Detection of Mutations in Mitochondrial DNA by Droplet Digital PCR
J. K. Sofronova1, Y. Y. Ilinsky1,2,3, K. E. Orishchenko1,2, E. G. Chupakhin1, E. A. Lunev1, and I. O. Mazunin1*
1Immanuil Kant Baltic Federal University, Institute of Chemistry and Biology, 236038 Kaliningrad, Russia; fax: +7 (4012) 595-595; E-mail: IMazunin@kantiana.ru2Federal Research Center Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, 630090 Novosibirsk, Russia; fax: +7 (383) 333-1278
3Novosibirsk State University, 630090 Novosibirsk, Russia; fax: +7 (383) 363-4333
* To whom correspondence should be addressed.
Received April 22, 2016; Revision received June 13, 2016
Mutations in mitochondrial DNA (mtDNA) may result in various pathological processes. Detection of mutant mtDNAs is a problem for diagnostic practice that is complicated by heteroplasmy – a phenomenon of the inferring presence of at least two allelic variants of the mitochondrial genome. Also, the level of heteroplasmy largely determines the profile and severity of clinical manifestations. Here we discuss detection of mutations in heteroplasmic mtDNA using up-to-date methods that have not yet been introduced as routine clinical assays. These methods can be used for detecting mutations in mtDNA to verify diagnosis of “mitochondrial disease”, studying dynamics of mutant mtDNA in body tissues of patients, as well as investigating structural features of mtDNAs. Original data on allele-specific discrimination of m.11778G>A mutation by droplet digital PCR are presented, which demonstrate an opportunity for simultaneous detection and quantitative assessment of mutations in mtDNAs.
KEY WORDS: mitochondria, mutations, mitochondrial diseases, heteroplasmy, droplet digital PCRDOI: 10.1134/S0006297916100011
Mitochondrial diseases comprise a group of diseases related to disturbed mitochondrial biogenesis due to mutations in nuclear DNA (nDNA) or the mitochondrial genome. Such diseases are characterized by variable symptoms, from mild headache and muscle weakness to subacute necrotizing encephalomyopathies [1]. Because this review will focus only on mitochondrial DNA (mtDNA) mutations, we briefly outline features of mitochondrial genetics. Mitochondrial inheritance occurs along maternal lineage, and the mitochondrial genome in humans consists of circular DNA containing 16,569 bp. On two DNA strands, 37 densely packed contiguous and overlapping genes are located [2]. In the mtDNA, the level of mutational events is very high, and the rate of spontaneous mutagenesis exceeds that of nDNA by about 20 times. On one hand, this determines a possibility for mutations to manifest in the first generation or even in an individual in whom it arose; on the other hand, harmfulness of mutations may be largely alleviated by multiple copies of mtDNAs, ranging from 2 to 10 per mitochondrion in cells, which contain up to several thousand mitochondria [3]. In humans, mitochondria can be found in all cell types except erythrocytes, and the amount of mtDNA per cell varies depending on the body tissue and the age of the host. The number of mtDNA copies ranges from 250-350 in leukocytes up to ≥6000 in myocardial muscle cells [4]. Mitotic segregation of mitochondria in ontogenesis may result in a situation when body tissues would differ qualitatively in terms of mitochondrial variants, which, in turn, places certain limitations while choosing biological samples for analysis.
According to a 2014 epidemiological study, the frequency of mitochondrial disorders with verified mutations in mtDNA in the European population was estimated to be ~1 per 5000 people [5]. However, there are missing data on the frequency of mitochondrial disorders based only on clinical symptoms, and diagnostics of such illnesses has some difficulties for the general practitioner due to nonspecific symptoms, especially at early stage of developing pathology as well as clinical picture similar to other diseases [6]. To promptly choose treatment strategy, molecular biology methods should be used to confirm or rule out mutations in mtDNA. Unfortunately, although under development, no therapeutic approaches now exist for treating mitochondrial disorders [7, 8].
