids Res., 40,
5368-5377.
126.Valton, J., Dupuy, A., Daboussi, F., Thomas,
S., Marechal, A., Macmaster, R., Melliand, K., Juillerat, A., and
Duchateau, P. (2012) Overcoming transcription activator-like effector
(TALE) DNA binding domain sensitivity to cytosine methylation, J.
Biol. Chem., 287, 38427-38432.
127.Deng, D., Yin, P., Yan, C., Pan, X., Gong, X.,
Qi, S., Xie, T., Mahfouz, M., Zhu, J. K., Yan, N., and Shi, Y. (2012)
Recognition of methylated DNA by TAL effectors, Cell Res.,
22, 1502-1504.
128.Koike-Yusa, H., Li, Y., Tan, E. P.,
Velasco-Herrera Mdel, C., and Yusa, K. (2014) Genome-wide recessive
genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA
library, Nat. Biotechnol., 32, 267-273.
129.Nissim, L., Perli, S. D., Fridkin, A.,
Perez-Pinera, P., and Lu, T. K. (2014) Multiplexed and programmable
regulation of gene networks with an integrated RNA and CRISPR/Cas
toolkit in human cells, Mol. Cell, 54, 698-710.
130.Porteus, M. H., and Baltimore, D. (2003)
Chimeric nucleases stimulate gene targeting in human cells,
Science, 300, 763.
131.Urnov, F. D., Miller, J. C., Lee, Y. L.,
Beausejour, C. M., Rock, J. M., Augustus, S., Jamieson, A. C., Porteus,
M. H., Gregory, P. D., and Holmes, M. C. (2005) Highly efficient
endogenous human gene correction using designed zinc-finger nucleases,
Nature, 435, 646-651.
132.Meng, X., Noyes, M. B., Zhu, L. J., Lawson, N.
D., and Wolfe, S. A. (2008) Targeted gene inactivation in zebrafish
using engineered zinc-finger nucleases, Nat. Biotechnol.,
26, 695-701.
133.Hwang, W. Y., Fu, Y., Reyon, D., Maeder, M. L.,
Tsai, S. Q., Sander, J. D., Peterson, R. T., Yeh, J. R., and Joung, J.
K. (2013) Efficient genome editing in zebrafish using a CRISPR-Cas
system, Nat. Biotechnol., 31, 227-229.
134.Li, H., Haurigot, V., Doyon, Y., Li, T., Wong,
S. Y., Bhagwat, A. S., Malani, N., Anguela, X. M., Sharma, R., Ivanciu,
L., Murphy, S. L., Finn, J. D., Khazi, F. R., Zhou, S., Paschon, D. E.,
Rebar, E. J., Bushman, F. D., Gregory, P. D., Holmes, M. C., and High,
K. A. (2011) In vivo genome editing restores haemostasis in a
mouse model of haemophilia, Nature, 475, 217-221.
135.Lombardo, A., Genovese, P., Beausejour, C. M.,
Colleoni, S., Lee, Y. L., Kim, K. A., Ando, D., Urnov, F. D., Galli,
C., Gregory, P. D., Holmes, M. C., and Naldini, L. (2007) Gene editing
in human stem cells using zinc finger nucleases and integrase-defective
lentiviral vector delivery, Nat. Biotechnol., 25,
1298-1306.
136.Wang, T., Wei, J. J., Sabatini, D. M., and
Lander, E. S. (2014) Genetic screens in human cells using the
CRISPR-Cas9 system, Science, 343, 80-84.
137.Gaj, T., Guo, J., Kato, Y., Sirk, S. J., and
Barbas, C. F., 3rd. (2012) Targeted gene knockout by direct delivery of
zinc-finger nuclease proteins, Nat. Methods, 9,
805-807.
138.Pruett-Miller, S. M., Reading, D. W., Porter,
S. N., and Porteus, M. H. (2009) Attenuation of zinc finger nuclease
toxicity by small-molecule regulation of protein levels, PLoS
Genet., 5, e1000376.
139.Bibikova, M., Beumer, K., Trautman, J. K., and
Carroll, D. (2003) Enhancing gene targeting with designed zinc finger
nucleases, Science, 300, 764.
140.Beumer, K., Bhattacharyya, G., Bibikova, M.,
Trautman, J. K., and Carroll, D. (2006) Efficient gene targeting in
Drosophila with zinc-finger nucleases, Genetics,
172, 2391-2403.
