REVIEW: mRNA-Based Therapeutics – Advances and Perspectives
O. V. Sergeeva1,2, V. E. Koteliansky1,2, and T. S. Zatsepin1,2,3*
1Lomonosov Moscow State University, Department of Chemistry, 119991 Moscow, Russia; fax: +7 (495) 939-3181; E-mail: olga.sergeeva.v@gmail.com2Skolkovo Institute of Science and Technology, 3 Nobel str., Skolkovo Innovation Center, 143026 Moscow, Russia; E-mail: t.zatsepin@skoltech.ru, V.Kotelianski@skoltech.ru
3Central Research Institute of Epidemiology, The Federal Service on Customers’ Rights Protection and Human Well-being Surveillance, 111123 Moscow, Russia; E-mail: tsz@yandex.ru
* To whom correspondence should be addressed.
Received February 29, 2016; Revision received March 27, 2016
In this review we discuss features of mRNA synthesis and modifications used to minimize immune response and prolong efficiency of the translation process in vivo. Considerable attention is given to the use of liposomes and nanoparticles containing lipids and polymers for the mRNA delivery. Finally we briefly discuss mRNAs which are currently in the clinical trials for cancer immunotherapy, vaccination against infectious diseases, and replacement therapy.
KEY WORDS: mRNA, in vivo delivery, therapy, nucleic acids, nanoparticlesDOI: 10.1134/S0006297916070075
Abbreviations: CRISPR-Cas9, system of clustered regularly interspaced short palindromic repeats and Cas9 nuclease; dsRNA, double stranded RNA; FDA, Food and Drug Administration (USA); HIV, human immunodeficiency virus; LNP, lipid nanoparticles; mRNA, messenger RNA; NA, nucleic acids; ORF, open reading frame; PEG, polyethylene glycol; siRNA, small interfering RNA; TALEN, transcription activator-like effector nuclease; TLRs, Toll-like receptors; UTR, untranslated region; ZFN, zinc finger nuclease.
Besides common small molecule drugs that are widely used in clinical
practice, monoclonal antibodies and peptides – so-called
biopharmaceuticals – were recently introduced. Also great
expectations are anticipated to gene therapy. Although for 30 years
gene therapy has been expected to become a breakthrough approach in the
treatment of many diseases, only recent considerable changes open the
application of nucleic acids (NA) therapy for human in the near
future.
There are two completely different approaches for NA based therapy from historical and methodological viewpoint: 1) inhibition or enhancement of gene expression; 2) excise or replacement of genes within the genome. In the late 1970s, Zamecnik et al. [1, 2] proposed a so-called antisense technology based on blocking of the mRNA translation by complementary synthetic oligonucleotides. After substantial development, this approach yielded in the chemically modified oligonucleotides, which have been approved by FDA as drugs [3, 4]. Upon discovery of previously unknown natural mechanisms, novel applications of the nucleic acids in therapy have been proposed. For example, after discovery of RNA interference in 1998 many pharmaceutical companies started development of small interfering RNAs (siRNAs) as the therapeutics. Despite the experience obtained within the 1990s during development of antisense oligonucleotides and ribozymes, the majority of the projects failed due to poor stability of RNAs in vivo and inefficient delivery. In the late 2000s most of these issues were partially resolved that lead to a renaissance in the field of therapeutic NAs [5]. Currently many siRNAs are at the different phases of the clinical trials [6]. Data of the last years give a hope that 3-5 NA drugs will be approved by FDA in near future.
Technologies allowing gene editing within the genome should be discussed separately due to significant differences in overall [7, 8]. First attempts in this field were made with meganucleases and restriction endonucleases containing zinc finger motifs (ZFN). Later fusion proteins consisting of bacterial effectors and FokI restriction endonuclease were created (TALEN – transcription activator-like effector nucleases). Despite significant success demonstrated in model systems, their wide in vivo use is limited by insufficient specificity and efficacy. In 2013, a novel approach for genome editing with the CRISPR-Cas9 system (clustered regularly interspaced short palindromic repeats and Cas9 nuclease) was proposed [9]. Superior efficiency of CRISPR-Cas9 in comparison to earlier developed systems resulted in the explosive growth of its application in various areas ranging from biomedicine to agricultural biotechnology [10]. Nonetheless, it should be emphasized that these systems are used mainly for in vitro and ex vivo engineering due to moderate efficacy. The reasons are probabilistic pattern of homology-directed recombination or nonhomologous ends joining as well as the low availability of tentative target genes because of poor understanding of chromatin structure and dynamics [11]. However there are examples of CRISPR-Cas9 application in vivo with the correction rate 0.4-6.0% [12, 13] that gives promise for the development of the efficient genome editing system in vivo in near future.
In the early 1990s the possibility of using plasmid DNA-encoding genes to induce antibody synthesis as well as to trigger T- and B-cell response was demonstrated, that led to the start of the DNA vaccines development [14, 15]. Moreover, it was shown that plasmids might be used for therapy of inherited and multifaceted diseases [16, 17]. In the therapeutic terms, mRNA delivery is a novel alternative approach for treatment of such diseases [18]. Compared to the use of plasmid DNA, several advantages can be emphasized: in case of mRNA delivery there is no risk of developing mutations and integration into genome; mRNA can be delivered to non-dividing cells as it does not need to get into the nucleus as DNA plasmids. In addition, it was shown that the efficacy of the cell transfection with mRNA in the liver in vivo might reach up to 100% while plasmid DNA was less efficient [19]. This data is very important for the future development of mRNA based replacement therapy. In comparison to protein pharmaceuticals mRNA gives more prolonged therapeutic effect. It is worth mentioning that mRNA can be used for the proteins and peptides expression within the intercellular space or for the generating pluripotent stem cells [20]. Furthermore, mRNA can encode nucleases such as ZFN, TALEN, and CRISPR-Cas9, which can be used for genome editing [21, 22]. Today marked progress in using mRNA as therapeutics at least in clinical trials is definitely noted. In this review, we will discuss this application for mRNAs translated in vitro and pay the main attention to a structural optimization and use of modified nucleosides as well as targeted delivery. Delivery approaches based upon physical methods [23] such as electroporation, micropipetting, ultrasound, laser radiation, and the gene gun system are beyond the scope of our review and will not be discussed. In addition, viral vectors [24, 25] as highly efficient system for delivering nucleic acids will not be covered. Also we will omit discussing mRNA transfection in vitro presented in a recent excellent review by Andreev et al. [26].
In vitro mRNA SYNTHESIS – ESSENTIAL
STRUCTURAL ELEMENTS AND THEIR OPTIMIZATION
Chemical automated synthesis of mRNA is impossible due to its length, which significantly exceeds common 100-150 nucleotides available by oligosynthesis, and the only way still relies on in vitro enzymatic synthesis or cell-free systems.
In eukaryotes, the structure of mature mRNAs can be divided into five major regions (Fig. 1): (i) cap – m7GpppN or m7Gp3N (N – any nucleotide); (ii) 5′-untranslated region (5′-UTR); (iii) protein-encoding open reading frame (ORF); (iv) 3′-untranslated region (3′-UTR); (v) 100-250 adenosine-containing region (3′-poly(A)-tail).
Fig. 1. a) General scheme depicting protein synthesis in vitro and the structure of mRNA; b) structure of 5′-cap in mRNA (B is a heterocyclic base); c) structure of modified cap-ends.
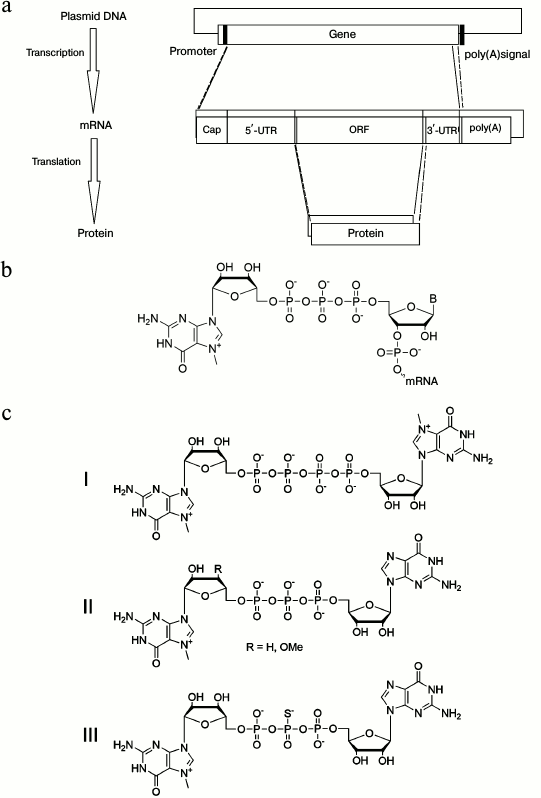
During posttranscriptional mRNA processing in the nucleus, the 5′-cap is incorporated into the mRNA molecule and plays an important role by stabilizing the structure of mRNAs as well as being involved in splicing, transport, and translation [27]. The 5′-cap is composed of an N7-methylguanosine (m7G) residue linked to the first transcribed nucleotide of mRNA via a triphosphate bridge (Fig. 1b). RNA polymerase can attach the m7GpppG to RNA in two orientations, making Gpppm7GpN as well as m7GpppGpN after linking the first nucleotide. This result presents in approximately half of in vitro-generated transcripts that would not be recognized by the ribosome inside cells. For solving this problem, several cap-analogs were synthesized that were unable to reverse bind, or it did not affect this process. For example, a symmetric cap-analog having both guanosine residues methylated at their N7-atoms, which are linked via tetraphosphate m7Gppppm7G (Fig. 1c, I), gave mRNA with high efficiency of the translation in vitro [28]. Alternatively, methylation or removal of the 3′-hydroxyl group of classic cap-region was proposed as well (Fig. 1c, II): such modification blocks elongation of the N7-methylated guanosine residue [29]. In addition to modifications blocking potential reverse orientation of the cap-region, the stability and efficacy of mRNA translation have been studied. For instance, introduction of a phosphorothioate cap group (Fig. 1c, III) results in downregulated binding between the cap-region and Dcp2 protein, which is involved in mRNA degradation, thereby increasing its stability without affecting its translational efficiency [30].
