REVIEW: Inactivation of Inflammasomes by Pathogens Regulates Inflammation
F. Yu. Garib1,2,3*, A. P. Rizopulu4, A. A. Kuchmiy5, and V. F. Garib6
1Lomonosov Moscow State University, Biological Faculty, 119991 Moscow, Russia2Russian Medical Academy of Postgraduate Education, Department of Immunology, 123995 Moscow, Russia
3Sechenov First Moscow State Medical University, Department of Immunology, 119991 Moscow, Russia; E-mail: fgarib@yandex.ru
4Committee on Science and High Technologies, State Duma of Russian Federation, 103265 Moscow, Russia; E-mail: annarizopulu@inbox.ru
5Ghent University, Inflammation Research Center, VIB, Zwijnaarde, B-9052, Belgium, Department of Internal Medicine, B-9000 Ghent, Belgium; E-mail: kuchmiyanna@gmail.com
6Medical University of Vienna, Department of Pathophysiology and Allergy Research, 1090 Vienna, Austria; E-mail: viktoriya.garib@meduniwien.ac.at
* To whom correspondence should be addressed.
Received July 8, 2016; Revision received July 28, 2016
Inflammatory response is initiated and sustained by the action of quintessential pro-inflammatory cytokines of immune system namely IL-1β and IL-18. The maturation process of those cytokines is ensured by caspase-1 enzymatic activity, that is in turn is tightly controlled by multiprotein complexes called inflammasomes. Inflammasomes are activated in cells of innate immune system in response to recognition of conservative parts of microbes (pathogen-associated molecular patterns) or by sensing molecular signs of tissue damage (damage-associated molecular patterns). Inflammasome activation apart of cytokines secretion leads to pro-inflammatory cell death, so-called pyroptosis. That culminates in release of cytoplasmatic content of cells including cytokines and alarmins that boost immune response against pathogens, as well as pyroptosis destroys replicative niches of intracellular pathogens. During co-evolution with the host, bacterial and viral pathogens developed a range of molecular inhibitors targeting each step of inflammasome activation. In current review, we will discuss the latest knowledge of inflammasomes’ signaling pathways and tricks that pathogens use to avoid immune recognition and clearance. Our better understanding of inflammasome inhibition by pathogens can lead to better therapeutic approaches for the treatment of infectious diseases.
KEY WORDS: inflammasome, IL-1β, IL-18, evasion, bacterial and viral pathogens, pyroptosisDOI: 10.1134/S0006297916110109
Abbreviations and definitions: Alarmins (or DAMPs) are endogenous molecules that are released upon tissue damage and activate innate immunity. The best-characterized alarmins are heat shock proteins (HSP), HMGB1, purine metabolites, adenosine diphosphate, uric acid crystals, etc. Inflammation is an immune response towards PAMPs and DAMPs that is mediated by proinflammatory cytokines IL-1β, IL-18, TNF, and IL-8, which is aiming to restrict dissemination and eventually eliminate pathogens. Immune evasion is a strategy used by pathogenic organisms to evade a host’s immune response to maximize their probability of being transmitted to a fresh host or to continue growing. Inflammasomes are large cytosolic complexes assembled by cytosolic receptors (NLR, AIM, PYRIN) in response to PAMPs and DAMPs that activate caspase-1 and -11 resulting in the production of pro-inflammatory cytokines IL-1β and IL-18 as well as in pyroptotic cell death. Damage-associated molecular patterns (DAMPs) are endogenous molecules released by stressed, damaged, or dying cells, which activate PRR and initiate a noninfectious inflammatory response. Pathogen-associated molecular patterns (PAMPs) are conservative molecular features of pathogens recognized by the innate immune system. Pattern recognition receptors (PRRs) are germ line encoded receptors of the innate immune system, located on the plasma membrane, endosomal membrane, or in cytosol, that detect and respond to exogenous and endogenous stress signals. Pyroptosis is a proinflammatory type of cell death characterized by formation of large pores in the cellular membranes and release of cytosolic contents that contains processed IL-1β and IL-18 and other bioactive substances (e.g. alarmins), as well as intracellular pathogens. NOD-like receptors (NLRs) represent a large family of intracellular PRRs characterized by the presence of a centrally located nucleotide binding and oligomerization domain (referred to as NBD; NOD or NACHT domain) and carboxy-terminal leucine-rich repeats (LRRs). A subset of NLRs can assemble “inflammasomes”. Toll-like receptors (TLRs) are membrane-bound PRRs that recognize PAMPs and DAMPs at the cell surface and inside of the endosomes in different cell types; activation of TLR signaling pathways induces expression of inflammatory and antiviral genes.
Immune recognition of infection, injured or stressed tissues induces
acute inflammatory response that is aiming to restore host homeostasis.
If the acute inflammatory response however fails to eliminate the
pathogen, infection disease can progress and result in lethal outcome.
To another hand, chronic inflammation itself can cause many diseases
such as allergies, autoimmunity, and many others. Bacteria have evolved
a set of intriguing mechanisms to counter immune responses, and
inhibition of cytokines production appears as dominating strategy used
by pathogenic bacteria for their survival [1, 2].
The innate immune system recognizes infections through a limited number of germ line encoded pattern recognition receptors (PRRs) located on the cell surface, in endosomes, and in cytosol. In total, there are more than 40 germ-line encoded PRRs [3, 4]. PRRs recognize conserved microbial components called pathogen-associated molecular patterns (PAMPs) [5]. PAMPs have conservative structure that makes them an attractive target for recognition by immune system. In addition, PRRs also recognize host-derived danger-associated molecular patterns (DAMPs), which are released in response to tissue stress and sterile inflammation [6-8].
PRRs are expressed by various immune cells originating from myeloid and lymphoid lineages and by non-immune epithelial cells. PAMP recognition by PRRs of innate immune system triggers a number of transcriptional and posttranslational programs, resulting in production of inflammatory mediators [9]. Toll-like receptors (TLRs) and C-type lectin receptors (CLRs) recognize microbial moieties including bacterial wall components and microbial nucleic acids at the cell surface and in endosomes [10]. Intracellular PRRs ensures that microbes evading extracellular surveillance encounter another line of recognition in the host cytosol. Such cytosolic PRRs include RIG-I-like receptors (RLRs) [12], the AIM2-like receptor (ALR) [13-15], nucleotide-binding domain and leucine-rich repeat containing (NLR) proteins [11], as well as an increasing range of intracellular sensors of nucleic acids, including OAS proteins and cGAS, safeguarding the intracellular environment. The ability of the innate immune system to detect pathogens in the cytosol allows for recognition of pathogens regardless of their life-cycle stage. Increased interest in cytosolic NLRs emerged during the last decade, as they can directly or indirectly recognize a large variety of cytoplasmic pathogens and are involved in a multitude of immune signaling pathways including multi-step production of key proinflammatory cytokines IL-1β and IL-18. We will discuss current knowledge about structural and functional features of NLRs in the next chapter [11, 16, 17].
