Molecular Genetic Markers of Intra- and Interspecific Divergence within Starfish and Sea Urchins (Echinodermata)
N. B. Petrov1*, I. P. Vladychenskaya1, A. L. Drozdov2,3, and O. S. Kedrova4
1Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119991 Moscow, Russia; E-mail: petr@belozersky.msu.ru2Zhirmunsky Institute of Marine Biology, Far Eastern Branch, Russian Academy of Sciences, 690041 Vladivostok, Russia; fax: (4232) 31-0900; E-mail: anatoliyld@mail.ru
3Far Eastern Federal University, 690091 Vladivostok, Russia
4Lomonosov Moscow State University, Faculty of Biology, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received April 21, 2016; Revision received June 21, 2016
A fragment of the mitochondrial COI gene from isolates of several echinoderm species was sequenced. The isolates were from three species of starfish from the Asteriidae family (Asterias amurensis and Aphelasterias japonica collected in the Sea of Japan and Asterias rubens collected in the White Sea) and from the sea urchin Echinocardium cordatum (family Loveniidae) collected in the Sea of Japan. Additionally, regions including internal transcribed spacers and 5.8S rRNA (ITS1 – 5.8S rDNA – ITS2) were sequenced for the three studied starfish species. Phylogenetic analysis of the obtained COI sequences together with earlier determined homologous COI sequences from Ast. forbesii, Ast. rubens, and Echinocardium laevigaster from the North Atlantic and E. cordatum from the Yellow and North Seas (GenBank) placed them into strictly conspecific clusters with high bootstrap support (99% in all cases). Only two exceptions – Ast. rubens DQ077915 sequence placed with the Ast. forbesii cluster and Aph. japonica DQ992560 sequence placed with the Ast. amurensis cluster – were likely results of species misidentification. The intraspecific polymorphism for the COI gene within the Asteriidae family varied within a range of 0.2-0.9% as estimated from the genetic distances. The corresponding intrageneric and intergeneric values were 10.4-12.1 and 21.8-29.8%, respectively. The interspecific divergence for the COI gene in the sea urchin of Echinocardium genus (family Loveniidae) was significantly higher (17.1-17.7%) than in the starfish, while intergeneric divergence (14.6-25.7%) was similar to that in asteroids. The interspecific genetic distances for the nuclear transcribed sequences (ITS1 – 5.8S rDNA – ITS2) within the Asteriidae family were lower (3.1-4.5%), and the intergeneric distances were significantly higher (32.8-35.0%), compared to the corresponding distances for the COI gene. These results suggest that the investigated molecular-genetic markers could be used for segregation and identification of echinoderm species.
KEY WORDS: molecular evolution, population, speciation, Echinodermata, cytochrome oxidase subunit I gene, internal transcribed spacersDOI: 10.1134/S0006297916090066
The main goal of molecular genetic studies of microevolution is investigation of biological diversity. For this purpose, new approaches for species identification are required that differ from traditional morphology-based methods. It is known that different parts of genomes evolve at different rates. The initial steps in studying microevolution require identification and structural characterization of genome regions undergoing rapid evolutionary changes and therefore exhibiting high levels of inter- and intraspecific polymorphism. These studies help understand the gene flow between geographically close or distant populations and evolutionary changes in geographically isolated populations. The results of such studies are now widely used for evaluation of biological diversity by the DNA barcoding methods [1]. The molecular genetic approaches developed could be used for identification of species and populations of various organisms, especially those used in industry or serving as indicators.
Studying biological diversity and mechanisms of speciation requires the use of various nuclear genome regions that would allow comparison of populations and species with different degrees of genetic isolation. Such regions might be noncoding repeats, in particular satellite DNAs of heterochromatin [2]. Other genetic markers widely used for identification and segregation of species are internal transcribed spacers of nuclear rRNA genes (ITS1 and ITS2) and a 5′-fragment of the mitochondrial cytochrome c oxidase subunit I gene (~650 bp). The latter has become a standard gene for DNA barcoding of various animal species [1], including echinoderms [3-5] and mollusks [6-10]. The applicability of ITS1 and ITS2 for species identification has yet to be estimated; however, nuclear ITS sequences in combination with two plastid DNA regions (rpL32-trnL and trnL-trnF) have been successfully used for revision of the widely distributed group of Allium saxatile onions [11].
