Sustained Early Disruption of Mitochondrial Function Contributes to Arsenic-Induced Prostate Tumorigenesis
B. Singh1, M. Kulawiec2, K. M. Owens2, A. Singh3, and K. K. Singh4,5*
1University of Alabama at Birmingham, Department of Genetics, 35294 Birmingham, AL, USA2Roswell Park Cancer Institute, Department of Cancer Genetics, 14263 Buffalo, NY, USA
3State University of New York, Department of Pediatrics, 12246 Buffalo, NY, USA
4University of Alabama at Birmingham, Center for Aging and UAB Comprehensive Cancer Center, Center for Free Radical Biology, Departments of Genetics, Pathology, Environmental Health, 35294 Birmingham, AL, USA; E-mail: kksingh@uab.edu
5Birmingham Veterans Affairs Medical Center, 35294 Birmingham, AL, USA
* To whom correspondence should be addressed.
Received April 8, 2016; Revision received May 19, 2016
Arsenic is a well-known human carcinogen that affects millions of people worldwide, but the underlying mechanisms of carcinogenesis are unclear. Several epidemiological studies have suggested increased prostate cancer incidence and mortality due to exposure to arsenic. Due to lack of an animal model of arsenic-induced carcinogenesis, we used a prostate epithelial cell culture model to identify a role for mitochondria in arsenic-induced prostate cancer. Mitochondrial morphology and membrane potential was impacted within a few hours of arsenic exposure of non-neoplastic prostate epithelial cells. Chronic arsenic treatment induced mutations in mitochondrial genes and altered mitochondrial functions. Human non-neoplastic prostate epithelial cells continuously cultured for seven months in the presence of 5 µM arsenite showed tumorigenic properties in vitro and induced tumors in SCID mice, which indicated transformation of these cells. Protein and mRNA expression of subunits of mtOXPHOS complex I were decreased in arsenic-transformed cells. Alterations in complex I, a main site for reactive oxygen species (ROS) production as well as increased expression of ROS-producing NOX4 in arsenic-transformed cells suggested a role of oxidative stress in tumorigenic transformation of prostate epithelial cells. Whole genome cGH array analyses of arsenic-transformed prostate cells identified extensive genomic instability. Our study revealed mitochondrial dysfunction induced oxidative stress and decreased expression of p53 in arsenic-transformed cells as an underlying mechanism of the mitochondrial and nuclear genomic instability. These studies suggest that early changes in mitochondrial functions are sustained during prolong arsenic exposure. Overall, our study provides evidence that arsenic disruption of mitochondrial function is an early and key step in tumorigenic transformation of prostate epithelial cells.
KEY WORDS: arsenic, mitochondria, mtOXPHOS, genomic instability, tumorigenic transformation, prostate cancerDOI: 10.1134/S0006297916100072
Abbreviations: BAC, bacterial artificial chromosomes; cGH, comparative genome hybridization (array analysis); mtOXPHOS, mitochondrial oxidative phosphorylation; ROS, reactive oxygen species.
Arsenic is a metalloid that is ubiquitously present in the environment.
Inorganic arsenic poses a global health problem affecting millions of
people worldwide [1-4]. Arsenic
contamination in drinking water is found throughout the world. It is
estimated that nearly 150 million people in more than 70 countries are
impacted from arsenic [5]. In Bangladesh, about 50
million people are at risk of developing disease due to drinking water
contaminated with arsenic [4, 6]. Arsenic exposure is quite extensive in rural areas
of the United States [7]. About 20 million people
in the United States drink excess arsenic [3, 7, 8]. In some places in the
United States such as New Mexico, Arizona, and Texas, arsenic
concentrations are reported to be very high (ranges from 680 to
1880 mg/liter or ~9 to 25 mM) [9].
Recently, a US based study showed a significant association between
arsenic levels in community water systems and prostate cancer incidence
[10]. Recent studies suggest that human exposure
to arsenic also occurs through medicines and food sources such as
juice, wine, rice, milk, meat, infants’ formula, rice cereals,
and other foods [11-16]. Some
of these food sources including wine and juice contain arsenic
contamination much above the World Health Organization (WHO) and U.S.
Environmental Protection Agency (USEPA) maximum contaminant level (MCL)
limit of 10 µg/liter or 10 parts per billion (ppb) total arsenic
in drinking water [8, 14, 15, 17]. Arsenic exposure leads
to cardiovascular and peripheral vascular disease and neurological,
neurobehavioral, and several other human diseases [18]. Exposure to arsenic also induces cancer. This
includes skin, lung, liver, urinary tract, pancreas, breast, and
prostate cancer [19-24]. At
least five epidemiological studies conducted both in the United States
and other parts of the world suggest that exposure to arsenic leads to
high incidence of prostate cancer in humans [25-29]. Additionally, a recent US prospective study also
suggests an increased prostate cancer incidence and mortality due to
low dose exposure to arsenic [30]. Unfortunately,
the molecular mechanism underlying arsenic-induced prostate
carcinogenicity is poorly understood.
Arsenic is one of the few human carcinogens that do not induce tumors in laboratory animals except at extremely high doses that do not recapitulate human exposure conditions [31-34]. Numerous attempts by different investigators to develop a mouse model of arsenic-induced prostate cancer have not been successful [31-36]. Thus, defining the mechanisms involved in arsenic-induced carcinogenesis has been difficult. In the absence of a suitable animal model of arsenic-induced prostate carcinogenesis, as an alternative we utilized a cell culture model of arsenic-induced prostate cancer in which a normal non-tumorigenic prostate epithelial cell line was exposed to a low dose of arsenic for seven months [35].
A few studies have demonstrated that arsenic accumulates in mitochondria via phosphate-transport proteins and the dicarboxylate carrier [37, 38]. When injected into rabbits, arsenic also accumulates in vivo in the mitochondria of kidneys [39]. To date, little is known about the role of mitochondria in arsenic-induced cancers. In this study, we utilized an epithelial cell culture model [35] to identify a role for mitochondria in arsenic-induced prostate cancer.
MATERIALS AND METHODS
Arsenic exposure time and dose. This study was performed using prostate epithelial RWPE-1 and CAsPE cells (a kind gift from Dr. M. P. Waalkes, NIEHS). Briefly, non-tumorigenic RWPE-1 prostate epithelial cells were continuously cultured for seven months in the presence of 5 µM of sodium arsenite. Seven months arsenic-treated RWPE-1 cells are named as CAsPE cells. Simultaneous passage matched control cells were also grown for the same period of time but without arsenite. Cells were collected and frozen every 4 weeks, and various analyses, including in vitro transformation assays and tumor development in SCID mouse, were performed. The length of exposure and a dose of arsenic were chosen to recapitulate the arsenic exposure conditions in the field [25, 36, 40-47]. To compare the cell growth rate, RWPE-1 and CAsPE cells were plated at a density of 10,000 cells/well in a six well plate and cells were counted every day up to 5 days.
Matrigel invasion assay. The Matrigel invasion assay was accomplished with BD BioCoat Matrigel Invasion Chambers (BD Biosciences, USA) as described earlier [48]. Briefly, RWPE-1 and CAsPE cells were seeded at 0.5·105 cells per chamber. Culture media with 10% FBS was used as the chemoattractant. Cells were allowed to migrate for 24 h, and then the membranes were stained with the Diff-Quik Stain Set (Dade Behring, USA). The invading cells were counted in six views per membrane under a microscope at 20× magnification. Cell counts were averaged and statistically analyzed.
Cell-culture migration assay. Cell-culture migration assay was carried out as described earlier [48]. Briefly, 1·106 RWPE-1 and CAsPE cells were seeded into six-well plates and reached confluence on the next day. The cells were wounded with yellow pipet tips. Pictures of the wounded lines at the same positions were taken at 0 and 24 h after wounding.