Mammalian cells possess components of innate immunity including a defense mechanism against viruses (Fig. 2). For instance, in the cytosol atypical RNA containing no cap- or 3′-poly(A)-region is recognized by RIG-I, MDA5, and RIG-I-like receptors. As a result, by acting via MAVS adaptor protein, they trigger a signaling cascade followed by release of type I interferons (IFN-α/β) and activation of several transcription factors such as NF-κB, IRF3, and IRF7 [31-33]. This signaling pathway is transmitted through activation of IkB kinase complex (IKK) and IKK-linked kinases (TBK1) and results in nuclear translocation of transcription factors NF-κB, IRF3, and IRF7, which then induce expression of type I interferons. Additionally, NF-κB activates expression of inflammatory cytokines such as TNF-α, IL-6, and IL-12 [34]. Type I interferons can be secreted into the intercellular space, where they bind to IFN receptors on surrounding cells with subsequently transduced signals informing the cell of detected foreign RNA and inducing immune response [35].
Fig. 2. Scheme depicting innate immune response to delivered artificial mRNA. dsRNA, double-stranded RNA; DUBA, deubiquitinase; IFN, interferon; IRF, interferon regulatory factor; NEMO, NF-κB essential modulator.
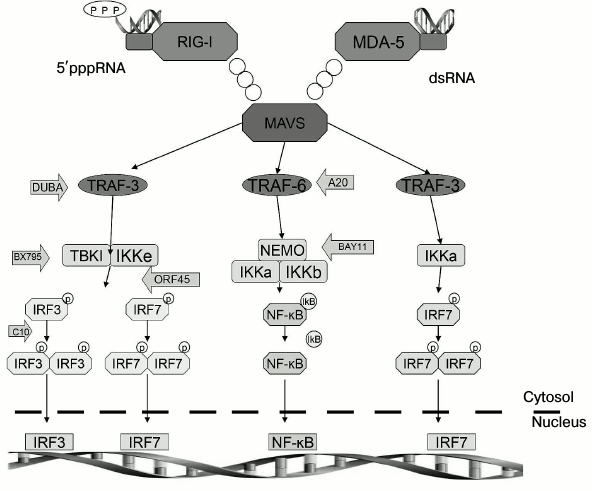
Recently it was demonstrated that NOD-like receptors are another representative of PRR family that are involved in the recognizing foreign mRNAs [36]. Mainly, they take part in regulating caspase 1 family proteins, whereas NLRP3 receptor is responsible for recognizing double-stranded (ds) RNAs. After that, the receptor forms a multi-protein complex with adaptor ASC and caspase 1 (the inflammasome), which is responsible for proteolytic maturation of IL-1β and IL-18 [37]. In addition, atypical dsRNAs can also activate autophosphorylation of protein kinases (PKR), which phosphorylates initiation factor eIF2a followed by inhibiting translation initiation and preventing replication of pathogens or synthesis of foreign proteins [38]. Thus, capping of mRNA at its 5′-end is necessary for minimizing activation of innate immunity. While performing mRNA capping in vitro, it is impossible to reach 100% efficacy of this reaction. Therefore, to reduce immunogenicity of mRNA, transcripts should be additionally treated with phosphatase to remove 5′-triphosphates [39].
The presence and length of the 3′-poly(A)-tail in mRNA also have crucial importance for efficient translation and increased stability of mRNAs. In mammalian cells, mRNAs containing 100-250 adenosine residues at the 3′-end are translated with the highest efficiency [40]. It was shown that by adding at least 150 nucleotides during polyadenylation reduces mRNA immunogenicity [41]. It is known that poly(A)-binding proteins (PABPs) and translation initiation factor eIF4G bind to poly(A)-tail. By acting synergistically, the cap and poly(A)-tail of mRNA circularize the mRNA by forming a complex cap/PABP/eIF4E-eIF4G-PABP-poly(A), which improves binding to the ribosome and protects mRNA from degradation.
Another approach to optimizing an mRNA sequence is based on replacing unstable regions in untranslated areas of the mRNA by sequences with the stable structure. For instance, the presence of adenosine/uridine-rich sequences within the 3′-UTR contributes to removal of the poly(A)-region upon degradation of mRNAs. Replacing the AU-rich regions by sequences from the 3′-UTR of stable mRNA significantly increased the half-life of the mRNA [42]. Additionally, iron-responsive elements (IRE) are found within the 3′-UTR, which can also regulate mRNA stability. Therefore, to increase stability and translational efficiency, the 3′-UTR derived from α- and β-globins are inserted when synthesizing mRNAs in vitro. The stabilizing effect can be enhanced by using two 3′-UTRs from β-globin in head-to-tail orientation [43, 44].
In addition, the order of codons also influences the efficiency of translation. Replacement of rare codons for synonymous codons is one of the ways to optimize the sequence of the coding region in mRNA. However, this approach should be used with care, as rare codons are necessary for correct folding in some cases.
Use of various modified nucleotides for in vitro mRNA translation dramatically alters its stability and immunogenicity. Modified nucleotides presented in mRNA influence on its recognition by Toll-like receptors (TLRs) involved in cellular immune response against foreign RNA. In humans and mice, 13 TLRs have been identified. In particular, TLR3, TLR7, and TLR8 localized in endosomes are responsible for recognizing foreign mRNA. For example, uridine-rich single-stranded RNA activates TLR7 [45], whereas double-stranded RNA acts on TLR3 [46]. It is known that mRNAs are able to form various secondary structures, hairpins, double-stranded regions, thereby making them proper ligands for both TLR7 and TLR3. Activated TLRs trigger signaling cascades via adaptor proteins (for TLR7 and TLR8 – via MyD88, for TLR3 – via TRIF) eventually resulting in activation of transcription factors NF-κB, IRF3, and IRF7 [14].
It was demonstrated that activation of TLR3, TLR7, and TLR8 can be minimized by inserting into RNA 2′-O-methylnucleosides, 5-methylcytidine (m5C), N6-methyladenosine (m6A), 5-methyluridine (m5U), pseudouridine, and 2-thiouridine [47]. The same modified nucleotides hinder recognition of mRNA by RIG-I receptors. Moreover, insertion of phosphorothioate groups into RNA results in two-fold decrease in RIG-I activation by single-stranded RNA [48] (Fig. 3).
Fig. 3. Modified nucleosides decreasing immunogenicity of mRNA [21]. B is a heterocyclic base.
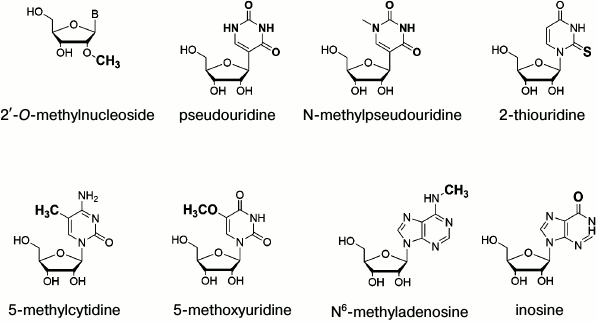
Interestingly, mRNA containing pseudouridine modifications exhibit higher translation activity compared to unmodified. It was found that pseudouridine residues increase biological stability of mRNA, perhaps by stabilizing secondary RNA structure. In addition, it should be noted that pseudouridine occurs naturally, and it does not increase mRNA toxicity [49]. Apart from pseudouridine, translational activity can also be upregulated by inserting N-methylpseudouridine and 5-methoxyuridine [50]. In particular, it was found that combination of the modified nucleotides, e.g. by substituting 25% of uridine and cytosine residues by 2-thiouridine and 5-methylcytosine, can reduce the mRNA recognition by pattern recognition receptors (PRRs) such as TLR3, TLR7, TLR8, RIG-I as well as by dsRNA-activated protein kinases (PKR) [51]. In the process of the modified mRNA design it is necessary to balance between decreased immunogenicity and potentially lowered mRNA stability. For instance, inserting N6-methyladenine decreases mRNA lifetime, as this modification results in binding of YTH family proteins, which stimulate RNA degradation [52]. Also HPLC purification of mRNA decreases its immunogenicity parameters due to removing dsRNA fragments. Along with inducing immune response, dsRNA can also be used for RNA interference for targeted downregulation of RNA translation [53].
In vivo delivery of mRNA. First attempts to use mRNA without any delivery vehicle revealed that this approach might be applied for vaccination with strongly induced immune response lacking side effects [54]. Although the vast majority of mRNA is degraded in the bloodstream and cell lysosomes, some amount of mRNA was still delivered to the cytosol followed by translation. It should be taken into consideration that positive results were obtained mainly after subcutaneous or intramuscular administration of non-optimized mRNA, whereas i.v. route results in rapid mRNA degradation by ribonucleases and in activation of the immune system. This approach is now widely used for the development of mRNA-containing vaccines currently going through various phases of clinical trials [55, 56].