The aim of this work was to estimate the levels of inter- and intraspecific polymorphism of echinoderm species from the White Sea and the Sea of Japan by comparing rapidly evolving fragments of nuclear and mitochondrial genomes. Total DNA was isolated from 96% ethanol-fixed tissues from various echinoderm species, and fragments of the mitochondrial COI gene and the nuclear rRNA ITS region were amplified and sequenced. The resulting sequences were clustered by building phylogenetic trees, and the polymorphism within and between clusters was analyzed.
MATERIALS AND METHODS
Isolates of the sea urchin Echinocardium cordatum and starfish species Asterias amurensis and Aphelasterias japonica were collected in regions of Peter the Great Bay (Sea of Japan). Isolates of the starfish Asterias rubens were collected in Kandalaksha Bay (White Sea) in the vicinities of the White Sea Biological Station of Moscow State University. Total DNA was isolated from the ambulacral feet or, in some cases, gonads fixed in 96% ethanol using the NucleoSpin® Tissue DNA isolation kit (Macherey-Nagel, Germany) as recommended by the manufacturer.
A fragment of the mitochondrial COI gene was amplified using either a set of primers designed for sea urchins and starfish based on a set of primers universal for all multicellular animals [12] or a set of echinoderm-specific primers [13] developed based on new data on echinoderm sequences. To amplify the ITS1, ITS2, and 5.8S rDNA nuclear sequences, we designed and tested a set of primers specific for the genes coding for 18S, 5.8S, and 28S rRNA. The location of the primers and the amplification strategy for ITS1, ITS2, and 5.8S rDNA are shown in Fig. 1. The sequences of all designed and used primers are shown in Table 1.
Fig. 1. Scheme for amplification of internal transcribed spacers and a gene for 5.8S rRNA (ITS1 – 5.8S rDNA – ITS2). Sites of primer annealing and direction of amplification are shown with arrows.
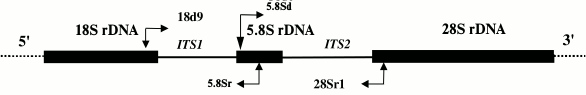
Table 1. Primers for amplification of the
COI gene and internal transcribed spacer region (ITS1
– 5.8S rDNA – ITS2)

Fragments of the mitochondrial COI gene were amplified by PCR using an Encyclo Plus PCR kit (Evrogen, Russia) as recommended by the manufacturer. The amplification reaction included initial denaturation at 94°C for 2 min, 30 cycles of denaturation at 94°C for 30 s, primer annealing at 45°C for 30 s, elongation at 72°C for 1 min, and final elongation at 72°C for 10 min. The conditions for the ITS amplification were the same except that primer annealing temperature was raised to 55°C (30 s).
PCR products were separated by electrophoresis in agarose gel. DNA bands of interest were cut out from the gel, purified using a DNA gel extraction kit (Cytokine, Russia), and sequenced (both chains) with an automated sequencer (Genom, Institute of Molecular Biology, Russian Academy of Sciences). The resulting sequences were deposited in GenBank under accession numbers KX592544-KX592561 (COI) and KX592562-KX592568 (ITS1, ITS2, and 5.8S rDNA).
Phylogenetic analysis, including building phylogenetic trees, clustering of sequences, and estimation of the inter- and intragroup levels of polymorphism, was performed using the MEGA6 suite of molecular genetic programs [14]. The evolutionary model for the analyzed sets of sequences was selected with the corresponding program from the MEGA6 suite; phylogenetic trees were constructed by the method of maximum likelihood based on the general model of reversible evolution [15]. The tree for the heuristic search was constructed using the NJ and BioNJ methods and the distance matrix calculated by the MCL method [15] with subsequent selection of the best topology. The inter- and intragroup distances were calculated using the Kimura two-parameter model [16].