SCID mouse xenograft and analysis of tumor growth. Xenograft experiments in SCID mice were carried out as described earlier [49]. SCID mice were injected subcutaneously with 1·106 RWPE-1 or CAsPE cells mixed with Matrigel (1 : 1 ratio) in both flanks of the mice using 26 G needles. Tumor growth was monitored externally using calipers for up to 10 weeks.
Histology and immunohistochemical analyses. Xenograft tumors were fixed in buffered formalin, embedded in paraffin, sectioned (5 µM), and stained with hematoxylin and eosin. Immunohistochemical analyses of tumor tissues were carried as described earlier [50]. Briefly, the slides were de-paraffinized by incubation in xylene and ascending grades of alcohol. Antigen retrieval was done by heating in citrate-based antigen unmasking solution (Vector Laboratories, USA) for 30 min at 98°C, incubated in 3% hydrogen peroxide for 10 min, blocked with serum for 30 min, incubated with anti-ki67 antibody (1 : 500 dilution; Santa Cruz Biotechnology, USA) or anti-PSA antibody (1 : 300 dilution; Santa Cruz Biotechnology) for 1 h at room temperature, followed by incubation with biotinylated secondary antibody for 30 min and another 30 min with Vectastain ABC kit (Vector Laboratories). Color was developed by incubating slides with peroxidase substrate solution followed by counterstaining with hematoxylin. All sections were examined with an Olympus BX50 microscope. The pictures were taken with an Olympus DP 70 camera connected to DP Controller software (Olympus, USA).
Mitochondrial staining and microscopy. RWPE-1 cells were grown on coverslips in six well plates overnight at 37°C, 95% humidity, and 5% carbon dioxide environment. Next day, the cells were treated with sodium arsenite (0.5 µM) for 8 h. Following washing with PBS, the cells were incubated with media containing 100 nM MitoTracker (Molecular Probes, USA) for 20 min. The cells were then fixed in 2% paraformaldehyde in PBS and stained with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories) prior to mounting the coverslips onto microscope slides. Confocal pictures were taken with a Leica confocal instrument. The pictures were analyzed with ImageJ software (NIH, http://rsbweb.nih.gov/ij/).
Mitochondrial genome analysis. The complete mitochondrial genome was amplified with 24 sets of overlapping primers [51] in RWPE-1 and CAsPE cells. All the amplicons were checked by agarose gel electrophoresis and directly sequenced by the Sanger method using 100 ng of PCR product. After the assembly of all sequences, the variations were identified by comparing the mtDNA sequences of RWPE-1 cells with CAsPE cells. The significance of mutations were analyzed by identifying the region where the mutations were found and comparing mutated sequences with lower organisms to determine the level of conservation of the amino acids across species.
Mitochondrial membrane potential measurement. Mitochondrial membrane potential of RWPE-1 and CAsPE cells was assessed using the fluorescent probe 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzamidazolocarbocyanin iodide (JC-1; Molecular Probes) following established protocols and recommendations [52]. Cells were incubated with JC-1 (2 µM) dye diluted in culture medium at 37°C for 15 min. The cells were washed with PBS and analyzed using a Becton Dickinson FACS Calibur Flow Cytometer (Franklin Lakes, USA). JC-1 was detected by using blue and yellow lasers to excite JC-1 monomers (FL1 channel) and aggregates (FL2 channel).
Western blot analyses. RWPE-1 and CAsPE cells were lysed in RIPA lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM PMSF, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS) with addition of Protease Inhibitor Cocktail (Thermo Fisher Scientific, USA). Fifteen micrograms of protein was size-fractionated on a 12% SDS-polyacrylamide gel and transferred onto a polyvinylidene difluoride membrane (Millipore Corp., USA) using the wet transfer system and blocked in 5% skim milk in PBST for 1 h. A premixed cocktail (7.2 µg/ml) containing primary monoclonal antibodies against subunits of OXPHOS complexes (Mitosciences, USA) was used to detect representative subunits from OXPHOS complex I, II, III, IV, and V. Mouse monoclonal primary antibody against p53 (Santa Cruz Biotechnology) was used to detect the expression of p53 in RWPE-1 and CAsPE cells. α-Tubulin (Santa Cruz Biotechnology) antibody or Ponceau S staining was used as a loading control. HRP-conjugated secondary antibodies (Vector Laboratories) and ECL reagent kits (GE Healthcare Biosciences, USA) were used for film development.
Whole genome cGH array analysis of arsenic-exposed prostate epithelial cells. The construction of the custom cGH (comparative genomic hybridization) BAC (bacterial artificial chromosomes) array was described previously [53]. The log2 ratios of the background corrected test/control were normalized using a loess correction. For each BAC, a median of the log2 ratios of all its replicates was computed and BACs with less than two successful replicates were excluded. The corrected log2 ratio for each BAC was then used to segment the array using Circular Binary Segmentation (CBS) using DNAcopy software. For each segment, the median absolute deviations (MAD) of the corrected log2 ratios were computed. Segments with a median corrected log2 ratio greater than 1× the median of the MAD (MMAD) were considered gained, and segments with a median corrected log2 ratio less than 2× MMAD were considered lost. In addition, all single BACs within a segment whose corrected log2 ratios exceed 2× the interquartile range of the segment are marked as outliers. Outliers with corrected log2 ratio exceeding 2× the global interquartile range are outlined as gains or losses accordingly. All BACs were assigned a call of gained (1), lost (–1), or normal (0) based on the call of the segment they are in, and the karyotype. The cGH data from arsenic-treated CAsPE cells was normalized with paired untreated RWPE cells grown to the same length of time under the same culture condition except without arsenic.
Gene expression analyses. The total RNA was isolated from RWPE-1 and CAsPE cells by the Trizol extraction method according to the manufacturer’s protocol (Invitrogen, USA) and reverse transcribed using a Superscript III First Strand kit (Invitrogen). RT PCR was performed to measure the expression levels of the NOX1, NOX2, NOX3, NOX4, NOX5, and NDUFA13 genes using GoTaq Green 2× Master Mix (Promega Corporation, USA) and gene-specific primers. β-Actin was used as an internal control.
Statistical analyses. All statistical analyses were performed with Sigma Plot 11.0 software (Systat Software, USA). Data were compared using two-tailed Student’s t-tests.
RESULTS
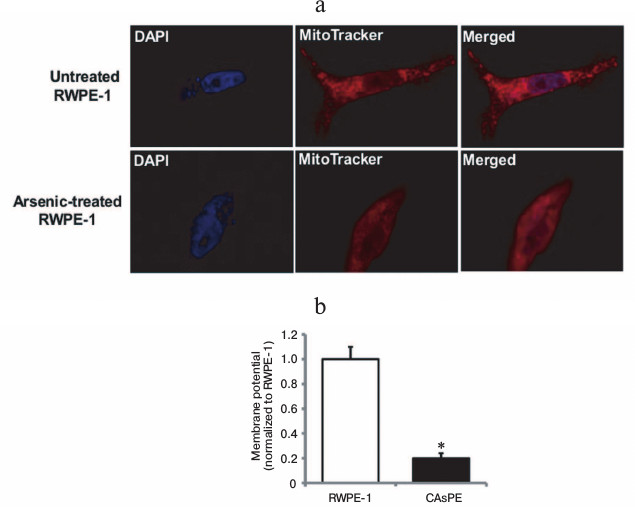
Mitochondria are early targets of arsenic exposure. To determine whether arsenic-mediated mitochondrial changes are early events, we treated non-tumorigenic RWPE-1 prostate epithelial cells with 0.5 µM sodium arsenite for 8 h and stained the cells with MitoTracker to observe the mitochondrial morphological and membrane potential changes. After 8 h of arsenic treatment, non-tumorigenic RWPE-1 prostate epithelial cells displayed diffused mitochondrial morphology that indicated change in mitochondrial membrane potential (Fig. 1a). Untreated control cells showed distinct mitochondrial morphological characteristics (Fig. 1a). We also analyzed the mitochondria in non-tumorigenic and arsenic-transformed cells [35] after long-term arsenic exposure. Our studies revealed a severe decrease in mitochondrial membrane potential in arsenic-transformed cells compared to paired arsenic-untreated non-tumorigenic control cells (Fig. 1b). These studies suggest that brief exposure to arsenic results in altered mitochondrial morphology and membrane potential and these mitochondrial alterations are sustained during long-term (seven months) exposure to arsenic.