As common direct route of delivery suffers from poor efficiency and does not provide a significant expression of target protein, it can be applied only for vaccination. In case of replacement therapy delivery vehicles must be used. Overall, application of synthetic NAs in clinical practice is determined by: (i) selection of chemical modifications of NAs that provide resistance to nucleases without affecting functionality of the NAs, and (ii) efficiency of targeted delivery to a tissue of interest and inside cells. Hence, the delivery process is a sophisticated task, because NA should first be delivered to a certain tissue, then passed through the cell membrane, and finally released into the cytosol. The ability to pass across the cell membrane was always considered as the main issue in targeted NA delivery. As NAs are large anionic hydrophilic molecules, they cannot penetrate directly through the hydrophobic cell membrane. Moreover, many defense mechanisms against pathogens intended to recognize foreign NAs have evolved in cells. Therefore, the optimal NA delivery method must provide its transfer across the membrane, protection from nucleases, and release from endosomes into cytosol for subsequent translation without activating immune system. For long time the use of mRNAs was shadowed by plasmids and siRNAs, so a large number of in vivo delivery methods were first studied for these NAs. However, any vehicles cannot be directly transferred for mRNA delivery – thorough optimization is always needed [57-59].
Common mRNA delivery may be improved just by adding Ca2+-containing solution prior to administering mRNA in vivo [60]. This significantly elevated the level and duration of mRNA translation. Historically, protamines (arginine-rich ~4 kDa proteins), which form complexes with NAs, were used as the first RNA delivery system [61]. In such complexes, mRNA is protected from nucleases [62], but its immunogenicity was increased [63]. The latter is an unfavorable factor for replacement therapy, but, undoubtedly, is a positive feature for developing mRNA vaccines. Currently, CureVac AG (Germany) has commercialized this approach.
For a long time, various lipids have been used to deliver different drugs in vivo. Liposomes, lipid nanoparticles, and lipoplexes consisting of cationic lipids are used for delivering NAs. Additionally, cationic nanoemulsions are also described [64]. Lipid nanoparticles (LNP) possess crucial differences from liposomes in terms of forming periodic multilamellar structure between lipids and NAs [65]. According to transmission electron microscopy data [66], such structures in case of mRNA are formed in LNP only partially and single liposomes with mRNA can be also found in LNP. At the beginning, simple liposomes containing DOTAP (1,2-dioleoyl-3-(trimethylammonium)propane) [67] or DOTAP-DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine) were used [68]. Later, more efficient systems such as lipid nanoparticles based on DLinDMA (1,2-dilinoleyloxy-3-dimethylaminopropane) and PEGylated lipids [69] or lipoplexes – polyplexes covered with lipids (discussed later) were developed. Cationic lipids per se are not quite efficient NA delivery vehicles. Therefore a number of helper lipids is needed – fusogenic phospholipids for improving transfection, PEGylated lipids for minimizing particle aggregation and decrease of nonspecific immune response, cholesterol for increasing of particle stability [70]. Besides reducing immune response, the proportion of PEGylated lipids contained in the mixture determines the size of particles, which should be ranged within 50-100 nm. On one hand this decreases nonspecific immune response, on the other hand it prevents nanoparticles from undergoing glomerular filtration in the kidneys and renal escape. Moreover, surface charge on nanoparticles is also an important parameter. While circulating in the blood, neutral charge is optimal to exclude non-specific interactions, whereas positive charge of nanoparticles is critical for disruption of endosome membranes and release of mRNA into the cytosol. Thus, pH-sensitive lipids with pKa 6.0-6.5 were developed [71], which are more effective for delivery due to neutral charge in the bloodstream followed by protonation upon lowered pH in endosomes that lead to membrane disruption. Later, biodegradable lipids containing ester or acetal groups were synthesized with significantly reduced toxicity [72].
Pardi et al. obtained important data on comparing expression kinetics in vivo of LNP packed mRNA applied via various administration routes [73]. In particular, subcutaneous or intramuscular vs. i.v. or intratracheal routes resulted in increasing halftime of protein synthesis, on average, by 3-4-fold (20-30 and 7-7.5 h, respectively). It is interesting to note that the intradermal route gave rise to two-fold increase in halftime compared to the subcutaneous route (29.6 and 14.7 h, respectively). In addition, it is important that the total amount of synthesized protein differed after administering mRNA via various routes at doses of 0.1 and 1 µg. For instance, after administering 1 µg mRNA via different routes, the final efficacy was comparable, but upon introducing it at the dose of 0.1 µg via the i.v. or intratracheal route it was either lower up to 2-3 orders of magnitude or undetectable, respectively. In the latter case low protein level was most likely due to rapid degradation of mRNA by nucleases, in spite of protection by lipid nanoparticles. Again, this example shows the ultimate need to develop novel modified nucleotides that could be incorporated into mRNAs, compatible with ribosomal translation and able to enhance nuclease resistance.
Despite the fact that upon the i.v. route liposomes and lipid nanoparticles are mainly delivered to the liver [74], there are methods allowing targeting of lipid nanoparticles containing mRNA to dendrocytes [75, 76], retina [77], lung [78], and some other cells. Improvement of mRNA LNP delivery relies on both designing novel lipids [79, 80] as well as optimizing their composition [81, 82]. On the other hand lipid conjugation with ligands of cellular receptors involved in receptor-mediated endocytosis is another important way for LNP optimization. It is worth mentioning the use of lipids containing N-acetylgalactosamine residues [83, 84] – ligands of asialoglycoprotein receptor in hepatocytes – ensures accelerated delivery of lipid nanoparticles. In addition, mannosylated liposomes containing RNA were able to transfect dendritic cells more efficiently due to interaction with mannose receptor in comparison to common liposomes [85]. Still this approach is rarely used due to the difficulties in the synthesis of such lipids.
A polyplex is a complex of mRNA with various polymers, which represents a valuable alternative to liposomes and lipid nanoparticles for efficient delivery of mRNA in vivo. Usually, cationic polymers are used to make stable complexes with NA for generating polyplexes. In particular, it was shown that mRNA inside a polyplex is significantly protected from ribonucleases and interactions with TLRs in vivo [86], and its expression was higher by an order of magnitude compared to free mRNA. Polyplexes have been known for more than 50 years [87, 88]; however, for a long time, due to high toxicity, clinical trials never proceeded further than phase I-II. Linear and branched polyethylenimine [89, 90], poly(2-(dimethylamino)ethyl methacrylate), and poly(L-lysine) [91] are classic examples of cationic polymers. Despite being frequently used for delivery of NA in vitro, their relatively high toxicity and poor transfection capacity in vivo do not allow considering them as optimal vehicles for delivery of mRNA. Recently, a variety of novel polymers have been proposed that are more efficient and less toxic, primarily due to their biodegradability. Among them, poly(β-amino esters) [92, 93] and polyaspartamides [94] should be noted. Interestingly, Kataoka et al. described how the structure of a polymer might influence translation efficiency [95]. In particular, it was demonstrated that by decreasing the number of aminoethylene units contained in various polyamines used to modify poly(β-benzyl-L-aspartate), it profoundly reduced translation of delivered mRNA via downregulated binding of translation initiation factor eIF4E. Apart from regulating translation, this further confirms that polymer-bound rather than free mRNA is transferred from endosomes into the cytosol. Special attention should be given to the report of Phua et al. [96], where translation kinetics for delivered polyplex-bound vs. free mRNA was examined in vitro and in vivo. It should be noted that if a polyplex was superior in the in vitro setting, then upon subcutaneous administration in vivo the half-life of translation of free mRNA was notably higher. This study shows that any novel vehicle must be thoroughly investigated, as unexpected results may be obtained in case of different routes of administration.
Also there are polyplexes containing functional conjugated molecules, e.g. cholesterol [97] or ligands for cell receptors such as sugar residues [98, 99] or their mimetics [100, 101]. For instance, introduction of a cholesterol residue significantly improved circulation of a polyplex in mice that lead not only to undetectable level of the antiangiogenic protein sFlt-1, but also to reduction of the tumor size in the pancreatic cancer xenograft model. As already discussed for lipids, application of receptor-mediated endocytosis allows significantly decrease the amount of administered mRNA with preserving level of translation. It should be emphasized that polyplexes can be used for delivering mRNA not only to dendritic cells, but for example, to bronchial epithelial cells in aerosol [102] or to the central nervous system [103, 104].
Moreover more complex constructs containing a polyplex with mRNA with subsequent formation of a lipid shell have been increasingly used [64, 88]. This lead to high efficiency of mRNA release from endosomes as well as minimizing toxicity of the entire delivery vehicle due to polymer masking.
Therapeutic applications of mRNA. Therapeutic use of mRNA is promising for cancer immunotherapy, vaccination against infectious diseases, inducing tolerance to type I allergy [105], and replacement therapy as well as regenerative medicine.
Antigenic epitopes bound to major histocompatibility complex (MHC) class I and II molecules are exposed on the surface of human dendritic cells, which can simultaneously trigger humoral and cell-mediated immune response. mRNA induces release of proinflammatory cytokines such as TNF-α, IL-1β, IL-12, IL-6, and IL-8. Moreover, after delivery of mRNA into peripheral blood cells, it also stimulates secretion of type I interferons (IFN-α and IFN-β), chemokines GRO, MCP-1, RANTES, and MDC [106]. Overall, such substances activate both innate and adaptive immune cells: the latter is achieved via stimulating maturation of antigen-presenting cells (dendritic cells, B-cells, activated macrophages). On one hand, this may complicate the use of mRNA in replacement therapy. On the other hand, such particular properties underlie the usage of mRNA for immunotherapy and vaccination upon its delivery to dendritic and T- and B-cells in vivo or ex vivo. At the present time, various anticancer mRNAs, which are able to stimulate immune response, have been examined in preclinical and clinical trials. The main advantage of this approach is an opportunity to develop personalized anticancer therapy by taking into consideration individual mutations.