RESULTS
Analysis of clustering of the COI and ITS genes. The obtained COI gene sequences were aligned with homologous COI sequences from other species of Atlantic and Pacific starfish (GenBank) and analyzed phylogenetically. COI sequences from the following species were used in analysis: Aphelasterias japonica (five sequences) and three of the most widely distributed species of the genus Asterias (family Asteriidae) – Ast. forbesii and Ast. rubens from the East and West Atlantic populations, and Ast. amurensis from several Pacific populations. Distolasterias nipon and Forcipulatida sp. (one sequence for each species) were used as an external group.
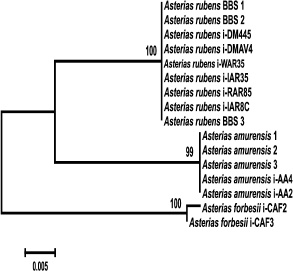
To identify monophyletic groups of sequences and to elucidate evolutionary relations between species of the genus Asterias, we constructed a phylogenetic tree for the COI sequences (Fig. 2). All three species formed a monophyletic group with a high bootstrap support (99%). Asterias rubens and Ast. amurensis clusters form a group with 76% bootstrap support; the Ast. forbesii cluster appeared to be equidistant from the two other clusters. In general, the grouping of sequences from all three Asterias species had 98% bootstrap support. Clusters of Ast. rubens and Ast. forbesii sequences were more homogenous than the Ast. amurensis cluster, in which two groups could be distinguished with bootstrap support of 90 and 86%, respectively Note that in this phylogenetic tree, the Pacific species Ast. amurensis and the North Atlantic species Ast. rubens were closer to each other than the two North Atlantic species, Ast. rubens and Ast. forbesii. The constructed phylogenetic tree had seven monophyletic groups. COI sequences from Ast. rubens specimens collected in the White Sea clustered within the homogenous group of the North Atlantic populations of this species.
Fig. 2. Phylogenetic tree for the 5′-region of the mitochondrial COI gene from starfish of the Asteriidae family. The tree with the maximum logarithm of likelihood (–2417.442) is shown. The bootstrap values are shown on the branches leading to the corresponding clusters. The lengths of branches are proportional to the number of substitutions. Forty-one sequences (length, 614 positions) were analyzed considering all three codon positions.
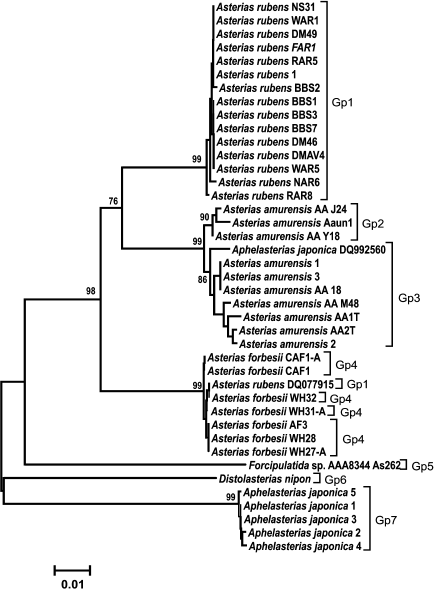
The constructed tree had two artefacts. One of Aph. japonica sequences (DQ992560) was assigned to the group formed by sequences from Ast. amurensis. GenBank sequence DQ077915 that had been deposited as a sequence from Ast. rubens was found in a cluster formed by sequences from Ast. forbesii (see “Discussion” for a possible explanation).