Fig. 1. Arsenic exposure alters mitochondrial morphology and mitochondrial membrane potential. a) RWPE-1 cells were treated with 0.5 µM sodium arsenite for 8 h, stained with MitoTracker red and DAPI, and images were obtained by fluorescence microscopy. b) Mitochondrial membrane potential (ΔΨm) in RWPE-1 and arsenic-treated CAsPE cells was determined using the fluorescent cationic dye JC-1. * Denotes p < 0.05.
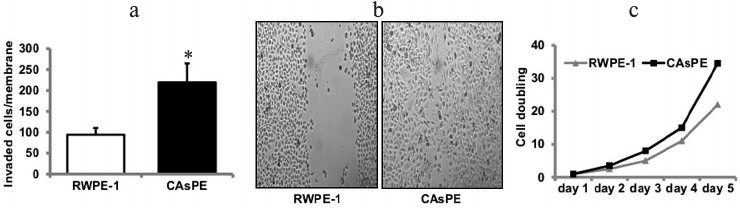
Exposure of prostate epithelial cells to arsenic induces tumorigenic transformation in vitro. The development of models for arsenic-induced prostate carcinogenesis is critical for understanding the mechanisms underlying the carcinogenic process. We utilized an in vitro prostate epithelial cell culture model to identify the carcinogenic mechanisms underlying the development of cancer induced by arsenic. RWPE-1 prostate epithelial cells were continuously treated with sodium arsenite (5 µM) for approximately seven months. Paired control cell lines grown simultaneously to seven months, but without arsenic treatment, were also included in the study. Medium was replaced each week with fresh medium, either with or without arsenite. We determined transformed phenotypes by monitoring Matrigel invasion after a treatment period of seven months. Arsenic treatment for seven months resulted in increased Matrigel invasion (Fig. 2a), faster wound healing (Fig. 2b), and increased growth rate (Fig. 2c) than the paired untreated RWPE-1 prostate cells (grown to seven months continuously without treatment). These studies suggest that exposure to arsenic leads to in vitro transformation of prostate epithelial cells.
Fig. 2. Arsenic induces transformation of prostate epithelial cells. Matrigel invasion (a) and wound-healing assay (b) of RWPE-1 cells treated continuously with 5 µM sodium-arsenite for seven months (marked CAsPE), as well as non-treated control (RWPE-1 grown continuously for seven months but without arsenic treatment) was performed as described [48]. c) The growth rate of RWPE-1 and CAsPE cells is expressed relative to the initial plating number (day 0). While the experiment was repeated three times, representative data are shown in the figure. * Denotes p < 0.05.
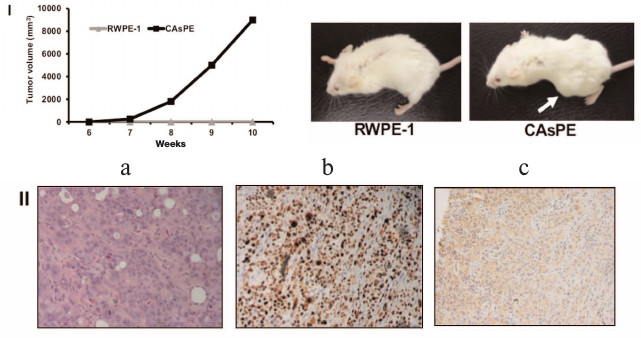
Arsenic-transformed prostate epithelial cells form tumors in vivo. Since arsenic-treated CAsPE cells showed features of a transformed phenotype, we sought to determine the ability of these cells to form tumors in mice. For this, one million cells were mixed with Matrigel and injected subcutaneously into the flanks of SCID mice and tumor formation was monitored for 10 weeks. Animals injected with RWPE-1 cells did not form tumors at all during the experiment (0/4). In contrast, all mice injected with CAsPE cells developed tumors (4/4), on average eight weeks after injection, as presented in Fig. 3, I. Together these results reveal that CAsPE cells manifest a gain of tumorigenicity in a normally non-tumorigenic cell. Histochemical analysis of primary tumors showed increased levels of the proliferation marker Ki-67 (Fig. 3, II, b) and human PSA (Fig. 3, II, c) staining, demonstrating that the tumors formed in mice were of human origin.
Fig. 3. Arsenic-transformed prostate epithelial cells form tumors in vivo. I) Cell lines were injected subcutaneously into the flanks of SCID mice, dimensions of the tumors were measured weekly, and tumor volume was calculated. Results are shown as means ± s.e.m. Representative animals are shown in the photos. II) Representative slides of sections of xenograft tumors (a) stained with hematoxylin and eosin, (b) stained with Ki-67, and (c) stained with human prostate-specific antigen PSA are presented.
Arsenic induces distinct changes in subunit gene expression involved in mtOXPHOS complex I. We measured changes in subunit gene expression comprising all five mtOXPHOS complexes. An analysis of mtOXPHOS gene expression shows that complex I subunit NDUFB8 was expressed at a much lower level in arsenic-transformed cells than in matched untreated control cells (Fig. 4, a and b). In contrast, expression of complex V subunit ATP5A, complex III subunit UQCRC2, complex IV subunit COX II, and complex II subunit SDHB was higher in arsenic-transformed cells (Fig. 4, a and b). Since complex I appeared to be severely affected, we analyzed the expression of NDUFA13 (also known as GRIM-19) and found that arsenic-transformed cells express significantly lower level of NDUFA13/GRIM-19 compared to non-treated paired control cells (Fig. 4c). This study suggests that arsenic targets complex I function.
Fig. 4. Arsenic alters gene expression. a) Western blot analysis of representative subunits of mitochondrial respiratory complexes I-V in RWPE-1 and CAsPE cells. A Ponceau S-stained blot serves as a loading control. b) Densitometric analysis of protein expression of subunits of mitochondrial respiratory complexes I-V in RWPE-1 and CAsPE cells is represented as a histogram. RNA was isolated from both, RWPE-1 prostate epithelial cells and arsenic-transformed CAsPE cells, and mRNA expression of NDUFA13 (c) and members of NOX family (d) was determined by RT-PCR after cDNA synthesis. β-Actin served as control.
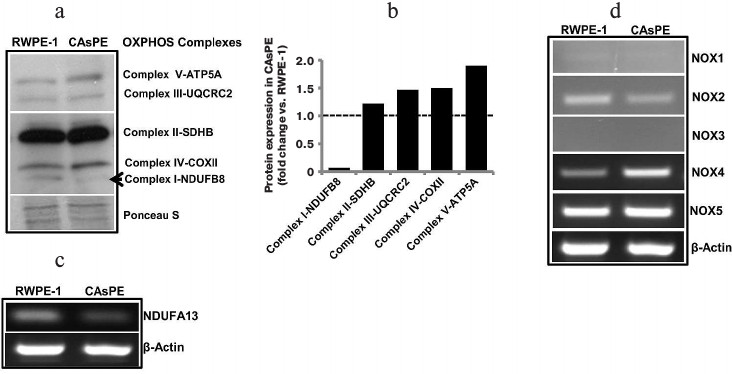
Arsenic exposure leads to increased expression of ROS-producing genes. Oxidative stress is known to be involved in malignant transformation of cells [54-57], and the NOX family of reactive oxygen species (ROS)-generating NADPH oxidases is one of the main ROS-producing enzymes in cells [58]. NOX enzymes transport electrons across the plasma membrane to generate superoxide and downstream ROS [58]. We analyzed expression of NOX genes (NOX1, NOX2, NOX3, NOX4, and NOX5) in arsenic-transformed CAsPE cells. mRNA expression of NOX4 was significantly increased in CAsPE cells compared to RWPE-1 cells (Fig. 4d). In contrast, expression of NOX1 and NOX3 was not detected, and expression of NOX2 was somewhat lower in CAsPE cells. However, no change was detected in NOX5 expression (Fig. 4d). We conclude that NOX4-induced oxidative stress plays an important role in arsenic-induced transformation. It is noteworthy that NOX4 localizes to mitochondria [59].