STRUCTURE AND FUNCTION OF INFLAMMASOMES
NLRs represent a large family of intracellular PRRs united by common structural features that are involved in a multitude of immune signaling pathways ranging from the regulation of MAP kinase and NF-κB signaling pathways over modulation of the expression of MHC II receptors and interferons [11]. Humans and rhesus macaque monkeys have 22 NLRs [18] whereas mice have 34 NLRs genes, originating from gene duplication.
The NLR family is characterized by the presence of three structural domains. Two of them (NACHT and LRRs) are common for nearly all the members of the NLR family. The central nucleotide-binding and oligomerization (NACHT) domain facilitates the activation of the signalling complex via ATP-dependent oligomerization. The carboxy-terminal leucine-rich repeats (LRRs) domain, present in all NLRs except NLRP10, recognizes microbial motifs through a protein–protein interaction [19, 20]. NLRs can be subdivided according to the type of the amino (N)-terminal effector domain. Members of NLRA subfamily have a caspase activation and recruitment domain (CARD) and an acid transactivation domain (AD). While members of the NLRB subfamily have a baculovirus inhibitory repeat (BIR)-like domain. A pyrin domain (PYD) is typical for members of the NLRP subfamily. Proteins containing either a CARD domain or a still undefined domain are grouped together in the NLRC/X subfamily. CARDs, PYD domains and BIR domains mediate homotypic protein–protein interactions for downstream signalling [20].
NLRs are also of particular interest, because some members of NLR family (due to their unique domain structure) are able to form inflammasomes in response to tissue stress or infection. Inflammasomes are multiprotein cytosolic complexes that facilitate the proximity-induced autoactivation of the pro-inflammatory cysteine protease caspase-1, which, in turn, converts key immune cytokines pro-IL-1β and pro-IL-18 to their active forms and induces proinflammatory type of cell death “pyroptosis” [11, 16]. Members of other molecular families AIM2 [13] and PYRIN [21] are also able to form active inflammasomes after ligand recognition.
IL-1β is a quintessential pro-inflammatory cytokine and pyrogen that regulates systemic and local responses by generating fever, activates lymphocytes, and promotes myeloid cells recruitment and activation at the site of infection. It has been shown to participate in host defence by inducing adoptive immune response skewed to T-helper 17 (Th17) and Th1 cell subsets, as well as controlling B-cell and humoral immune responses. IL-18 cytokine lacks the pyrogenic activity of IL-1β but plays an important role in T-cell polarization by skewing T-cell responses toward either a Th1 or Th2 profile, depending on the inflammatory context. IL-18 also promotes IL-17 expression by already-committed Th17 cells. Unlike most cytokines, IL-1β and IL-18 are not released through the classic endoplasmic reticulum–Golgi route but are produced as cytosolic precursor proteins waiting for their maturation by caspase-1 [22].
Inflammasome activation usually coincides with pro-inflammatory route of cell death called pyroptosis that leads to the formation of large pores in the cell membranes and to the release of inner content of the cells. As a result, processed IL-1β and IL-18 [23] as well as other intracellular bioactive substances (so-called “alarmins”) such as IL-1α, HMGB1, FBF2, and IL-33 release at the site of inflammation [24]. Pyroptosis contributes to the host defence by eliminating intracellular replicating niches of pathogens and by exposing intracellular pathogens for phagocytes. The concomitant release of alarmins is boosting immune responses and accelerates pathogen clearance. Secreted alarmins are helping to clear pathogen and to present intracellular pathogens to adaptive immune system [25, 26]. Recently the molecular mechanism of pyroptosis has been unraveled; it appeared that caspase-1/-11 cleaves gasdermin D (Gsdmd), and its N-terminal fragment [27] induces formation of large membrane pores [28] that is followed by release of cell content and cell death. There are special cases of IL-1β secretion without pyroptotic cell death limited to some cell types, such as human monocytes [29], dendritic cells [30], and neutrophils [31], occurring by unknown secretory mechanism, as well as expulsion of enterocytes into the gut lumen as a special caspase-1-dependent type of cell death [32].
There are several inflammasomes that have been described to date, and each is named after the specific PRR contained in their sensory part, namely NLRP1, NLRP3, NLRP6, NLRP12, NLRC4, AIM2 and PYRIN (Fig. 1) [11, 24, 33]. Molecules NRLP3, AIM2, and PYRIN require adaptor protein ASC (apoptosis-associated speck-like protein containing a caspase activation and recruitment domain) to recruit caspase-1 for inflammasome assembly. When ASC adaptor protein is recruited into the complex, it actually allows for assembly of supramolecular complexes microscopically seen as “speck” [34]. The other inflammasome-forming NLRs such as NLRC4 and NLRP1b can directly interact with caspase-1 using their CARD domain
(Fig. 1). NLRP1b and NLRC4 however can still recruit ASC while ASC-containing NLRC4 and NLRP1b complexes induce caspase-1 autoproteolysis and cytokine maturation [35, 36]; NLRC4 and NLRP1b complexes lacking ASC trigger caspase-1-dependent pyroptosis.
Fig. 1. Schematic view of currently characterized inflammasomes. The nature of a pathogenic stimulus determines type of PRRs for its recognition. There are several canonical inflammasomes – NLRP1, NLRP3, NLRC4, AIM2 and PYRIN – that have been described to date, and each is named after the specific PRR contained in its sensory part. Inflammasomes are defined as intracellular multiprotein complexes that facilitate the proximity-induced autoactivation of the pro-inflammatory cysteine protease caspase-1. Active caspase-1 subsequently cleaves interleukin (IL)-1β and IL-18, an event essential for extracellular secretion of the bioactive forms of these major pro-inflammatory cytokines. Additionally active caspase-1 induces pro-inflammatory pyroptotic cell death by processing Gasdermin D (Gsdmd) that destroys cell membrane. As a result, processed IL-1β and IL-18 together with other alarmins, IL-1α, HMGB1, etc., can be secreted to inflammatory locus. Cellular membrane destruction results in pathogens release from intracellular niches that makes them accessible for clearance by phagocytes and exposing their antigens for antigen-presenting cells. The NLRP1 inflammasome is activated by the protease activity of lethal toxin (LT) of Bacillus anthracis. Unlike its human homolog, murine Nlrp1b lacks a PYD motif. The canonical NLRP3 inflammasome is triggered by vast variety of stimuli. For optimal activation of NLRP3 the priming step is required when the NLRP3 itself and pro-IL-1β have to be transcriptionally upregulated and synthesized in NF-κB-dependent manner (signal 1). NLRP3 becomes activated after stimuli recognition and de-ubiquitinylation (signal 2). NLRC4 inflammasome is activated in two steps. Cytosolic flagellin first activates PKCδ and other kinases that may phosphorylate NLRC4 Ser533. Secondly, NAIP-mediated recognition of bacterial components promotes NAIP-NLRC4 oligomerization and inflammasome activation. The AIM2 inflammasome recognizes cytosolic dsDNA. Toxin-induced modifications of cellular Rho GTPases are sensed by PYRIN, which results in PYRIN inflammasome activation. Compared to its human homolog, murine pyrin lacks the N-terminal PRY and SPRY domains. The noncanonical NLRP3 inflammasome is stimulated by intracellular LPS downstream of caspase-11 in mice and caspase-4 and -5 in humans. Abbreviations: BBOX, B-box-type zinc finger; CARD, caspase recruitment domain; FIIND, domain with function to find; HIN200, hematopoietic interferon-inducible nuclear protein with a 200-a.a. repeat; LRR, leucine-rich repeat; NACHT, nucleotide-binding and oligomerization domain; PRY, SPRY associated; PYD, pyrin domain; SPRY, Dictyostelium discoideum dual specificity kinase SplA and ryanodine receptor; TLR, Toll-like receptors; TNF, tumor necrosis factor; MTX, mitochondria.