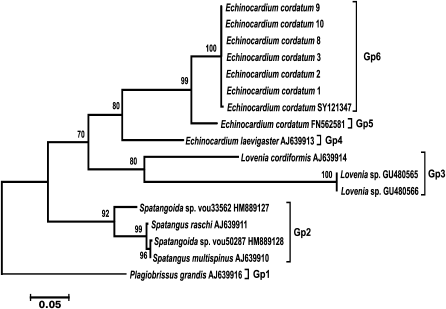
A set of sea urchin-specific primers was used for amplification of COI genes from isolates of the heart-shaped sea urchin Echinocardium cordatum collected in various regions of the Sea of Japan (Vostok Bay, Ussuri Bay, Patroklus Bay, and Troitsa Bay). The phylogenetic tree constructed using amplified COI sequences together with E. cordatum COI sequences deposited in GenBank and COI sequences from other sea urchins of the Loveniidae family is shown in Fig. 3. All COI sequences from E. cordatum isolates from the Sea of Japan and from E. cordatum specimens collected at the Korean coast of the Yellow Sea (SY121347) formed a single cluster (Gp6) with high bootstrap support. The COI sequence of E. cordatum from the North Sea (FN562581) was located at a considerable distance from this cluster (Gp5). Another species of the same genius, E. laevigaster (AJ639913) from the Atlantic coast of England, was even more distant from the first two groups (Gp4) and formed with them a group with low bootstrap support. Groups Gp3 and Gp2 were formed by sequences from two other species of the same genus with bootstrap support of 80 and 92%, respectively. The COI sequence from another family of spatangoid sea urchins was at the base of the phylogenetic tree. These results demonstrate that nucleotide sequences of the mitochondrial COI gene form specific or, as in the case of Ast. amurensis, population clusters.
Fig. 3. Phylogenetic tree for the 5′-region of the mitochondrial COI gene from sea urchins of the Loveniidae family. The tree with the maximum logarithm of likelihood (–2555.0476) is shown. The bootstrap values are shown on the branches leading to the corresponding clusters. The lengths of branches are proportional to the number of substitutions. Seventeen sequences (length, 656 positions) were analyzed considering all three codon positions.
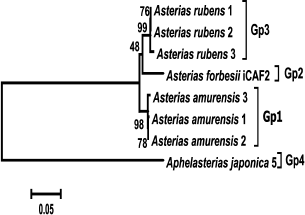
DNA sequences for the ITS1 and ITS2 nuclear spacers and 5.8S rDNA (total length, 1365-1466 nucleotides) were amplified and sequenced for three isolates of Ast. amurensis (1, 2, and 3), three isolates of Ast. rubens (BBS1, BBS2, and BBS3), and one isolate of Aph. japonica. These sequences were aligned together with a homologous sequence from the Ast. forbesii CAF2 isolate and analyzed phylogenetically. Similarly to COI genes, ITS1, ITS2, and 5.8S rDNA sequences from each of the species (Ast. rubens and Ast. amurensis) formed monophyletic groups with high bootstrap support (99 and 98%, respectively). The Ast. forbesii sequence was assigned to the Ast. rubens cluster with low bootstrap support (48%) (Fig. 4).
Fig. 4. Phylogenetic tree of the ITS1, ITS2, and 5.8S rDNA sequences from starfish of the Asteriidae family. The tree with the maximum logarithm of likelihood (–4008.5599) is shown. The bootstrap values are shown on the branches leading to the corresponding clusters. The lengths of branches are proportional to the number of substitutions. Eight sequences (length, 1600 positions) were analyzed.
Analysis of 16 ITS1 sequences (Fig. 5) showed with 100% bootstrap support that sequences from Ast. rubens isolates from the White Sea (BBS1, BBS2, and BBS3) belonged to a homogenous group of the North Atlantic population of this species. Asterias amurensis ITS1 sequences also formed a cluster with high bootstrap support (99%) that was a sister cluster to Ast. rubens. The ITS1 sequences of two Ast. forbesii isolates form a group (100% bootstrap) at the base of the tree.
Fig. 5. Phylogenetic tree of the ITS1 sequences from starfish of the Asteriidae family. The tree with the maximum logarithm of likelihood (–862.2724) is shown. The bootstrap values are shown on the branches leading to the corresponding clusters. The lengths of branches are proportional to the number of substitutions. Sixteen sequences (length, 488 positions) were analyzed.
