Phylogenomic Analysis of Type 1 NADH:Quinone Oxidoreductase
G. E. Novakovsky1, D. V. Dibrova2, and A. Y. Mulkidjanian1,2,3*
1Lomonosov Moscow State University, Faculty of Bioengineering and Bioinformatics, 119991 Moscow, Russia2Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119991 Moscow, Russia
3Osnabrueck University, Department of Physics, 49069 Osnabrueck, Germany; E-mail: amulkid@uos.de, armen.mulkidjanian@uni-osnabrueck.de
* To whom correspondence should be addressed.
Received November 10, 2015; Revision received March 22, 2016
We performed phylogenomic analysis of the catalytic core of NADH:quinone oxidoreductases of type 1 (NDH-1). Analysis of phylogenetic trees, as constructed for the core subunits of NDH-1, revealed fundamental differences in their topologies. In the case of four putatively homologous ion-carrying membrane subunits, the trees for the NuoH and NuoN subunits contained separate archaeal clades, whereas subunits NuoL and NuoM were characterized by multiple archaeal clades spread among bacterial branches. Large, separate clades, which united sequences belonging to different archaeal subdomains, were also found for cytoplasmic subunits NuoD and NuoB, homologous to the large and small subunits of nickel-iron hydrogenases. A smaller such clade was also shown for subunit NuoC. Based on these data, we suggest that the ancestral NDH-1 complex could be present already at the stage of the Last Universal Cellular Ancestor (LUCA). Ancestral forms of membrane subunits NuoN and NuoH and cytoplasmic subunits NuoD, NuoB, and, perhaps NuoC, may have formed a membrane complex that operated as an ion-translocating membrane hydrogenase. After the complex attained the ability to reduce membrane quinones, gene duplications could yield the subunits NuoL and NuoM, which enabled translocation of additional ions.
KEY WORDS: proton bioenergetics, molecular evolution, respiration, protein complexes, quinone reduction, phylogenetic analysisDOI: 10.1134/S0006297916070142
The NADH:quinone oxidoreductase (EC 1.6.5.2, also known as mitochondrial complex I or NADH-dehydrogenase of type 1, NDH-1) is the largest enzyme complex in the respiratory chain of mitochondria. It couples electron transfer from NADH molecules in the cytoplasm to ubiquinone molecules in the membrane with the translocation of protons from the mitochondrial matrix into the intermembrane space. In many prokaryotes, homologs of complex I couple reduction of different quinones with ion translocation across the membrane [1, 2].
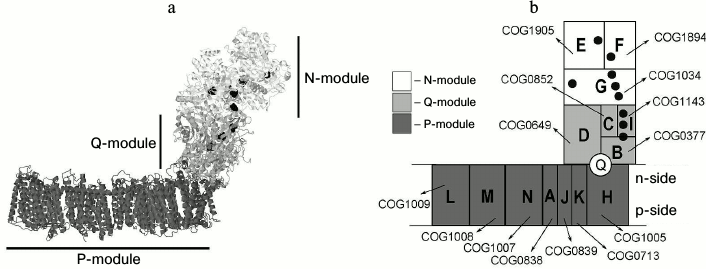
Prokaryotic NDH-1 complexes are generally built of a cytoplasmic quinone-reducing module (Q-module, four subunits) and a membrane-embedded proton translocation module (P-module, seven subunits). In some prokaryotes (specifically, in α-proteobacteria that are believed to be the ancestors of mitochondria), the NDH-1 has an additional NADH dehydrogenase module (N-module, three subunits). Generally, NDH-1 is an L-shaped complex with a peripheral cytoplasmic “arm” (consisting of N- and Q-modules) and a large membrane-embedded “anchor” (P-module) (Fig. 1). Mitochondrial complex I, in addition to the named 14 key subunits, has about 30 additional subunits [3, 4] whose functions remain mostly obscure.
Fig. 1. a) Structure of NDH-1 from Thermus thermophilus HB8 (PDB ID 4HEA) with modules indicated [17]. The iron-sulfur clusters are shown as spheres. b) Schematic representation of the 14 core NDH-1 subunits; subunits are designated as on panel (a). The “Clusters of Orthologous Group” (COG) number for each subunit is shown; the quinone-binding site is depicted at the interface of the NuoD, NuoB, and NuoH subunits [17]. The iron-sulfur clusters are shown as black circles.
The N-module consists of three proteins, which are usually called NuoE, NuoF, and NuoG following the Escherichia coli nomenclature (where Nuo is an abbreviation of the NADH:ubiquinone oxidoreductase). The NuoF subunit has a FMN (flavin mononucleotide) coenzyme and one 4Fe-4S iron-sulfur cluster N3. This subunit also has a NADH-binding site. The small NuoE subunit has one 2Fe-2S cluster N1a and appears to be a ferredoxin paralog [5]. The NuoG subunit is the largest in this module. It carries a 2Fe-2S cluster N1b, as well as three 4Fe-4S clusters N4, N5 and N7 [6]. The C-terminal domain of NuoG is homologous to molybdopterin-containing enzymes (such as formate dehydrogenase), whereas the N-terminal domain is a paralog of the subunits of soluble NAD+ hydrogenases [5].
The N-module is present not in all prokaryotic lineages; specifically, it is absent from cyanobacteria (and, correspondingly, from chloroplasts) [7]. In addition, this module is not found in some archaea (Euryarchaeota), in which a homolog of complex I, which is called F420H2 dehydrogenase, uses, instead of NADH, 2-hydroxyphenazine as an electron donor [8]; here, a single FpoF subunit takes the function of an electron-donor module. Some prokaryotes have 12-subunit NDH-1 complexes (the NuoE and NuoF subunits are absent) [9]. Flavodoxin was proposed to be an electron donor for these complexes [10].
The Q-module consists of four subunits. It accepts electrons from the N-module and transfers them via iron–sulfur clusters to a quinone-like acceptor (menaquinone in the majority of bacteria and archaea, plastoquinone in cyanobacteria/chloroplasts, ubiquinone in α-proteobacteria and mitochondria, methanophenazine in methanogenic archaea, etc.). This module is composed of a NuoC subunit, a ferredoxin-like NuoI subunit, and two proteins (NuoB and NuoD subunits) that are paralogous to the small and large subunits of soluble NiFe-hydrogenases [11]. The NuoI subunit carries two 4Fe-4S clusters N6a and N6b, which are involved in electron transfer to the 4Fe-4S cluster N2 of NuoB subunit [5]. The electrons from N2 go to a quinone molecule in the quinone-binding site. This site is situated at the junction of the cytoplasm and membrane parts of the complex, ~2.5 nm away from the lipid bilayer [12].
The remaining seven subunits (NuoA, NuoH, NuoJ, NuoK, NuoL, NuoM, and NuoN) form the proton translocating P-module. Its three subunits (NuoL, NuoM, and NuoN) have similar sequences, and they are probably paralogs. The NuoH subunit, which connects the cytoplasmic and membrane segments of the complex, has distant structural similarity with the NuoL/M/N subunits [13, 14]. In addition, the NuoL/M/N subunits were found to be distant paralogs of subunits A and D of Na+/H+-antiporters (also denoted as Mrp/Pha/Sha, or Mnh in some organisms) [15]. This family is considered as secondary active transporters that use the energy of transmembrane proton gradient to pump sodium ions (or potassium ions as in Rhizobium meliloti [16]) out of the cell.