141.Morton, J., Davis, M. W., Jorgensen, E. M., and
Carroll, D. (2006) Induction and repair of zinc-finger
nuclease-targeted double-strand breaks in Caenorhabditis elegans
somatic cells, Proc. Natl. Acad. Sci. USA, 103,
16370-16375.
142.Takasu, Y., Kobayashi, I., Beumer, K., Uchino,
K., Sezutsu, H., Sajwan, S., Carroll, D., Tamura, T., and Zurovec, M.
(2010) Targeted mutagenesis in the silkworm Bombyx mori using
zinc finger nuclease mRNA injection, Insect Biochem. Mol. Biol.,
40, 759-765.
143.Foley, J. E., Yeh, J. R., Maeder, M. L., Reyon,
D., Sander, J. D., Peterson, R. T., and Joung, J. K. (2009) Rapid
mutation of endogenous zebrafish genes using zinc finger nucleases made
by Oligomerized Pool ENgineering (OPEN), PLoS One, 4,
e4348.
144.Doyon, Y., McCammon, J. M., Miller, J. C.,
Faraji, F., Ngo, C., Katibah, G. E., Amora, R., Hocking, T. D., Zhang,
L., Rebar, E. J., Gregory, P. D., Urnov, F. D., and Amacher, S. L.
(2008) Heritable targeted gene disruption in zebrafish using designed
zinc-finger nucleases, Nat. Biotechnol., 26, 702-708.
145.Geurts, A. M., Cost, G. J., Freyvert, Y.,
Zeitler, B., Miller, J. C., Choi, V. M., Jenkins, S. S., Wood, A., Cui,
X., Meng, X., Vincent, A., Lam, S., Michalkiewicz, M., Schilling, R.,
Foeckler, J., Kalloway, S., Weiler, H., Menoret, S., Anegon, I., Davis,
G. D., Zhang, L., Rebar, E. J., Gregory, P. D., Urnov, F. D., Jacob, H.
J., and Buelow, R. (2009) Knockout rats via embryo microinjection of
zinc-finger nucleases, Science, 325, 433.
146.Mashimo, T., Takizawa, A., Voigt, B., Yoshimi,
K., Hiai, H., Kuramoto, T., and Serikawa, T. (2010) Generation of
knockout rats with X-linked severe combined immunodeficiency (X-SCID)
using zinc-finger nucleases, PLoS One, 5, e8870.
147.Meyer, M., De Angelis, M. H., Wurst, W., and
Kuhn, R. (2010) Gene targeting by homologous recombination in mouse
zygotes mediated by zinc-finger nucleases, Proc. Natl. Acad. Sci.
USA, 107, 15022-15026.
148.Carbery, I. D., Ji, D., Harrington, A., Brown,
V., Weinstein, E. J., Liaw, L., and Cui, X. (2010) Targeted genome
modification in mice using zinc-finger nucleases, Genetics,
186, 451-459.
149.Zou, J., Maeder, M. L., Mali, P.,
Pruett-Miller, S. M., Thibodeau-Beganny, S., Chou, B. K., Chen, G., Ye,
Z., Park, I. H., Daley, G. Q., Porteus, M. H., Joung, J. K., and Cheng,
L. (2009) Gene targeting of a disease-related gene in human induced
pluripotent stem and embryonic stem cells, Cell Stem Cell,
5, 97-110.
150.Hockemeyer, D., Soldner, F., Beard, C., Gao,
Q., Mitalipova, M., DeKelver, R. C., Katibah, G. E., Amora, R.,
Boydston, E. A., Zeitler, B., Meng, X., Miller, J. C., Zhang, L.,
Rebar, E. J., Gregory, P. D., Urnov, F. D., and Jaenisch, R. (2009)
Efficient targeting of expressed and silent genes in human ESCs and
iPSCs using zinc-finger nucleases, Nat. Biotechnol., 27,
851-857.
151.Perez, E. E., Wang, J., Miller, J. C.,
Jouvenot, Y., Kim, K. A., Liu, O., Wang, N., Lee, G., Bartsevich, V.
V., Lee, Y. L., Guschin, D. Y., Rupniewski, I., Waite, A. J.,
Carpenito, C., Carroll, R. G., Orange, J. S., Urnov, F. D., Rebar, E.
