Previously Unclassified Mutation of mtDNA m.3472T>C: Evidence of Pathogenicity in Leber’s Hereditary Optic Neuropathy
N. L. Sheremet1*, T. A. Nevinitsyna1, N. V. Zhorzholadze1, I. A. Ronzina1, Y. S. Itkis2, T. D. Krylova2, P. G. Tsygankova2, V. A. Malakhova2, E. Y. Zakharova2, A. V. Tokarchuk3, A. A. Panteleeva3, E. M. Karger3, K. G. Lyamzaev3*, and S. E. Avetisov1
1Research Institute of Eye Diseases, 119021 Moscow, Russia; E-mail: sheremet_n@mail.ru2Research Center for Medical Genetics, 115478 Moscow, Russia
3Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: lyamzaev@gmail.com
* To whom correspondence should be addressed.
Received March 21, 2016; Revision received April 8, 2016
Leber’s hereditary optic neuropathy (LHON) refers to a group of mitochondrial diseases and is characterized by defects of the mitochondrial electron transport chain and decreased level of oxidative phosphorylation. The list of LHON primary mtDNA mutations is regularly updated. In this study, we describe the homoplasmic nucleotide substitution m.3472T>C in the MT-ND1 (NADH-ubiquinone oxidoreductase chain 1) gene and specific changes in cell metabolism in a patient with LHON and his asymptomatic sister. To confirm the presence of mutation-related mitochondrial dysfunction, respiration of skin fibroblasts and platelets from the patient and his sister was studied, as well as the mitochondrial potential and production of reactive oxygen species in the skin fibroblasts. In addition, based on characteristics of the toxic effect of paraquat, a new approach was developed for detecting the functional activity of complex I of the mitochondrial respiratory chain.
KEY WORDS: Leber’s hereditary optic neuropathy, mtDNA mutations, respiratory chain complex I, fibroblastsDOI: 10.1134/S0006297916070117
Abbreviations: LHON, Leber’s hereditary optic neuropathy; MLPA method, Multiplex Ligase-dependent Probe Amplification respirometry; mtDNA, mitochondrial DNA; NGS, Next-Generation Sequencing; ROS, reactive oxygen species.
Leber’s hereditary optic neuropathy (LHON) is a disease associated
with mutations of mitochondrial DNA (mtDNA) leading to a painless
bilateral loss of the central vision. The diagnosis can be confirmed
using molecular genetics methods for analyzing a patient’s blood,
which reveals the mutation in mtDNA. About 95% of LHON cases are
associated with one of three major primary mutations (m.11778G>A,
m.3460G>A, m.14484T>C), but in addition 14 rare primary mutations
have been described that are also related with LHON pathogenesis [1]. Moreover, there are also nucleotide substitutions
that are assigned to candidate mutations. Their contribution to the
disease has not been proven due to insufficient number of studies.
However, there are many reports about new primary and potential mtDNA
mutations associated with LHON that are not yet included in genetic
databases, but it seems that the list of mutations will be updated [2, 3]. Studies on the
mitochondrial genome of such patients are rather difficult because
mtDNA is highly polymorphic [4]. To proclaim a
nucleotide substitution a pathogenic mutation, it is necessary to
establish a genotype–phenotype correlation and the presence of
the mutation in at least two unrelated patients [2]. Measurements of the activities of the respiratory
chain complexes in cell cultures obtained from the patients and
bioinformatics studies are also very important.
Mutations in mtDNA lead to defects in a structure of mtDNA-encoded proteins (subunits of the mitochondrial respiratory chain complexes I-IV, ATP-synthase complex) and mitochondrial transport RNAs. In LHON, complex I is affected that results in the decrease of the transmembrane potential on the inner mitochondrial membrane, which is necessary for functioning of H+-ATP-synthase (complex V) synthesizing ATP from ADP and Pi [5, 6]. LHON patient’s cells suffer not only from energy insufficiency, but also from excess production of reactive oxygen species (ROS), which damage neighboring structures, thus aggravating the respiratory dysfunction [6-10]. These changes can be detected by investigating patients’ skin fibroblasts and blood cells and also hybrid cells carrying mutant mtDNA.