Analysis of crystal structures has shown that the P-module is crossed by eight hydrophilic half-channels that seem to be involved in the translocation of four protons across the membrane in response to the two-electron reduction of a quinone [1, 13, 17]. Each of the large membrane subunits, NuoN, NuoM, and NuoL, contains two half-channels accounting, seemingly, for translocation of one proton. The fourth proton seems to be translocated by the joint action of the NuoH subunit (cytoplasmic half-channel) and small membrane subunits that together form the outer half-channel [1, 13, 17]. To get electrons from the iron–sulfur cluster N2, which is located ~2.5 nm away from the lipid bilayer, the quinone head protrudes from the membrane by 2.0-2.5 nm, going through the cavities inside the NuoH and NuoB subunits [2, 6]. The molecular mechanism of coupling between the two-electron quinone reduction and the translocation of four protons remains obscure.
Evolution of NADH:quinone oxidoreductases. NADH:quinone oxidoreductases have a modular structure, so they could have emerged from an interaction between enzymes that already played functional roles in the cell [14, 18, 19]. An evolutionary scenario proposed by Friedrich and coauthors [19] suggests that NADH:quinone oxidoreductase may have originated from a combination of a two-subunit enzyme, the precursor of soluble NiFe-hydrogenases, which consisted of homologs of NuoD and NuoB subunits, a ferredoxin-like protein (NuoI homolog), a quinone-reducing protein (NuoH homolog), an ion-translocating protein (homolog (ortholog) of either NuoL or NuoM), and a protein of unknown function (NuoC homolog) [20]. Their combination may have resulted in a six-subunit transmembrane complex – a potential common ancestor of NDH-1-related enzymes and so-called membrane-associated hydrogenases [21, 22]. These ancestral complexes contained a catalytic NiFe-site typical for membrane-associated hydrogenases. Later, this ancestral complex was assumed to acquire several additional membrane subunits (NuoA/J/K). The membrane ion-translocating protein probably triplicated and gave origin to the modern NuoL/NuoM/NuoN subunits. In this scenario, such an 11-subunit complex was a common ancestor of all NDH-1-related enzymes (in archaea, prokaryotes, and eukaryotes). The recruitment of additional electron-transport subunits may have occurred later in different lineages.
An alternative scenario was proposed by Hägerhäll and coauthors [18] and took into account the homology between the NuoN/M/L/K subunits and proteins of the Mrp-antiporter family. In this scenario, soluble NiFe-hydrogenases were suggested to form a complex with the Na+/H+-antiporter composed of subunits that are homologous to corresponding subunits of the NDH-1 complex. Also, some further proteins could have joined to this hypothetical membrane complex as ancestors of the NuoI and NuoH subunits, yielding the last common ancestor of NiFe membrane-bound hydrogenases and NDH-1-related complexes. Later, this scheme was supported with additional data; an 11-subunit complex, built of subunits of P- and Q-modules (including NuoA and NuoJ subunits) and having a NiFe-binding site, was suggested as the last common ancestor of NDH-1 and NiFe-membrane-bound hydrogenases [7]. Both the modern membrane-bound hydrogenases (through the loss of several subunits; hydrogenases usually contain fewer subunits than NDH-1) and the modern NDH-1-related enzymes (through the loss of a NiFe-site and acquisition of various electron-donor modules) were suggested to originate from it.
The recruitment of the ancestral antiporter module was also suggested in the scenario proposed by Adams and coauthors [23, 24]. In this scenario, the addition of an Mrp-antiporter module did not occur upon the formation of an 11-subunit complex, but on the stage of an ancestral membrane hydrogenase that resembled modern Ech hydrogenases [22]; according to this scenario, the 11-subunit complex evolved from this ancestral form. In fact, this scenario differs from the scenario proposed by Hägerhäll and coauthors in the nature of the primordial enzyme complex: whether it was a 6-subunit membrane hydrogenase, resembling modern Ech-hydrogenases, or an 11-subunit version of NDH-1.
According to the most recent scenario of Pereira and coauthors, the family of Mrp-antiporters might have developed from the last common ancestor of NDH-1 and membrane-bound hydrogenases, and not vice versa [14]. This scenario is based on a structural similarity between the NuoH subunit and NuoN/M/L subunits (which are homologous to the Mrp-antiporter subunits). As a homolog of NuoH is suggested to be a part of a so-called universal adaptor (as the authors suggest, a complex of homologs of subunits NuoB/NuoD/NuoH and NuoL that are common to all NDH-1-related complexes and all membrane-bound hydrogenases), this subunit was suggested to be a part of the last common ancestor of corresponding hydrogenases and NDH-1-related complexes. It was proposed that NuoN/M/L might have evolved from NuoH, and afterwards could have given rise to the Mrp-antiporter family.
Here we have applied phylogenomic analysis to reconstruct the evolution of NDH-1-related complexes. This method allows clarifying the phylogenetic relations between proteins through considering genomic level information, specifically, by using clusters of orthologous groups (COGs), which group genes according to their evolutionary relatedness (orthology) [25-27]. Earlier, by using such approach, we reconstructed the evolution of rotary membrane ATPases [28-30], cytochrome bc complexes [31] and photosynthetic machinery [32].
Our analysis of NDH-1 subunits has revealed the evolutionary primacy of the NuoN and NuoH subunits as compared to the two other ion-translocation subunits NuoM and NuoL. Our data indicate that the ancestor of NDH-1 could be built from an ancestral form of soluble NiFe-hydrogenases (homologous to modern NuoB/D/C) and a pair of ion-translocation membrane subunits (orthologs of NuoH and NuoN). The key point in the evolution of this membrane hydrogenase could be the emergence of a mechanism of quinone “escape” from the lipid bilayer. This trait would make it possible to reduce a hydrophobic quinone by the iron–sulfur cluster at 2-3-nm distance from the lipid bilayer. The free energy that is released during the electron transfer from a low-potential electron donor (NADH or ferredoxin) to a relatively high-potential acceptor (quinone) is sufficient for translocation of several ions across the membrane, which could have prompted the multiplication of ion-translocation subunits.
METHODS
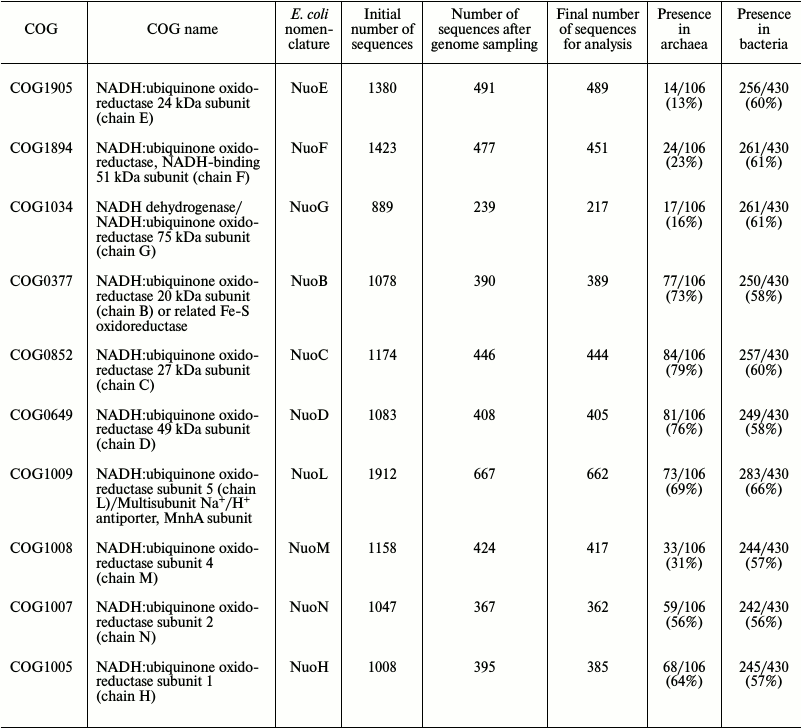
Searching for NDH-1 sequences and their alignment. Upon phylogenomic analysis, we used the version of COGs (Clusters of Orthologous Groups [25, 26]) available at ftp://ftp.ncbi.nih.gov/pub/wolf/COGs/Prok1202/. The following COGs were used: NuoE (COG1905), NuoF (COG1894), NuoG (COG1034); NuoB (COG0377), NuoC (COG0852), NuoD (COG0649), NuoI (COG1143); NuoH (COG1005), NuoL (COG1009), NuoM (COG1008), NuoN (COG1007), NuoA (COG0838), NuoJ (COG0839), NuoK (COG0713). A total of 1511 prokaryotic genomes were present in this COGs release. A representative sample from these genomes was made by applying the following two criteria: (i) if several strains of a single species were present in the database, only one of them was randomly sampled (for some model organisms, we took the strains nonrandomly, e.g. Escherichia coli K12 DH10B and Mycobacterium tuberculosis CDC1551), and (ii) for species of overrepresented bacterial phyla (namely, Firmicutes, Actinobacteria, and Proteobacteria), only a single species per family was sampled. For further analysis we used only proteins belonging to the sampled genomes, thus the number of sequences in each COG was reduced: for example, COG1005 originally contained 1008 proteins, whereas only 395 sequences remained after genome sampling. The representative sample thus contained 536 genomes (430 bacterial genomes and 106 archaeal genomes). In addition, after construction of multiple alignments, the number of sequences was finally reduced manually: some sequences that were apparently erroneously assigned to particular COGs were deleted, as they did not align with other sequences (table).