One of the first attempts of usage mRNA for the therapy of metastatic prostate cancer has an approach, when patients were administered autologous dendritic cells ex vivo transfected with mRNA encoding prostate-specific antigen (PSA). This resulted in detection of PSA-specific T-cells in peripheral blood (9 out of 9 examined patients), thereby allowing the use of this approach for modulating T-cell immune response in humans [107]. Nonetheless, a recent clinical trial with mRNA vaccine against prostate cancer (stage I/IIa) [108] has already been based on applying several mRNAs encoding four different antigens, and the data obtained suggest a fifth candidate antigen. Thus, this approach becomes significantly more complicated and begins to resemble use of total tumor-derived RNA for stimulation of polyclonal tumor-specific T-cell immunity [109, 110]. However, the use of mRNA vaccines undoubtedly wins in terms of safety. For instance, this approach was applied to stimulate T-cell immune response in prostate cancer (upregulated T-cell response was observed in 12 out of 19 patients) and melanoma (10 out of 19 patients) [111, 112]. Alternatively, mRNA can be administered directly via the intradermal route. A two-component vaccine containing mRNA encoding antigen supplemented with mRNA–protamine complex for enhancing immune response was developed by CureVac AG (Germany) based on studies on stimulating immune response via direct mRNA vaccination containing granulocyte–monocyte colony stimulating factor [113]. It was demonstrated during preclinical studies that this vaccine produced good results, and it is currently being used in clinical trials with prostate cancer patients [114]. In this review, we just outline a few examples from such a broad field, which, undoubtedly, have great potential for rapid introduction into clinical practice [115].
Perhaps one of the most prominent events in medical history was discovery of vaccination against infectious diseases, which can prevent later infection or ameliorate its course. Currently used live attenuated virus vaccines protect against poliomyelitis, rubella, smallpox, and other dangerous infections. Apart from them, safer recombinant vaccines containing pathogen-derived antigens are used as well. In some cases, pathogen-derived proteins administered to the host must be matured via processing by endoproteases. As an alternative, vaccines based on mRNA might also trigger both humoral and cell-mediated immune response. Compared to antibodies recognizing one viral subtype, which is developed after a standard vaccination, T-cell-mediated immune response might be triggered by various viral subtypes, thereby making mRNA vaccines potentially more universal. Moreover, in the case of pandemics, mRNA can be quickly adjusted to new mutant viruses and manufactured in sufficient amount for vaccination of many people.
In 1993 it was demonstrated for the first time that mRNA encoding influenza virus protein induced virus-specific T-cell immune response in mice [116]. This study laid the foundation for applying artificially synthesized mRNA in vaccination. In the case of influenza, a two-component vaccine containing an auxiliary mRNA and hemagglutinin-encoding mRNA was designed [117]. An alternative one-component anti-influenza mRNA vaccine was found to be effective as well [118]. Here, it is interesting to note an unsuccessful attempt to create anti-HIV mRNA-based vaccine. In particular, patients were administered dendritic cells containing mRNA that coded for HIV proteins and triggered antigen-specific CD8+ and CD4+ T-cell immune response [119, 120]. However, no antiviral effect was found in the vaccinated patients [120].
The possibility of using mRNA in replacement therapy depends on development of methods for chemical modification of mRNA for improving its stability and reducing immunogenicity. It was demonstrated that mRNA encoding surfactant protein-B (SP-B) delivered in aerosol into the lungs resulted in its marked expression and increased mice survival [31]. Delivery of mRNA encoding transcription factor FOXP2 efficiently downregulated immune response in asthma model [121]. Also mRNA-based therapy might be used in therapy of cardiovascular diseases, primarily in regenerative cardiology [122]. For instance, after myocardial infarction, mice administered mRNA encoding Vascular Endothelial Growth Factor (VEGF) via the intramyocardial route resulted in significant vessel regeneration and increased long-term survival [123]. However, it should be kept in mind that this approach currently has serious limitations related to poor stability and delivery of mRNA as well as high immunogenicity.
In addition we will describe several interesting approaches based on mRNA, some of which so far have been performed only in vitro. As expression of transcription factors can reprogram fibroblast differentiation [124], delivery of appropriate mRNAs can induce pluripotent stem cells [125]. Interestingly, in this case immunogenicity of mRNA had a positive effect, as TLR3 activation is necessary for efficient induction of pluripotency genes in the cells [126]. Moreover, use of mRNAs encoding nucleases required for genome editing (ZFN, TALEN, CRISPR-Cas9) may be another promising approach. Nonspecific insertion of DNA fragments resulting from a prolonged translation of nucleases from DNA vectors now represents a major problem during genome editing. There are studies reporting successful use of mRNA encoding nucleases in vivo [127-129], and most likely this approach will be applied more often in the future. In addition, mRNA encoding transposase Sleeping Beauty and piggyBac in genome editing has been reported [130-132]. So far, such studies are far from preclinical trials, but they may become a powerful tool in molecular and cell biology.
For managing protein expression, a large number of methods relying on nucleic acids have been described. Synthetic nucleic acids (NA) have been extensively investigated for use in clinical practice. More than 70 NAs are currently passing through different phases of clinical trials, and two of them have already been approved by the FDA. Now major efforts are directed to applying antisense oligonucleotides, siRNAs, and aptamers. Despite such a variety of NA-based technologies, use of synthetic mRNAs will undoubtedly find its niche in therapy. Obviously, in the near future mRNA vaccines will come into medical practice, especially in cancer immunotherapy. In addition, use of mRNAs holds promise for reprograming somatic cells towards pluripotent stem cells [133].
The possibility of rapid and short-term increase in concentration of a target protein is a crucial point for the use of mRNAs in replacement therapy. Usually, the highest expression level is observed within the first 24 h after administration, and existing alternative approaches do not allow achieving such kinetics for protein accumulation and downregulation. However, efficient application of mRNAs for expression of therapeutic proteins or replacement therapy does not seem easily attainable at this time. Despite the data discussed above, there is a need to search for novel modified nucleosides to stabilize mRNA in vivo as well as to develop more efficient means for targeted delivery to lower the amount of applied mRNA and associated side effects. Based on these data, polymer and lipid vehicles, notably conjugated with ligands of cell receptors, seem to be the most promising delivery systems. In the latter, sugar residues including mannose and N-acetylgalactosamine as well as folic acid derivatives hold the greatest promise. Similar to other NAs, release of mRNA from endosomes still represents a key problem for a delivery system at the cell level. This may be solved only by creating pH-dependent lipids or polymers able to break the endosomal membrane more efficiently than current ones. Therefore, if the above-mentioned problems could be solved, there is no doubt that mRNA will find applications for local short-term expression of therapeutic proteins and replacement therapy. Also we want to emphasize that a significant part of the studies on mRNA therapeutics is carried out in pharmaceutical companies – Moderna Therapeutics (USA), Arcturus Therapeutics (USA), PhaseRx (USA), Acuitas Therapeutics (USA). However their results are not available in the press now.
This study was supported by the Russian Science Foundation (project No. 14-34-00017).
REFERENCES
1.Zamecnik, P. C., and Stephenson, M. L. (1978)
Inhibition of Rous sarcoma virus replication and cell transformation by
a specific oligodeoxynucleotide, Proc. Natl. Acad. Sci. USA,
75, 280-284.
2.Stephenson, M. L., and Zamecnik, P. C. (1978)
Inhibition of Rous sarcoma viral RNA translation by a specific
oligodeoxyribonucleotide, Proc. Natl. Acad. Sci. USA, 75,
285-288.
3.Bennett, C. F., and Swayze, E. E. (2010) RNA
targeting therapeutics: molecular mechanisms of antisense
oligonucleotides as a therapeutic platform, Annu. Rev. Pharmacol.
Toxicol., 50, 259-293.
4.Lundin, K. E., Gissberg, O., and Smith, C. I.
(2015) Oligonucleotide therapies: the past and the present, Hum.
Gene Ther., 26, 475-485.
5.Dobrovolskaia, M. A., and McNeil, S. E. (2015)
Immunological and hematological toxicities challenging clinical
translation of nucleic acid-based therapeutics, Expert Opin. Biol.
Ther., 15, 1023-1048.
6.Bobbin, M. L., and Rossi, J. J. (2016) RNA
interference (RNAi)-based therapeutics: delivering on the promise?
Annu. Rev. Pharmacol. Toxicol., 56, 103-122.
7.Prakash, V., Moore, M., and Yanez-Munoz, R. J.
(2016) Current progress in therapeutic gene editing for monogenic
diseases, Mol. Ther., 24, 465-474.
8.LaFountaine, J. S., Fathe, K., and Smyth, H. D.
(2015) Delivery and therapeutic applications of gene editing
technologies ZFNs, TALENs, and CRISPR/Cas9, Int. J. Pharm.,
494, 180-194.
9.Maeder, M. L., and Gersbach, C. A. (2016)
Genome-editing technologies for gene and cell therapy, Mol.
Ther., 24, 430-446.
10.Lander, E. S. (2016) The heroes of CRISPR,
Cell, 164, 18-28.
11.Lee, C. M., Cradick, T. J., Fine, E. J., and Bao,
G. (2016) Nuclease target site selection for maximizing on-target
activity and minimizing off-target effects in genome editing, Mol.
Ther., 24, 475-487.
12.Yin, H., Xue, W., Chen, S., Bogorad, R. L.,
Benedetti, E., Grompe, M., Koteliansky, V., Sharp, P. A., Jacks, T.,
and Anderson, D. G. (2014) Genome editing with Cas9 in adult mice
corrects a disease mutation and phenotype, Nat. Biotechnol.,
32, 551-553.
13.Yin, H., Song, C. Q., Dorkin, J. R., Zhu, L. J.,
Li, Y., Wu, Q., Park, A., Yang, J., Suresh, S., Bizhanova, A., Gupta,
A., Bolukbasi, M. F., Walsh, S., Bogorad, R. L., Gao, G., Weng, Z.,
Dong, Y., Koteliansky, V., Wolfe, S. A., Langer, R., Xue, W., and
Anderson, D. G. (2016) Therapeutic genome editing by combined viral and
non-viral delivery of CRISPR system components in vivo, Nat.
Biotechnol., 34, 328-333.
14.Prazeres, D. M., and Monteiro, G. A. (2014)
Plasmid biopharmaceuticals, Microbiol. Spectr., 2, doi:
10.1128/microbiolspec.PLAS-0022-2014.