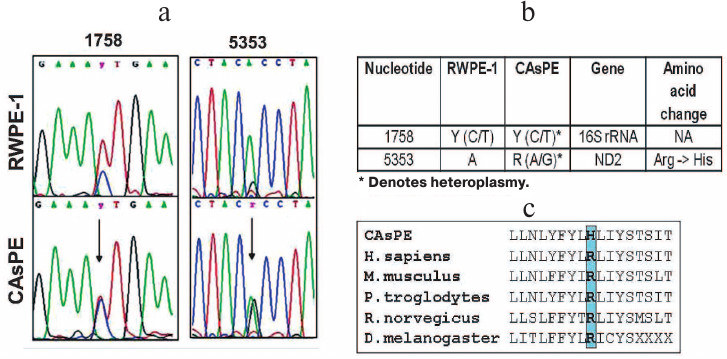
Arsenic induces mitochondrial genome instability. Since arsenic affected mitochondria, localized oxidative stress may damage the mitochondrial genome, and mitochondrial genomic alteration has been shown to be involved in the development of tumors [60, 61]. We designed primers to amplify the entire mitochondrial genome and sequenced the PCR products. Our study revealed mutations in the 16S rRNA gene (1758 bp) and ND2 (5353 bp) gene in arsenic-transformed CAsPE cells when compared with the RWPE-1 untreated control cells (Fig. 5, a and b). Interestingly, both of these mutations were found to be heteroplasmic, i.e. the CAsPE cells contained both wild type and mutant mtDNA (Fig. 5a). The ND2 gene mutation changes amino acid arginine to histidine, which is conserved across species (Fig. 5c). ND2 encodes a protein that is part of the multiprotein mtOXPHOS complex I. Mutations in the 16S rRNA gene do not lead to amino acid change (Fig. 5b). Together these observations suggest that arsenic induces mutation in the mitochondrial genome, and mtOXPHOS complex I is a sustained target of arsenic-induced transformation of prostate epithelial cells.
Fig. 5. Arsenic induces mutations in mtDNA. mtDNA was isolated from RWPE-1 and CAsPE cells. The whole mitochondrial genome (16.6 kb) was amplified and sequenced with 24 sets of primers as described earlier [51]. a) Two novel heteroplasmic mutations were identified in CAsPE cells at a higher percentage than in RWPE-1 cells. b) One of the mutations was found in 16S rRNA and another one in the ND2 gene. c) The ND2 gene mutation changes the conserved amino acid arginine to histidine.
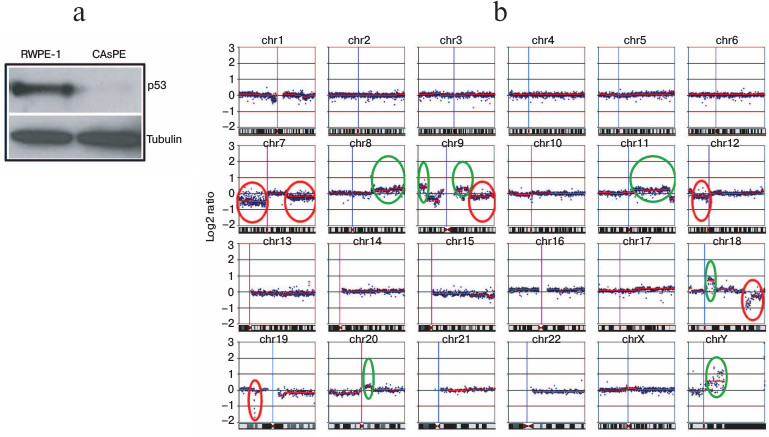
Arsenic inhibits p53 expression and induces nuclear genomic instability. p53 is a well-known tumor suppressor gene. It is described as guardian of the nuclear as well as the mitochondrial genome [62, 63]. Loss of expression of tumor suppressor gene p53 is frequently associated with nuclear genome instability and malignant transformation of cells [64-66]. We tested the expression of p53 in arsenic-transformed and arsenic-untreated non-tumorigenic cells. We noticed a severe loss of p53 expression in the arsenic-transformed cells (Fig. 6a).
To compare the differences in DNA aberrations between arsenic-transformed and arsenic-unexposed cells, we performed whole genome cGH array analysis. Nuclear genomic changes were detected in arsenic-transformed cells compared to arsenic-untreated non-tumorigenic cells. Arsenic-transformed cells had significant changes compared to arsenic-unexposed cells (Fig. 6b). We identified amplification (circled in green) and deletion (circled in red) of chromosomal regions in arsenic-transformed cells (Fig. 6b). The most frequent DNA gains, which were strikingly different in arsenic-exposed cells compared with arsenic-unexposed cells, included those at loci in chromosome 8q, 9p, 9q, 11q, 18q, 20q, and chromosome Yq (Fig. 6b). A higher frequency of DNA losses in arsenic-transformed cells compared with arsenic-unexposed cells was also observed in several chromosomes such as loci at 7p, 7q, 9q, 12p, 18q, and 19p (Fig. 6b).
We also performed cGH analysis of arsenic-transformed breast epithelial cells. Interestingly, we observed amplification of only a single locus at chromosome 5 (unpublished data). These data suggest that arsenic exposure leads to genomic instability. However, arsenic-mediated genomic changes (chromosomal amplification or deletion) may be cell-type specific.
Fig. 6. Arsenic induces nuclear genomic instability. a) Western blot analysis of p53 protein expression in RWPE-1 and CAsPE cells. b) Comparative genome hybridization (cGH) array analysis of RWPE-1 and CAsPE cells treated with arsenic for seven months was carried out. DNA aberrations are denoted for each chromosome. Significant amplifications of chromosomal regions are circled in green, and deletions of chromosomal regions are circled in red.
DISCUSSION
Otto Warburg described altered mtOXPHOS in the mitochondria of tumor cells and hypothesized that decreased mtOXPHOS in cancer cells leads to increased glycolytic energy production [67]. Decreased mitochondrial oxidative phosphorylation (mtOXPHOS) has also been identified as a new hallmark of cancer [67-71]. We utilized a prostate epithelial cell culture model to identify effects of arsenic on mitochondria and a role for arsenic-induced disruption of mitochondrial function in the development of prostate cancer.
Mitochondria are dynamic organelles in a cell. Mitochondria undergo constant fission and fusion processes and maintain their morphology, membrane potential, and number in a cellular system [72, 73]. Any change in fission and fusion processes and thus change in morphology of mitochondria in a cell is an important indicator of physiological changes in this organelle. Aberrant mitochondrial morphology and membrane potential in RWPE-1 cells within a few hours of arsenic exposure not only indicate physiological changes in mitochondria in these cells after arsenic exposure but also support the notion that arsenic induces mitochondrial changes at an early stage (Fig. 1a). Mitochondrial morphological changes have been attributed to development of cancer and a number of other human disorders [74]. Decreased mitochondrial membrane potential (Fig. 1b) as well as altered expression of mtOXPHOS complex subunit proteins (Fig. 4a) in arsenic-transformed CAsPE prostate epithelial cells further supports the conclusion that arsenic targets mitochondria.