Data on the numbers of sequences in each examined COG. The initial
number of sequences in each COG, the number of sequences that remained
after removal of overrepresented taxons, and the final number of
sequences (after removal of sequences upon correction of alignments)
are given. Additionally, the numbers of genomes of archaea and bacteria
that contain representatives of a given COG (of 106 archaeal and 430
bacterial genomes that were considered) are provided
Protein sequences from the chosen organisms and their COGs were downloaded from the NCBI Genomes database (ftp://ftp.ncbi.nlm.nih.gov/genomes/) on the August 1, 2014.
Multiple sequence alignments were constructed using the Muscle program [33, 34] and manually inspected in JalView v.2.8.2 [35] to identify defects (badly aligned proteins, abnormally short and long sequences, etc.). The Alnalyser program (D. V. Dibrova, unpublished) was used for checking homogeneity of sequences in the alignment from their domain structure and overlap of transmembrane segments. The Alnalyser program maps predicted domains (according to the Pfam database [36]) and transmembrane segments (according to the TMHMM program [37]) on multiple sequences alignment. These data were taken into account upon correcting alignment defects.
Phylogenomic analysis. Phylogenetic trees were constructed from the alignments by the MEGA v.6.06 program [38] using the neighbor-joining method [39] with the JTT evolutionary model [40] and under assumption of uniform rates for alignment positions. The number of bootstrap replicas [41] was 100.
The leaves on phylogenetic trees were rendered according to taxonomy of organisms to which they belong: the names of archaeal proteins are rendered in black and the names of bacterial proteins in gray.
RESULTS
While analyzing the phylogenetic trees of NDH-1 subunits, we searched for well-separated characteristic archaeal and bacterial clades. Such clades on a phylogenetic tree could be considered as evidence for antiquity of a protein and its possible presence in the Last Universal Cellular Ancestor, LUCA [42], see also “Discussion”.
Phylogenomic analysis of the N-module. We analyzed all the three subunits of this module. The overall topology of the phylogenetic tree for the NuoE subunit is shown in Fig. 2a; the detailed and zoomable version of the tree is given in Supplement to this paper on the site of this journal http://protein.bio.msu.ru/biokhimiya and Springer site link.springer.com. The tree was built using an alignment of 489 protein sequences from COG1905 (the number of sequences was reduced according to the procedure described in “Methods”). Four hundred twenty nine positions were chosen for building the tree. The phylogenetic tree of NuoE contains only a few archaeal sequences, and they are scattered and do not form a single and separate clade. The lengths of branches that separate individual archaeal clades do not differ significantly from the lengths of other branches. It is likely that orthologs of NuoE were acquired by archaea from bacteria through horizontal gene transfer.
Fig. 2. Phylogenetic trees for subunits of the N-module; the detailed and zoomable version of the trees is given in the Supplement. Archaeal sequences are rendered in black, and bacterial sequences are rendered in gray. a) Phylogenetic tree for the NuoE subunit; b) phylogenetic tree for the NuoF subunit; c) phylogenetic tree for the NuoG subunit.
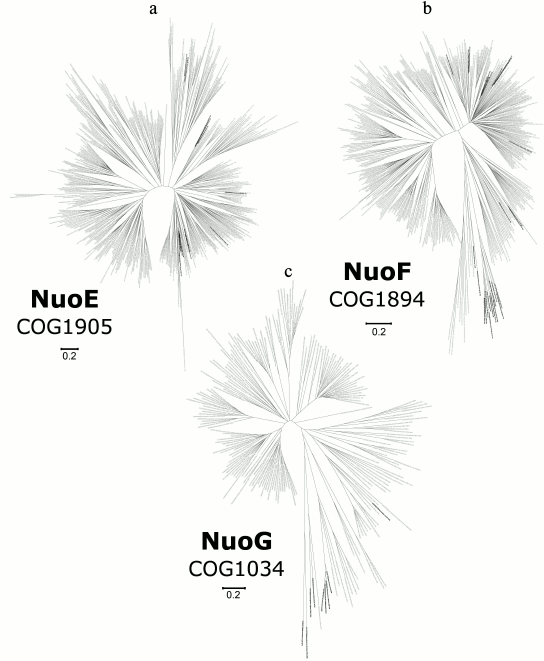
A phylogenetic tree for the NuoF subunit is shown in Fig. 2b; the detailed and zoomable version of the tree is given in the Supplement. An alignment of 451 sequences from COG1894 was used to build the tree by relying on 400 key positions. Again, the tree does not show either a distinct or a large archaeal clade: archaeal sequences are present in a small number, and lengths of corresponding branches are similar with the lengths of other branches. According to the distribution of archaeal sequences on this tree, we concluded that a series of events of horizontal transfer of NuoF orthologs to archaea from bacteria played a large role in their evolution.
A phylogenetic tree for the NuoG subunit is shown in Fig. 2c; the detailed and zoomable version of the tree is given in the Supplement. The tree was constructed from the alignment of 217 sequences from COG1034 (413 key positions in the alignment were chosen). Archaea on the NuoG subunit tree are represented by a small number of sequences, which do not group separately from bacterial proteins. It seems plausible that NuoG orthologs appeared in archaea by horizontal transfer.
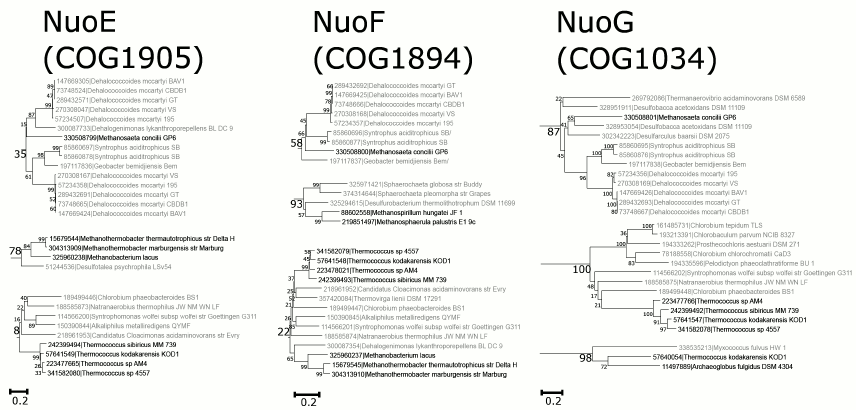
Only seven archaea species out of 106 encode orthologs of all three subunits of the N-module. Two of them belong to Methanobacteria, one species belongs to Methanomicrobia, whereas four species belong to Thermococci. The sequences from Thermococcus sp. 4557, Thermococcus kodakarensis KOD1, and Thermococcus sibiricus MM 739 show similarity with sequences from Clostridia (Fig. 3; the detailed and zoomable version of the tree is given in the Supplement), so it is tempting to suggest a single horizontal transfer event of the N-module genes from Clostridia to the ancestor of these archaea. For another species, Methanosaeta concilii GP6, which belongs to Methanomicrobia, the most similar sequences were found among Proteobacteria. For other archaeal organisms, the closest homologs of the N-module proteins belong to different bacterial taxa, which may point to a series of independent events of horizontal transfer from bacteria.