15.Meunier, M., Chemaly, M., and Dory, D. (2016) DNA
vaccination of poultry: the current status in 2015, Vaccine,
34, 202-211.
16.Fewell, J. G., MacLaughlin, F., Mehta, V., Gondo,
M., Nicol, F., Wilson, E., and Smith, L. C. (2001) Gene therapy for the
treatment of hemophilia B using PINC-formulated plasmid delivered to
muscle with electroporation, Mol. Ther., 3, 574-583.
17.Nikol, S., Baumgartner, I., Van Belle, E., Diehm,
C., Visona, A., Capogrossi, M. C., Ferreira-Maldent, N., Gallino, A.,
Wyatt, M. G., Wijesinghe, L. D., Fusari, M., Stephan, D., Emmerich, J.,
Pompilio, G., Vermassen, F., Pham, E., Grek, V., Coleman, M., and
Meyer, F. (2008) Therapeutic angiogenesis with intramuscular NV1FGF
improves amputation-free survival in patients with critical limb
ischemia, Mol. Ther., 16, 972-978.
18.Sahin, U., Kariko, K., and Ureci, O. (2014)
mRNA-based therapeutics – developing a new class of drugs,
Nat. Rev. Drug Discov., 13, 759-780.
19.Matsui, A., Uchida, S., Ishii, T., Itaka, K., and
Kataoka, K. (2015) Messenger RNA-based therapeutics for the treatment
of apoptosis-associated diseases, Sci. Rep., 5,
15810.
20.Warren, L., Manos, P. D., Ahfeldt, T., Loh, Y.
H., Li, H., Lau, F., Ebina, W., Mandal, P. K., Smith, Z. D., Meissner,
A., Daley, G. Q., Brack, A. S., Collins, J. J., Cowan, C., Schlaeger,
T. M., and Rossi, D. J. (2010) Highly efficient reprogramming to
pluripotency and directed differentiation of human cells with synthetic
modified mRNA, Cell Stem Cell, 7, 618-630.
21.Uchida, S., Kataoka, K., and Itaka, K. (2015)
Screening of mRNA chemical modification to maximize protein expression
with reduced immunogenicity, Pharmaceutics, 7,
137-151.
22.Mahiny, A. J., Dewerth, A., Mays, L. E.,
Alkhaled, M., Mothes, B., Malaeksefat, E., Loretz, B., Rottenberger,
J., Brosch, D. M., Reautschnig, P., Surapolchai, P., Zeyer, F., Schams,
A., Carevic, M., Bakele, M., Griese, M., Schwab, M., Nurnberg, B.,
Beer-Hammer, S., Handgretinger, R., Hartl, D., Lehr, C. M., and
Kormann, M. S. (2015) In vivo genome editing using
nuclease-encoding mRNA corrects SP-B deficiency, Nat.
Biotechnol., 33, 584-586.
23.Mehier-Humbert, S., and Guy, R. H. (2005)
Physical methods for gene transfer: improving the kinetics of gene
delivery into cells, Adv. Drug Deliv. Rev., 57,
733-753.
24.Zhang, X., and Godbey, W. T. (2006) Viral vectors
for gene delivery in tissue engineering, Adv. Drug Deliv. Rev.,
58, 515-534.
25.Kotterman, M. A., and Schaffer, D. V. (2014)
Engineering adeno-associated viruses for clinical gene therapy, Nat.
Rev. Genet., 15, 445-451.
26.Andreev, D. E., Terenin, I. M., Dmitriev, S. E.,
and Shatsky, I. N. (2016) Pros and cons of pDNA and mRNA transfection
to study mRNA translation in mammalian cells, Gene, 578,
1-6.
27.Gallie, D. R. (1991) The cap and poly(A) tail
function synergistically to regulate mRNA translational efficiency,
Genes Dev., 5, 2108-2116.
28.Kuhn, A. N., Diken, M., Kreiter, S., Selmi, A.,
Kowalska, J., Jemielity, J., Darzynkiewicz, E., Huber, C., Tureci, O.,
and Sahin, U. (2010) Phosphorothioate cap analogs increase stability
and translational efficiency of RNA vaccines in immaturedendritic cells
and induce superior immune responses in vivo, Gene Ther.,
17, 961-971.
29.Stepinski, J., Waddell, C., Stolarski, R.,
Darzynkiewicz, E., and Rhoads, R. E. (2001) Synthesis and properties of
mRNAs containing the novel “anti-reverse” cap analogs
7-methyl(3′-O-methyl)GpppG and 7-methyl
(3′-deoxy)GpppG, RNA, 7, 1486-1495.
30.Grudzien-Nogalska, E., Jemielity, J., Kowalska,
J., Darzynkiewicz, E., and Rhoads, R. E. (2007) Phosphorothioate cap
analogs stabilize mRNA and increase translational efficiency in
mammalian cells, RNA, 13, 1745-1755.
31.Loo, Y. M., Fornek, J., Crochet, N., Bajwa, G.,
Perwitasari, O., Martinez-Sobrido, L., Akira, S., Gill, M. A.,
Garcia-Sastre, A., Katze, M. G., and Gale, M., Jr. (2008) Distinct
RIG-I and MDA5 signaling by RNA viruses in innate immunity, J.
Virol., 82, 335-345.
32.Pichlmair, A., Schulz, O., Tan, C. P., Rehwinkel,
J., Kato, H., Takeuchi, O., Akira, S., Way, M., Schiavo, G., and Reis e
Sousa, C. (2009) Activation of MDA5 requires higher-order RNA
structures generated during virus infection, J. Virol.,
83, 10761-10769.
33.Lu, C., Xu, H., Ranjith-Kumar, C. T., Brooks, M.
T., Hou, T. Y., Hu, F., Herr, A. B., Strong, R. K., Kao, C. C., and Li,
P. (2010) The structural basis of 5′-triphosphate double-stranded
RNA recognition by RIG-I C-terminal domain, Structure,
18, 1032-1043.
34.Yoneyama, M., and Fujita, T. (2010) Recognition
of viral nucleic acids in innate immunity, Rev. Med. Virol.,
20, 4-22.
35.Sen, G. C., and Sarkar, S. N. (2007) The
interferon-stimulated genes: targets of direct signaling by
interferons, double-stranded RNA, and viruses, Curr. Top. Microbiol.
Immunol., 316, 233-250.
36.Andries, O., De Filette, M., De Smedt, S. C.,
Demeester, J., Van Poucke, M., Peelman, L., and Sanders, N. N. (2013)
Innate immune response and programmed cell death following
carrier-mediated delivery of unmodified mRNA to respiratory cells,
J. Control. Release, 167, 157-166.
37.Kanneganti, T. D., Body-Malapel, M., Amer, A.,
Park, J. H., Whitfield, J., Franchi, L., Taraporewala, Z. F., Miller,
D., Patton, J. T., Inohara, N., and Nunez, G. (2006) Critical role for
cryopyrin/Nalp3 in activation of caspase-1 in response to viral
infection and double-stranded RNA, J. Biol. Chem., 281,
36560-36568.
38.Nallagatla, S. R., Hwang, J., Toroney, R., Zheng,
X., Cameron, C. E., and Bevilacqua, P. C. (2007)
5′-Triphosphate-dependent activation of PKR by RNAs with short
stem-loops, Science, 318, 1455-1458.
39.Pardi, N., Muramatsu, H., Weissman, D., and
Kariko, K. (2013) In vitro transcription of long RNA containing
modified nucleosides, Methods Mol. Biol., 969, 29-42.
40.Brawerman, G. (1981) The role of the poly(A)
sequence in mammalian messenger RNA, CRC Crit. Rev. Biochem.,
10, 1-38.
41.Koski, G. K., Kariko, K., Xu, S., Weissman, D.,
Cohen, P. A., and Czerniecki, B. J. (2004) Cutting edge: innate immune
system discriminates between RNA containing bacterial versus eukaryotic
structural features that prime for high-level IL-12 secretion by
dendritic cells, J. Immunol., 172, 3989-3993.
42.Caput, D., Beutler, B., Hartog, K., Thayer, R.,
Brown-Shimer, S., and Cerami, A. (1986) Identification of a common
nucleotide sequence in the 3′-untranslated region of mRNA
molecules specifying inflammatory mediators, Proc. Natl. Acad. Sci.
USA, 83, 1670-1674.
43.Holtkamp, S. (2006) Modification of
antigen-encoding RNA increases stability, translational efficacy, and
T-cell stimulatory capacity of dendritic cells, Blood,
108, 4009-4017.
44.Kariko, K., Kuo, A., and Barnathan, E. (1999)
Overexpression of urokinase receptor in mammalian cells following
administration of the in vitro transcribed encoding mRNA,
Gene Ther., 6, 1092-1100.
45.Diebold, S. S., Massacrier, C., Akira, S.,
Paturel, C., Morel, Y., and Reis e Sousa, C. (2006) Nucleic acid
agonists for Toll-like receptor 7 are defined by the presence of
uridine ribonucleotides, Eur. J. Immunol., 36,
3256-3267.
46.Alexopoulou, L., Holt, A. C., Medzhitov, R., and
Flavell, R. A. (2001) Recognition of double-stranded RNA and activation
of NF-kappaB by Toll-like receptor 3, Nature, 413,
732-738.
47.Kariko, K., Buckstein, M., Ni, H., and Weissman,
D. (2005) Suppression of RNA recognition by Toll-like receptors: the
impact of nucleoside modification and the evolutionary origin of RNA,
Immunity, 23, 165-175.
48.Nallagatla, S. R., and Bevilacqua, P. C. (2008)
Nucleoside modifications modulate activation of the protein kinase PKR
in an RNA structure-specific manner, RNA, 14,
1201-1213.