On the other hand, consultation with prospective mothers is another important medical aspect of mitochondrial genetics [9]. Multiple copies of mtDNA underlie the heteroplasmy phenomenon, i.e. when a mixture of various mtDNAs is found in the body. A prospective mother may be clinically healthy but bear high-level mutant mtDNA, so that with high probability pathology may develop in her baby. A disease may clinically manifest and progress upon reaching a certain quantitative level of mutant mtDNA, known as a threshold effect [10]. Current methods reveal mutant mtDNA when its level comprises 5-10% of total mtDNA, which is sufficient for performing early diagnostics of mitochondrial disorders.
In this review, we discuss methods routinely used for detecting pathogenic mutations in mtDNA as well as up-to-date approaches that have not yet been introduced into broad clinical practice. In addition, droplet digital PCR (ddPCR) is discussed as an assay for counting absolute number of mtDNA copies. Accuracy of determining the level of heteroplasmy is required for examining dynamics of accumulated mutant mtDNA as well as assessing efficacy of currently developed systems for editing mitochondrial genome [11-14].
DETECTION OF MUTATIONS IN MITOCHONDRIAL DNA
Diagnostics of mitochondrial disorders includes clinical and biochemical examination. Clinical manifestations of mitochondrial diseases vary greatly, although the following features can be highlighted: simultaneously affect on several organs or organ systems; acute episodes observed at early stage of developing disease or during overt clinical picture; onset of disease symptoms varies with patient’s age. When the clinical picture indicates mitochondria-related nature of illness, differential diagnosis must be performed by adhering to the following strategy: a thorough analysis of family history of disease to distinguish between mitochondrial vs. nuclear inheritance; assessment of mitochondrial disease biomarkers (level of lactate, pyruvate, alanine, and fibroblast growth factor 21); immunocytochemistry examination and assessment of activity of complexes involved in oxidative phosphorylation [15, 16]. Note that fibroblast growth factor 21 regulating lipid and glucose metabolism is a new marker for mitochondrial encephalomyopathies [17]. Expression of this gene increases in cells of skeletal muscles as a compensatory mechanism in response to impaired functioning of the respiratory chain in mitochondria [18]. Based on clinical and biochemical diagnostics, patients are referred for molecular genetic analysis to detect any of the known mutations. According to [19], as few as 5-20% patients were found to bear predicted mutations in their mitochondrial genome. The remainder is accounted for by nuclear mutations, improperly selected biological sample, or emergence of previously unknown mutation at initial stages of embryogenesis.
Methods for detecting mutations in mtDNA do not significantly differ from those used to determine the primary sequence of any DNA. Key diagnostic approaches are based on restriction enzymes and assays involving polymerase chain reaction (PCR). As mentioned above, in medical practice heteroplasmy is an important feature when dealing with mtDNA. Wild type variants may be present along with mutant types of mtDNA. If the percentage of mutant forms in an examined sample is low, there is a probability that the outcome would give rise to a false negative result. Analytical complexity increases if a mutant variant is present as a single nucleotide substitution, but not an insertion or deletion. Gel electrophoresis of amplification products reveals deleted and duplicated regions in mtDNA, and in the case of heteroplasmy, two amplification products will be found. Prior to gel electrophoresis, amplification products may be treated with restriction endonucleases, a method known as Restriction Fragment Length Polymorphisms (RFLP). Certain nucleotide substitutions are known to alter recognition pattern for restriction sites in DNA. RFLP analysis provides quite high resolving power revealing as low as 5% mutant mtDNA [20]. However, as RFLP analysis does not determine primary sequence in mtDNA, there is a probability for both false positive and false negative results, which may be due to impairment or emergence of a recognition site for a restriction enzyme resulting from synonymous mutations, i.e. without altering amino acid sequence.
Using multiplex ligation-dependent probe amplification (MLPA) [21], MRC-Holland (The Netherlands) developed a kit (SALSA MLPA P125 Mitochondria probemix) for detecting qualitative and quantitative structural reorganization in mtDNA; the kit contains 37 various probes resulting in amplification products ranging from 136 to 427 bp in length. Each probe consists of two oligonucleotides. In case a sample sequence is complimentary to a probe, then they bind via contiguous hybridization, subsequently being cross-linked by ligase. After the amplification step, probes would have a unique length, which is used upon their separation in capillary electrophoresis. Relative level of heteroplasmy in mtDNA with deletions is determined using peak analysis plotted after capillary electrophoresis. Thus, this assay assesses number of deletions along the entire sequence of mtDNA [22].