Fig. 3. Separate clades with archaeal sequences from the trees of the N-module, see text for details; the bootstrap values are indicated; the detailed and zoomable version of the trees is given in the Supplement.
Phylogenetic analysis of the Q-module. For this module, three subunits (NuoB/D/C) of the four were analyzed: due to a significant contamination of COG1143 with highly divergent proteins, we were unable to analyze the NuoI (ferredoxin-like) subunit. The problem arose from the fact that NuoI is a relatively short ferredoxin-like protein and belongs to a protein family that is very large and poorly conserved. During construction of the COG database, there was an effort to divide the proteins of this family into separate COGs, but, because of insufficiency of sequence data, the resulting COGs, including COG1143, apparently were contaminated by either distantly related or unrelated sequences.
A phylogenetic tree for the NuoB subunit is shown in Fig. 4a; the detailed and zoomable version of the tree is given in the Supplement. We used 142 positions from the alignment of 389 sequences from COG0377 to build this tree. The NuoB tree has evident separate clades, which consist of proteins from distinct prokaryotic taxa including a proteobacterial clade, two actinobacterial clades, Bacteroides clades, and a cyanobacterial clade. There is a large clade, with a relatively long branch, which includes sequences from Crenarchaea, Euryarchaea, and Korarchaea; in addition, four separate Euryarchaeal clades are scattered over the tree.
Fig. 4. Phylogenetic trees for the subunits of the Q-module; the detailed and zoomable version of the trees is given in the Supplement. The main archaeal clade, which is separated from bacterial sequences, is shown by gray ovals. Black arrows point to small archaeal clades from Thermoplasmatales, the clades with archaeal proteins from Halobacteria are shown with solid gray lines, and the clades of bacterial proteins from Thermotogae are marked with dashed arrows. a) Phylogenetic tree for the NuoB subunit; b) phylogenetic tree for the NuoC subunit; c) phylogenetic tree for the NuoD subunit.
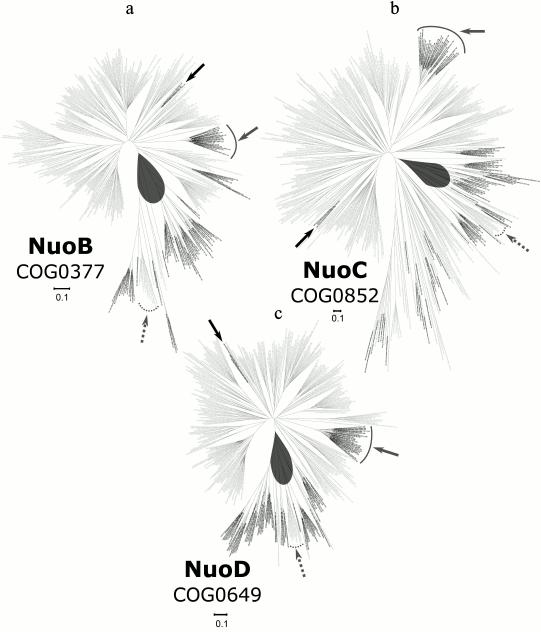
A phylogenetic tree for the NuoC subunit is shown in Fig. 4b; the detailed and zoomable version of the tree is given in the Supplement. For building the tree, we used 174 positions from the alignment of 444 sequences from COG0852. The distribution of archaea on the NuoC tree is quite similar with the phylogenetic trees of other subunits of the Q-module, but the number of archaeal sequences is larger in the case of NuoC. Most of these archaeal subunits belong to Euryarchaeota; one clade, besides Euryarchaea, also contains sequences of Crenarchaea and Korarchaea.
A phylogenetic tree for the NuoD subunit is shown in Fig. 4c; the detailed and zoomable version of the tree is given in the Supplement. The 263 positions of the alignment of 405 sequences from COG0649 were used for the tree construction. The tree of NuoD has three separate Euryarchaeal clades and one large clade with Crenarchaea, Euryarchaea, and Korarchaea representatives, where the sequences from Euryarchaea form a separate group. Euryarchaeal sequences are also present on other branches of the tree, which is, most likely, due to a number of horizontal gene transfer events. In total, the tree topology resembles the tree for the NuoB subunit.
The phylogenetic trees of the analyzed subunits of the Q-module exhibit distinct archaeal clades. At the same time, some archaeal sequences are located on separate branches and do not belong to the major archaeal clade. This pattern suggests that horizontal transfer of genes from bacteria to archaea likely occurred after divergence of bacteria and archaea. For instance, each tree from Fig. 4 contains a clade being composed of proteins of the three archaeal species from the Thermoplasmatales order (Picrophilus torridus DSM 9790, Thermoplasma acidophilum DSM 1728, and Thermoplasma volcanium GSS1; marked with a black arrow). We cannot trace any specific clustering between this clade and a particular bacterial sequence group; however, its position inside the bacterial part of the tree is undoubtable. The same is true for the halobacterial clade, which is located outside of the main archaeal clade in each tree (these clades are shown in Fig. 4 with solid gray arc and solid gray arrows; see the Supplement).
However, it should be noted that the obtained trees show also the horizontal gene transfer from archaea to bacteria: bacterial sequences are found inside the main archaeal clade on all trees. These sequences form one separate clade of Thermotogae group members (shown in Fig. 4 by dashed gray arcs and arrows) and also are represented by single sequences from Aquificae (Desulfurobacterium thermolithotrophum DSM 11699, Thermovibrio ammonificans HB-1) and Magnetococcus marinus MC-1. In addition, T. ammonificans HB-1, and M. marinus MC-1 organisms have typical proteins in bacterial clades, which points indirectly to the direction of horizontal transfer in those clades – from archaea to bacteria.
Phylogenetic analysis of the P-module. Four subunits from the seven were analyzed, namely NuoL, NuoM, NuoN, and NuoH. Sequences of the A, J, and K subunits were too short for individual phylogenomic analysis (the average lengths of those proteins in our sample were 128, 186, and 103 amino acids respectively; the numbers of key positions in the alignment blocks were 70, 35, and 85).
A phylogenetic tree for the NuoL subunit is shown in Fig. 5a; the detailed and zoomable version of the tree is given in the Supplement. It was constructed using 140 key positions of the alignment of 662 sequences from COG1009. This COG contains both the NuoL subunit and one of the Mrp subunits, thus we expected that the tree would split into two separate parts. Apparently, there is no such pattern on the tree: it has at least three equidistant groups of proteins, and the NuoL group is among them (it is shown in Fig. 5 by a black polygonal line). There is one archaeal clade inside the NuoL part of the tree, but it is not separated from the bacterial clades and, in fact, is located in one of them. The second part of the tree does not contain a separate archaeal clade.
Fig. 5. Phylogenetic trees for the subunits of the P-module; the detailed and zoomable version of the trees is given in the Supplement. The main archaeal clade, which is separated from bacterial sequences, where present, is shown in a gray oval. The clades with archaeal proteins from Halobacteria are shown with solid gray lines and solid gray arrows. a) Phylogenetic tree for the NuoL/MnhA subunit (the part of this tree, which probably stands for NuoL clades, as shown by genomic neighborhood analysis, is marked with broken line); b) phylogenetic tree for the NuoM subunit; c) phylogenetic tree for the NuoN subunit; d) phylogenetic tree for the NuoH subunit.