Analysis of intra- and intergroup polymorphism. To determine the levels of intra- and intergroup polymorphism of the studied species, their sequences were clustered into groups based on the results of phylogenetic reconstruction, and the inter- and intragroup genetic distances were determined. We found that the levels of intergroup polymorphism for the COI gene in starfish species of the Asteriidae family varied from 0.003 to 0.009 (as estimated from genetic distances within the corresponding groups), while the levels of intergroup polymorphism varied from 0.104 to 0.298 (as estimated from genetic distances between specific and generic clusters) (Table 2).
Table 2. Intergroup divergence matrix for
the mitochondrial COI genes from starfish of the Asteriidae
family
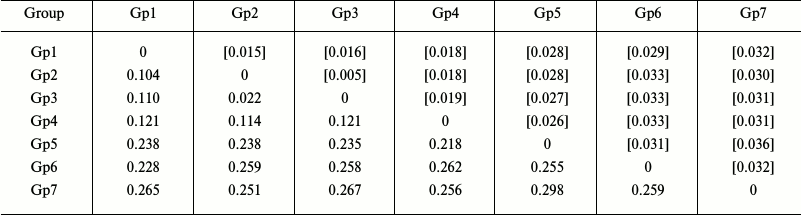
Notes: Distribution between groups was based on sequence clustering in
the phylogenetic tree (Fig. 2): Gp1, Ast.
rubens, 53 isolates; Gp2, Ast. amurensis, 6 isolates; Gp3,
Ast. amurensis, 12 isolates; Gp4, Ast. forbesii, 20
isolates; Gp5, Forcipulatida sp., 1 isolate; Gp6, Aph.
japonica, 5 isolates; Gp7, Distolasterias nipon, 1 isolate.
Below the diagonal, intergroup divergence mean values expressed in
number of nucleotide substitutions per site (after accounting for
intragroup divergence); above diagonal (in brackets), standard errors.
A total of 98 sequences were analyzed considering all three codon
positions. Genetic distances were calculated using the two-parameter
model of nucleotide substitutions [16].
The genetic distances between species of the genus Asterias were within the range 0.104-0.121; the corresponding values of the intergeneric divergence varied from 0.218 to 0.298 (Table 2). Therefore, the values for the intraspecific (0.003-0.009) and interspecific (0.104-0.121) genetic distances for the genus Asterias did not overlap.
Evaluation of the intragroup genetic distances for sea urchins of the Loveniidae family (Table 3) showed a considerably higher level of interspecific divergence of the COI gene in the genus Echinocardium (0.171-0.177) compared to starfish. Besides, the genetic distance (0.068) between the COI sequences from an E. cordatum specimen from the North Sea (Gp5) and specimens from the Sea of Japan and the Yellow Sea (Gp6) considerably exceeded the intraspecific genetic distances in starfish. The levels of the intrageneric divergence (0.146-0.257) were similar to those in starfish.
Table 3. Intergroup divergence matrix for
the mitochondrial COI genes from sea urchins of the Loveniidae
family (order Spatangoida)
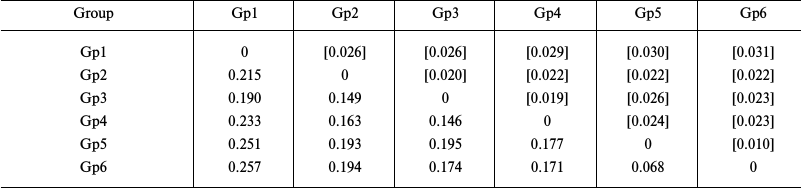
Notes: Distribution between groups was based on sequence clustering in
the phylogenetic tree (Fig. 3): Gp1,
Plagiobrissus grandis AJ639916, 1 isolate; Gp2, genus
Spatangus and Spatangoida sp., 2 isolates each; Gp3,
genus Lovenia, 3 isolates; Gp4, Echinocardium
laevigaster, 1 isolate; Gp5, E. cordatum, 1 isolate; Gp6,
E. cordatum, 6 isolates. Below diagonal, intergroup divergence
mean values expressed in number of nucleotide substitutions per site
(after accounting for intragroup divergence); above diagonal (in
brackets), standard errors. Seventeen sequences were analyzed
considering all three codon positions. Genetic distances were
calculated using the two-parameter model of nucleotide substitutions
[16].