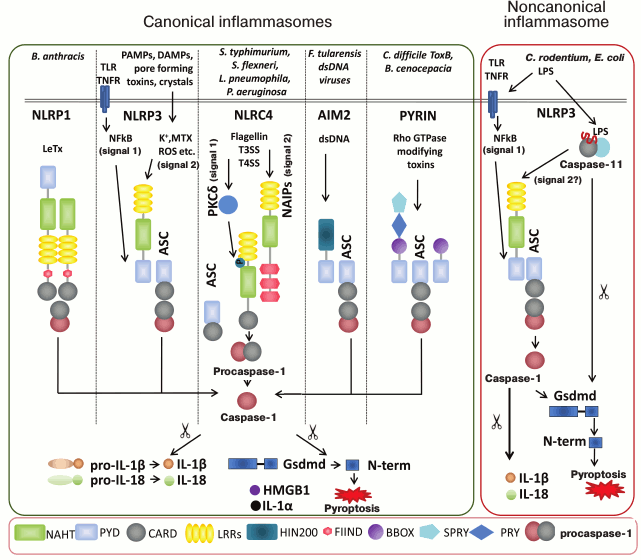
The first identified inflammasome NLRP1b is responsible for sensing of lethal toxin secreted by Bacillus anthracis (Fig. 1, left panel). Macrophages from Nlrp1b-deficient mice fail to activate caspase-1 and are defective at IL-1β secretion and pyroptosis in response to LeTx. The functions of Nlrp1b are well studied in mice, however molecular mechanisms triggered by human NLRP1 still remain controversial. There is no ligand for NLRP1 being identified so far [37].
The Nlrp3 inflammasome is expressed in variety of myeloid cells, and it can be activated by bacterial, viral, and fungal pathogens, pore-forming toxins, crystals, aggregates such as β-amyloid, and DAMPs such as ATP and hyaluronan [38]. It is generally accepted that NLRP3 is able to recognize such great variety of antigens by indirect mechanisms (so-called guard theory) [39]. Several models for indirect NLRP3 activation have been proposed: 1) NLRP3 can sense changes in K+ ion efflux; 2) NLRP3 can sense production of reactive oxygen species (ROS) due to mitochondrial instability and impaired phagocytosis of mitochondria (mitophagy); 3) lysosomal instability and release of lysosomal cathepsins into the cytosol can activate NLRP3 inflammasome [11]. NLRP3 has two-step regulation to ensure secure immune responses. First, the priming step, when NF-κB-activating stimulus is inducing IL-1β production and transcription of optimal amounts of NLRP3. Second step that actually permits NLRP3 inflammasome assembly includes Nlrp3-activating agent together with Nlrp3 de-ubiquitination [40]. Recently, it was demonstrated that serine-threonine kinase NEK7 plays crucial role for activation of NLRP3 inflammasome through a yet unknown mechanism [41, 42]. It should be also noted that in non-primed immune cells there is baseline activity of the NLRP3 inflammasome, which can trigger an immediate production of IL-18 (Fig. 1, left panel) [43].
Additionally, invasion of Gram-negative pathogens (Citrobacter rodentium, Escherichia coli, Vibrio cholerae, Salmonella typhimurium and others) and LPS transfection induce non-canonical inflammasome. Canonical inflammasomes convert procaspase-1 into the catalytically active enzyme, whereas an undefined noncanonical inflammasome promotes activation of procaspase-11 (Fig. 1, right panel) [44]. Recently, Shi et al. [45] demonstrated that N-terminal CARD domain of murine caspase-11 can recognize polysaccharide [45]. Activated caspase-11 cleaves Gsdmd [27, 28], which leads to pyroptosis as well as to NLRP3 inflammasome activation (unknown signal 2).
NLRC4 expression is attributed to epithelial and myeloid cells at mucosal sites. NLRC4 inflammasome detects limited spectrum of stimuli as bacterial flagellin and components of bacterial type III secretion systems inherent for facultative intracellular pathogens such as Salmonella typhimurium, Shigella flexneri, Pseudomonas aeruginosa, Burkholderia thailandensis, and Legionella pneumophila [46, 47]. Biphasic activation mechanism exists for the Nlrc4 inflammasome in which Ser533 phosphorylation prepares Nlrc4 for subsequent activation [48]. NLRC4 does not directly interact with ligands, in this way Naip5 serves as a sensor for flagellin and Naip2 is a sensor for sensor T3SS. After ligand recognition, Naips initiate NLRC4 inflammasome assembly, and crystal structure of such complex was recently reported [49]. Interestingly, in humans, there is only one sensory protein (NAIP), which is homologous to murine Naip1 that detects T3SS needle protein.
In addition, several NLRs were proposed to form inflammasomes, whereas the mechanism of their activation remains to be further verified. For instance, it is believed that molecule NLRP6 is able to form an inflammasome in response to colitogenic microbiota and intestinal viruses; however, cognate ligand and activation mechanism have not been shown [50, 51]. Recently, it was found that NLRP12-containing inflammasomes play a protective role during Yersinia pestis infection [52]. NLRP7-containing inflammasome was reported to ensure IL-1β processing and secretion in human THP-1 cells (macrophage) in response to microbial lipopeptides [53], whereas its close homolog NLRP2 overexpressed in vitro is able to spontaneously form inflammasome [38].
ALRs that contain an N-terminal pyrin domain (PYD) and one or two C-terminal hematopoietic IFN-inducible nuclear proteins with 200-a.a. (HIN-200) domains also can form canonical inflammasomes (Fig. 1, right panel). The HIN-200 domain assembles into an oligonucleotide/oligosaccharide-binding fold (OB-fold), which facilitates DNA binding. AIM2 preferentially recognizes dsDNA from vaccinia virus and cytomegalovirus and the bacterial pathogens Francisella tularensis and Listeria monocytogenes [13], whereas IFI16 can bind dsDNA and ssDNA (e.g. HIV) [54]. Another unclassified PRR PYRIN can detect modifications of cellular Rho GTPases induced by toxins from bacterial pathogens Clostridium difficile and Burkholderia cenocepacia. This is the only example of animal PRR that can recognize the consequences of virulence factor (guard theory has been demonstrated only in plants) in contrast to direct recognition of microbial moieties by PRRs [21, 39].