J., Ando, D., Gregory, P. D., Riley, J. L., Holmes, M. C., and June, C.
H. (2008) Establishment of HIV-1 resistance in CD4+ T-cells by genome
editing using zinc-finger nucleases, Nat. Biotechnol.,
26, 808-816.
152.Goldberg, A. D., Banaszynski, L. A., Noh, K.
M., Lewis, P. W., Elsaesser, S. J., Stadler, S., Dewell, S., Law, M.,
Guo, X., Li, X., Wen, D., Chapgier, A., DeKelver, R. C., Miller, J. C.,
Lee, Y. L., Boydston, E. A., Holmes, M. C., Gregory, P. D., Greally, J.
M., Rafii, S., Yang, C., Scambler, P. J., Garrick, D., Gibbons, R. J.,
Higgs, D. R., Cristea, I. M., Urnov, F. D., Zheng, D., and Allis, C. D.
(2010) Distinct factors control histone variant H3.3 localization at
specific genomic regions, Cell, 140, 678-691.
153.Connelly, J. P., Barker, J. C., Pruett-Miller,
S., and Porteus, M. H. (2010) Gene correction by homologous
recombination with zinc finger nucleases in primary cells from a mouse
model of a generic recessive genetic disease, Mol. Ther.,
18, 1103-1110.
154.Cost, G. J., Freyvert, Y., Vafiadis, A.,
Santiago, Y., Miller, J. C., Rebar, E., Collingwood, T. N., Snowden,
A., and Gregory, P. D. (2010) BAK and BAX deletion using zinc-finger
nucleases yields apoptosis-resistant CHO cells, Biotechnol.
Bioeng., 105, 330-340.
155.Liu, P. Q., Chan, E. M., Cost, G. J., Zhang,
L., Wang, J., Miller, J. C., Guschin, D. Y., Reik, A., Holmes, M. C.,
Mott, J. E., Collingwood, T. N., and Gregory, P. D. (2010) Generation
of a triple-gene knockout mammalian cell line using engineered
zinc-finger nucleases, Biotechnol. Bioeng., 106,
97-105.
156.Carlson, D. F., Tan, W., Lillico, S. G.,
Stverakova, D., Proudfoot, C., Christian, M., Voytas, D. F., Long, C.
R., Whitelaw, C. B., and Fahrenkrug, S. C. (2012) Efficient
TALEN-mediated gene knockout in livestock, Proc. Natl. Acad. Sci.
USA, 109, 17382-17387.
157.Gupta, A., Hall, V. L., Kok, F. O., Shin, M.,
McNulty, J. C., Lawson, N. D., and Wolfe, S. A. (2013) Targeted
chromosomal deletions and inversions in zebrafish, Genome Res.,
23, 1008-1017.
158.Bedell, V. M., Wang, Y., Campbell, J. M.,
Poshusta, T. L., Starker, C. G., Krug, R. G., 2nd, Tan, W., Penheiter,
S. G., Ma, A. C., Leung, A. Y., Fahrenkrug, S. C., Carlson, D. F.,
Voytas, D. F., Clark, K. J., Essner, J. J., and Ekker, S. C. (2012)
In vivo genome editing using a high-efficiency TALEN system,
Nature, 491, 114-118.
159.Huang, P., Xiao, A., Zhou, M., Zhu, Z., Lin,
S., and Zhang, B. (2011) Heritable gene targeting in zebrafish using
customized TALENs, Nat. Biotechnol., 29, 699-700.
160.Sander, J. D., Cade, L., Khayter, C., Reyon,
D., Peterson, R. T., Joung, J. K., and Yeh, J. R. (2011) Targeted gene
disruption in somatic zebrafish cells using engineered TALENs, Nat.
Biotechnol., 29, 697-698.
161.Cade, L., Reyon, D., Hwang, W. Y., Tsai, S. Q.,
Patel, S., Khayter, C., Joung, J. K., Sander, J. D., Peterson, R. T.,
and Yeh, J. R. (2012) Highly efficient generation of heritable
zebrafish gene mutations using homo- and heterodimeric TALENs,
Nucleic Acids Res., 40, 8001-8010.