Increased Matrigel invasion, wound healing, and growth rate of arsenic-exposed prostate epithelial cells provide in vitro evidences for the transformation of these cells after seven-month arsenic treatment (Fig. 2). Formation of tumors in SCID mice confirms that arsenic treatment of prostate epithelial cells results in development of tumors in vivo (Fig. 3). These studies are consistent with previous reports describing tumorigenic transformation of prostate epithelial cell by arsenic exposure [35, 46]. Although these studies identified arsenic-induced transformation of prostate epithelial cells, identification of the mechanism underlying arsenic-induced transformation of the cells is not clear. Mitochondria play a key role in cellular metabolism, cell death, and reactive oxygen species production, and these pathways are often disrupted in tumors [49, 75]. We focused our efforts to identify the changes and consequences of altered mitochondrial functions in arsenic-induced transformation of prostate epithelial cells.
Exposure to arsenic leads to generation of ROS, resulting in increased oxidative stress [76-78]. Reactive oxygen species produced during metabolism has been shown to cause DNA damage in the nucleus [42, 79-81] as well as in mitochondria [43, 80]. Mitochondrial OXPHOS complex I is a major site for ROS production in cells [82]. Dysfunctional complex I was reported to increase ROS production [82]. Uncontrolled generation of ROS leads to oxidative stress and plays a causative role in the development of cancer [54-57]. Decreased expression of complex I subunits NDUFB8 (Fig. 4, a and b) and NDUFA13/GRIM-19 (Fig. 4c) indicates dysfunctional mtOXPHOS complex I. NDUFA13/GRIM-19 is essential for the assembly and function of mtOXPHOS complex I [83]. Decreased expression of the nuclear-genome-encoded mtOXPHOS complex I subunits as well as induction of mutation in mitochondrial-genome-encoded ND2 gene that code a protein subunit of mtOXPHOS complex I (Fig. 5) indicate that complex I is a prime mitochondrial target of arsenic. Moreover, increased expression of mitochondria-localized NOX4 [59], an oncogene involved in generation of ROS, also supports the role of mitochondria and associated oxidative stress in arsenic-mediated oncogenic transformation of prostate epithelial cells.
Inhibition of tumor suppressor gene(s) or amplification of oncogene(s) are critical events that lead to the transformation of cells and cancer development [84]. Recently, Ngalame et al. reported that arsenic induces overexpression of oncogene KRAS through regulation of microRNAs, which plays a key role in arsenic-induced malignant phenotype [85, 86]. Notably, mitochondrial impairment-induced ROS generation has been reported as an essential factor for KRAS-mediated tumorigenicity [87]. These studies indicate that mitochondria might play an important role in regulation of oncogenes and tumorigenesis. Our observation of significant loss of p53 in arsenic-transformed prostate epithelial cells indicates a role of this protein in neoplastic transformation after arsenic treatment (Fig. 6a). p53 is a tumor suppressor gene that regulates cell cycle, apoptosis, and genomic stability [62, 65]. We have earlier shown that p53 functions as a mito-checkpoint protein and regulates mtDNA copy number and mitochondrial biogenesis [88]. The mito-checkpoint can transiently block cell cycle progression when cells contain damaged/dysfunctional mitochondria. If the mito-checkpoint fails, cells may accumulate genetic abnormalities in the nucleus.
Our group has demonstrated that mitochondrial loss-of-function in breast epithelial cells induces an increase in chromosomal rearrangements resulting in transformation of the cells [89]. We also demonstrated that mitochondrial dysfunction downregulates p53 and induces supernumerary centrosome amplification, and direct inhibition of mtOXPHOS complex I in cells recapitulate this abnormal phenotype, suggesting a role for mtOXPHOS and specifically complex I in maintaining centrosomal homeostasis [90]. It is therefore conceivable that on downregulation of p53, a mito-checkpoint protein triggers nuclear genomic instability due to arsenic-induced disruption of mitochondrial function [88, 91]. Other mechanisms of arsenic-induced nuclear genome instability have also been identified. Arsenic targets the RING finger domains of histone E3 ubiquitin ligase and alters a histone epigenetic mark [92]. Arsenic also inhibits recruitment of BRCA1 and RAD51 proteins to DNA double-strand break sites that diminish DNA double-strand break repair, which ultimately leads to nuclear genome instability [92].
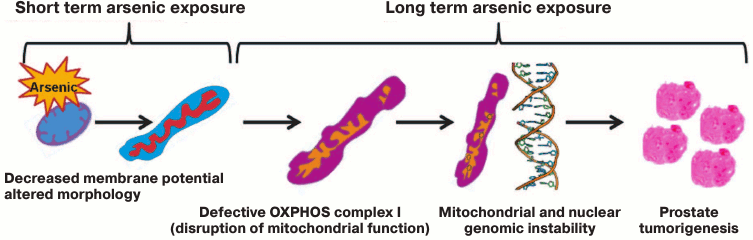
Cellular transformation is a multistage process, and genomic instability is one of the prerequisites for transformation/cancer development [93]. Prevalent mutations in mtDNA in arsenic-treated cells indicate that mitochondria are early and important targets of arsenic. Indeed, we have previously shown that mutations in mtDNA increase the frequency of mutations in the nuclear genome [94, 95]. Thus, mitochondrial genome instability that we demonstrated upon arsenic treatment may push cells towards nuclear genome instability leading to tumorigenic transformation. Decreased expression of p53, a guardian of the nuclear and mitochondrial genome [62, 63], and increased expression of NOX4, a ROS-producing oncogene that also localizes to mitochondria [59], may further exacerbate the mitochondria-mediated effects on nuclear genomic instability. Our cGH array analyses with arsenic-exposed cells indicating widespread chromosomal amplification as well as deletion across the nuclear genome confirm increase in nuclear genome instability in arsenic-exposed cells (Fig. 6b). Overall, our study provides evidences that arsenic exposure alters mtOXPHOS complex I and disrupts mitochondrial function that contributes to tumorigenic transformation of prostate epithelial cells (Fig. 7).
Fig. 7. Mitochondrial model for arsenic-induced prostate carcinogenesis. Our results indicate that arsenic alters mitochondrial function of prostate epithelial cells as early as 8 h after exposure. Inhibition of complex I of mitochondrial OXPHOS is a prime target of arsenic toxicity. Arsenic inhibits expression of subunits of OXPHOS complex I including expression of GRIM-19 (NDUFA13), a cell death regulatory protein that is also essential for the assembly and function of OXPHOS complex I. Moreover, arsenic induces mutations in the ND2 gene, a subunit of complex I. Altered complex I function as well as increased expression of NOX4 are responsible for generation of ROS, and thus oxidative stress in arsenic-exposed cells play a direct or indirect role in nuclear DNA damage and genomic instability. p53, a guardian of the nuclear genome, also acts as a mito-checkpoint and can transiently block cell cycle progression when cells contain damaged/dysfunctional mitochondria [62, 88]. Therefore, absence or decreased expression of p53 further aggravates both nuclear and mitochondrial genome instability. Both p53 and NOX4 protein also localize and function in mitochondria [59, 88]. Thus, dysfunctional mitochondria via known pathways/proteins like p53 and NOX4 and other yet unknown pathways exacerbate nuclear genomic instability, a prerequisite for transformation of cells and tumorigenesis. Our model suggests that mitochondria are the early target for arsenic, and disruption of mitochondrial function plays a role in arsenic-induced prostate tumorigenesis.
Acknowledgements
We are highly indebted to Dr. Michael P. Waalkes, NIEHS, Research Triangle Park, North Carolina for providing RWPE-1 and CAsPE cells used in this study. We thank Marija Vujcic, Hongjun Wei, Drs. Changchun Ren and Ayyasamy Vanniarajan for their assistance.
This study was supported by grant from Veterans Administration 1I01BX001716.