High-throughput sequencing (NGS, Next Generation Sequencing) allows determining nucleotide sequence in mtDNA together with simultaneously assessed level of heteroplasmy [23]. NGS can reveal single mutations as well as deletions/duplications during the same reaction (in the same test tube) simultaneously in an entire mtDNA molecule including precise mapping of loci with developing structural reorganizations [24]. Despite being attractive, NGS technologies are far from routine laboratory practice due to high cost and requirement for highly qualified personnel.
Practical comparison of methods used to detect deletions and single nucleotide insertions into mtDNA were discussed in earlier publications [25-27].
DIGITAL DETECTION OF QUALITATIVE AND QUANTITATIVE CHANGES IN
mtDNA
Digital PCR can amplify a number of DNA copies, wherein the reaction simultaneously proceeds inside several thousands of nanoliter microspheres, thereby allowing increasing data reliability. It is common for real-time PCR to execute it in triplicate, followed by calculation of the arithmetic mean. By carrying out digital PCR, several thousand reactions undergo simultaneously, which substantially reduces inaccuracy. Digital PCR has now become more common as an accurate assay for quantitative assessment of nucleic acids.
Droplet digital PCR is a variant of digital PCR proposed by Bio-Rad (USA) [28]. The essence of the method is amplification of extremely low amounts of DNA followed by analyzing relative number of microspheres with/without template. Use of high DNA concentration results in methodological error so that template enters all microspheres and PCR proceeds in all “droplets”, making impossible quantitative estimation.
The components for reaction include commercial PCR mixture and custom-selected probes and/or oligonucleotide primers. In the first stage, a reaction mixture is separated in specially prepared oil solution into 15-25 thousand droplets with volume of 1 nl. After the amplification reaction, fluorescent signal is registered at a final point by using a special module working on the principle of flow cytometry, and the results are expressed as absolute number of DNA copies.
At the time of preparation of the manuscript (June 2016), four studies [29-32] were published wherein ddPCR was used to examine mtDNA. Investigation of mtDNA with PCR can be divided into two types: (i) calculating number of mtDNA copies per haploid chromosome set and (ii) assessing level of heteroplasmy in mutant mtDNA. Similar to PCR, ddPCR used to count mtDNA copies in the cells require the presence of a conservative nuclear gene of known copy number. It was demonstrated many times that change in amount of mtDNA represents a molecular marker of disease. During such assays, accuracy is necessary for creating a mathematic model for dynamics of mutant mtDNA in molecular pathogenesis of a disease.
Estimation of mutant mtDNA amount is a more complicated task, because it requires performing efficient allelic discrimination. Based on ddPCR, a precise method for quantitative measurement of deletions in mtDNA with extremely high sensitivity threshold (1 : 107) was developed [31]. This method is known as 3D (Digital Deletion Detection), which includes three steps: template enrichment, amplification, and analysis. In the first step (template enrichment), DNA samples initially collected at high concentration are selectively treated with restriction endonuclease that cut all wild type mtDNAs, thereby, leaving intact only mtDNAs containing deletions. Then, the digestion products serve as template for amplification with TaqMan-probes selected to bind to regions flanking a deletion in the mtDNA. As a result, absolute quantitative data on the amount of mtDNA with deletions contained in the examined sample is obtained. Thus, an advantage of the 3D technique is based on initial concentration of mutant mtDNA so that their subsequent detection is performed immediately without preliminary amplification, thereby excluding PCR artifacts.
By using allele-specific hydrolyzed probes and allele-specific oligonucleotide primers, it is possible to measure number of mtDNA copies differing in one oligonucleotide substitution. We demonstrated feasibility of this approach in case of mutation m.11778G>A, which is associated with Leber’s disease (OMIM # 535000). For this, fragments from the gene encoding NADH-ubiquinone oxidoreductase chain 4 (MT-ND4) containing a region with mutation (m.11778G) and the wild type (m.11778A) variant were cloned into pUC19 plasmid. The sequences of oligonucleotide primers and hydrolyzed probes are shown in the table.
Primers and probes used for ddPCR
Note: “+”, before nucleotide with LNA-modification.