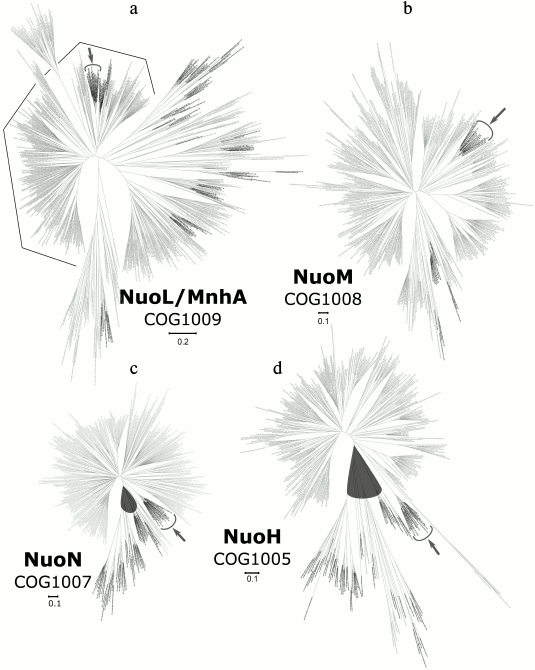
A phylogenetic tree for the NuoM subunit is shown in Fig. 5b; the detailed and zoomable version of the tree is given in the Supplement. It was constructed relying on 204 key positions of the alignment of 417 sequences from COG1008. This tree contains only a few archaeal sequences and does not exhibit an individual archaeal clade. Thus, it is likely that a horizontal gene transfer event from bacteria to archaea accounts for the archaeal sequences.
A phylogenetic tree for the NuoN subunit is shown in Fig. 5c; the detailed and zoomable version of the tree is given in the Supplement. The alignment of the 362 sequences from COG1007 was used to build the tree (relying on 162 key positions). As can be seen from the NuoN tree, all archaeal sequences group together in one clade, which, however, has long internal branches. Their length may reflect some archaea-specific traits. In general, most of the branches on the tree are similar in length, except the branches in the archaeal clade.
A phylogenetic tree for the NuoH subunit is shown in Fig. 5d; the detailed and zoomable version of the tree is given in the Supplement. It was constructed by relying on 164 key positions of the alignment of 385 sequences of COG1005. Similarly to the NuoN tree, it has a single separate and large archaeal clade (except one small haloarchaeal clade; haloarchaea are well known for acquiring a huge number of bacterial sequences through horizontal transfer [43]). The main archaeal clade contains several bacterial sequences.
The data presented above show that the NuoN and NuoH trees both contain large, distinct archaeal clades, which are separated from the bacterial part of the tree. However, for the NuoL and NuoM trees, no such separate archaeal clades could be identified.
DISCUSSION
The main result of this work is identification of distinct bacterial and archaeal clades for the NuoN and NuoH subunits of the NDH-1 complex. This finding suggests that the ancestors of these membrane proteins could be present already in the Last Universal Cellular Ancestor (LUCA).
The modern view on the LUCA was prompted by sequencing of complete genomes, which revealed a set of RNA- and protein-coding genes that are common to all free-living organisms (so-called “ubiquitous” genes [44, 45]). These genes are believed to be present in the LUCA and serve as a primary evidence of its very existence. Ubiquitous genes are characterized by distinct archaeal/bacterial forms [44, 45]. Therefore, distinct bacterial and archaeal clades for “non-ubiquitous” genes – which are shared by the majority of organisms but not by all of them – may indicate that the ancestors of these genes may have been also present in the LUCA. However, for non-ubiquitous genes a possibility of an early horizontal gene transfer between domains (from bacteria to archaea or vice versa) followed by a further spread of domain-specific gene forms could not be fully excluded.
Our analysis indicates that the ancestral NuoH and NuoN subunits could be present in the LUCA; see Figs. 5c and 5d and the Supplement. Taking into account the possibility of an early horizontal gene transfer between domains, we cannot fully exclude the emergence of the ancestral NuoH and NuoN subunits within ancient archaea or bacteria followed by their prompt transfer to the other domain. In contrast, the NuoL and NuoM subunits seem to be of a more recent, probably, bacterial origin, whereby they could be acquired by archaea as a result of horizontal gene transfer from bacteria.
Separate distinct archaeal clades were also found for subunits NuoB, NuoC, and NuoD of the Q-module (Fig. 4 and the Supplement). However, there are small branches of archaeal sequences of these proteins among bacterial clades, which could be due to late horizontal transfers of their genes from bacteria. The tree of the NuoC subunit has a particularly large number of archaeal branches in one of its bacterial clades. It is tempting to speculate that this clade corresponds to a paralog of the NuoC subunit that actively spreads among bacteria and archaea via horizontal gene transfer. In summary, the NuoB, NuoD, and NuoC subunits may have been present in the LUCA.
According to our data (Figs. 2 and 3), the subunits of the N-module have bacterial origin and were acquired by several archaea through horizontal gene transfer.
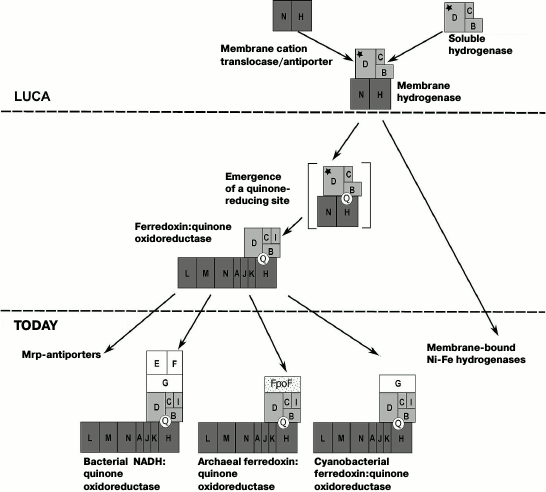
The data do not support those evolutionary models that suggest the emergence of the NDH-1 complex via combining a multisubunit membrane antiporter with a soluble hydrogenase [7, 18, 23]. Our results are better compatible with scenarios where the ancestral form of NDH-1 was suggested to be a primitive complex with only two membrane subunits and a cytoplasmic oxidoreductase module that contained NiFe-clusters [14, 19, 22]. Our analysis indicates that the primordial membrane subunits could have been the ancestral forms of NuoN and NuoH subunits (but not of the NuoL subunit, as assumed in [19, 22], and not of the NuoL and NuoH subunits, as assumed in [14]). The ancestors of NuoN and NuoH subunits could interact with hydrogenase, which was composed of the ancestral forms of the NuoB and NuoD subunits and, probably, of the NuoC subunit. This complex might have already been in the LUCA. The NuoL and NuoM subunits could emerge from duplication of the NuoN subunit followed by horizontal transfer of the genes among prokaryotic groups, as can be suggested from the topology of the corresponding trees. The resulting evolutionary scenario is shown in Fig. 6.
Fig. 6. Proposed evolutionary scenario for NDH-1. On the evolutionary stage of the LUCA, the ancestral NDH-1 complex could contain up to five main subunits and may have coupled the oxidation of ferredoxin by water protons (yielding molecular hydrogen) with (electroneutral) export of sodium ions from the cell. The following stages of the enzyme evolution imply the emergence of a quinone-binding site followed by duplications of the ancestral NuoN gene, which should have been essential for increasing the number of translocated ions and, accordingly, the overall efficiency of the membrane complex.