The biology of inflammasomes is one of the most appealing and rapidly developing areas in molecular immunology. The emerging role of inflammasomes in the induction, regulation and maintenance of both innate and adaptive immune responses, and mutations in NLR genes associated with a wide range of autoinflammatory and autoimmune disorders in humans make them desirable targets for therapeutic intervention and drug development [33, 55, 56].
FUNCTIONS OF INFLAMMASOMES MODULATED BY BACTERIAL VIRULENCE
FACTORS
Given the importance of inflammasomes in controlling replication and dissemination of microbial pathogens, it is not surprising that bacteria have evolved a set of mechanisms to antagonize inflammasome assembly [57-59]. In particular Yersinia spp., Pseudomonas aeruginosa, Vibrio parahaemolyticus, Chlamydia trachomatis, Francisella tularensis, Shigella flexneri, Legionella pneumophila, and Coxiella burnetii developed a range of virulence factors helping them to preserve intracellular replicative niches [60-62].
Bacterial dissemination and evasion relies on specialized secretion systems and pore-forming toxins that facilitate penetration of multiple virulence factors into the host cells (Table 1).
Table 1. Inhibition of inflammasomes by
bacterial pathogens for immune evasion of microbes
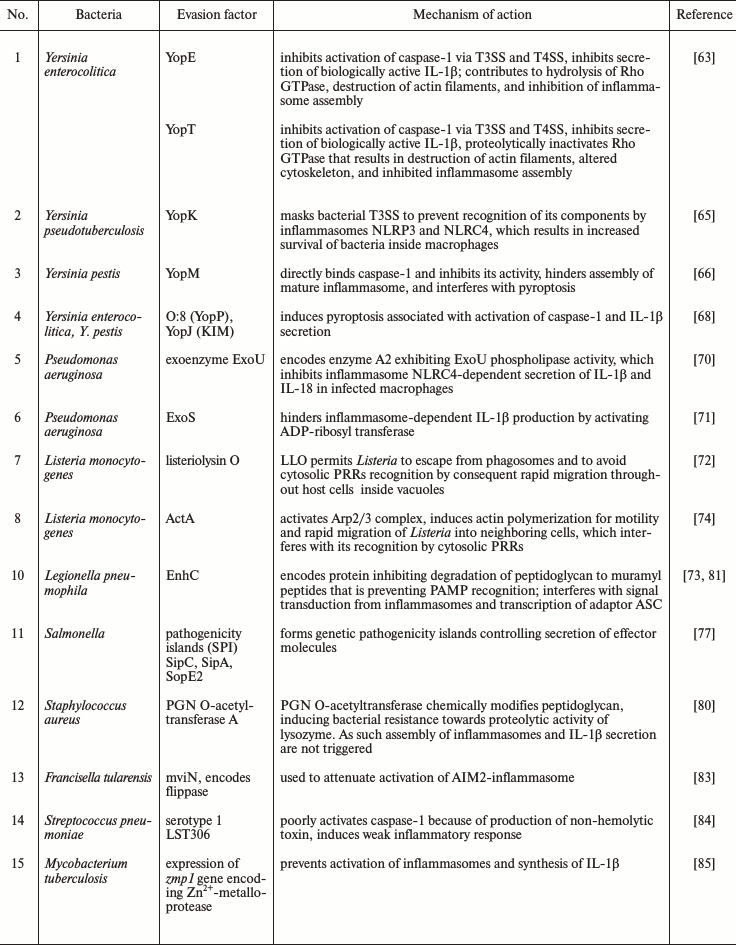
The enteropathogenic bacteria Yersinia enterocolitica inject virulence factors Yop (Yersinia outer membrane proteins) into the cytosol of host cells using specialized type III and IV secretion systems (T3SS) (a so-called bacterial syringe). T3SS and T4SS are sophisticated molecular complexes, which are composed of a series of basal rings that span the bacterial inner and outer membranes, connected to a hollow needle allowing to deliver effector proteins across the bacterial and host membranes. As such YopE and YopT are injected through TSS injectosome and inhibit Rho GTPase family members, and this event subsequently leads to caspase-1 inactivation and absence of secretion of IL-1β [63].
Rho GTPases belong to cell signaling G-proteins, which regulate intracellular actin dynamics and play an important role in cell proliferation, apoptosis, gene expression, and other vital cell functions [64]. Yop E can keep Rho GTPase family members in the inactive GDP-bound state inducing collapse of cytoskeleton and blocking inflammasome assembly by unclear mechanism. YopT inactivates Rho GTPases by proteolytically removing the C-terminal prenyl membrane anchor. YopT also inhibits phagocytosis in macrophages by downregulating their oxidative burst. This leads to destruction of actin filaments and alteration of cytoskeleton with subsequent macrophage death [63].
In turn, Yersinia pseudotuberculosis, to prevent its recognition by inflammasomes NLRP3 and NLRC4, uses effector protein YopK for masking T3SS [65]. Thus, YopK ensures survival of bacteria inside macrophages. Taking together the presence of diverse Yop proteins acting at different levels indicates that inflammasomes are very important for coping Yersinia.
Inhibition of pyroptosis may play an exceptional role in increasing virulence of certain pathogenic microorganisms, such as Yersinia pestis. The latter secretes effector molecule YopM, which directly binds caspase-1 and thus inhibits pyroptosis [66]. In addition, it was also demonstrated that YopM interacts with several host cell proteins to reduce inflammation and can serve as attractive target for antiinflammatory therapeutics development [67].
Not all virulence factors of Yersinia are inhibiting inflammasomes. Paradoxically, some of them can activate pyroptosis for survival purposes. For instance, YopJ, when injected by T3SS of Y. pestis, Y. enterocolitica, and Y. pseudotuberculosis into macrophages, can demonstrate a range of cytotoxicity determining the success of infection. The peak cytotoxic activity is observed in isoforms derived from Y. enterocolitica O:8 (YopP) and Y. pestis KIM (YopJ (KIM)). YopJ (KIM) facilitates macrophage death associated with activated caspase-1 and IL-1β secretion, i.e. pyroptosis [68, 69], and thus facilitates infection dessimination. Though, in all cases only the combination of virulence factors that can inhibit and activate inflammasomes results in optimal spread of Yersinia. To summarize, Yop proteins prevent immune recognition of bacterial TSS and contribute to bacteria dissemination by both routes suppressing activity of inflammasomes and pyroptosis induction.
Pseudomonas aeruginosa isolates expressing the virulence factor exoenzyme (Exo)U use a different strategy to inhibit caspase-1 activation in human phagocytes. The precise molecular mechanism used by enzyme ExoU with phospholipase A2 activity to inhibit NLRC4 inflammasome-dependent secretion of IL-1β and IL-18 in infected macrophages is unknown [70]. ExoS is another virulence factor of Pseudomonas that interferes with inflammasome-dependent IL-1β production by not fully characterized process involving activation of ADP-ribosyltransferase [71].