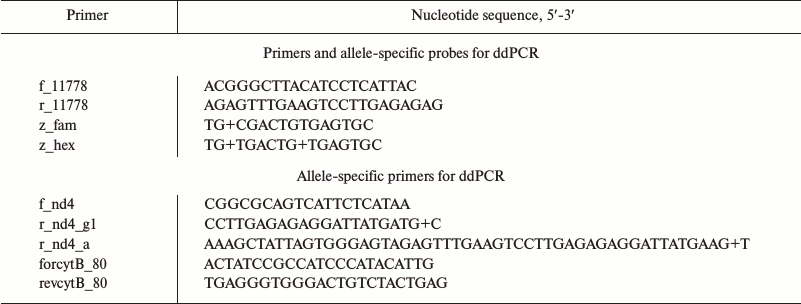
The ddPCR was performed according to the manufacturer’s protocol, with some optimization procedure. Distribution of droplet clusters points at specificity of interaction between hydrolyzed probe and matching templates (Fig. 1). During the experiment, diplex PCR was applied: one of the allele-specific probes was complimentary to wild type m.11778G mtDNA, whereas another to mutant m.11778A mtDNA. Allele-specific probes were also used for detecting rare mutations in mtDNA [32].
Fig. 1. Clusters with droplet distribution after amplification of templates p.11778A and p.11778G with allele-specific probes. Top, left-to-right: clusters with droplet distribution after amplification with allele-specific probe detecting mutation m.11778G: template p.11778G, mixture of templates p.11778G + p.11778A, 1 : 1 ratio, template p.11778A. Bottom, left-to-right: clusters of droplet distribution after amplification with allele-specific probe detecting mutation m.11778A.
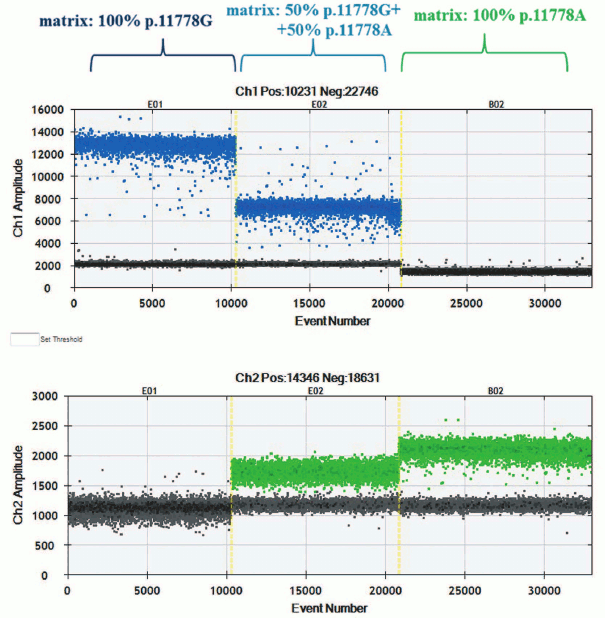
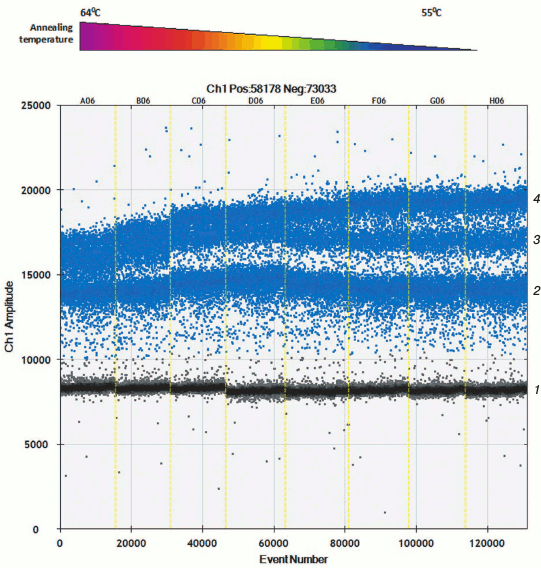
Apart from hydrolyzed allele-specific probes for allelic discrimination, allele-specific primers and intercalating dye can be used. Only two detection channels are used in present-day equipment for performing ddPCR, which allow applying probes with dye FAM and VIC/HEX. Selecting allele-specific probes allows measuring either amount of mutant and wild type mtDNA or total amount of mtDNA and one of the allelic variants. To assess level of heteroplasmy m.11778A>G as well as to calculate total amount of mtDNA, a set of primers was developed that results in amplification products of different length. Using software, this allows allocation of amplicons into independent droplet clusters due to the differences in amount of intercalating dye bound to them. During the optimization process, it was found that droplet clusters were separated more efficiently at lowered temperature during the annealing step (Fig. 2).