Our analysis is compatible with the already suggested [18, 19] late recruitment of the N-module. Thereby, the ancestral NDH-1 form probably did not work as a NADH-oxidoreductase. More likely, it interacted with low-potential ferredoxins and/or the H2/2H+ redox pair. The similar reaction in several modern prokaryotes is catalyzed by the ferredoxin:NAD+-oxidoreductase (RNF), whereby the oxidation of ferredoxin is coupled with the translocation of one sodium ion out of the cell [46, 47]. The latter trait might indicate the antiquity of the RNF in the view of the concept of evolutionary primacy of sodium-dependent bioenergetics [28, 30, 48-52]. The concept suggests that the ability to use Na+ as a coupling ion, which is currently found only in some, usually anaerobic, bacteria and archaea [53], was the primordial form of membrane bioenergetics. This ability might be very useful upon the earlier stages of evolution, when membranes would not be able to hold a large transmembrane proton gradient needed for ATP synthesis (the permeability of the lipid bilayer for protons is 106-108 times higher than for sodium ions [54]). Arguably, sodium-dependent bioenergetics could naturally evolve from an interaction of several membrane enzymes that pumped sodium ions out of the cells [51]. Indeed, it is well established that several indispensable cell systems, the translation system in the first place, are activated by potassium ions and inhibited by sodium ions [51, 55], which demands the K+/Na+ ratio > 1 within the cell. The dependence of key cell systems on K+ ions supports the earlier suggestion on the origin of the first living organisms in potassium-enriched environments [56]. There are several geochemical models of potassium-rich habitats for the first cells [55, 57-59]; regardless of the specific scenario, the subsequent inevitable invasion of the ocean by cellular organisms could proceed only on a condition of presence of sodium export pumps in such organisms. Many such sodium export pumps might have preceded modern energy-converting enzymes; some relict Na+-translocating forms are still preserved in modern organisms [51, 52]. Only with increase in the tightness of cell membranes for protons and elevation of oxygen level in the atmosphere, the proton-dependent bioenergetics, as more beneficial under oxidizing conditions [48], could oust the Na+-dependent bioenergetics, which, however, still persists in some (usually anaerobic) bacteria and archaea [51-53].
Thus, the ability of a two-subunit membrane module of the ancestral NDH-1 complex to translocate ions as well as the possible nature of these ions deserve consideration. The modern NDH-1 complexes couple electron transfer from NADH to quinone with translocation of several protons across the membrane. However, the coupling of a redox reaction with proton translocation was unlikely to result in energy conservation at the stage of the LUCA, when membranes, most probably, were built of single-chain lipids [49, 54, 60, 61]; such membranes are permeable for protons [62], so that no energy could be stored.
Recently, Hirst and Roberts showed that deactivation of mitochondrial complex I by using the approach of Kotlyar and Vinogradov [63] turned the membrane module of complex I into a Na+/H+ antiporter [64]. The deactivation of the cytoplasmic module (as could be judged from the structure) could affect the membrane part only by affecting the connection between the Q-module and the NuoH-subunit, which alone accounts for the interaction between the cytoplasmic and membrane parts of the complex [13]. As the mitochondrial NDH-1 complex, at least according to available data [64], was not shown to translocate sodium ions in its active state, the sodium ions in a deactivated complex may pass through either the NuoH-subunit or the interface of the NuoH and NuoN subunits. It is tempting to speculate that the deactivation of complex I “releases” some charged groups of the NuoH subunit on the cytoplasmic side of membrane, so that these groups attain a capability to bind sodium ions.
The function of a Na+/H+ antiporter may have been present already in the ancestral NDH-1 complex, which would explain the antiquity of the NuoH and NuoN subunits. If the ancient NDH-1 complex coupled a redox reaction in the cytoplasmic module with pumping of sodium ions (just as it happens in the case of RNF [46, 47]), the translocation of protons inside the cell would kinetically facilitate the pumping of sodium ions, making it electroneutral. Under the assumption of single-chain lipids and proton-leaky primordial membranes [49, 54, 60, 61], those protons that entered the cell via the ancestral NDH-1 would be able to promptly equilibrate with the environment, thus preventing the acidification of cytoplasm.
Two recently published works independently noted the structural similarity between the NuoH and NuoN subunits and suggested their origin by some ancestral membrane protein duplication [13, 14]. Our analysis does not exclude the possibility that this duplication may have occurred even before the LUCA stage. In this case the initial NDH-1 form had only one type of membrane subunit (the ancestor of NuoN and NuoH), which could function as a sodium transporter. The duplication of the gene would result in a different subunit, which improved the kinetic effectiveness of the redox-dependent sodium export pump (that participated in maintenance of [K+]/[Na+] > 1 in a primal cell) by facilitating proton translocation in the reverse direction.
As shown in Fig. 6, in one of the lineages, a membrane quinone managed to replace protons as an acceptor of electrons in the ancestral hydrogenase moiety. Structurally, such a change would require a unique mechanism of “protruding” of the hydrophobic quinone out of the lipid bilayer by about 2.5 nm through subunits NuoH and NuoB in order to reach the closest to the membrane redox center N2. Consequently, the catalytic Ni-Fe center of hydrogenase would be lost. From the thermodynamics viewpoint, the recruiting of quinones as electron acceptors would increase the amount of released energy per one transferred electron from 100-150 meV in the case of a hydrogen-producing enzyme to ~400 meV for the ubiquinone-reducing NADH-dehydrogenases of modern aerobic organisms. However, the number of ions that are translocated by a redox-pump is determined by thermodynamics of corresponding redox reactions – to avoid short-circuiting. Therefore, the recruitment of an electron acceptor with a higher redox potential should lead to an increase in the number of translocated ions, which, apparently, was achieved by duplications of the NuoN subunit. Ultimately, these duplications yielded a modern-type NADH:ubiquinone oxidoreductase that contained up to four homologs (if the NuoH subunit is also counted) of the initial membrane protein. In organisms with proton-dependent energetics, NDH-1 now couples reduction of quinones with translocation of protons across the membrane [65-68]. In organisms with sodium-dependent energetics, the nature of translocated ions still needs further clarification. It cannot be ruled out that some NDH-1-related complexes of prokaryotes translocate sodium ions [69, 70].
In summary, the data obtained are compatible with the evolutionary scenario shown in Fig. 6. According to this scenario, the ancestral form of NDH-1 may have been similar to membrane-bound electrogenic hydrogenases that make the class 4 hydrogenases (related to the Ech hydrogenase) [14, 19, 22]. Our analysis indicates that, of the three homologous membrane subunits NuoL, NuoM, and NuoN, subunit NuoN appears to be the closest to the ancestral form.
Our phylogenomic analysis indicates that the ancestral form of NDH-1 already contained the ancestors of the membrane subunits NuoN and NuoH as well as the cytoplasmic subunits NuoD, NuoB, and (probably) NuoC. This ancestral form, supposedly capable of coupling the electron transfer from ferredoxin to hydrogen with an (electroneutral) export of one sodium ion, may have been present already in the LUCA. The subsequently attained ability to reduce membrane quinones should have increased the effectiveness of the enzyme and, accordingly, the number of translocated ions, which would prompt the multiplication of the ion-translocation subunits.
We would like to thank Drs. Y. Bertsova and A. Bogachev for useful discussions.
This work was supported by the Russian Science Foundation (projects No. 14-50-00029, G. E. Novakovsky, phylogenomic analysis of separate subunits of NDH-1; No. 14-14-00592, D. V. Dibrova and A. Y. Mulkidjanian, construction of the evolution scheme for the whole enzyme and its modules).
REFERENCES
1.Sazanov, L. A. (2015) A giant molecular proton
pump: structure and mechanism of respiratory complex I, Nat. Rev.
Mol. Cell Biol., 16, 375-388.
2.Brandt, U. (2006) Energy converting NADH:quinone
oxidoreductase (complex I), Annu. Rev. Biochem., 75,
69-92.