Intracellular bacteria such as Mycobacterium tuberculosis, Listeria monocytogenes, and Legionella pneumophila escaping immune recognition by preventing phagosome maturation, either by suppressing cellular bactericidal functions, or by escaping from phagosomes into the cytosol of neighboring cells [72, 73].
To exit from a phagolysosome, L. monocytogenes releases its major virulence factor listeriolysin O (it is hemolysin that is triggering membrane damage). Then within the cytosol, the bacteria evade recognition by cytosolic PRRs by activation of cellular Arp2/3 complex by another virulence factor ActA. Cellular Arp2/3 complex contributes to actin polymerization on the bacterial surface and allows Listeria to rapidly move throughout different cells. This accelerated motility of bacteria bearing an actin tail is known as comet-like movement. This evading mechanism is also used by Shigella flexneri and Burkholderia pseudomallei [74].
Some bacteria affect MyD88 signaling downstream of TLR activation by subjecting MyD88to proteasome degradation, disturbing in this manner induction of innate immune reactions, and particularly IL-1β synthesis [75, 76].
Virulence of Salmonella typhimurium relies on expression of products of genes encoded by pathogenicity islands (SPI) SPI-1 and SPI-2, that are embody various types of T3SS. Salmonella uses SPI-1 T3SS for entering enterocytes and intestinal lamina propria, whereas SPI-2 T3SS is used during infection of macrophages and helps to form vacuolar replication niches. During systemic infection in vivo, Salmonella can downregulate expression of its own flagellin and SPI-1 T3SS. This allows Salmonella to escape recognition of its flagellin by TLR5 and NLRC4 and avoid recognition of T3SS subunit PrgJ coded by SPI-1 via NLRC4. Of note, the analogous subunit of SPI-2 T3SS is not recognized by NLRC4. Compensatory genetic mutations that are preventing Salmonella flagellin from binding to TLR5, but at the same time, preserving flagellin function, are essential for bacterial evasion [77].
Salmonella is also able to infect B cells. In contrast to vacuolar localization in macrophages, it is localized inside late endosomes of B cells. Bacteria infect B-cell precursors in bone marrow that can serve as a bacterial reservoir for a long time. NLRC4 inflammasome can be active in B cells, however, Salmonella induces phosphorylation of transcription factor Yap in infected B cells, facilitating its interaction with Hck and subsequently preventing nuclear translocation of Yap and NLRC4 gene expression. The ability of Salmonella to inhibit inflammasome formation prevents death of B cells, and they become an ideal niche for bacteria survival, persistence and spread [78, 79].
It is known that production of IL-1β by phagocytes efficiently protects the host from Staphylococcus aureus infection. If bacterial cell wall peptidoglycan is present in particulated form, it may activate NLRP3 inflammasome with subsequent IL-1β secretion. To evade recognition by NLRP3, S. aureus produces enzyme PGN O-acetyltransferase A, which chemically modifies bacterial peptidoglycan and increases it resistance to proteolytic activity of lysozyme. As a result, after macrophages engulf S. aureus with modified peptidoglycan, the latter is not recognized by cytosolic receptors [80].
Legionella pneumophila causes Legionnaires disease and interferes with transcription of inflammasome adaptor ASC, thereby suppressing assembly of NLRC4 inflammasome and promoting their proliferation both in vitro and in vivo [81]. Legionella pneumophila also applies a strategy for hiding antigenic determinants by using protein SdhA, which is important for preserving replicative activity and preventing exit of the pathogen into the cytosol for subsequent recognition [82].
Francisella tularensis uses lipid-II flippase encoded by the mviN gene (virulence factor) to dampen activation of the AIM2 inflammasome [83]. Flippases are transmembrane lipid transporters proteins that transfer phospholipid molecules between the two leaflets that compose a cellular membrane. It is believed that in the absence of mviN, the bacterium F. tularensis has reduced virulence and is more susceptible for lysis and release of dsDNA into the cytosol that enhances AIM2 inflammasome-mediated IL-1β secretion and macrophage pyroptosis.
Streptococcus pneumoniae is a causative agent of pneumonia, sepsis, and meningitis. The pore-forming toxin pneumolysin is a key virulence factor of St. pneumoniae, which is recognized by the NLRP3 inflammasome. Among more than 90 serotypes, serotype 1 (especially MLST306) of St. pneumoniae is the major cause of invasive infection. In contrast to other serotypes, these bacteria are not recognized by NLRP3 because they produce non-hemolytic pneumolysin, and, therefore, they induce weak inflammatory response [84].
Gram-positive bacteria, particularly Mycobacterium tuberculosis, also prevent activation of inflammasomes and production of IL-1β in human myeloid cells. Mycobacterium is expressing Zn2+-metalloprotease (encoded by zmp1 gene) required for inhibiting inflammasome assembly. Mycobacterium bovis mutants lacking Zmp1 failed to prevent caspase-1 activation, which resulted in enhanced maturation of Mycobacterium-containing vacuoles and rapid mycobacterial clearance from infected macrophages and in the lungs of aerosol-infected mice [85].
FUNCTIONS OF INFLAMMASOMES MODULATED BY VIRAL PATHOGENS
Modulation of inflammasome function is not limited to bacterial pathogens. Viruses provide some well-characterized mechanisms by which microbial pathogens control inflammasomes and thereby restrict immune response (Table 2). As such, during coevolution, viruses that bear genes of caspase inhibitors have been naturally selected, because they inhibited inflammation and exhibit better viral replication [61, 86].
Table 2. Evasion mechanisms used by viral
pathogens to inhibit inflammasome
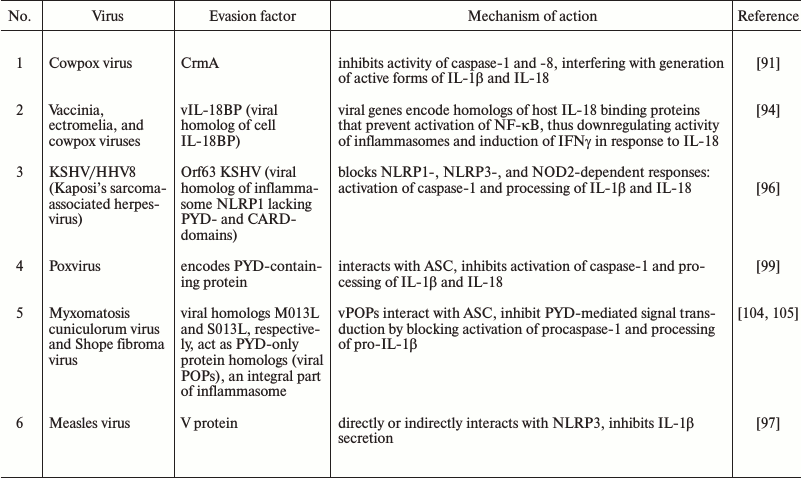
To increase viability of infected cells and facilitate replication, viruses often use inhibitors that can directly target the enzymatic activity of caspases. There are four main classes of viral inhibitors that are able to hijack caspases: serpins, p35 family members, inhibitors of apoptotic proteins, and viral FLICE-inhibiting proteins. For instance, baculovirus p35 protein is able to bind and inhibit a large variety of caspases including caspase-1 [87, 88].