MATERIALS AND METHODS
The patient, 19-year-old male, with clinical features of LHON and his 27-year-old sister were examined. mtDNA sequence analysis, functional studies of skin fibroblasts and blood platelets mitochondria and bioinformatics analysis were performed.
Clinical examination. Anamnesis morbi, anamnesis vitae including family history of the disease were studied in detail; routine ophthalmologic examination was also performed. The visual acuity was assessed using the LogMAR scale and the Sivtsev–Golovin table; color vision was assessed using polychromatic tables of E. B. Rabkin for studies on color perception [11]; the visual field was studied using static and kinetic perimetry (neurologic and semi-automatic kinetic programs Octopus 900; Interzeag AG, Switzerland); parameters of the retina and optic nerve head were assessed using optical coherence tomography (RTVue-100; Optovue, USA) with subsequent analysis of the peripapillar retinal nerve fibers layer and optic nerve disk and assessment of the ganglion cell complex including the ganglion cells and the inner plexiform layers. Electrophysiological parameters were also determined: Flash and Pattern Visual Evoked Potential (programs “VEP flash” and “VEP standard”, Tomey EP-1000; Multifocal, Germany), the threshold of retinal electric sensitivity and lability of the visual analyzer (Lametesk, Russia), and the critical frequency of flickering fusion (Lametesk).
Molecular genetic analysis. Total DNA was isolated from venous blood (4 ml into standard EDTA tubes) taken from a patient with supposed LHON and from his non-affected sister. Three primary mutations (m.11778G>A, m.3460G>A, m.14484T>C) were analyzed using a MLPA method with subsequent analysis of the fragment lengths in 9% polyacrylamide gel. The MLPA method — Multiplex Ligase-dependent Probe Amplification of nucleotide sequences — is based on the ability of ligases to form a chemical bond between nucleotides in the place of a DNA chain break; this method allows researchers to provide simultaneous detection of multiple mutations in the same tube [12]. The whole mtDNA sequence analysis was performed using semiconductor Next-Generation Sequencing method (NGS). The samples were prepared in the order listed: PCR of two long fragments of mtDNA (8.5 kb each) covering the whole mtDNA sequence was conducted using a polymerase of Phusion® High-Fidelity DNA Polymerase (New England Biolabs, Great Britain), with the subsequent isolation from the agarose gel on innuPREP Gel Extraction Kit columns (Analytikjena, Germany) according to the manufacture’s protocol. A mixture of two mtDNA fragments (50 ng each) was used to prepare libraries for sequencing using the commercial kit NEBNext® Fast DNA Fragmentation & Library Prep Set for Ion Torrent™ (New England Biolabs) for subsequent parallel sequencing on the IonTorrent device (Life Technologies, Thermo Fisher Scientific, USA). The sequence data alignment (GenBank NC_012920) was done in the IGV online program (BROAD Institute). The detected nucleotide substitutions were compared with the database on mutations and polymorphisms of mtDNA Mitomap. This study did not include a search for large rearrangements of mtDNA.
Cell cultures. Fibroblasts were cultivated from the patient’s and his sister’s skin biopsy as described earlier [13]. Studies were performed using intact cells from the patient, his sister, and control cultures and also cells permeabilized with digitonin, what allowed to use different substrates for the respiratory chain to study the mitochondrial respiration.
The cells were cultured in medium containing antibiotics (gentamycin 10 mg/ml, penicillin 5000 U/ml, streptomycin 5000 µg/ml), HEPES, DMEM with glucose (4.5 g/liter), and fetal bovine serum (FBS). The rate of oxygen consumption by fibroblasts (intact and permeabilized) and by platelets was measured with an Oxygraph-2k (Oroboros Corp., Austria) with the DatLab 5.0 program [14].
The experiment with intact cells was performed as follows: after measurement of the basal respiration of the cells (on endogenous substrates) (R), 5 mM pyruvate and 0.5 mM malate were added and then 2.5 µM oligomycin to inhibit complex V of the respiratory chain (L). The maximum respiration was achieved using the uncoupler of oxidative phosphorylation carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP) (E), then 0.5 µM rotenone was added to inhibit complex I, then complex III was inhibited with 2.5 µM antimycin A to determine the antimycin-resistant respiration [15].