REFERENCES
1.Banerjee, M., Sarkar, J., Das, J. K., Mukherjee,
A., Sarkar, A. K., Mondal, L., and Giri, A. K. (2007) Polymorphism in
the ERCC2 codon 751 is associated with arsenic-induced premalignant
hyperkeratosis and significant chromosome aberrations,
Carcinogenesis, 28, 672-676.
2.De Chaudhuri, S., Mahata, J., Das, J. K.,
Mukherjee, A., Ghosh, P., Sau, T. J., Mondal, L., Basu, S., Giri, A.
K., and Roychoudhury, S. (2006) Association of specific p53
polymorphisms with keratosis in individuals exposed to arsenic through
drinking water in West Bengal, India, Mutat. Res., 601,
102-112.
3.Frazer, L. (2005) Metal attraction: an ironclad
solution to arsenic contamination? Environ. Health Perspect.,
113, A398-401.
4.Pearce, F. (2003) Arsenic’s fatal legacy
grows worldwide, New Scientist, 179, 1-3.
5.Ravenscroft, P., Brammer, H., and Richards, K.
(2009) Arsenic Pollution: A Global Synthesis, Wiley-Blackwell,
Oxford.
6.Mandal, B. K., Chowdhury, T. R., Samanta, G., Basu,
G. K. Chowdhury, P. P., Chanda, C. R., Lodh, D., Karan, N. K., Dhar, R.
K., Tamili, D., Das, D., Saha, K. C., and Chakraborti, C. (1996)
Arsenic in groundwater in seven districts of West Bengal, India –
the biggest arsenic calamity in the world, Curr. Sci.,
70, 976-986.
7.Davey, J. C., Bodwell, J. E., Gosse, J. A., and
Hamilton, J. W. (2007) Arsenic as an endocrine disruptor: effects of
arsenic on estrogen receptor-mediated gene expression in vivo
and in cell culture, Toxicol. Sci., 98, 75-86.
8.US Environmental Protection Agency (2010) Arsenic
in drinking water
(http://water.epa.gov/lawsregs/rulesregs/sdwa/arsenic/index.cfm).
9.Camacho, L. M., Gutierrez, M., Alarcon-Herrera, M.
T., Villalba Mde, L., and Deng, S. (2011) Occurrence and treatment of
arsenic in groundwater and soil in northern Mexico and southwestern
USA, Chemosphere, 83, 211-225.
10.Bulka, C. M., Jones, R. M., Turyk, M. E.,
Stayner, L. T., and Argos, M. (2016) Arsenic in drinking water and
prostate cancer in Illinois counties: An ecologic study, Environ.
Res., 148, 450-456.
11.Gundert-Remy, U., Damm, G., Foth, H., Freyberger,
A., Gebel, T., Golka, K., Rohl, C., Schupp, T., Wollin, K. M., and
Hengstler, J. G. (2015) High exposure to inorganic arsenic by food: the
need for risk reduction, Arch. Toxicol., 89,
2219-2227.
12.Mathews, V. V., Paul, M. V., Abhilash, M., Manju,
A., Abhilash, S., and Nair, R. H. (2013) Myocardial toxicity of acute
promyelocytic leukemia drug-arsenic trioxide, Eur. Rev. Med.
Pharmacol. Sci., 17 (Suppl. 1), 34-38.
13.Shibata, T., Meng, C., Umoren, J., and West, H.
(2016) Risk assessment of arsenic in rice cereal and other dietary
sources for infants and toddlers in the U.S., Int. J. Environ. Res.
Public Health, 13, pii: E361.
14.Wilson, D. (2015) Arsenic content in American
wine, J. Environ. Health, 78, 16-22.
15.Wilson, D. (2015) Arsenic consumption in the
United States, J. Environ. Health, 78, 8-14, quiz 44.
16.Wilson, D., Hooper, C., and Shi, X. (2012)
Arsenic and lead in juice: apple, citrus, and apple-base, J.
Environ. Health, 75, 14-20, quiz 44.
17.World Health Organization (2010) Exposure to
arsenic: a major public health concern
(http://www.who.int/ipcs/features/arsenic.pdf).
18.Tchounwou, P. B., Patlolla, A. K., and Centeno,
J. A. (2003) Carcinogenic and systemic health effects associated with
arsenic exposure – a critical review, Toxicol. Pathol.,
31, 575-588.
19.Chappell, W. R., Beck, B. D., Brown, K. G.,
Chaney, R., Cothern, R., Cothern, C. R., Irgolic, K. J., North, D. W.,
Thornton, I., and Tsongas, T. A. (1997) Inorganic arsenic: a need and
an opportunity to improve risk assessment, Environ. Health
Perspect., 105, 1060-1067.
20.Dantzig, P. I. (2009) Breast cancer,
dermatofibromas and arsenic, Indian J. Dermatol., 54,
23-25.
21.Karagas, M. R., Gossai, A., Pierce, B., and
Ahsan, H. (2015) Drinking water arsenic contamination, skin lesions,
and malignancies: a systematic review of the global evidence, Curr.
Environ. Health Rep., 2, 52-68.
22.Saint-Jacques, N., Parker, L., Brown, P., and
Dummer, T. J. (2014) Arsenic in drinking water and urinary tract
cancers: a systematic review of 30 years of epidemiological evidence,
Environ. Health, 13, 44.
23.Steinmaus, C., Ferreccio, C., Yuan, Y., Acevedo,
J., Gonzalez, F., Perez, L., Cortes, S., Balmes, J. R., Liaw, J., and
Smith, A. H. (2014) Elevated lung cancer in younger adults and low
concentrations of arsenic in water, Am. J. Epidemiol.,
180, 1082-1087.
24.Wang, W., Cheng, S., and Zhang, D. (2014)
Association of inorganic arsenic exposure with liver cancer mortality:
a meta-analysis, Environ. Res., 135, 120-125.
25.Benbrahim-Tallaa, L., and Waalkes, M. P. (2008)
Inorganic arsenic and human prostate cancer, Environ. Health
Perspect., 116, 158-164.
26.Chen, C. J., Kuo, T. L., and Wu, M. M. (1988)
Arsenic and cancers, Lancet, 1, 414-415.
27.Chen, C. J., and Wang, C. J. (1990) Ecological
correlation between arsenic level in well water and age-adjusted
mortality from malignant neoplasms, Cancer Res., 50,
5470-5474.
28.Lewis, D. R., Southwick, J. W.,
Ouellet-Hellstrom, R., Rench, J., and Calderon, R. L. (1999) Drinking
water arsenic in Utah: a cohort mortality study, Environ. Health
Perspect., 107, 359-365.
29.Wu, M. M., Kuo, T. L., Hwang, Y. H., and Chen, C.
J. (1989) Dose–response relation between arsenic concentration in
well water and mortality from cancers and vascular diseases, Am. J.
Epidemiol., 130, 1123-1132.
30.Garcia-Esquinas, E., Pollan, M., Umans, J. G.,
Francesconi, K. A., Goessler, W., Guallar, E., Howard, B., Farley, J.,
Best, L. G., and Navas-Acien, A. (2013) Arsenic exposure and cancer
mortality in a US-based prospective cohort: the strong heart study,
Cancer Epidemiol. Biomarkers Prev., 22, 1944-1953.
31.Shi, H., Shi, X., and Liu, K. J. (2004) Oxidative
mechanism of arsenic toxicity and carcinogenesis, Mol. Cell.
Biochem., 255, 67-78.
32.Smith, A. H., Goycolea, M., Haque, R., and Biggs,
M. L. (1998) Marked increase in bladder and lung cancer mortality in a
region of Northern Chile due to arsenic in drinking water, Am. J.
Epidemiol., 147, 660-669.
33.Waalkes, M. P., Ward, J. M., Liu, J., and Diwan,
B. A. (2003) Transplacental carcinogenicity of inorganic arsenic in the
drinking water: induction of hepatic, ovarian, pulmonary, and adrenal
tumors in mice, Toxicol. Appl. Pharmacol., 186, 7-17.