Estimation of genetic distances for the sequences of transcribed nuclear markers (ITS1 – 5.8S rDNA – ITS2) in starfish of the Asteriidae family showed significantly lower level of intraspecific divergence (0.031-0.045) and higher level of intrageneric divergence (0.328-0.350) compared to the values obtained using the mitochondrial COI gene (Table 4).
Table 4. Intergroup divergence matrix for
sequences of nuclear spacers ITS1, ITS2, and 5.8S rDNA
from starfish from the Asteriidae family
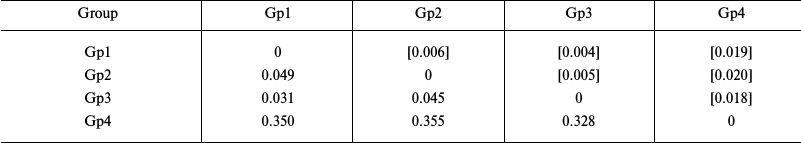
Notes: Distribution between groups was based on sequence clustering in
the phylogenetic tree (Fig. 4): Gp1, Ast.
amurensis, 3 isolates; Gp2, Ast. forbesii, 1 isolate; Gp3,
Ast. rubens, 3 isolates; Gp4, Aph. japonica, 1 isolate.
Below diagonal, intergroup divergence mean values expressed in number
of nucleotide substitutions per site (after accounting for intragroup
divergence); above diagonal (in brackets), standard errors. Eight
sequences were analyzed; total number of positions (including gaps) was
1601. Genetic distances were calculated using the two-parameter model
of nucleotide substitutions [16].
DISCUSSION
Genetic analysis showed (Fig. 2) that COI gene sequences from different populations of the same starfish species form monophyletic clusters. Thus, sequences of three studied species of the genus Asterias form monophyletic groups with high bootstrap values (99%). Note that geographically distant North Atlantic (American), East Atlantic (European), and White Sea populations of Ast. rubens form a homogenous cluster in which no subgroups could be distinguished. The observed two artifacts (assignment of Aph. japonica DQ592560 sequence to the Ast. amurensis cluster and assignment of Ast. rubens DQ077915 sequence to the Ast. forbesii cluster) were most probably due to species misidentification. It is evident that the COI sequence from Aph. japonica DQ592560 belongs to the Ast. amurensis cluster, since sequences of all five other Aph. japonica specimens formed a single cluster positioned at the base of the phylogenetic tree and distant from the Ast. amurensis cluster. The sequence Ast. rubens DQ077915 most probably belongs to Ast. forbesii. Since species of the genus Asterias are difficult to be distinguished based on morphological features only, they could be easily misidentified, especially when their areas overlap. Indeed, two Asterias species were found in the North Atlantics. Asterias forbesii inhabits the shelf zone of the North American coast from Cape Hatteras to Cape Cod. The area of Ast. rubens lies north of the area of Ast. forbesii. The European population of Ast. rubens inhabits the Atlantic coast shelf from Iceland to West France. The areas of North American populations of Ast. forbesii and Ast. rubens overlap in a large shelf region around Cape Cod [17]. Therefore, the data (including the artefacts) show that the fragment of the COI gene could be used for species identification of starfish.