The experiment with the permeabilized cells was performed according to the protocol proposed by Ye and Hoppel [13] for successive measurement of oxygen consumption rates using substrates and inhibitors for respiratory chain complexes I, II, and IV: the cells were supplemented with 5 mM pyruvate and 0.5 mM malate, and then 0.5 µg/ml digitonin to permeabilize the plasma membrane, then 1 mM ADP, 10 mM glutamate, 10 mM succinate, 0.05 µM FCCP were added, then 0.5 µM rotenone, and later 2.5 µM antimycin A.
The level of ROS in the intact fibroblasts without exogenous substrates or mitochondrial inhibitors was determined using fluorescent probes DCF-DA [16] and MitoSOX [17]. The membrane potential of the mitochondria was assessed with the fluorescent probe TMRM [18] (Sigma, USA). The cells were incubated with DCF-DA (4 µM) for 30 min, MitoSOX (2.5 µM) for 30 min, or with TMRM (100 nM) for 15 min. Then the cells were taken from the substrate with a solution of trypsin with Versene, and cells fluorescence was measured using a flow cytofluorimeter (Beckman Coulter Cytomics FC 500; Beckman, USA). In the experiments, fibroblasts cultures obtained from three healthy persons were used for controls.
Cell death under the influence of the prooxidant paraquat was determined by fluorimetry using CellTiterBlue® reagent (Promega, USA) containing resazurin dye capable of penetrating into the cells and being reduced to a fluorescent resorufin within a living and metabolically active cell. To determine a number of living cells, the CellTiterBlue® reagent was added into the cell culture that was incubated at 37°C for 5 h (the incubation time was chosen experimentally considering the low metabolic activity of fibroblasts), and then the fluorescent signal of resorufin was measured (λex = 560 nm; λem = 590 nm) (Fluoroskan Ascent, Thermo, USA).
RESULTS AND DISCUSSION
The 19-year-old male patient applied to the Institute of Eye Diseases complaining of a dramatic painless bilateral vision loss that had arisen 2 months earlier.
At the examination, his visual acuity was as follows: OD – 0.04 (1.41 logMAR in the Freiburg test [19]) and OS – 0.04 (1.44 logMAR), a pronounced dyschromatopsy and centrocecal scotoma of both eyes were revealed. Biomicro- and ophthalmoscopy revealed no changes. Optic coherent tomography showed an increased thickness of the peripapillar retinal nerve fibers layer in the upper temporal quadrant and thinning of the macular ganglion cell complex, what is typical for the early stage of LHON [20]. Changes were found in electrophysiological parameters: a decrease in electrosensitivity of the retina inner layers and in the visual nerve conductivity, an increase in the P100 peak latency, a decrease in visual analyzer lability and the peak P100 amplitude, as well as a decrease in critical frequency of flickering fusion. MRI of the brain and of the orbits detected no changes.
At the first examination and during subsequent observation, any other etiology of visual loss (traumatic, toxic, inflammatory, demyelinating, vascular, or compression) was excluded. The observed clinical features corresponded to LHON, but none of the three major mutations (m.11778G>A, m.3460G>A, m.14484T>C) was found. Therefore, an enhanced molecular genetic analysis was performed to confirm the influence of hereditary factors on the vision decrease. To be sure in the diagnosis, functional parameters of cell respiration were studied on fibroblasts and platelets of the patient and his sister.
The sequencing of mtDNA revealed the nucleotide substitution of thymine by cytosine in position 3472 of mtDNA in the homoplasmic state, which led to substitution of phenylalanine by leucine in position 56 (p.F56L) in the subunit I of NADH dehydrogenase (respiratory chain complex I). According to GenBank, the frequency of such substitution is very low – 0.01%. No other pathogenic or undescribed changes were detected in the patient’s mtDNA. Based on a totality of mtDNA polymorphisms detected in the analysis, the patient was assigned to haplogroup H2a1. In the 27-year-old patient’s sister without any clinical or subclinical manifestations of optical neuropathy, the same nucleotide homoplasmic transition was also found. According to the Mitomap database, the detected substitution was not included in the list of confirmed primary or candidate LHON mutation. Analysis of the detected nucleotide substitution in the PolyPhen-2 program predicting the functional effect showed a high value of 0.897. Phenylalanine 56 in the protein subunit I of NADH dehydrogenase is located in a very conservative region according to the UniProtKB database. All these data allowed us to classify the m.3472T>C substitution as probably pathogenic.