The family of poxviruses encodes serpins that target innate immune effectors inside the cell. The first viral inhibitor described was the cowpox virus serpin protein, CrmA (cytokine response modifier A), which blocks caspase-1 and -8 by direct binding, thereby preventing generation of active forms of IL-1β and IL-18 and inhibiting apoptosis induced by tumor necrosis factor (TNF) or Fas ligand. CrmA inhibits caspase-1 activity with an inhibition constant of 0.01 nM, making it one of the most effective caspase-1 inhibitors. This effect is achieved by acting as a pseudosubstrate for caspase-1 that subsequently results in cleavage of CrmA at catalytic pocket and formation of a permanent covalent bond with the active site cysteine of caspase-1 to render the protease inactive [61, 87, 89, 90].
CrmA homologs in orthopoxviruses such as vaccinia, ectromelia, and rabbitpox virus directly target the enzymatic activity of caspase-1.
The importance of the CrmA gene was demonstrated in vivo as the deletion of CrmA lead to attenuated virulence of poxvirus in intranasally and intracranially infected BALB/c and C57BL/6 mice [91].
Similarly, when rabbits were infected with myxoma virus lacking Serp2 (the closest homolog of CrmA), viral titers in vivo were dramatically decreased. In contrast, deletion of the CrmA homologs SPI-1 and SPI-2 in vaccinia virus failed to affect virulence in intranasally infected BALB/c mice. Ectromelia virus has serpin SPI-2, which also directly inhibits activation of caspase-1 in vitro [92].
As it has been discussed before, IL-18 is a multifunctional proinflammatory cytokine that: induces production of IFNγ by T cells, regulates growth and differentiation of Th1, activates NK cells, regulates apoptosis, induces synthesis of IFNβ by B cells, reduces IgE secretion, and stimulates production of multiple cytokines such as TNF, IL-1α, IL-8, MIP-a, IL-5, IL-6, GM-CSF, and G-CSF. Moreover, IL-18 can trigger Th2 responses by eliciting production of IL-4, IL-5, IL-9, and IL-13, that mediates allergic inflammation. It has been demonstrated that when IL-18 was overexpressed in murine lungs, the application of anti-CD4+ antibody or deleting IL-13 gene ameliorated ovalbumin-induced airway inflammation [93].
In mice and humans, there are circulating IL-18-binding proteins (IL-18BP) that block IL-18 activity. Several poxvirus genes code proteins similar to IL-18BP. Vaccinia, ectromelia, and cowpox viruses promote secretion of soluble viral scavenger receptor IL-18BPs (vIL-18BP) by infected cells that can modulate inflammation. Ectromelia virus vIL-18BP blocks activation of NF-κB and induction of IFNγ in response to IL-18. Moreover, molluscum contagiosum virus (MCV) that induces virus-filled skin lesions in immunocompromised persons and HIV-infected patients also expresses vIL-18BP (gene MC54L), which interferes with productive inflammatory response [94].
Additionally, the vaccinia genome encodes a scavenger receptor named virus-encoded IL-1β receptor (vIL-1R), the protein product of which neutralizes secreted IL-1β that compensates for absence of serpins [95].
To directly target the enzymatic activity of caspase-1 and interfere with ligation of the IL-1β and IL-18 receptors, viruses deploy molecules that prevent inflammasome assembly altogether to further enhance virulence. The most evident example is protein Orf63 from Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV8). KSHV is causative agent inducing severe malignant neoplasms in humans, such as primary lymphoma and rare Castleman’s disease. Orf63 is a viral Nlrp1 homolog that lacks the pyrin and caspase recruitment domain (CARD) motifs found at the amino-terminal and carboxyl-terminal of human Nlrp1, respectively. Thus, it inhibits NLRP1 and NLRP3 inflammasomes by dominant negative mechanism. In addition, protein Orf63 can interact with NOD2 and inhibit its activity. Transcriptional downregulation of KSHV Orf63 expression reduced the rate of viral replication as a consequence of enhanced NLRP1- and NLRP3-mediated IL-1β secretion and pyroptosis induction in KSHV-infected human monocytes and 293T cells [96].
Recent studies demonstrated that measles virus V protein directly targets NLRP3. In human macrophage-like THP-1 cell line, the infection with measles virus can induce caspase-1-dependent production of IL-1β in an NLRP3-dependent manner. However, when V protein was removed from virus, the antagonism against innate immune reactions was reduced that results in higher level IL-1β production. THP-1 cells stably expressing V protein do not secrete IL-1β mediated by the NLRP3-inflammasome. V protein coimmunoprecipitates with C-terminal domain of NLRP3 that proves their direct interaction. The NLRP3-inflammasome was located inside cytoplasmic granules of THP-1 cells stably expressing V protein, but during activation of the inflammasome, it relocated to the perinuclear area for binding V protein. These results point to the ability of measles virus V protein to directly or indirectly interact with NLRP3, alter its location, and suppress secretion of IL-1β [97].
NS1 derived from serotype H1N1 influenza virus A/PR/8/34 (PR8) can block assembly of inflammasomes in addition to its ability to suppress type I interferon production. In vitro experiments demonstrated that the N-terminal RNA-binding and dimerization domain of the NS1 protein is crucial for this activity. It is assumed that NS1 is able to directly bind NLRP3, which needs to be further clarified [98].
M13L and S013L molecules that are encoded by rabbit myxomatosis cuniculorum virus and Shope fibroma virus, respectively, represent viral PYD-only proteins (vPOPs) that are homologs to endogenous PYD domains of inflammasomes. They interfere with inflammasome assembly at the level of ASC complex [99] by molecular inhibitory mechanism similar to the action of endogenous POPs as well as CARD-only proteins (COPs) in humans. Namely, homotypic interactions among Pyrin domains and caspase recruitment domains (CARDs) in inflammasome-complex components mediate oligomerization into filamentous assemblies. And in the presence of caspase-recruitment domain protein INCA (COPs) it caps caspase-1 filaments, thereby exerting potent inhibition on caspase-1 (CARD) polymerization in vitro and inflammasome activation in cells [100]. There are no viral homologs of COPs known so far though they may be identified in the near future.