Fig. 2. Allele-specific interaction of primers with templates p.11778A and p.11778G with thermal gradient. Clusters of droplet distribution after multiplex ddPCR with allele-specific primers. Left-to-right: thermal gradient from 64 to 55°C. Separate clusters are designated on the right: 1) cluster of “negative” droplets containing no specific template; 2) cluster of amplification products after using 88-bp-long cytB primers; 3) cluster of amplification products with allele-specific primers specific to mutation m.11778G, 122-bp-long; 4) cluster of amplification products, 88- and 122-bp-long.
Droplet digital PCR has not been introduced into laboratory practice yet; however, this approach fully meets the requirements of data reproducibility and low threshold sensitivity applied to systems for detecting level of heteroplasmy.
Acknowledgements
The study was performed with financial support from the Ministry of Education and Science (Federal Targeted Program “2014-2020 Research and Development on Priority Development Fields in Science and Technology Complex in Russia”, Grant Agreement ID RFMEFI57514X0108).
REFERENCES
1.Chinnery, P. F., Howell, N., Andrews, R. M., and
Turnbull, D. M. (1999) Clinical mitochondrial genetics, J. Med.
Genet., 36, 425-436.
2.Anderson, S., Bankier, A. T., Barrell, B. G., De
Bruijn, M. H., Coulson, A. R., Drouin, J., Eperon, I. C., Nierlich, D.
P., Roe, B. A., Sanger, F., Schreier, P. H., Smith, A. J., Staden, R.,
and Young, I. G. (1981) Sequence and organization of the human
mitochondrial genome, Nature, 290, 457-465.
3.Chinnery, P. F., and Schon, E. A. (2003)
Mitochondria, J. Neurol. Neurosurg. Psychiatry, 74,
1188-1199.
4.Wachsmuth, M., Hubner, A., Li, M., Madea, B., and
Stoneking, M. (2016) Age-related and heteroplasmy-related variation in
human mtDNA copy number, PLoS Genet., 12, e1005939.
5.Gorman, G. S., Schaefer, A. M., Ng, Y., Gomez, N.,
Blakely, E. L., Alston, C. L., Feeney, C., Horvath, R., Yu-Wai-Man, P.,
Chinnery, P. F., Taylor, R. W., Turnbull, D. M., and McFarland, R.
(2015) Prevalence of nuclear and mitochondrial DNA mutations related to
adult mitochondrial disease, Ann. Neurol., 77,
753-759.
6.Pfeffer, G., and Chinnery, P. F. (2013) Diagnosis
and treatment of mitochondrial myopathies, Ann. Med., 45,
4-16.
7.Pfeffer, G., Horvath, R., Klopstock, T., Mootha, V.
K., Suomalainen, A., Koene, S., Hirano, M., Zeviani, M., Bindoff, L.
A., Yu-Wai-Man, P., Hanna, M., Carelli, V., McFarland, R., Majamaa, K.,
Turnbull, D. M., Smeitink, J., and Chinnery, P. F. (2013) New
treatments for mitochondrial disease-no time to drop our standards,
Nat. Rev. Neurol., 9, 474-481.
8.Hyslop, L. A., Blakeley, P., Craven, L.,
Richardson, J., Fogarty, N. M., Fragouli, E., Lamb, M., Wamaitha, S.
E., Prathalingam, N., Zhang, Q., O’Keefe, H., Takeda, Y., Arizzi,
L., Alfarawati, S., Tuppen, H. A., Irving, L., Kalleas, D., Choudhary,
M., Wells, D., Murdoch, P., Turnbull, D. M., Niakan, K. K., and
Herbert, M. (2016) Towards clinical application of pronuclear transfer
to prevent mitochondrial DNA disease, Nature, doi:
10.1038/nature18303.