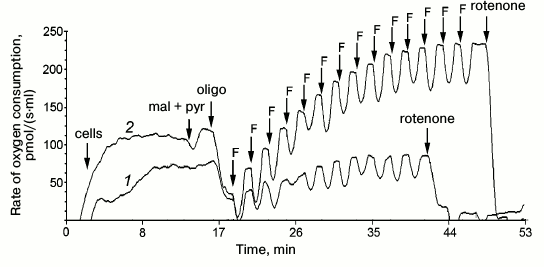
Platelets and fibroblasts of the patient and his sister were studied to evaluate the pathogenicity of the m.3472T>C substitution. The rate of oxygen consumption of intact platelets and fibroblasts of the patient (Fig. 1) and of his sister on endogenous substrate was significantly lower than in the control samples. Moreover, in both cases the ratios R/E, L/E, and (R – L)/E were increased (R is cell respiration on endogenous substrates; E is maximal respiration rate stimulated by uncoupler of oxidative phosphorylation; L is rate of oxygen consumption by cells upon addition of inhibitor of ATP synthase oligomycin; the ratio (R – L)/E characterizes the contribution of respiration coupled with synthesis of ATP) (table). In the fibroblasts of the patient and his sister, the ratio (R – L)/E was more than twofold higher than normal, and similar changes, although less pronounced, were observed in the platelets. These data indicated that the reserve respiration power of the cells of the patient and his sister was strongly decreased on the endogenous substrates, i.e. the respiratory chain functioned with rate close to maximal, and the major part of the released energy was expended for synthesis of ATP. Concurrently, the relative contribution of oligomycin-resistant respiration (L/E), i.e. of respiration uncoupled with synthesis of ATP, was significantly increased, what suggested decreased efficiency of oxidative phosphorylation.
Fig. 1. Rate of oxygen consumption by intact fibroblasts of the patient (1) and of control culture (2) on addition of 5 mM pyruvate and 0.5 mM malate (pyr + mal), 2.5 µM oligomycin (oligo); titration by 0.05 µM FCCP (F); 0.5 µM rotenone, 2.5 µM antimycin A.
Rates of respiration of intact fibroblasts and platelets of the patient,
his sister, and of the control group
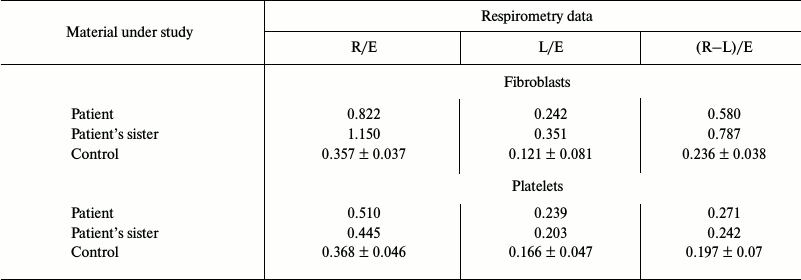
Note: R is cell respiration on endogenous substrates; E is maximal rate
of respiratory chain functioning; L is rate of oxygen consumption after
addition of oligomycin (an inhibitor of ATP synthase). Data for the
control group are presented as average values obtained from
measurements of respiration of samples taken from seven healthy
donors.