49.Kariko, K., Muramatsu, H., Welsh, F. A., Ludwig,
J., Kato, H., Akira, S., and Weissman, D. (2008) Incorporation of
pseudouridine into mRNA yields superior nonimmunogenic vector with
increased translational capacity and biological stability, Mol.
Ther., 16, 1833-1840.
50.Li, B., Luo, X., and Dong, Y. (2016) Effects of
chemically modified messenger RNA on protein expression, Bioconj.
Chem., 27, 849-853.
51.Kormann, M. S., Hasenpusch, G., Aneja, M. K.,
Nica, G., Flemmer, A. W., Herber-Jonat, S., Huppmann, M., Mays, L. E.,
Illenyi, M., Schams, A., Griese, M., Bittmann, I., Handgretinger, R.,
Hartl, D., Rosenecker, J., and Rudolph, C. (2011) Expression of
therapeutic proteins after delivery of chemically modified mRNA in
mice, Nat. Biotechnol., 29, 154-157.
52.Wang, X., Zhao, B. S., Roundtree, I. A., Lu, Z.,
Han, D., Ma, H., Weng, X., Chen, K., Shi, H., and He, C. (2015)
N6-methyladenosine modulates messenger RNA translation efficiency,
Cell, 161, 1388-1399.
53.Kariko, K., Muramatsu, H., Ludwig, J., and
Weissman, D. (2011) Generating the optimal mRNA for therapy: HPLC
purification eliminates immune activation and improves translation of
nucleoside-modified, protein-encoding mRNA, Nucleic Acids Res.,
39, e142.
54.Rittig, S. M., Haentschel, M., Weimer, K. J.,
Heine, A., Muller, M. R., Brugger, W., Horger, M. S., Maksimovic, O.,
Stenzl, A., Hoerr, I., Rammensee, H. G., Holderried, T. A., Kanz, L.,
Pascolo, S., and Brossart, P. (2011) Intradermal vaccinations with RNA
coding for TAA generate CD8+ and CD4+ immune
responses and induce clinical benefit in vaccinated patients, Mol.
Ther., 19, 990-999.
55.Deering, R. P., Kommareddy, S., Ulmer, J. B.,
Brito, L. A., and Geall, A. J. (2014) Nucleic acid vaccines: prospects
for non-viral delivery of mRNA vaccines, Expert Opin. Drug
Deliv., 11, 885-899.
56.Brito, L. A., Kommareddy, S., Maione, D.,
Uematsu, Y., Giovani, C., Berlanda Scorza, F., Otten, G. R., Yu, D.,
Mandl, C. W., Mason, P. W., Dormitzer, P. R., Ulmer, J. B., and Geall,
A. J. (2015) Self-amplifying mRNA vaccines, Adv. Genet.,
89, 179-233.
57.Loomis, K. H., Kirschman, J. L., Bhosle, S.,
Bellamkonda, R. V., and Santangelo, P. J. (2016) Strategies for
modulating innate immune activation and protein production of in
vitro transcribed mRNAs, J. Mat. Chem. B, 4,
1619-1632.
58.Islam, M. A., Reesor, E. K. G., Xu, Y., Zope, H.
R., Zetter, B. R., and Shia, J. (2015) Biomaterials for mRNA delivery,
Biomater. Sci., 3, 1519-1533.
59.Kauffman, K. J., Webber, M. J., and Anderson, D.
G. (2015) Materials for non-viral intracellular delivery of messenger
RNA therapeutics, J. Control. Release, pii:
S0168-3659(15)30283-2.
60.Van Lint, S., Goyvaerts, C., Maenhout, S.,
Goethals, L., Disy, A., Benteyn, D., Pen, J., Bonehill, A., Heirman,
C., Breckpot, K., and Thielemans, K. (2012) Preclinical evaluation of
TriMix and antigen mRNA-based antitumor therapy, Cancer Res.,
72, 1661-1671.
61.Amos, H. (1961) Protamine enhancement of RNA
uptake by cultured chick cells, Biochem. Biophys. Res. Commun.,
5, 1-4.
62.Choi, Y. S., Lee, J. Y., Suh, J. S., Kwon, Y. M.,
Lee, S. J., Chung, J. K., Lee, D. S., Yang, V. C., Chung, C. P., and
Park, Y. J. (2010) The systemic delivery of siRNAs by a cell
penetrating peptide, low molecular weight protamine,
Biomaterials, 31, 1429-1443.
63.Kallen, K. J., Heidenreich, R., Schnee, M.,
Petsch, B., Schlake, T., Thess, A., Baumhof, P., Scheel, B., Koch, S.
D., and Fotin-Mleczek, M. (2013) A novel, disruptive vaccination
technology: self-adjuvanted RNActive® vaccines, Hum.
Vaccin. Immunother., 9, 2263-2276.
64.Brito, L. A., Chan, M., Shaw, C. A., Hekele, A.,
Carsillo, T., Schaefer, M., Archer, J., Seubert, A., Otten, G. R.,
Beard, C. W., Dey, A. K., Lilja, A., Valiante, N. M., Mason, P. W.,
Mandl, C. W., Barnett, S. W., Dormitzer, P. R., Ulmer, J. B., Singh,
M., O’Hagan, D. T., and Geall, A. J. (2014) A cationic
nanoemulsion for the delivery of next-generation RNA vaccines, Mol.
Ther., 22, 2118-2129.
65.Safinya, C. R. (2001) Structures of
lipid–DNA complexes: supramolecular assembly and gene delivery,
Curr. Opin. Struct. Biol., 11, 440-448.
66.Mockey, M., Bourseau, E., Chandrashekhar, V.,
Chaudhuri, A., Lafosse, S., Le Cam, E., Quesniaux, V. F., Ryffel, B.,
Pichon, C., and Midoux, P. (2007) mRNA-based cancer vaccine: prevention
of B16 melanoma progression and metastasis by systemic injection of
MART1 mRNA histidylated lipopolyplexes, Cancer Gene Ther.,
14, 802-814.
67.Hess, P. R., Boczkowski, D., Nair, S. K., Snyder,
D., and Gilboa, E. (2006) Vaccination with mRNAs encoding
tumor-associated antigens and granulocyte-macrophage colony-stimulating
factor efficiently primes CTL responses, but is insufficient to
overcome tolerance to a model tumor/self antigen, Cancer Immunol.
Immunother., 55, 672-683.
68.Pollard, C., Rejman, J., De Haes, W., Verrier,
B., Van Gulck, E., Naessens, T., De Smedt, S., Bogaert, P., Grooten,
J., Vanham, G., and De Koker, S. (2013) Type I IFN counteracts the
induction of antigen-specific immune responses by lipid-based delivery
of mRNA vaccines, Mol. Ther., 21, 251-259.
69.Geall, A. J., Verma, A., Otten, G. R., Shaw, C.
A., Hekele, A., Banerjee, K., Cu, Y., Beard, C. W., Brito, L. A.,
Krucker, T., O’Hagan, D. T., Singh, M., Mason, P. W., Valiante,
N. M., Dormitzer, P. R., Barnett, S. W., Rappuoli, R., Ulmer, J. B.,
and Mandl, C. W. (2012) Nonviral delivery of self-amplifying RNA
vaccines, Proc. Natl. Acad. Sci. USA, 109,
14604-14609.
70.Cheng, X., and Lee, R. J. (2016) The role of
helper lipids in lipid nanoparticles (LNPs) designed for
oligonucleotide delivery, Adv. Drug Deliv. Rev., 99,
129-137.
71.Tanaka, H., Sato, Y., Harashima, H., and Akita,
H. (2016) Cellular environment-responsive nanomaterials for use in gene
and siRNA delivery: molecular design for biomembrane destabilization
and intracellular collapse, Expert Opin. Drug Deliv., 21,
1-13.
72.Maier, M. A., Jayaraman, M., Matsuda, S., Liu,
J., Barros, S., Querbes, W., Tam, Y. K., Ansell, S. M., Kumar, V., Qin,
J., Zhang, X., Wang, Q., Panesar, S., Hutabarat, R., Carioto, M.,
Hettinger, J., Kandasamy, P., Butler, D., Rajeev, K. G., Pang, B.,
Charisse, K., Fitzgerald, K., Mui, B. L., Du, X., Cullis, P., Madden,
T. D., Hope, M. J., Manoharan, M., and Akinc, A. (2013) Biodegradable
lipids enabling rapidly eliminated lipid nanoparticles for systemic
delivery of RNAi therapeutics, Mol. Ther., 21,
1570-1578.
73.Pardi, N., Tuyishime, S., Muramatsu, H., Kariko,
K., Mui, B. L., Tam, Y. K., Madden, T. D., Hope, M. J., and Weissman,
D. (2015) Expression kinetics of nucleoside-modified mRNA delivered in
lipid nanoparticles to mice by various routes, J. Control.
Release, 217, 345-351.
74.Naseri, N., Valizadeh, H., and Zakeri-Milani, P.
(2015) Solid lipid nanoparticles and nanostructured lipid carriers:
structure, preparation and application, Adv. Pharm. Bull.,
5, 305-313.
75.Midoux, P., and Pichon, C. (2015) Lipid-based
mRNA vaccine delivery systems, Expert Rev. Vaccines, 14,
221-234.
76.Warashina, S., Nakamura, T., Sato, Y., Fujiwara,
Y., Hyodo, M., Hatakeyama, H., and Harashima, H. (2016) A lipid
nanoparticle for the efficient delivery of siRNA to dendritic cells,
J. Control. Release, 225, 183-191.
77.Wang, Y., Rajala, A., and Rajala, R. V. (2015)
Lipid nanoparticles for ocular gene delivery, J. Funct.
Biomater., 6, 379-394.
78.Kim, Y. D., Park, T. E., Singh, B., Maharjan, S.,
Choi, Y. J., Choung, P. H., Arote, R. B., and Cho, C. S. (2015)
Nanoparticle-mediated delivery of siRNA for effective lung cancer
therapy, Nanomedicine, 10, 1165-1188.