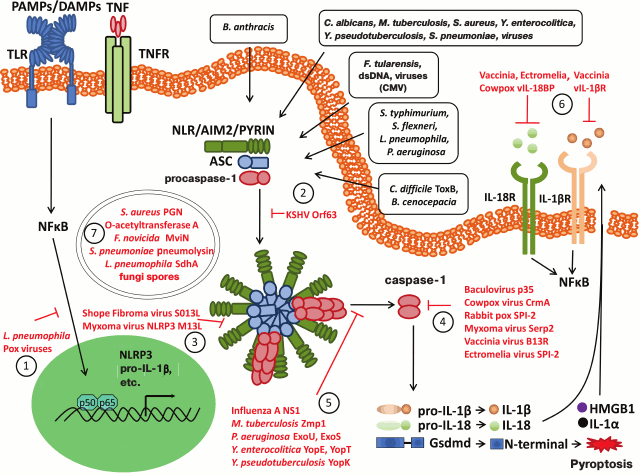
Inflammatory response is an effective way to protect the body against infections and to induce adaptive immune response. Assembly of multiprotein inflammasome complexes serves as a platform for recruiting and activation of caspase-1, that processes major pro-inflammatory cytokines into biologically active forms (IL-1β and IL-18), and their secretion initiates and regulates the inflammatory process. Activation of inflammasomes can trigger proinflammatory cell death pyroptosis to enhance inflammation and to eliminate disease-causing intracellular microbial pathogens. Bacterial and viral pathogens can inhibit inflammasomes at all stages of activation (Fig. 2). For instance: (1) at the level of gene expression – by inhibiting activation of NF-κB and downregulating production of inflammasome components as well as pro-IL-1β and pro-IL-18; (2) at the PRR level, e.g. by blocking NLRP1 via KSHV protein (dominant negative NLRP1 inhibitor); (3) at the level of ASC complexes – via protein traps consisting of pyrin-only decoy proteins (vPOPs) derived from orthopoxviruses; (4) by directly inhibiting activity of caspase-1 via serpins CrmA and homologous proteins derived from myxoma and vaccinia viruses, which competitively inhibit the catalytic site of caspase-1; (5) by indirect inhibition of caspase-1 via effector proteins YopE and YopT as well as ExoS derived from Yersinia and Pseudomonas bacteria, respectively, which inhibit cytoskeletal changes mediated by Rho GTPase, whereas Yersinia spp. virulence factor YopK masks bacterial T3SS proteins; (6) at the cytokine level – via viral analogs of host endogenous receptors vIL-1bR and vIL-18BR by preventing activation of IL-1βR and IL-18R, etc.
Fig. 2. Modulation of inflammasome pathways by bacterial and viral factors. Viral and bacterial pathogens have evolved a number of mechanisms (shown in red) to interfere with inflammasome assembly and activity. Those pathogenic factors can repress inflammasomes or even escape inflammasome recognition at the different levels of the pathway: 1) at the level of gene expression: inhibition of NF-κB translocation and transactivation; 2) at the level of sensory molecule: KSHV Orf63 encodes a viral Nlrp1 decoy protein that interacts with human Nlrp1 and Nlrp3 to prevent assembly of their respective inflammasomes; 3) at the level of ASC speck: pyrin-only decoy proteins (vPOPs) from orthopoxviruses are blocking ASC speck formation. In the presence of vPOPs capsase-1 filaments are capped to prevent recruitment of pro-caspase-1 thereby repressing inflammasome activation; 4) direct inhibition of enzymatic activity of caspase-1: serpins such as CrmA and homologous protein produced by myxoma and vaccinia virus are inhibiting caspase-1 as competitive inhibitor to its substrate; 5) indirect inhibitors of inflammasome activation: the N-terminal RNA-binding domain of influenza virus NS1 protein inhibits caspase-1 activation and secretion of IL-1β and IL-18 through an unknown mechanism. The Yersinia effector proteins YopE and YopT and the Pseudomonas virulence factor ExoS may prevent caspase-1 activation indirectly by interfering with Rho GTPase-mediated cytoskeletal changes, whereas Yersinia YopK masks the bacterial type III secretion system. Pseudomonas ExoU makes use of phospholipase A2 activity to block caspase-1 activation in inflammasomes, whereas Francisella mviN prevents activation of the AIM2 inflammasome; 6) at the level of effectors: interfere with downstream activation of IL-1β and IL-18 receptors through scavenger receptors vIL-1bR and vIL-18BR; 7) antigenic stealth: during infection, some pathogens use stealth to avoid inflammasome activation, for example L. pneumophila employs an antigen masking strategy through its SdhA protein, which is important for maintaining the L. pneumophila replication vacuole and preventing cytosolic recognition of antigens by the inflammasome.
The evasion strategies used by pathogens are not limited to inflammasomes inhibition. For example, for invasion and efficient proliferation, bacterial and viral pathogens use regulatory T cells to suppress immune response [101-103]. Further understanding of the molecular mechanisms by which these and other microbes interfere with inflammasome pathways (both stimulating and inhibiting) may open up new windows of opportunity to develop much needed therapies against infectious agents. On the other hand, use of unique and specific inflammasome inhibitors derived from pathogens would help to develop therapeutic approaches for treatment of inflammatory and autoimmune diseases.
REFERENCES
1.Janeway, C. A., Jr. (1988) Frontiers of the immune
system, Nature, 333, 804-806.
2.Medzhitov, R. (2008) Origin and physiological roles
of inflammation, Nature, 454, 428-435.
3.Kumar, H., Kawai, T., and Akira, S. (2009) Pathogen
recognition in the innate immune response, Biochem. J.,
420, 1-16.
4.Murphy, K. W. (2017) Janeway’s immunobiology,
in Janeway’s Immunobiology, 9th Edn., Garland
Science/Taylor & Francis Group, New York, pp. 35-54.
5.Medzhitov, R. (2013) Pattern recognition theory and
the launch of modern innate immunity, J. Immunol., 191,
4473-4474.
6.Matzinger, P. (2002) The danger model: a renewed
sense of self, Science, 296, 301-305.
7.Barton, G. M., and Medzhitov, R. (2003) Toll-like
receptor signaling pathways, Science, 300, 1524-1525.
8.Van Gorp, H., Kuchmiy, A., Van Hauwermeiren, F.,
and Lamkanfi, M. (2014) NOD-like receptors interfacing the immune and
reproductive systems, FEBS J., 281, 4568-4582.
9.Takeuchi, O., and Akira, S. (2010) Pattern
recognition receptors and inflammation, Cell, 140,
805-820.
10.Drickamer, K., and Taylor, M. E. (2015) Recent
insights into structures and functions of C-type lectins in the immune
system, Curr. Opin. Struct. Biol., 34, 26-34.
11.Lamkanfi, M., and Dixit, V. M. (2014) Mechanisms
and functions of inflammasomes, Cell, 157, 1013-1022.
12.Yoneyama, M., Kikuchi, M., Natsukawa, T.,
Shinobu, N., Imaizumi, T., Miyagishi, M., Taira, K., Akira, S., and
Fujita, T. (2004) The RNA helicase RIG-I has an essential function in
double-stranded RNA-induced innate antiviral responses, Nat.
Immunol., 5, 730-737.