162.Moore, F. E., Reyon, D., Sander, J. D.,
Martinez, S. A., Blackburn, J. S., Khayter, C., Ramirez, C. L., Joung,
J. K., and Langenau, D. M. (2012) Improved somatic mutagenesis in
zebrafish using transcription activator-like effector nucleases
(TALENs), PLoS One, 7, e37877.
163.Wood, A. J., Lo, T. W., Zeitler, B., Pickle, C.
S., Ralston, E. J., Lee, A. H., Amora, R., Miller, J. C., Leung, E.,
Meng, X., Zhang, L., Rebar, E. J., Gregory, P. D., Urnov, F. D., and
Meyer, B. J. (2011) Targeted genome editing across species using ZFNs
and TALENs, Science, 333, 307.
164.Li, T., Huang, S., Zhao, X., Wright, D. A.,
Carpenter, S., Spalding, M. H., Weeks, D. P., and Yang, B. (2011)
Modularly assembled designer TAL effector nucleases for targeted gene
knockout and gene replacement in eukaryotes, Nucleic Acids Res.,
39, 6315-6325.
165.Liu, J., Li, C., Yu, Z., Huang, P., Wu, H.,
Wei, C., Zhu, N., Shen, Y., Chen, Y., Zhang, B., Deng, W. M., and Jiao,
R. (2012) Efficient and specific modifications of the Drosophila
genome by means of an easy TALEN strategy, J. Genet. Genom.,
39, 209-215.
166.Watanabe, T., Ochiai, H., Sakuma, T., Horch, H.
W., Hamaguchi, N., Nakamura, T., Bando, T., Ohuchi, H., Yamamoto, T.,
Noji, S., and Mito, T. (2012) Non-transgenic genome modifications in a
hemimetabolous insect using zinc-finger and TAL effector nucleases,
Nat. Commun., 3, 1017.
167.Tesson, L., Usal, C., Menoret, S., Leung, E.,
Niles, B. J., Remy, S., Santiago, Y., Vincent, A. I., Meng, X., Zhang,
L., Gregory, P. D., Anegon, I., and Cost, G. J. (2011) Knockout rats
generated by embryo microinjection of TALENs, Nat. Biotechnol.,
29, 695-696.
168.Liu, H., Chen, Y., Niu, Y., Zhang, K., Kang,
Y., Ge, W., Liu, X., Zhao, E., Wang, C., Lin, S., Jing, B., Si, C.,
Lin, Q., Chen, X., Lin, H., Pu, X., Wang, Y., Qin, B., Wang, F., Wang,
H., Si, W., Zhou, J., Tan, T., Li, T., Ji, S., Xue, Z., Luo, Y., Cheng,
L., Zhou, Q., Li, S., Sun, Y. E., and Ji, W. (2014) TALEN-mediated gene
mutagenesis in rhesus and cynomolgus monkeys, Cell Stem Cell,
14, 323-328.
169.Li, M., Suzuki, K., Kim, N. Y., Liu, G. H., and
Izpisua Belmonte, J. C. (2014) A cut above the rest: targeted genome
editing technologies in human pluripotent stem cells, J. Biol.
Chem., 289, 4594-4599.
170.Hockemeyer, D., Wang, H., Kiani, S., Lai, C.
S., Gao, Q., Cassady, J. P., Cost, G. J., Zhang, L., Santiago, Y.,
Miller, J. C., Zeitler, B., Cherone, J. M., Meng, X., Hinkley, S. J.,
Rebar, E. J., Gregory, P. D., Urnov, F. D., and Jaenisch, R. (2011)
Genetic engineering of human pluripotent cells using TALE nucleases,
Nat. Biotechnol., 29, 731-734.
171.Nakayama, T., Fish, M. B., Fisher, M.,
Oomen-Hajagos, J., Thomsen, G. H., and Grainger, R. M. (2013) Simple
and efficient CRISPR/Cas9-mediated targeted mutagenesis in Xenopus
tropicalis, Genesis, 51, 835-843.
172.Yang, D., Xu, J., Zhu, T., Fan, J., Lai, L.,
Zhang, J., and Chen, Y. E. (2014) Effective gene targeting in rabbits
using RNA-guided Cas9 nucleases, J. Mol. Cell Biol., 6,
97-99.