3.Carroll, J., Fearnley, I. M., Skehel, J. M.,
Shannon, R. J., Hirst, J., and Walker, J. E. (2006) Bovine complex I is
a complex of 45 different subunits, J. Biol. Chem., 281,
32724-32727.
4.Vinothkumar, K. R., Zhu, J., and Hirst, J. (2014)
Architecture of mammalian respiratory complex I, Nature,
515, 80-84.
5.Sazanov, L. A., and Hinchliffe, P. (2006) Structure
of the hydrophilic domain of respiratory complex I from Thermus
thermophilus, Science, 311, 1430-1436.
6.Sazanov, L. A. (2014) The mechanism of coupling
between electron transfer and proton translocation in respiratory
complex I, J. Bioenerg. Biomembr., 46, 247-253.
7.Moparthi, V. K., and Hägerhäll, C. (2011)
The evolution of respiratory chain complex I from a smaller last common
ancestor consisting of 11 protein subunits, J. Mol. Evol.,
72, 484-497.
8.Baumer, S., Ide, T., Jacobi, C., Johann, A.,
Gottschalk, G., and Deppenmeier, U. (2000) The F420H2
dehydrogenase from Methanosarcina mazei is a
redox-driven proton pump closely related to NADH dehydrogenases, J.
Biol. Chem., 275, 17968-17973.
9.Finel, M. (1998) Does NADH play a central role in
energy metabolism in Helicobacter pylori? Trends
Biochem. Sci., 23, 412-413.
10.Weerakoon, D. R., and Olson, J. W. (2007) The
Campylobacter jejuni NADH:ubiquinone oxidoreductase
(complex I) utilizes flavodoxin rather than NADH, J. Bacteriol.,
190, 915-925.
11.Bohm, R., Sauter, M., and Bock, A. (1990)
Nucleotide sequence and expression of an operon in Escherichia
coli coding for formate hydrogenlyase components, Mol.
Microbiol., 4, 231-243.
12.Efremov, R. G., Baradaran, R., and Sazanov, L. A.
(2010) The architecture of respiratory complex I, Nature,
465, 441-445.
13.Sazanov, L. A., Baradaran, R., Efremov, R. G.,
Berrisford, J. M., and Minhas, G. (2013) A long road towards the
structure of respiratory complex I, a giant molecular proton pump,
Biochem. Soc. Trans., 41, 1265-1271.
14.Marreiros, B. C., Batista, A. P., Duarte, A. M.,
and Pereira, M. M. (2013) A missing link between complex I and group 4
membrane-bound [NiFe] hydrogenases, Biochim. Biophys. Acta,
1827, 198-209.
15.Swartz, T. H., Ikewada, S., Ishikawa, O., Ito,
M., and Krulwich, T. A. (2005) The Mrp system: a giant among monovalent
cation/proton antiporters? Extremophiles, 9, 345-354.
16.Putnoky, P., Kereszt, A., Nakamura, T., Endre,
G., Grosskopf, E., Kiss, P., and Kondorosi, A. (1998) The pha
gene cluster of Rhizobium meliloti involved in pH
adaptation and symbiosis encodes a novel type of K+ efflux
system, Mol. Microbiol., 28, 1091-1101.
17.Baradaran, R., Berrisford, J. M., Minhas, G. S.,
and Sazanov, L. A. (2013) Crystal structure of the entire respiratory
complex I, Nature, 494, 443-448.
18.Mathiesen, C., and Hägerhäll, C. (2003)
The “antiporter module” of respiratory chain complex I
includes the MrpC/NuoK subunit – a revision of the modular
evolution scheme, FEBS Lett., 549, 7-13.
19.Friedrich, T., and Scheide, D. (2000) The
respiratory complex I of bacteria, archaea and eukarya and its module
common with membrane-bound multisubunit hydrogenases, FEBS
Lett., 479, 1-5.
20.Albracht, S. P., Mariette, A., and De Jong, P.
(1997) Bovine-heart NADH:ubiquinone oxidoreductase is a monomer with 8
Fe-S clusters and 2 FMN groups, Biochim. Biophys. Acta,
1318, 92-106.
21.Tersteegen, A., and Hedderich, R. (1999)
Methanobacterium thermoautotrophicum encodes two
multisubunit membrane-bound [NiFe] hydrogenases. Transcription of the
operons and sequence analysis of the deduced proteins, Eur. J.
Biochem., 264, 930-943.
22.Hedderich, R. (2004) Energy-converting [NiFe]
hydrogenases from archaea and extremophiles: ancestors of complex I,
J. Bioenerg. Biomembr., 36, 65-75.
23.Schut, G. J., Boyd, E. S., Peters, J. W., and
Adams, M. W. (2013) The modular respiratory complexes involved in
hydrogen and sulfur metabolism by heterotrophic hyperthermophilic
archaea and their evolutionary implications, FEMS Microbiol.
Rev., 37, 182-203.
24.Schut, G. J., Zadvornyy, O., Wu, C. H., Peters,
J. W., Boyd, E. S., and Adams, M. W. (2016) The role of geochemistry
and energetics in the evolution of modern respiratory complexes from a
proton-reducing ancestor, Biochim. Biophys. Acta, pii:
S0005-2728.
25.Tatusov, R. L., Koonin, E. V., and Lipman, D. J.
(1997) A genomic perspective on protein families, Science,
278, 631-637.
26.Galperin, M. Y., Makarova, K. S., Wolf, Y. I.,
and Koonin, E. V. (2015) Expanded microbial genome coverage and
improved protein family annotation in the COG database, Nucleic
Acids Res., 43 (Database issue), D261-269.
27.Kristensen, D. M., Kannan, L., Coleman, M. K.,
Wolf, Y. I., Sorokin, A., Koonin, E. V., and Mushegian, A. (2010) A
low-polynomial algorithm for assembling clusters of orthologous groups
from intergenomic symmetric best matches, Bioinformatics,
26, 1481-1487.
28.Dibrova, D. V., Galperin, M. Y., and
Mulkidjanian, A. Y. (2010) Characterization of the N-ATPase, a
distinct, laterally transferred Na+-translocating form of
the bacterial F-type membrane ATPase, Bioinformatics, 26,
1473-1476.
29.Mulkidjanian, A. Y., Makarova, K. S., Galperin,
M. Y., and Koonin, E. V. (2007) Inventing the dynamo machine: the
evolution of the F-type and V-type ATPases, Nat. Rev.
Microbiol., 5, 892-899.
30.Mulkidjanian, A. Y., Galperin, M. Y., Makarova,
K. S., Wolf, Y. I., and Koonin, E. V. (2008) Evolutionary primacy of
sodium bioenergetics, Biol. Direct, 3, 13.
31.Dibrova, D. V., Cherepanov, D. A., Galperin, M.
Y., Skulachev, V. P., and Mulkidjanian, A. Y. (2013) Evolution of
cytochrome bc complexes: from membrane-anchored dehydrogenases
of ancient bacteria to triggers of apoptosis in vertebrates,
Biochim. Biophys. Acta, 1827, 1407-1427.
32.Mulkidjanian, A. Y., Koonin, E. V., Makarova, K.
S., Mekhedov, S. L., Sorokin, A., Wolf, Y. I., Dufresne, A., Partensky,
F., Burd, H., Kaznadzey, D., Haselkorn, R., and Galperin, M. Y. (2006)
The cyanobacterial genome core and the origin of photosynthesis,
Proc. Natl. Acad. Sci. USA, 103, 13126-13131.
33.Edgar, R. C. (2004) MUSCLE: a multiple sequence
alignment method with reduced time and space complexity, BMC
Bioinformatics, 5, 113.
34.Edgar, R. C. (2004) MUSCLE: multiple sequence
alignment with high accuracy and high throughput, Nucleic Acids
Res., 32, 1792-1797.