9.Bredenoord, A. L., Dondorp, W., Pennings, G., and
De Wert, G. (2010) Avoiding transgenerational risks of mitochondrial
DNA disorders: a morally acceptable reason for sex selection? Hum.
Reprod., 25, 1354-1360.
10.Ylikallio, E., and Suomalainen, A. (2012)
Mechanisms of mitochondrial diseases, Ann. Med., 44,
41-59.
11.Bacman, S. R., Williams, S. L., Pinto, M.,
Peralta, S., and Moraes, C. T. (2013) Specific elimination of mutant
mitochondrial genomes in patient-derived cells by mitoTALENs, Nat.
Med., 19, 1111-1113.
12.Gammage, P. A., Rorbach, J., Vincent, A. I.,
Rebar, E. J., and Minczuk, M. (2014) Mitochondrially targeted ZFNs for
selective degradation of pathogenic mitochondrial genomes bearing
large-scale deletions or point mutations, EMBO Mol. Med.,
6, 458-466.
13.Jo, A., Ham, S., Lee, G. H., Lee, Y. I., Kim, S.,
Lee, Y. S., Shin, J. H., and Lee, Y. (2015) Efficient mitochondrial
genome editing by CRISPR/Cas9, Biomed Res. Int., doi:
10.1155/2015/305716.
14.Orishchenko, K. E., Sofronova, Yu. K., Chupakhin,
E. G., Lunev, E. A., and Mazunin, I. O. (2016) Delivery of Cas9 into
mitochondria, Genes Cells, 11, in press.
15.Chinnery, P. F. (2016) Mitochondrial disease in
adults: what’s old and what’s new? EMBO Mol. Med.,
12, 1503-1512.
16.Koopman, W. J., Beyrath, J., Fung, C. W., Koene,
S., Rodenburg, R. J., Willems, P. H., and Smeitink, J. A. (2016)
Mitochondrial disorders in children: toward development of
small-molecule treatment strategies, EMBO Mol. Med., 8,
311-327.
17.Suomalainen, A., Elo, J. M., Pietilainen, K. H.,
Hakonen, A. H., Sevastianova, K., Korpela, M., Isohanni, P.,
Marjavaara, S. K., Tyni, T., Kiuru-Enari, S., Pihko, H., Darin, N.,
Ounap, K., Kluijtmans, L. A., Paetau, A., Buzkova, J., Bindoff, L. A.,
Annunen-Rasila, J., Uusimaa, J., Rissanen, A., Yki-Jarvinen, H.,
Hirano, M., Tulinius, M., Smeitink, J., and Tyynismaa, H. (2011) FGF-21
as a biomarker for muscle-manifesting mitochondrial respiratory chain
deficiencies: a diagnostic study, Lancet Neurol., 9,
806-818.
18.Ji, K., Zheng, J., Lv, J., Xu, J., Ji, X., Luo,
Y. B., Li, W., Zhao, Y., and Yan, C. (2015) Skeletal muscle increases
FGF21 expression in mitochondrial disorders to compensate for energy
metabolic insufficiency by activating the mTOR-YY1-PGC1α pathway,
Free Radic. Biol. Med., 84, 161-170.
19.Wong, L. J., Scaglia, F., Graham, B. H., and
Craigen, W. J. (2010) Current molecular diagnostic algorithm for
mitochondrial disorders, Mol. Genet. Metab., 100,
111-117.
20.Ma, Y., Fang, F., Yang, Y., Zou, L., Zhang, Y.,
Wang, S., Xu, Y., Pei, P., and Qi, Y. (2009) The study of mitochondrial
A3243G mutation in different samples, Mitochondrion, 9,
139-143.
21.Kozlowski, P., Jasinska, A. J., and Kwiatkowski,
D. J. (2008) New applications and developments in the use of multiplex
ligation-dependent probe amplification, Electrophoresis,
23, 4627-4636.