173.Xiao, A., Wang, Z., Hu, Y., Wu, Y., Luo, Z.,
Yang, Z., Zu, Y., Li, W., Huang, P., Tong, X., Zhu, Z., Lin, S., and
Zhang, B. (2013) Chromosomal deletions and inversions mediated by
TALENs and CRISPR/Cas in zebrafish, Nucleic Acids Res.,
41, e141.
174.Jao, L. E., Wente, S. R., and Chen, W. (2013)
Efficient multiplex biallelic zebrafish genome editing using a CRISPR
nuclease system, Proc. Natl. Acad. Sci. USA, 110,
13904-13909.
175.Wang, Y., Li, Z., Xu, J., Zeng, B., Ling, L.,
You, L., Chen, Y., Huang, Y., and Tan, A. (2013) The CRISPR/Cas system
mediates efficient genome engineering in Bombyx mori, Cell
Res., 23, 1414-1416.
176.DiCarlo, J. E., Norville, J. E., Mali, P.,
Rios, X., Aach, J., and Church, G. M. (2013) Genome engineering in
Saccharomyces cerevisiae using CRISPR-Cas systems, Nucleic
Acids Res., 41, 4336-4343.
177.Yu, Z., Ren, M., Wang, Z., Zhang, B., Rong, Y.
S., Jiao, R., and Gao, G. (2013) Highly efficient genome modifications
mediated by CRISPR/Cas9 in Drosophila, Genetics,
195, 289-291.
178.Gratz, S. J., Cummings, A. M., Nguyen, J. N.,
Hamm, D. C., Donohue, L. K., Harrison, M. M., Wildonger, J., and
O’Connor-Giles, K. M. (2013) Genome engineering of
Drosophila with the CRISPR RNA-guided Cas9 nuclease,
Genetics, 194, 1029-1035.
179.Bassett, A. R., Tibbit, C., Ponting, C. P., and
Liu, J. L. (2013) Highly efficient targeted mutagenesis of
Drosophila with the CRISPR/Cas9 system, Cell Rep.,
4, 220-228.
180.Jiang, W., Zhou, H., Bi, H., Fromm, M., Yang,
B., and Weeks, D. P. (2013) Demonstration of CRISPR/Cas9/sgRNA-mediated
targeted gene modification in Arabidopsis, tobacco, sorghum and
rice, Nucleic Acids Res., 41, e188.
181.Li, W., Teng, F., Li, T., and Zhou, Q. (2013)
Simultaneous generation and germline transmission of multiple gene
mutations in rat using CRISPR-Cas systems, Nat. Biotechnol.,
31, 684-686.
182.Li, D., Qiu, Z., Shao, Y., Chen, Y., Guan, Y.,
Liu, M., Li, Y., Gao, N., Wang, L., Lu, X., Zhao, Y., and Liu, M.
(2013) Heritable gene targeting in the mouse and rat using a CRISPR-Cas
system, Nat. Biotechnol., 31, 681-683.
183.Li, J. F., Norville, J. E., Aach, J.,
McCormack, M., Zhang, D., Bush, J., Church, G. M., and Sheen, J. (2013)
Multiplex and homologous recombination-mediated genome editing in
Arabidopsis and Nicotiana benthamiana using guide RNA and
Cas9, Nat. Biotechnol., 31, 688-691.
184.Nekrasov, V., Staskawicz, B., Weigel, D.,
Jones, J. D., and Kamoun, S. (2013) Targeted mutagenesis in the model
plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease,
Nat. Biotechnol., 31, 691-693.
185.Pattanayak, V., Lin, S., Guilinger, J. P., Ma,
E., Doudna, J. A., and Liu, D. R. (2013) High-throughput profiling of
off-target DNA cleavage reveals RNA-programmed Cas9 nuclease
specificity, Nat. Biotechnol., 31, 839-843.
186.Mali, P., Yang, L., Esvelt, K. M., Aach, J.,
Guell, M., DiCarlo, J. E., Norville, J. E., and Church, G. M. (2013)
RNA-guided human genome engineering via Cas9, Science,
339, 823-826.
187.Cheng, A. W., Wang, H., Yang, H., Shi, L.,
Katz, Y., Theunissen, T. W., Rangarajan, S., Shivalila, C. S., Dadon,
D. B., and Jaenisch, R. (2013) Multiplexed activation of endogenous
genes by CRISPR-on, an RNA-guided transcriptional activator system,
Cell Res., 23, 1163-1171.