34.Wen, G., Calaf, G. M., Partridge, M. A.,
Echiburu-Chau, C., Zhao, Y., Huang, S., Chai, Y., Li, B., Hu, B., and
Hei, T. K. (2008) Neoplastic transformation of human small airway
epithelial cells induced by arsenic, Mol. Med., 14,
2-10.
35.Achanzar, W. E., Brambila, E. M., Diwan, B. A.,
Webber, M. M., and Waalkes, M. P. (2002) Inorganic arsenite-induced
malignant transformation of human prostate epithelial cells, J.
Natl. Cancer Inst., 94, 1888-1891.
36.Benbrahim-Tallaa, L., Webber, M. M., and Waalkes,
M. P. (2005) Acquisition of androgen independence by human prostate
epithelial cells during arsenic-induced malignant transformation,
Environ. Health Perspect., 113, 1134-1139.
37.Indiveri, C., Capobianco, L., Kramer, R., and
Palmieri, F. (1989) Kinetics of the reconstituted dicarboxylate carrier
from rat liver mitochondria, Biochim. Biophys. Acta, 977,
187-193.
38.Wohlrab, H. (1986) Molecular aspects of inorganic
phosphate transport in mitochondria, Biochim. Biophys. Acta,
853, 115-134.
39.Vahter, M., and Marafante, E. (1989)
Intracellular distribution and chemical forms of arsenic in rabbits
exposed to arsenate, Biol. Trace Elem. Res., 21,
233-239.
40.Chilakapati, J., Wallace, K., Ren, H., Fricke,
M., Bailey, K., Ward, W., Creed, J., and Kitchin, K. (2010) Genome-wide
analysis of BEAS-2B cells exposed to trivalent arsenicals and
dimethylthioarsinic acid, Toxicology, 268, 31-39.
41.Ghosh, P., Basu, A., Singh, K. K., and Giri, A.
K. (2008) Evaluation of cell types for assessment of cytogenetic damage
in arsenic exposed population, Mol. Cancer, 7, 45.
42.Hei, T. K., Liu, S. X., and Waldren, C. (1998)
Mutagenicity of arsenic in mammalian cells: role of reactive oxygen
species, Proc. Natl. Acad. Sci. USA, 95, 8103-8107.
43.Liu, S. X., Davidson, M. M., Tang, X., Walker, W.
F., Athar, M., Ivanov, V., and Hei, T. K. (2005) Mitochondrial damage
mediates genotoxicity of arsenic in mammalian cells, Cancer
Res., 65, 3236-3242.
44.Pi, J., He, Y., Bortner, C., Huang, J., Liu, J.,
Zhou, T., Qu, W., North, S. L., Kasprzak, K. S., Diwan, B. A.,
Chignell, C. F., and Waalkes, M. P. (2005) Low level, long-term
inorganic arsenite exposure causes generalized resistance to apoptosis
in cultured human keratinocytes: potential role in skin
co-carcinogenesis, Int. J. Cancer, 116, 20-26.
45.Singh, K. K., and Vujcic, M. (2007) Genetic
mechanisms of arsenic induced carcinogenesis, in Natural Arsenic in
Ground Waters-Health Impact, Remediation and Management (Bundschuh,
J., Bhattacharya, P., Armienta, M. A., Matschullat, J., and Garcia, M.
E., eds.) Taylor and Francis Group, N.Y., pp. 427-435.
46.Tokar, E. J., Diwan, B. A., and Waalkes, M. P.
(2010) Arsenic exposure transforms human epithelial stem/progenitor
cells into a cancer stem-like phenotype, Environ. Health
Perspect., 118, 108-115.
47.Vujcic, M., Shroff, M., and Singh, K. K. (2007)
Genetic determinants of mitochondrial response to arsenic in yeast
Saccharomyces cerevisiae, Cancer Res., 67,
9740-9749.
48.Singh, B., Li, X., Owens, K. M., Vanniarajan, A.,
Liang, P., and Singh, K. K. (2015) Human REV3 DNA polymerase zeta
localizes to mitochondria and protects the mitochondrial genome,
PLoS One, 10, e0140409.
49.Ayyasamy, V., Owens, K. M., Desouki, M. M.,
Liang, P., Bakin, A., Thangaraj, K., Buchsbaum, D. J., LoBuglio, A. F.,
and Singh, K. K. (2011) Cellular model of Warburg effect identifies
tumor promoting function of UCP2 in breast cancer and its suppression
by genipin, PLoS One, 6, e24792.
50.Singh, B., Owens, K. M., Bajpai, P., Desouki, M.
M., Srinivasasainagendra, V., Tiwari, H. K., and Singh, K. K. (2015)
Mitochondrial DNA polymerase POLG1 disease mutations and germline
variants promote tumorigenic properties, PLoS One, 10,
e0139846.
51.Rieder, M. J., Taylor, S. L., Tobe, V. O., and
Nickerson, D. A. (1998) Automating the identification of DNA variations
using quality-based fluorescence re-sequencing: analysis of the human
mitochondrial genome, Nucleic Acids Res., 26,
967-973.
52.De Biasi, S., Gibellini, L., and Cossarizza, A.
(2015) Uncompensated polychromatic analysis of mitochondrial membrane
potential using JC-1 and multilaser excitation, Curr. Protoc.
Cytom., 72, 1-11.
53.Cowell, J. K., Wang, Y. D., Head, K., Conroy, J.,
McQuaid, D., and Nowak, N. J. (2004) Identification and
characterization of constitutional chromosome abnormalities using
arrays of bacterial artificial chromosomes, Br. J. Cancer,
90, 860-865.
54.Iwabuchi, T., Yoshimoto, C., Shigetomi, H., and
Kobayashi, H. (2015) Oxidative stress and antioxidant defense in
endometriosis and its malignant transformation, Oxid. Med. Cell.
Longev., 2015, 848595.
55.Khandrika, L., Kumar, B., Koul, S., Maroni, P.,
and Koul, H. K. (2009) Oxidative stress in prostate cancer, Cancer
Lett., 282, 125-136.
56.Mahalingaiah, P. K., Ponnusamy, L., and Singh, K.
P. (2015) Chronic oxidative stress leads to malignant transformation
along with acquisition of stem cell characteristics, and epithelial to
mesenchymal transition in human renal epithelial cells, J. Cell.
Physiol., 230, 1916-1928.
57.Reuter, S., Gupta, S. C., Chaturvedi, M. M., and
Aggarwal, B. B. (2010) Oxidative stress, inflammation, and cancer: how
are they linked? Free Radic. Biol. Med., 49,
1603-1616.
58.Bedard, K., and Krause, K. H. (2007) The NOX
family of ROS-generating NADPH oxidases: physiology and
pathophysiology, Physiol. Rev., 87, 245-313.
59.Graham, K. A., Kulawiec, M., Owens, K. M., Li,
X., Desouki, M. M., Chandra, D., and Singh, K. K. (2010) NADPH oxidase
4 is an oncoprotein localized to mitochondria, Cancer Biol.
Ther., 10, 223-231.
60.Kulawiec, M., Owens, K. M., and Singh, K. K.
(2009) Cancer cell mitochondria confer apoptosis resistance and promote
metastasis, Cancer Biol. Ther., 8, 1378-1385.
61.Kulawiec, M., Owens, K. M., and Singh, K. K.
(2009) mtDNA G10398A variant in African-American women with breast
cancer provides resistance to apoptosis and promotes metastasis in
mice, J. Hum. Genet., 54, 647-654.
62.Lane, D. P. (1992) Cancer. p53, guardian of the
genome, Nature, 358, 15-16.
63.Park, J. H., Zhuang, J., Li, J., and Hwang, P. M.
(2016) p53 as guardian of the mitochondrial genome, FEBS Lett.,
590, 924-934.
64.Agapova, L. S., Ilyinskaya, G. V., Turovets, N.