22.Mayorga, L., Laurito, S. R., Loos, M. A., Eiroa,
H. D., De Pinho, S., Lubieniecki, F., Arroyo, H. A., Pereyra, M. F.,
Kauffman, M. A., and Roque, M. (2016) Mitochondrial DNA deletions
detected by multiplex ligation-dependent probe amplification,
Mitochondrial DNA A DNA MappSeq. Anal., 27, 2864-2867
23.Vasta, V., Ng, S. B., Turner, E. H., Shendure,
J., and Hahn, S. H. (2009) Next generation sequence analysis for
mitochondrial disorders, Genome Med., 23, 100.
24.Palculict, M. E., Zhang, V. W., Wong, L. J., and
Wang, J. (2016) Comprehensive mitochondrial genome analysis by
massively parallel sequencing, Methods Mol. Biol., 1351,
3-17.
25.Moraes, C. T., Atencio, D. P., Oca-Cossio, J.,
and Diaz, F. (2003) Techniques and pitfalls in the detection of
pathogenic mitochondrial DNA mutations, J. Mol. Diagn.,
5, 197-208.
26.Kurelac, I., Lang, M., Zuntini, R., Calabrese,
C., Simone, D., Vicario, S., Santamaria, M., Attimonelli, M., Romeo,
G., and Gasparre, G. (2012) Searching for a needle in the haystack:
comparing six methods to evaluate heteroplasmy in difficult sequence
context, Biotechnol. Adv., 30, 363-371.
27.Sobenin, I. A., Mitrofanov, K. Y., Zhelankin, A.
V., Sazonova, M. A., Postnov, A. Y., Revin, V. V., Bobryshev, Y. V.,
and Orekhov, A. N. (2014) Quantitative assessment of heteroplasmy of
mitochondrial genome: perspectives in diagnostics and methodological
pitfalls, Biomed Res. Int., 292017.
28.Hindson, B. J., Ness, K. D., Masquelier, D. A.,
Belgrader, P., Heredia, N. J., Makarewicz, A. J., Bright, I. J.,
Lucero, M. Y., Hiddessen, A. L., Legler, T. C., Kitano, T. K., Hodel,
M. R., Petersen, J. F., Wyatt, P. W., Steenblock, E. R., Shah, P. H.,
Bousse, L. J., Troup, C. B., Mellen, J. C., Wittmann, D. K., Erndt, N.
G., Cauley, T. H., Koehler, R. T., So, A. P., Dube, S., Rose, K. A.,
Montesclaros, L., Wang, S., Stumbo, D. P., Hodges, S. P., Romine, S.,
Milanovich, F. P., White, H. E., Regan, J. F., Karlin-Neumann, G. A.,
Hindson, C. M., Saxonov, S., and Colston, B. W. (2011) High-throughput
droplet digital PCR system for absolute quantitation of DNA copy
number, Anal. Chem., 83, 8604-8610.
29.Podlesniy, P., Figueiro-Silva, J., Llado, A.,
Antonell, A., Sanchez-Valle, R., Alcolea, D., Lleo, A., Molinuevo, J.
L., Serra, N., and Trullas, R. (2013) Low cerebrospinal fluid
concentration of mitochondrial DNA in preclinical Alzheimer’s
disease, Ann. Neurol., 74, 655-668.
30.Wachsmuth, M., Hubner, A., Li, M., Madea, B., and
Stoneking, M. (2016) Age-related and heteroplasmy-related variation in
human mtDNA copy number, PLoS Genet., 12, e1005939.
31.Taylor, S. D., Ericson, N. G., Burton, J. N.,
Prolla, T. A., Silber, J. R., Shendure, J., and Bielas, J. H. (2014)
Targeted enrichment and high-resolution digital profiling of
mitochondrial DNA deletions in human brain, Aging Cell,
13, 29-38.
32.Rebolledo-Jaramillo, B., Su, M. S., Stoler, N.,
McElhoe, J. A., Dickins, B., Blankenberg, D., Korneliussen, T. S.,
Chiaromonte, F., Nielsen, R., Holland, M. M., Paul, I. M., Nekrutenko,
A., and Makova, K. D. (2014) Maternal age effect and severe germ-line
bottleneck in the inheritance of human mitochondrial DNA, Proc.
Natl. Acad. Sci. USA, 111, 15474-15479.