79.Li, B., Luo, X., Deng, B., Wang, J., McComb, D.
W., Shi, Y., Gaensler, K. M., Tan, X., Dunn, A. L., Kerlin, B. A., and
Dong, Y. (2015) An orthogonal array optimization of lipid-like
nanoparticles for mRNA delivery in vivo, Nano Lett.,
15, 8099-8107.
80.Li, B., Luo, X., Deng, B., Giancola, J. B.,
McComb, D. W., Schmittgen, T. D., and Dong, Y. (2016) Effects of local
structural transformation of lipid-like compounds on delivery of
messenger RNA, Sci. Rep., 6, 22137.
81.Kauffman, K. J., Dorkin, J. R., Yang, J. H.,
Heartlein, M. W., DeRosa, F., Mir, F. F., Fenton, O. S., and Anderson,
D. G. (2015) Optimization of lipid nanoparticle formulations for mRNA
delivery in vivo with fractional factorial and definitive
screening designs, Nano Lett., 15, 7300-7306.
82.Fenton, O. S., Kauffman, K. J., McClellan, R. L.,
Appel, E. A., Dorkin, J. R., Tibbitt, M. W., Heartlein, M. W., DeRosa,
F., Langer, R., and Anderson, D. G. (2016) Bioinspired alkenyl amino
alcohol ionizable lipid materials for highly potent in vivo mRNA
delivery, Adv. Mater., 28, 2939-2943.
83.Akinc, A., Querbes, W., De, S., Qin, J.,
Frank-Kamenetsky, M., Jayaprakash, K. N., Jayaraman, M., Rajeev, K. G.,
Cantley, W. L., Dorkin, J. R., Butler, J. S., Qin, L., Racie, T.,
Sprague, A., Fava, E., Zeigerer, A., Hope, M. J., Zerial, M., Sah, D.
W., Fitzgerald, K., Tracy, M. A., Manoharan, M., Koteliansky, V.,
Fougerolles, A., and Maier, M. A. (2010) Targeted delivery of RNAi
therapeutics with endogenous and exogenous ligand-based mechanisms,
Mol. Ther., 18, 1357-1364.
84.Kumar, V., Qin, J., Jiang, Y., Duncan, R. G.,
Brigham, B., Fishman, S., Nair, J. K., Akinc, A., Barros, S. A., and
Kasperkovitz, P. V. (2014) Shielding of lipid nanoparticles for siRNA
delivery: impact on physicochemical properties, cytokine induction, and
efficacy, Mol. Ther. Nucleic Acids, 3, e210.
85.Markov, O. V., Mironova, N. L., Shmendel, E. V.,
Serikov, R. N., Morozova, N. G., Maslov, M. A., Vlassov, V. V., and
Zenkova, M. A. (2015) Multicomponent mannose-containing liposomes
efficiently deliver RNA in murine immature dendritic cells and provide
productive anti-tumour response in murine melanoma model, J.
Control. Release, 213, 45-56.
86.Crowley, S. T., Poliskey, J. A., Baumhover, N.
J., and Rice, K. G. (2015) Efficient expression of stabilized mRNA
PEG-peptide polyplexes in liver, Gene Ther., 22,
993-999.
87.Lachelt, U., and Wagner, E. (2015) Nucleic acid
therapeutics using polyplexes: a journey of 50 years (and beyond),
Chem. Rev., 115, 11043-11078.
88.Rozenkrants, A. A., and Sobolev, A. S. (2015)
Polyethylenimine polyplex nanoparticles and their behavior in cells and
body tissues, Izv. Akad. Nauk Ser. Khim., 2749-2755.
89.Rejman, J., Tavernier, G., Bavarsad, N.,
Demeester, J., and De Smedt, S. C. (2010) mRNA transfection of cervical
carcinoma and mesenchymal stem cells mediated by cationic carriers,
J. Control. Release, 147, 385-391.
90.Demoulins, T., Milona, P., Englezou, P. C.,
Ebensen, T., Schulze, K., Suter, R., Pichon, C., Midoux, P., Guzman, C.
A., Ruggli, N., and McCullough, K. C. (2015) Polyethylenimine-based
polyplex delivery of self-replicating RNA vaccines,
Nanomedicine, 12, 711-722.
91.Bettinger, T., Carlisle, R. C., Read, M. L.,
Ogris, M., and Seymour, L. W. (2001) Peptide-mediated RNA delivery: a
novel approach for enhanced transfection of primary and post-mitotic
cells, Nucleic Acids Res., 29, 3882-3891.
92.Su, X., Fricke, J., Kavanagh, D. G., and Irvine,
D. J. (2011) In vitro and in vivo mRNA delivery using
lipid-enveloped pH-responsive polymer nanoparticles, Mol.
Pharm., 8, 774-787.
93.Mockey, M., Bourseau, E., Chandrashekhar, V.,
Chaudhuri, A., Lafosse, S., Le Cam, E., Quesniaux, V. F., Ryffel, B.,
Pichon, C., and Midoux, P. (2007) mRNA-based cancer vaccine: prevention
of B16 melanoma progression and metastasis by systemic injection of
MART1 mRNA histidylated lipopolyplexes, Cancer Gene Ther.,
14, 802-814.
94.Uchida, H., Itaka, K., Nomoto, T., Ishii, T.,
Suma, T., Ikegami, M., Miyata, K., Oba, M., Nishiyama, N., and Kataoka,
K. (2014) Modulated protonation of side chain aminoethylene repeats in
N-substituted polyaspartamides promotes mRNA transfection, J. Am.
Chem. Soc., 136, 12396-12405.
95.Uchida, H., Itaka, K., Uchida, S., Ishii, T.,
Suma, T., Miyata, K., Oba, M., Nishiyama, N., and Kataoka, K. (2016)
Synthetic polyamines to regulate mRNA translation through the
preservative binding of eukaryotic initiation factor 4E to the cap
structure, J. Am. Chem. Soc., 138, 1478-1481.
96.Phua, K. K., Leong, K. W., and Nair, S. K. (2013)
Transfection efficiency and transgene expression kinetics of mRNA
delivered in naked and nanoparticle format, J. Control. Release,
166, 227-233.
97.Uchida, S., Kinoh, H., Ishii, T., Matsui, A.,
Tockary, T. A., Takeda, K. M., Uchida, H., Osada, K., Itaka, K., and
Kataoka, K. (2016) Systemic delivery of messenger RNA for the treatment
of pancreatic cancer using polyplex nanomicelles with a cholesterol
moiety, Biomaterials, 82, 221-228.
98.Perche, F., Benvegnu, T., Berchel, M., Lebegue,
L., Pichon, C., Jaffres, P. A., and Midoux, P. (2011) Enhancement of
dendritic cells transfection in vivo and of vaccination against
B16F10 melanoma with mannosylated histidylated lipopolyplexes loaded
with tumor antigen messenger RNA, Nanomedicine, 7,
445-453.
99.Perche, F., Gosset, D., Mevel, M., Miramon, M.
L., Yaouanc, J. J., Pichon, C., Benvegnu, T., Jaffres, P. A., and
Midoux, P. (2011) Selective gene delivery in dendritic cells with
mannosylated and histidylated lipopolyplexes, J. Drug Target.,
19, 315-325.
100.Srinivas, R., Karmali, P. P., Pramanik, D.,
Garu, A., Mahidhar, Y. V., Majeti, B. K., Ramakrishna, S., Srinivas,
G., and Chaudhuri, A. (2010) Cationic amphiphile with shikimic acid
headgroup shows more systemic promise than its mannosyl analogue as DNA
vaccine carrier in dendritic cell based genetic immunization, J.
Med. Chem., 53, 1387-1391.
101.Srinivas, R., Garu, A., Moku, G., Agawane, S.
B., and Chaudhuri, A. (2012) A long-lasting dendritic cell DNA
vaccination system using lysinylated amphiphiles with mannose-mimicking
head-groups, Biomaterials, 33, 6220-6229.
102.Johler, S. M., Rejman, J., Guan, S., and
Rosenecker, J. (2015) Nebulisation of IVT mRNA complexes for
intrapulmonary administration, PLoS One, 10,
e0137504.
103.Uchida, S., Itaka, K., Uchida, H., Hayakawa,
K., Ogata, T., Ishii, T., Fukushima, S., Osada, K., and Kataoka, K.
(2013) In vivo messenger RNA introduction into the central
nervous system using polyplex nanomicelle, PLoS One, 8,
e56220.
104.Baba, M., Itaka, K., Kondo, K., Yamasoba, T.,
and Kataoka, K. (2015) Treatment of neurological disorders by
introducing mRNA in vivo using polyplex nanomicelles, J.
Control. Release, 201, 41-48.
105.Weiss, R., Scheiblhofer, S., Roesler, E.,
Weinberger, E., and Thalhamer, J. (2012) mRNA vaccination as a safe
approach for specific protection from type I allergy, Expert Rev.
Vaccines, 11, 55-67.
106.Scheel, B., Teufel, R., Probst, J., Carralot,
J. P., Geginat, J., Radsak, M., Jarrossay, D., Wagner, H., Jung, G.,
Rammensee, H. G., Hoerr, I., and Pascolo, S. (2005) Toll-like
receptor-dependent activation of several human blood cell types by
protamine-condensed mRNA, Eur. J. Immunol., 35,
1557-1566.
107.Heiser, A., Coleman, D., Dannull, J., Yancey,
D., Maurice, M. A., Lallas, C. D., Dahm, P., Niedzwiecki, D., Gilboa,
E., and Vieweg, J. (2002) Autologous dendritic cells transfected with
prostate-specific antigen RNA stimulate CTL responses against
metastatic prostate tumors, J. Clin. Invest., 109,
409-417.