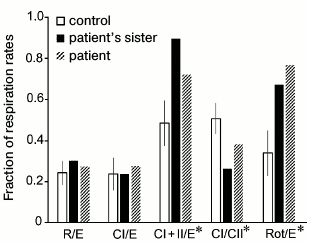
Results of measurement of cellular respiration of permeabilized fibroblasts are presented in graphs (Figs. 2 and 3). These results were compared with data obtained on seven control fibroblast cultures using the STATISTICA 10 software. The ratio of respiration rates on the mixture of substrates of respiratory chain complexes I and II and of maximal respiration rate on the same substrates (CI + II/E) was significantly increased in the cells of the patient and his sister. This ratio was 0.72 for the patient and 0.9 for his sister, whereas the normal values were from 0.37 to 0.59; this indicated low ability of the mitochondria to use substrates of complexes I and II for oxidative phosphorylation. This was also associated with an increased contribution of complex II-derived respiration. CI/CII ratio was decreased in the cells of the patient and his sister (0.38 and 0.26, respectively, normal values 0.41-0.59), which also implies complex I pathology or a compensatory complex II activation [21]. The rotenone-sensitive uncoupled respiration, i.e. that depends on complex I activity (Rot/E), was significantly altered in the patient (Fig. 2) and in his sister (0.78 and 0.69, respectively, normal values from 0.19 to 0.51), thus definitely indicating complex I defect.
Fig. 2. Data of respirometry of permeabilized fibroblasts of the patient (1) in comparison with control fibroblasts (2) on addition of 5 mM pyruvate and 0.5 mM malate (pyr + mal), 0.5 µg/ml digitonin, 1 mM ADP, 10 mM glutamate (glut), 10 mM succinate (succ); titration by 0.05 µM FCCP (F); 0.5 µM rotenone, 2.5 µM antimycin A (ant).
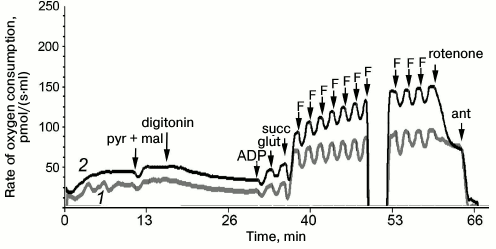
Fig. 3. Ratios of respiration rates in permeabilized fibroblasts of the patient, his sister, and in the control cultures. Experimental conditions were the same as in Fig. 2. R is the base respiration of cells on endogenous substrates; E is maximal rate of respiratory chain functioning; CI is rate of oxygen consumption by cells after addition of glutamate (complex I substrate); CII is rate of oxygen consumption by cells after addition of succinate (complex II substrate); Rot is rate of oxygen consumption by cells after addition of rotenone (complex I inhibitor). * Ratios with significant difference (p < 0.05) between the control group and the fibroblast cultures with deficiency in respiratory chain complex I.
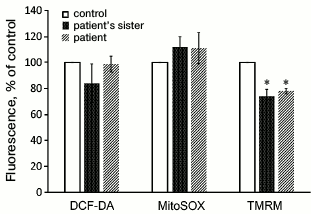
It was also found that the membrane potential of mitochondria in the cells with the m.3472T>C substitution was significantly lower than in the cell lines without mutations in complex I (Fig. 4). The level of oxidative stress measured with DCF-DA or the mitochondria-targeted dye MitoSOX did not show significant difference between the fibroblasts from the patient and his sister and from the control donors (Fig. 4).
Fig. 4. Oxidative stress level (DCF-DA, MitoSOX) and membrane potential of mitochondria (TMRM) in cultures of fibroblasts obtained from skin biopsy of healthy control donors and of the patient and his sister (n = 3, * p < 0.05).
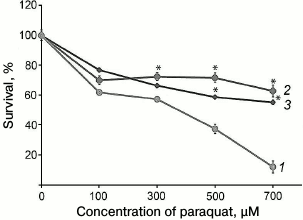
In the present study, we developed a new approach for determination of complex I function, and it can be used for rapid screening of multitude cell lines. This approach is based on a specific feature of the prooxidant effect of paraquat (N,N′-dimethyl-4,4′-bipyridinium dichloride), which is widely used in studies for inducing oxidative stress in cells. It was shown earlier that for manifestation of cytotoxic effect of paraquat, it must be reduced in mitochondria using up complex I activity, which gives paraquat the ability to interact with oxygen, producing superoxide [22]. Thus, the toxic effect of paraquat is directly associated with complex I activity. This observation was used for testing fibroblasts cultures obtained from patients with suspected LHON. It was found that cells with the m.3472T>C substitution were significantly more resistant to the paraquat, which suggested decreased ability of complex I to reduce paraquat (Fig. 5).