Analysis of relations at a higher taxonomic level showed that Ast. amurensis (North Pacific) and Ast. rubens (North Atlantic) are the closest species within the Asterias genus despite significant geographical remoteness of their areas. The proximity of these two species is emphasized by the number of common specific features: out of 36 phylogenetically informative sites, 13 supported clustering (vs. 7 sites for clustering of Ast. rubens and Ast. forbesii). At the same time, the North Atlantic species Ast. forbesii is equidistant from the other two species and localizes to the base of a group combining the three species of this genus. Positioning of sequences from species of two other genera from the Sea of Japan (Distolasterias and Aphelasterias) at the base of the tree indicates considerable distance between these genera and Asterias. It should be noted that reliability of the estimation of relations between taxa higher than species based on the COI gene sequence might be questionable because of gene saturation with mutations due to the old evolutionary age of the taxa.
Identification of animal species and populations is based on the levels and character of the intra- and interspecific divergence of marker sequences. By now, extensive, but not sufficient, information has been accumulated on the divergence of the mitochondrial COI gene. In most cases, the intraspecific divergence of this gene stays below 1% and rarely exceeds 2% (such increased levels are mostly found in animals of the same species but from geographically distant regions or can be explained by the presence of cryptic species) [18]. The level of divergence can differ in different groups, although similar groups usually display similar values. Thus, for mollusks from the Vesicomyidae family, the levels of the intraspecific and interspecific divergence of the COI gene were estimated as 0.2-0.8 and 3.9-10.3%, respectively [6-8]. Similar values were found for mollusks of the Veneridae family [9]. In echinoderms, the levels of the COI gene intraspecific divergence vary within the 0.0-3.0% range (average, 0.62%); the levels of interspecific divergence vary within the 0.0-27.06% range (average, 15.33%) [3-5]. Analysis of the COI gene divergence for 22,266 populations and species of animals from various groups revealed the following p-distance values for taxa of different rank: 0.89 ± 0.16 – population; 3.78 ± 1.18 – for subspecies, semispecies, and sibling species; 11.06 ± 0.53 – morphologically distinct species; 16.60 ± 0.69 – species from different genera within a family; 20.57 ± 0.40 – families within the same order [19].
The values for the COI gene divergence in starfish of the Asteriidae family and sea urchins of the Loveniidae family determined in this study lie within the above-mentioned ranges. Moreover, the genetic distance between COI sequences of E. cordatum specimens from the Northern Sea and the Sea of Japan (6.8%) raises some doubt if these animals belong to the same species, although we cannot neglect the fact that their populations are very geographically distant [18].
We failed to confirm the applicability of the rRNA nuclear spacers for identification of animal species and populations. Apparently, the usefulness of genetic distances for this purpose is questionable because of the low levels of interspecific divergence (3.1-4.9%). It has been shown that the presence of species-specific compensatory replacements in the ITS2 sequence that could be identified by analysis of the secondary structure might serve as a more reliable criterion in this case [20].
In conclusion, our results show that sequences of the transcribed nuclear spacers (ITS) and mitochondrial COI gene can be used as reliable markers for identification of echinoderm species from the Sea of Japan and the White Sea.
Acknowledgements
This work was supported by the Russian Science Foundation (project No. 14-50-00029, Scientific foundations for creation of the National depositary bank of living systems) and by the Far East Program (projects Nos. 15-I-6-014o and 15-I-6-007o).
REFERENCES
1.Shneyer, V. S. (2007) On the species-specificity of
DNA: fifty years later, Biochemistry (Moscow), 72,
1377-1384.
2.Shubina, E. A., Ponomareva, E. V., Klimov, A. V.,
Klimova, A. V., and Kedrova, O. S. (2015) Repetitive DNA sequences as
an indicator of the level of genetic isolation in fish, Mol. Biol.
(Moscow), 49, 405-416.
3.Ward, R. D., Holmes, B. H., and O’Hara, T. D.
(2008) DNA barcoding discriminates echinoderm species, Mol. Ecol.
Resour., 8, 1202-1211.
4.Minin, K. V., Petrov, N. B., and Vladychenskaya, I.
P. (2015) Sea urchins of the genus Gracilechinus Fell &
Pawson, from the Pacific Ocean: morphology and evolutionary history,
Marine Biol. Res., 11, 253-268.