A., Ivanov, A. V., Chumakov, P. M., and Kopnin, B. P. (1996) Chromosome
changes caused by alterations of p53 expression, Mutat. Res.,
354, 129-138.
65.Kirsch, D. G., and Kastan, M. B. (1998)
Tumor-suppressor p53: implications for tumor development and prognosis,
J. Clin. Oncol., 16, 3158-3168.
66.Van Oijen, M. G., and Slootweg, P. J. (2000)
Gain-of-function mutations in the tumor suppressor gene p53,
Clin. Cancer Res., 6, 2138-2145.
67.Warburg, O. (1956) On respiratory impairment in
cancer cells, Science, 124, 269-270.
68.Alberio, S., Mineri, R., Tiranti, V., and
Zeviani, M. (2007) Depletion of mtDNA: syndromes and genes,
Mitochondrion, 7, 6-12.
69.Hanahan, D., and Weinberg, R. A. (2011) Hallmarks
of cancer: the next generation, Cell, 144, 646-674.
70.Modica-Napolitano, J. S., Kulawiec, M., and
Singh, K. K. (2007) Mitochondria and human cancer, Curr. Mol.
Med., 7, 121-131.
71.Pedersen, P. L. (2007) Warburg, me and hexokinase
2: multiple discoveries of key molecular events underlying one of
cancers’ most common phenotypes, the “Warburg
Effect”, i.e. elevated glycolysis in the presence of oxygen,
J. Bioenerg. Biomembr., 39, 211-222.
72.Legros, F., Lombes, A., Frachon, P., and Rojo, M.
(2002) Mitochondrial fusion in human cells is efficient, requires the
inner membrane potential, and is mediated by mitofusins, Mol. Biol.
Cell, 13, 4343-4354.
73.Westrate, L. M., Drocco, J. A., Martin, K. R.,
Hlavacek, W. S., and MacKeigan, J. P. (2014) Mitochondrial
morphological features are associated with fission and fusion events,
PLoS One, 9, e95265.
74.Wallace, D. C. (2005) A mitochondrial paradigm of
metabolic and degenerative diseases, aging, and cancer: a dawn for
evolutionary medicine, Annu. Rev. Genet., 39,
359-407.
75.Wallace, D. C. (2005) Mitochondria and cancer:
Warburg addressed, Cold Spring Harb. Symp. Quant. Biol.,
70, 363-374.
76.Huang, C., Ke, Q., Costa, M., and Shi, X. (2004)
Molecular mechanisms of arsenic carcinogenesis, Mol. Cell.
Biochem., 255, 57-66.
77.Rossman, T. G. (2003) Mechanism of arsenic
carcinogenesis: an integrated approach, Mutat. Res., 533,
37-65.
78.Tchounwou, P. B., Centeno, J. A., and Patlolla,
A. K. (2004) Arsenic toxicity, mutagenesis, and carcinogenesis –
a health risk assessment and management approach, Mol. Cell.
Biochem., 255, 47-55.
79.Liu, S. X., Athar, M., Lippai, I., Waldren, C.,
and Hei, T. K. (2001) Induction of oxy-radicals by arsenic: implication
for mechanism of genotoxicity, Proc. Natl. Acad. Sci. USA,
98, 1643-1648.
80.Singh, K. P., Kumari, R., Treas, J., and DuMond,
J. W. (2011) Chronic exposure to arsenic causes increased cell
survival, DNA damage, and increased expression of mitochondrial
transcription factor A (mtTFA) in human prostate epithelial cells,
Chem. Res. Toxicol., 24, 340-349.
81.Yamanaka, K., Kato, K., Mizoi, M., An, Y.,
Takabayashi, F., Nakano, M., Hoshino, M., and Okada, S. (2004) The role
of active arsenic species produced by metabolic reduction of
dimethylarsinic acid in genotoxicity and tumorigenesis, Toxicol.
Appl. Pharmacol., 198, 385-393.
82.Koopman, W. J., Nijtmans, L. G., Dieteren, C. E.,
Roestenberg, P., Valsecchi, F., Smeitink, J. A., and Willems, P. H.
(2010) Mammalian mitochondrial complex I: biogenesis, regulation, and
reactive oxygen species generation, Antioxid. Redox Signal.,
12, 1431-1470.
83.Huang, G., Lu, H., Hao, A., Ng, D. C., Ponniah,
S., Guo, K., Lufei, C., Zeng, Q., and Cao, X. (2004) GRIM-19, a cell
death regulatory protein, is essential for assembly and function of
mitochondrial complex I, Mol. Cell. Biol., 24,
8447-8456.
84.Polsky, D., and Cordon-Cardo, C. (2003) Oncogenes
in melanoma, Oncogene, 22, 3087-3091.
85.Ngalame, N. N., Tokar, E. J., Person, R. J., and
Waalkes, M. P. (2014) Silencing KRAS overexpression in
arsenic-transformed prostate epithelial and stem cells partially
mitigates malignant phenotype, Toxicol. Sci., 142,
489-496.
86.Ngalame, N. N., Tokar, E. J., Person, R. J., Xu,
Y., and Waalkes, M. P. (2014) Aberrant microRNA expression likely
controls ras oncogene activation during malignant transformation
of human prostate epithelial and stem cells by arsenic, Toxicol.
Sci., 138, 268-277.
87.Weinberg, F., Hamanaka, R., Wheaton, W. W.,
Weinberg, S., Joseph, J., Lopez, M., Kalyanaraman, B., Mutlu, G. M.,
Budinger, G. R., and Chandel, N. S. (2010) Mitochondrial metabolism and
ROS generation are essential for Kras-mediated tumorigenicity, Proc.
Natl. Acad. Sci. USA, 107, 8788-8793.
88.Kulawiec, M., Ayyasamy, V., and Singh, K. K.
(2009) p53 regulates mtDNA copy number and mito-checkpoint pathway,
J. Carcinog., 8, 8.
89.Kulawiec, M., Safina, A., Desouki, M. M., Still,
I., Matsui, S., Bakin, A., and Singh, K. K. (2008) Tumorigenic
transformation of human breast epithelial cells induced by
mitochondrial DNA depletion, Cancer Biol. Ther., 7,
1732-1743.
90.Donthamsetty, S., Brahmbhatt, M., Pannu, V.,
Rida, P. C., Ramarathinam, S., Ogden, A., Cheng, A., Singh, K. K., and
Aneja, R. (2014) Mitochondrial genome regulates mitotic fidelity by
maintaining centrosomal homeostasis, Cell Cycle, 13,
2056-2063.
91.Minocherhomji, S., Tollefsbol, T. O., and Singh,
K. K. (2012) Mitochondrial regulation of epigenetics and its role in
human diseases, Epigenetics, 7, 326-334.
92.Zhang, F., Paramasivam, M., Cai, Q., Dai, X.,
Wang, P., Lin, K., Song, J., Seidman, M. M., and Wang, Y. (2014)
Arsenite binds to the RING finger domains of RNF20-RNF40 histone E3
ubiquitin ligase and inhibits DNA double-strand break repair, J. Am.
Chem. Soc., 136, 12884-12887.
93.Kennedy, S. R., Loeb, L. A., and Herr, A. J.
(2012) Somatic mutations in aging, cancer and neurodegeneration,
Mech. Ageing Dev., 133, 118-126.
94.Delsite, R. L., Rasmussen, L. J., Rasmussen, A.
K., Kalen, A., Goswami, P. C., and Singh, K. K. (2003) Mitochondrial
impairment is accompanied by impaired oxidative DNA repair in the
nucleus, Mutagenesis, 18, 497-503.
95.Rasmussen, A. K., Chatterjee, A., Rasmussen, L.
J., and Singh, K. K. (2003) Mitochondria-mediated nuclear mutator
phenotype in Saccharomyces cerevisiae, Nucleic Acids
Res., 31, 3909-3917.