Fig. 5. Cytotoxic effect of paraquat on fibroblasts obtained from skin biopsy of healthy control donors (1) and of the patient (2) and his sister (3) (* p < 0.05).
It is interesting that the study of the patient sister`s cells demonstrated more prominent functional changes. Nevertheless, LHON manifests only in about 50% of men and 10% of women carrying a pathogenic mutation of mtDNA [23]; therefore, the absence of visual loss is a typical situation for women carriers of mtDNA mutations in families with LHON. Obviously, the presence of the mutation is not sufficient for manifestation of the disease [23]. Now the influence of a nuclear and a mitochondrial genome and of a hormonal status on the LHON onset is actively studied [24-26], as well as the influence of such external factors as smoking, stress, alcohol and drug consumption [27, 28]. Moreover, recent studies of Giordano et al. [29] revealed that the absence of clinical features in individuals carrying the same mutation as family members with LHON could be associated with increased amount of mtDNA and activation of mitochondrial biogenesis (increased synthesis of mitochondrial proteins) in all cells of the body.
The only case of the m.3472T>C homoplasmic substitution in gene MT-ND1 in a patient with clinical features of LHON was published in 2014 [30]. The same substitution was found in the patient’s mother blood cells, but she did not have clinical signs of the disease. The authors of that study carefully analyzed the case and showed that the substitution could be considered as a primary mtDNA mutation responsible for LHON. To confirm the mitochondrial genesis of the disease, the authors studied hybrid cells carrying the patient’s mtDNA. Functional investigations revealed a decrease in membrane potential and in complex I activity, although the ROS level in the hybrid cells was within normal values.
At this moment, the m.3472T>C substitution had been detected in four individuals among 30,589 examined throughout the world. Two of these four individuals suffered from diseases that are commonly associated with mutations in mtDNA (LHON in the patient described in [30] and a case of schizophrenia [31]).
Thus, our study confirms that the m.3472T>C substitution in mtDNA could be associated with risk of LHON. It was also found that this mutation led to dysfunction of respiratory chain complex I and decreased membrane potential, but ROS level in the cells was within normal range, which is in agreement with previously published data [30].
CONCLUSIONS
1) A homoplasmic nucleotide substitution of thymine by cytosine in position 3472 in the MT-ND1 gene has been described in a patient with clinical features of LHON. This mutation leads to substitution of phenylalanine by leucine in position 56 in the protein subunit of NADH dehydrogenase.
2) Functional studies of fibroblasts and platelets from carriers of the m.3472T>C substitution were performed. A dysfunction of cellular respiration, in particular, a mitochondrial respiratory chain complex I deficiency, and a decrease in mitochondrial membrane potential were confirmed.
3) Analysis of nucleotide substitution m.3472T>C using the PolyPhen-2 software and of the amino acid substitution p.F56L in the ND1 subunit of NADH dehydrogenase in the UniProtKB database allows us to assign this substitution as probably pathogenic.
4) Considering the earlier described LHON case with the same nucleotide substitution and the results of the present study, the mutation m.3472T>C in the MT-ND1 gene can be classified as pathogenic.
This work was supported by the Russian Science Foundation (project No. 14-24-00107) and by the Russian Foundation for Basic Research (project No. 15-04-09050).
REFERENCES
1.http://www.mitomap.org/bin/view.pl/MITOMAP/MutationsLHON.
2.Yu-Wai-Man, P., Votruba, M., Moore, A. T., and
Chinnery, P. F. (2014) Treatment strategies for inherited optic
neuropathies: past, present and future, Eye (London), 28,
521-537.
3.Achilli, A., Iommarini, L., Olivieri, A., Pala, M.,
Hooshiar Kashani, B., Reynier, P., La Morgia, C., Valentino, M. L.,
Liguori, R., Pizza, F., Barboni, P., Sadun, F., De Negri, A. M.,
Zeviani, M., Dollfus, H., Moulignier, A., Ducos, G., Orssaud, C.,
Bonneau, D., Procaccio, V., Leo-Kottler, B., Fauser, S., Wissinger, B.,
Amati-Bonneau, P., Torroni, A., and Carelli, V. (2012) Rare primary
mitochondrial DNA mutations and probable synergistic variants in
Leber’s hereditary optic neuropathy, PLoS One, 7,
e42242.