35.Waterhouse, A. M., Procter, J. B., Martin, D. M.,
Clamp, M., and Barton, G. J. (2009) Jalview Version 2 – a
multiple sequence alignment editor and analysis workbench,
Bioinformatics, 25, 1189-1191.
36.Sonnhammer, E. L., Eddy, S. R., and Durbin, R.
(1997) Pfam: a comprehensive database of protein domain families based
on seed alignments, Proteins, 28, 405-420.
37.Krogh, A., Larsson, B., Von Heijne, G., and
Sonnhammer, E. L. (2001) Predicting transmembrane protein topology with
a Hidden Markov Model: application to complete genomes, J. Mol.
Biol., 305, 567-580.
38.Tamura, K., Stecher, G., Peterson, D., Filipski,
A., and Kumar, S. (2013) MEGA6: Molecular Evolutionary Genetics
Analysis version 6.0, Mol. Biol. Evol., 30,
2725-2729.
39.Saitou, N., and Nei, M. (1987) The
neighbor-joining method: a new method for reconstructing phylogenetic
trees, Mol. Biol. Evol., 4, 406-425.
40.Jones, D. T., Taylor, W. R., and Thornton, J. M.
(1992) The rapid generation of mutation data matrices from protein
sequences, Comput. Appl. Biosci., 8, 275-282.
41.Felsenstein, J. (1985) Confidence limits on
phylogenies: an approach using the bootstrap, Evolution,
39, 783-791.
42.Philippe, H., and Forterre, P. (1999) The rooting
of the universal tree of life is not reliable, J. Mol. Evol.,
49, 509-523.
43.Nelson-Sathi, S., Dagan, T., Landan, G., Janssen,
A., Steel, M., McInerney, J. O., Deppenmeier, U., and Martin, W. F.
(2012) Acquisition of 1000 eubacterial genes physiologically
transformed a methanogen at the origin of Haloarchaea, Proc. Natl.
Acad. Sci. USA, 109, 20537-20542.
44.Koonin, E. V. (2000) How many genes can make a
cell: the minimal-gene-set concept, Annu. Rev. Genom. Hum.
Genet., 1, 99-116.
45.Koonin, E. V. (2003) Comparative genomics,
minimal gene-sets and the last universal common ancestor, Nat. Rev.
Microbiol., 1, 127-136.
46.Biegel, E., and Muller, V. (2010) Bacterial
Na+-translocating ferredoxin:NAD+ oxidoreductase,
Proc. Natl. Acad. Sci. USA, 107, 18138-18142.
47.Biegel, E., Schmidt, S., Gonzalez, J. M., and
Muller, V. (2011) Biochemistry, evolution and physiological function of
the Rnf complex, a novel ion-motive electron transport complex in
prokaryotes, Cell. Mol. Life Sci., 68, 613-634.
48.Mulkidjanian, A. Y., Dibrov, P., and Galperin, M.
Y. (2008) The past and present of the sodium energetics: may the
sodium-motive force be with you, Biochim. Biophys. Acta,
1777, 985-992.
49.Dibrova, D. V., Chudetsky, M. Y., Galperin, M.
Y., Koonin, E. V., and Mulkidjanian, A. Y. (2012) The role of energy in
the emergence of biology from chemistry, Orig. Life Evol.
Biosph., 42, 459-468.
50.Mulkidjanian, A. Y., Galperin, M. Y., and Koonin,
E. V. (2009) Co-evolution of primordial membranes and membrane
proteins, Trends Biochem. Sci., 34, 206-215.
51.Dibrova, D. V., Galperin, M. Y., Koonin, E. V.,
and Mulkidjanian, A. Y. (2015) Ancient systems of sodium/potassium
homeostasis as predecessors of membrane bioenergetics, Biochemistry
(Moscow), 80, 495-516.
52.Klimchuk, O. I., Dibrova, D. V., and
Mulkidjanian, A. Y. (2016) Phylogenomic analysis identifies a
sodium-translocating decarboxylating oxidoreductase in Thermotogae,
Biochemistry (Moscow), 81, 481-490.
53.Skulachev, V. P. (1984) Sodium bioenergetics,
Trends Biochem. Sci., 9, 483-485.
54.Deamer, D. W. (1987) Proton permeation of lipid
bilayers, J. Bioenerg. Biomembr., 19, 457-479.
55.Mulkidjanian, A. Y., Bychkov, A. Y., Dibrova, D.
V., Galperin, M. Y., and Koonin, E. V. (2012) Origin of first cells at
terrestrial, anoxic geothermal fields, Proc. Natl. Acad. Sci.
USA, 109, E821-830.
56.Macallum, A. B. (1926) The paleochemistry of the
body fluids and tissues, Physiol. Rev., 6, 316-357.
57.Mulkidjanian, A. Y., Bychkov, A. Y., Dibrova, D.
V., Galperin, M. Y., and Koonin, E. V. (2012) Open questions on the
origin of life at anoxic geothermal fields, Orig. Life Evol.
Biosph., 42, 507-516.
58.Galimov, E. M., Natochin, Y. V., Ryzhenko, B. N.,
and Cherkasova, E. V. (2012) Chemical composition of the primary
aqueous phase of the Earth and origin of life, Geochem. Int.,
50, 1048-1068.
59.Maruyama, S., Ikoma, M., Genda, H., Hirose, K.,
Yokoyama, T., and Santosh, M. (2013) The naked planet Earth: most
essential pre-requisite for the origin and evolution of life,
Geosci. Frontiers, 4, 141-165.
60.Deamer, D. W. (1997) The first living systems: a
bioenergetic perspective, Microbiol. Mol. Biol. Rev., 61,
239-261.
61.Mulkidjanian, A. Y., and Galperin, M. Y. (2010)
Evolutionary origins of membrane proteins, in Structural
Bioinformatics of Membrane Proteins (Frishman, D., ed.) Springer,
pp. 1-28.
62.Chen, I. A., and Szostak, J. W. (2004) Membrane
growth can generate a transmembrane pH gradient in fatty acid vesicles,
Proc. Natl. Acad. Sci. USA, 101, 7965-7970.
63.Kotlyar, A. B., and Vinogradov, A. D. (1990) Slow
active/inactive transition of the mitochondrial NADH-ubiquinone
reductase, Biochim. Biophys. Acta, 1019, 151-158.
64.Roberts, P. G., and Hirst, J. (2012) The deactive
form of respiratory complex I from mammalian mitochondria is a
Na+/H+ antiporter, J. Biol. Chem.,
287, 34743-34751.
65.Wikström, M. (1984) Two protons are pumped
from the mitochondrial matrix per electron transferred between NADH and
ubiquinone, FEBS Lett., 169, 300-304.
66.Galkin, A., Drose, S., and Brandt, U. (2006) The
proton pumping stoichiometry of purified mitochondrial complex I
reconstituted into proteoliposomes, Biochim. Biophys. Acta,
1757, 1575-1581.
67.Galkin, A. S., Grivennikova, V. G., and
Vinogradov, A. D. (1999) H+/2e–
stoichiometry in NADH-quinone reductase reactions catalyzed by bovine
heart submitochondrial particles, FEBS Lett., 451,
157-161.
68.Bogachev, A. V., Murtazina, R. A., and Skulachev,
V. P. (1996) H+/e– stoichiometry for NADH
dehydrogenase I and dimethyl sulfoxide reductase in anaerobically grown
Escherichia coli cells, J. Bacteriol., 178,
6233-6237.
69.Mayer, F., and Muller, V. (2014) Adaptations of
anaerobic archaea to life under extreme energy limitation, FEMS
Microbiol. Rev., 38, 449-472.
70.Castro, P. J., Silva, A. F., Marreiros, B. C.,
Batista, A. P., and Pereira, M. M. (2016) Respiratory complex I: a dual
relation with H+ and Na+? Biochim. Biophys.
Acta, 1857, 928-937.
Supplementary Figures (PDF)