5.Wares, J. P. (2001) Biogeography of
Asterias: North Atlantic climate change and speciation, Biol.
Bull., 201, 95-103.
6.Goffredi, S. K., Hurtado, L. A., Hallam, S., and
Vrijenhoek, R. C. (2003) Evolutionary relationships of deep-sea vent
and cold seep clams (Mollusca: Vesicomyidae) of the
“pacifica/lepta” species complex, Marine
Biol., 142, 311-320.
7.Audzijonyte, A., Krylova, E. M., Sahling, H., and
Vrijenhoek, R. C. (2012) Molecular taxonomy reveals broad trans-oceanic
distributions and high species diversity of deep-sea clams (Bivalvia:
Vesicomyidae: Pliocardiinae) in chemosynthetic environments, System.
Biodivers., 10, 403-415.
8.Krylova, E. M., Kamenev, G. M., Vladychenskaya, I.
P., and Petrov, N. B. (2015) Vesicomyinae (Bivalvia: Vesicomyidae) of
the Kuril–Kamchatka Trench and adjacent abyssal regions,
Deep-Sea Res. Part II, 111, 198-209.
9.Chen, J., Li, Q., Kong, L., and Yu, H. (2011) How
DNA barcodes complement taxonomy and explore species diversity: the
case study of a poorly understood marine fauna, PLoS One,
6, e21326.
10.Ekimova, E., Korshunova, T. A., Shepetov, D. M.,
Neretina, T. V., Sanamyan, N. P., and Martynov, A. V. (2015)
Integrative systematics of northern and Arctic nudibranchs of the genus
Dendronotus (Mollusca, Gastropoda), with descriptions of three
new species, Zool. J. Linn. Soc., 173, 841-886.
11.Seregin, A. P., Anackov, G., and Friesen, N.
(2015) Molecular and morphological revision of the Allium
saxatile group (Amaryllidaceae): geographical isolation as the
driving force of underestimated speciation, Bot. J. Linn. Soc.,
178, 67-101.
12.Folmer, O., Black, M., Hoeh, W., Lutz, R., and
Vrijenhoek, R. (1994) DNA primers for amplification of mitochondrial
cytochrome c oxidase subunit I from diverse metazoan
invertebrates, Mol. Mar. Biol. Biotechnol., 5,
294-299.
13.Hoareau, T. B., and Boissin, E. (2010) Design of
phylum-specific hybrid primers for DNA barcoding: addressing the need
for efficient COI amplification in the Echinodermata, Mol.
Ecol. Resour., 10, 960-967.
14.Tamura, K., Stecher, G., Peterson, D., Filipski,
A., and Kumar, S. (2013) MEGA6: Molecular Evolutionary Genetics
Analysis version 6.0, Mol. Biol. Evol., 30,
2725-2729.
15.Nei, M., and Kumar, S. (2000) Molecular
Evolution and Phylogenetics, Oxford University Press, New York.
16.Kimura, M. A. (1980) Simple method for estimating
evolutionary rate of base substitutions through comparative studies of
nucleotide sequences, J. Mol. Evol., 16, 111-120.
17.Franz, D. R., Worley, E. K., and Merrill, A. S.
(1981) Distribution patterns of common sea stars of the Middle Atlantic
Continental shelf of the Northwest Atlantic (Gulf of Maine to Cape
Hatteras), Biol. Bull. Mar. Biol. Lab., 160, 394-418.
18.Avise, J. C., and Walker, D. (1999) Species
realities and numbers in sexual vertebrates: perspectives from an
asexually transmitted genome, Proc. Natl. Acad. Sci. USA,
96, 992-995.
19.Kartavtsev, Yu. F. (2013) Genetic divergence of
species and other taxa. Geographic speciation and genetic paradigm of
Neo-Darwinism in action, Usp. Sovrem. Biol., 133,
419-451.
20.Muller, T., Philippi, N., Dandekar, T., Schultz,
J., and Wolf, M. (2007) Distinguishing species, RNA, 13,
1469-1472.