4.Ruiz-Pesini, E., Lott, M. T., Procaccio, V., Poole,
J. C., Brandon, M. C., Mishmar, D., Yi, C., Kreuziger, J., Baldi, P.,
and Wallace, D. C. (2007) An enhanced MITOMAP with a global mtDNA
mutational phylogeny, Nucleic Acids Res., 35,
D823-D828.
5.Carelli, V., Ross-Cisneros, F. N., and Sadun, A. A.
(2004) Mitochondrial dysfunction as a cause of optic neuropathies,
Prog. Retin. Eye Res., 23, 53-89.
6.Baracca, A., Solaini, G., Sgarbi, G., Lenaz, G.,
Baruzzi, A., Schapira, A. H. V., Martinuzzi, A., and Carelli, V. (2005)
Severe impairment of complex I-driven adenosine triphosphate synthesis
in Leber hereditary optic neuropathy cybrids, Arch. Neurol.,
62, 730-736.
7.Handy, D. E., and Loscalzo, J. (2012) Redox
regulation of mitochondrial function, Antioxid. Redox Signal.,
16, 1323-1367.
8.Beretta, S., Mattavelli, L., Sala, G., Tremolizzo,
L., Schapira, A. H. V., Martinuzzi, A., Carelli, V., and Ferrarese, C.
(2004) Leber hereditary optic neuropathy mtDNA mutations disrupt
glutamate transport in hybrid cell lines, Brain, 127,
2183-2192.
9.Lenaz, G. (1998) Role of mitochondria in oxidative
stress and ageing, Biochim. Biophys. Acta, 1366,
53-67.
10.Wong, A., Cavelier, L., Collins-Schramm, H. E.,
Seldin, M. F., McGrogan, M., Savontaus, M. L., and Cortopassi, G. A.
(2002) Differentiation-specific effects of LHON mutations introduced
into neuronal NT2 cells, Human Mol. Genet., 11,
431-438.
11.Rabkin, E. B. (1971) Polychromatic Tables to
Study the Color Perception [in Russian], Meditsina, Moscow.
12.https://www.mlpa.com.
13.Ye, F., and Hoppel, C. L. (2013) Measuring
oxidative phosphorylation in human skin fibroblasts, Anal.
Biochem., 437, 52-58.
14.Sjovall, F., Ehinger, J. K. H., Marelsson, S. E.,
Morota, S., Frostner, E. A., Uchino, H., Lundgren, J., Arnbjornsson,
E., Hansson, M. J., Fellman, V., and Elmer, E. (2013) Mitochondrial
respiration in human viable platelets – methodology and
influence of gender, age and storage, Mitochondrion, 13,
7-14.
15.Pesta, D., and Gnaiger, E. (2012) High-resolution
respirometry: OXPHOS protocols for human cells and permeabilized fibers
from small biopsies of human muscle, Methods Mol. Biol.,
810, 25-58.
16.Eruslanov, E., and Kusmartsev, S. (2010)
Identification of ROS using oxidized DCFDA and flow-cytometry,
Methods Mol. Biol., 594, 57-72.
17.Mukhopadhyay, P., Rajesh, M., Yoshihiro, K.,
Hasko, G., and Pacher, P. (2007) Simple quantitative detection of
mitochondrial superoxide production in live cells, Biochem. Biophys.
Res. Commun., 358, 203-208.
18.Rasola, A., and Geuna, M. (2001) A flow cytometry
assay simultaneously detects independent apoptotic parameters,
Cytometry, 45, 151-157.
19.Bach, M. (1996) The freiburg visual acuity
test – automatic measurement of visual acuity, Optom.
Vis. Sci., 73, 49-53.
20.Avetisov, S. E., Sheremet, N. L., Fomin, A. V.,
Galoyan, N. S., Khanakova, N. A., Zhorzholadze, N. V., Loginova, A. N.,
Chukhrova, N. A., and Polyakov, A. V. (2014) Morphological changes in
retina and optical nerve head in patients with Leber’s hereditary
optic neuropathy, Vestn. Oftalmol., 130, 4-11.
21.Yen, M. Y., Lee, H. C., Liu, J. H., and Wei, Y.