108.Kubler, H., Scheel, B., Gnad-Vogt, U., Miller,
K., Schultze-Seemann, W., Vom Dorp, F., Parmiani, G., Hampel, C.,
Wedel, S., Trojan, L., Jocham, D., Maurer, T., Rippin, G.,
Fotin-Mleczek, M., Von der Mulbe, F., Probst, J., Hoerr, I., Kallen, K.
J., Lander, T., and Stenzl, A. (2015) Self-adjuvanted mRNA vaccination
in advanced prostate cancer patients: a first-in-man phase I/IIa study,
J. Immunother. Cancer, 3, 26.
109.Su, Z., Dannull, J., Heiser, A., Yancey, D.,
Pruitt, S., Madden, J., Coleman, D., Niedzwiecki, D., Gilboa, E., and
Vieweg, J. (2003) Immunological and clinical responses in metastatic
renal cancer patients vaccinated with tumor RNA-transfected dendritic
cells, Cancer Res., 63, 2127-2133.
110.Dannull, J., Su, Z., Rizzieri, D., Yang, B. K.,
Coleman, D., Yancey, D., Zhang, A., Dahm, P., Chao, N., Gilboa, E., and
Vieweg, J. (2005) Enhancement of vaccine-mediated antitumor immunity in
cancer patients after depletion of regulatory T-cells, J. Clin.
Invest., 115, 3623-3633.
111.Mu, L. J., Kyte, J. A., Kvalheim, G., Aamdal,
S., Dueland, S., Hauser, M., Hammerstad, H.,Waehre, H., Raabe, N., and
Gaudernack, G. (2005) Immunotherapy with allotumour mRNA-transfected
dendritic cells in androgen-resistant prostate cancer patients, Br.
J. Cancer, 93, 749-756.
112.Kyte, J. A., Mu, L., Aamdal, S., Kvalheim, G.,
Dueland, S., Hauser, M., Gullestad, H. P., Ryder, T., Lislerud, K.,
Hammerstad, H., and Gaudernack, G. (2006) Phase I/II trial of melanoma
therapy with dendritic cells transfected with autologous tumor-mRNA,
Cancer Gene Ther., 13, 905-918.
113.Weide, B., Carralot, J. P., Reese, A., Scheel,
B., Eigentler, T. K., Hoerr, I., Rammensee, H. G., Garbe, C., and
Pascolo, S. (2008) Results of the first phase I/II clinical vaccination
trial with direct injection of mRNA, J. Immunother., 31,
180-188.
114.Fotin-Mleczek, M., Duchardt, K. M., Lorenz, C.,
Pfeiffer, R., Ojkic-Zrna, S., Probst, J., and Kallen, K. J. (2011)
Messenger RNA-based vaccines with dual activity induce balanced TLR-7
dependent adaptive immune responses and provide antitumor activity,
J. Immunother., 34, 1-15.
115.Pascolo, S. (2008) Vaccination with messenger
RNA (mRNA), Handb. Exp. Pharmacol., 183, 221-235.
116.Martinon, F., Krishnan, S., Lenzen, G., Magne,
R., Gomard, E., Guillet, J. G., Levy, J. P., and Meulien, P. (1993)
Induction of virus-specific cytotoxic T lymphocytes in vivo by
liposome-entrapped mRNA, Eur. J. Immunol., 23,
1719-1722.
117.Petsch, B., Schnee, M., Vogel, A. B., Lange,
E., Hoffmann, B., Voss, D., Schlake, T., Thess, A., Kallen, K. J.,
Stitz, L., and Kramps, T. (2012) Protective efficacy of in vitro
synthesized, specific mRNA vaccines against influenza A virus
infection, Nat. Biotechnol., 30, 1210-1216.
118.Brazzoli, M., Magini, D., Bonci, A., Buccato,
S., Giovani, C., Kratzer, R., Zurli, V., Mangiavacchi, S., Casini, D.,
Brito, L. M., De Gregorio, E., Mason, P. W., Ulmer, J. B., Geall, A.
J., and Bertholet, S. (2015) Induction of broad-based immunity and
protective efficacy by self-amplifying mRNA vaccines encoding influenza
virus hemagglutinin, J. Virol., 90, 332-344.
119.Allard, S. D., De Keersmaecker, B., De Goede,
A. L., Verschuren, E. J., Koetsveld, J., Reedijk, M. L., Wylock, C., De
Bel, A. V., Vandeloo, J., Pistoor, F., Heirman, C., Beyer, W. E.,
Eilers, P. H., Corthals, J., Padmos, I., Thielemans, K., Osterhaus, A.
D., Lacor, P., Van der Ende, M. E., Aerts, J. L., Van Baalen, C. A.,
and Gruters, R. A. (2012) A phase I/IIa immunotherapy trial of
HIV-1-infected patients with Tat, Rev and Nef expressing dendritic
cells followed by treatment interruption, Clin. Immunol.,
142, 252-268.
120.Van Gulck, E., Vlieghe, E., Vekemans, M., Van
Tendeloo, V. F., Van De Velde, A., Smits, E., Anguille, S., Cools, N.,
Goossens, H., Mertens, L., De Haes, W., Wong, J., Florence, E., Vanham,
G., and Berneman, Z. N. (2012) mRNA-based dendritic cell vaccination
induces potent antiviral T-cell responses in HIV-1-infected patients,
AIDS, 26, 1-12.
121.Mays, L. E., Ammon-Treiber, S., Mothes, B.,
Alkhaled, M., Rottenberger, J., Muller-Hermelink, E. S., Grimm, M.,
Mezger, M., Beer-Hammer, S., Von Stebut, E., Rieber, N., Nurnberg, B.,
Schwab, M., Handgretinger, R., Idzko, M., Hartl, D., and Kormann, M. S.
(2013) Modified Foxp3 mRNA protects against asthma through an
IL-10-dependent mechanism, J. Clin. Invest., 123,
1216-1228.
122.Lui, K. O., Zangi, L., and Chien, K. R. (2014)
Cardiovascular regenerative therapeutics via synthetic paracrine factor
modified mRNA, Stem Cell Res., 13, 693-704.
123.Zangi, L., Lui, K. O., Von Gise, A., Ma, Q.,
Ebina, W., Ptaszek, L. M., Spater, D., Xu, H., Tabebordbar, M.,
Gorbatov, R., Sena, B., Nahrendorf, M., Briscoe, D. M., Li, R. A.,
Wagers, A. J., Rossi, D. J., Pu, W. T., and Chien, K. R. (2013)
Modified mRNA directs the fate of heart progenitor cells and induces
vascular regeneration after myocardialinfarction, Nat.
Biotechnol., 31, 898-907.
124.Takahashi, K., Tanabe, K., Ohnuki, M., Narita,
M., Ichisaka, T., Tomoda, K., and Yamanaka, S. (2007) Induction of
pluripotent stem cells from adult human fibroblasts by defined factors,
Cell, 131, 861-872.
125.Yakubov, E., Rechavi, G., Rozenblatt, S., and
Givol, D. (2010) Reprogramming of human fibroblasts to pluripotent stem
cells using mRNA of four transcription factors, Biochem. Biophys.
Res. Commun., 394, 189-193.
126.Lee, J., Sayed, N., Hunter, A., Au, K. F.,
Wong, W. H., Mocarski, E. S., Pera, R. R., Yakubov, E., and Cooke, J.
P. (2012) Activation of innate immunity is required for efficient
nuclear reprogramming, Cell, 151, 547-558.
127.Doyon, Y., McCammon, J. M., Miller, J. C.,
Faraji, F., Ngo, C., Katibah, G. E., Amora, R., Hocking T. D., Zhang,
L., Rebar, E. J., Gregory, P. D., Urnov, F. D., and Amacher, S. L.
(2008) Heritable targeted gene disruption in zebrafish using designed
zinc-finger nucleases, Nat. Biotechnol., 26, 702-708.
128.Tesson, L., Usal, C., Menoret, S., Leung, E.,
Niles, B. J., Remy, S., Santiago, Y., Vincent, A. I., Meng, X., Zhang,
L., Gregory, P. D., Anegon, I., and Cost, G. J. (2011) Knockout rats
generated by embryo microinjection of TALENs, Nat. Biotechnol.,
29, 695-696.
129.Wang, H., Yang, H., Shivalila, C. S., Dawlaty,
M. M., Cheng, A. W., Zhang, F., and Jaenisch, R. (2013) One-step
generation of mice carrying mutations in multiple genes by
CRISPR/Cas-mediated genome engineering, Cell, 153,
910-918.
130.Wilber, A., Frandsen, J. L., Geurts, J. L.,
Largaespada, D. A., Hackett, P. B., and McIvor, R. S. (2006) RNA as a
source of transposase for sleeping beauty-mediated gene insertion and
expression in somatic cells and tissues, Mol. Ther., 13,
625-630.
131.Furushima, K., Jang, C. W., Chen, D. W., Xiao,
N., Overbeek, P. A., and Behringer, R. R. (2012) Insertional
mutagenesis by a hybrid piggyBac and sleeping beauty transposon in the
rat, Genetics, 192, 1235-1248.
132.Bire, S., Gosset, D., Jegot, G., Midoux, P.,
Pichon, C., and Rouleux-Bonnin, F. (2013) Exogenous mRNA delivery and
bioavailability in gene transfer mediated by piggyBac transposition,
BMC Biotechnol., 13, 75.
133.Warren, L., Manos, P. D., Ahfeldt, T., Loh, Y.
H., Li, H., Lau, F., Ebina, W., Mandal, P. K., Smith, Z. D., Meissner,
A., Daley, G. Q., Brack, A. S., Collins, J. J., Cowan, C., Schlaeger,
T. M., and Rossi, D. J. (2010) Highly efficient reprogramming to
pluripotency and directed differentiation of human cells with synthetic
modified mRNA, Cell Stem Cell, 7, 618-630.