H. (1996) Compensatory elevation of complex II activity in
Leber’s hereditary optic neuropathy, Br. J. Ophthalmol.,
80, 78-81.
22.Cocheme, H. M., and Murphy, M. P. (2008) Complex
I is the major site of mitochondrial superoxide production by paraquat,
J. Biol. Chem., 283, 1786-1798.
23.Yu-Wai-Man, P., Griffiths, P. G., and Chinnery,
P. F. (2011) Mitochondrial optic neuropathies – disease
mechanisms and therapeutic strategies, Prog. Retin. Eye Res.,
30, 81-114.
24.Ji, Y., Jia, X., Li, S., Xiao, X., Guo, X., and
Zhang, Q. (2010) Evaluation of the X-linked modifier loci for Leber
hereditary optic neuropathy with the G11778A mutation in Chinese,
Mol. Vis., 16, 416-424.
25.Bu, X. D., and Rotter, J. I. (1991) X
chromosome-linked and mitochondrial gene control of Leber hereditary
optic neuropathy: evidence from segregation analysis for dependence on
X chromosome inactivation, Proc. Natl. Acad. Sci. USA,
88, 8198-8202.
26.Kirkman, M. A., Yu-Wai-Man, P., Korsten, A.,
Leonhardt, M., Dimitriadis, K., De Coo, I. F., Klopstock, T., and
Chinnery, P. F. (2009) Gene–environment interactions in Leber
hereditary optic neuropathy, Brain, 132, 2317-2326.
27.Amaral-Fernandes, M. S., Marcondes, A. M.,
Miranda, P. M., Maciel-Guerra, A. T., and Sartorato, E. L. (2011)
Mutations for Leber hereditary optic neuropathy in patients with
alcohol and tobacco optic neuropathy, Mol. Vis., 17,
3175-3179.
28.Giordano, L., Deceglie, S., D’Adamo, P.,
Valentino, M. L., La Morgia, C., Fracasso, F., Roberti, M., Cappellari,
M., Petrosillo, G., Ciaravolo, S., Parente, D., Giordano, C., Maresca,
A., Iommarini, L., Del Dotto, V., Ghelli, A. M., Salomao, S. R.,
Berezovsky, A., Belfort, R., Jr., Sadun, A. A., Carelli, V., Loguercio
Polosa, P., and Cantatore, P. (2015) Cigarette toxicity triggers
Leber’s hereditary optic neuropathy by affecting mtDNA copy
number, oxidative phosphorylation and ROS detoxification pathways,
Cell Death Dis., 6, e2021.
29.Giordano, C., Iommarini, L., Giordano, L.,
Maresca, A., Pisano, A., Valentino, M. L., Caporali, L., Liguori, R.,
Deceglie, S., Roberti, M., Fanelli, F., Fracasso, F., Ross-Cisneros, F.
N., D’Adamo, P., Hudson, G., Pyle, A., Yu-Wai-Man, P., Chinnery,
P. F., Zeviani, M., Salomao, S. R., Berezovsky, A., Belfort, R.,
Ventura, D. F., Moraes, M., Moraes, M., Barboni, P., Sadun, F., De
Negri, A., Sadun, A. A., Tancredi, A., Mancini, M., D’Amati, G.,
Polosa, P. L., Cantatore, P., and Carelli, V. (2014) Efficient
mitochondrial biogenesis drives incomplete penetrance in Leber’s
hereditary optic neuropathy, Brain, 137, 335-353.
30.Martinez-Romero, I., Herrero-Martin, M. D.,
Llobet, L., Emperador, S., Martin-Navarro, A., Narberhaus, B., Ascaso,
F. J., Lopez-Gallardo, E., Montoya, J., and Ruiz-Pesini, E. (2014) New
MT-ND1 pathologic mutation for Leber hereditary optic neuropathy,
Clin. Exp. Ophthalmol., 42, 856-864.
31.Ueno, H., Nishigaki, Y., Kong, Q. P., Fuku, N.,
Kojima, S., Iwata, N., Ozaki, N., and Tanaka, M. (2009) Analysis of
mitochondrial DNA variants in Japanese patients with schizophrenia,
Mitochondrion, 9, 385-393.