13.Hornung, V., Ablasser, A., Charrel-Dennis, M.,
Bauernfeind, F., Horvath, G., Caffrey, D. R., Latz, E., and Fitzgerald,
K. A. (2009) AIM2 recognizes cytosolic dsDNA and forms a
caspase-1-activating inflammasome with ASC, Nature, 458,
514-518.
14.Hornung, V., Hartmann, R., Ablasser, A., and
Hopfner, K. P. (2014) OAS proteins and cGAS: unifying concepts in
sensing and responding to cytosolic nucleic acids, Nat. Rev.
Immunol., 14, 521-528.
15.Hornung, V., and Latz, E. (2010) Intracellular
DNA recognition, Nat. Rev. Immunol., 10, 123-130.
16.Schroder, K., and Tschopp, J. (2010) The
inflammasomes, Cell, 140, 821-832.
17.Nedospasov, S. (2012) Innate Immunity and Its
Mechanisms [in Russian], Nauchnyi Mir, Moscow, pp. 21-23.
18.Zhang, P., Dixon, M., Zucchelli, M., Hambiliki,
F., Levkov, L., Hovatta, O., and Kere, J. (2008) Expression analysis of
the NLRP gene family suggests a role in human preimplantation
development, PLoS One, 3, e2755.
19.Kufer, T. A., and Sansonetti, P. J. (2011) NLR
functions beyond pathogen recognition, Nat. Immunol., 12,
121-128.
20.Ting, J. P., Lovering, R. C., Alnemri, E. S.,
Bertin, J., Boss, J. M., Davis, B. K., Flavell, R. A., Girardin, S. E.,
Godzik, A., Harton, J. A., Hoffman, H. M., Hugot, J. P., Inohara, N.,
Mackenzie, A., Maltais, L. J., Nunez, G., Ogura, Y., Otten, L. A.,
Philpott, D., Reed, J. C., Reith, W., Schreiber, S., Steimle, V., and
Ward, P. A. (2008) The NLR gene family: a standard nomenclature,
Immunity, 28, 285-287.
21.Xu, H., Yang, J., Gao, W., Li, L., Li, P., Zhang,
L., Gong, Y. N., Peng, X., Xi, J. J., Chen, S., Wang, F., and Shao, F.
(2014) Innate immune sensing of bacterial modifications of Rho GTPases
by the Pyrin inflammasome, Nature, 513, 237-241.
22.Dinarello, C. A. (2009) Immunological and
inflammatory functions of the interleukin-1 family, Annu. Rev.
Immunol., 27, 519-550.
23.Liu, T., Yamaguchi, Y., Shirasaki, Y., Shikada,
K., Yamagishi, M., Hoshino, K., Kaisho, T., Takemoto, K., Suzuki, T.,
Kuranaga, E., Ohara, O., and Miura, M. (2014) Single-cell imaging of
caspase-1 dynamics reveals an all-or-none inflammasome signaling
response, Cell Rep., 8, 974-982.
24.De Vasconcelos, N. M., Van Opdenbosch, N., and
Lamkanfi, M. (2016) Inflammasomes as polyvalent cell death platforms,
Cell. Mol. Life Sci., 73, 2335-2347.
25.Aachoui, Y., Leaf, I. A., Hagar, J. A., Fontana,
M. F., Campos, C. G., Zak, D. E., Tan, M. H., Cotter, P. A., Vance, R.
E., Aderem, A., and Miao, E. A. (2013) Caspase-11 protects against
bacteria that escape the vacuole, Science, 339,
975-978.
26.Ceballos-Olvera, I., Sahoo, M., Miller, M. A.,
Barrio, L. D., and Re, F. (2011) Inflammasome-dependent pyroptosis and
IL-18 protect against Burkholderia pseudomallei lung
infection while IL-1β is deleterious, PLoS Pathog.,
7, e1002452.
27.Kayagaki, N., Stowe, I. B., Lee, B. L.,
O’Rourke, K., Anderson, K., Warming, S., Cuellar, T., Haley, B.,
Roose-Girma, M., Phung, Q. T., Liu, P. S., Lill, J. R., Li, H., Wu, J.,
Kummerfeld, S., Zhang, J., Lee, W. P., Snipas, S. J., Salvesen, G. S.,
Morris, L. X., Fitzgerald, L., Zhang, Y., Bertram, E. M., Goodnow, C.
C., and Dixit, V. M. (2015) Caspase-11 cleaves gasdermin D for
non-canonical inflammasome signalling, Nature, 526,
666-671.
28.Ding, J., Wang, K., Liu, W., She, Y., Sun, Q.,
Shi, J., Sun, H., Wang, D. C., and Shao, F. (2016) Pore-forming
activity and structural autoinhibition of the gasdermin family,
Nature, 535, 111-116.
29.Gaidt, M. M., Ebert, T. S., Chauhan, D., Schmidt,
T., Schmid-Burgk, J. L., Rapino, F., Robertson, A. A., Cooper, M. A.,
Graf, T., and Hornung, V. (2016) Human monocytes engage an alternative
inflammasome pathway, Immunity, 44, 833-846.
30.Zanoni, I., Tan, Y., Di Gioia, M., Broggi, A.,
Ruan, J., Shi, J., Donado, C. A., Shao, F., Wu, H., Springstead, J. R.,
and Kagan, J. C. (2016) An endogenous caspase-11 ligand elicits
interleukin-1 release from living dendritic cells, Science,
352, 1232-1236.
31.Chen, K. W., Gross, C. J., Sotomayor, F. V.,
Stacey, K. J., Tschopp, J., Sweet, M. J., and Schroder, K. (2014) The
neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation
without pyroptosis during acute Salmonella challenge, Cell
Rep., 8, 570-582.
32.Sellin, M. E., Muller, A. A., Felmy, B.,
Dolowschiak, T., Diard, M., Tardivel, A., Maslowski, K. M., and Hardt,
W. D. (2014) Epithelium-intrinsic NAIP/NLRC4 inflammasome drives
infected enterocyte expulsion to restrict Salmonella replication
in the intestinal mucosa, Cell Host Microbe, 16,
237-248.
33.Saavedra, P. H., Demon, D., Van Gorp, H., and
Lamkanfi, M. (2015) Protective and detrimental roles of inflammasomes
in disease, Semin. Immunopathol., 37, 313-322.
34.Lu, A., Magupalli, V. G., Ruan, J., Yin, Q.,
Atianand, M. K., Vos, M. R., Schroder, G. F., Fitzgerald, K. A., Wu,
H., and Egelman, E. H. (2014) Unified polymerization mechanism for the
assembly of ASC-dependent inflammasomes, Cell, 156,
1193-1206.
35.Case, C. L., and Roy, C. R. (2011) Asc modulates
the function of NLRC4 in response to infection of macrophages by
Legionella pneumophila, MBio, 2, pii:
e00117-11.
36.Van Opdenbosch, N., Gurung, P., Vande Walle, L.,
Fossoul, A., Kanneganti, T. D., and Lamkanfi, M. (2014) Activation of
the NLRP1b inflammasome independently of ASC-mediated caspase-1
autoproteolysis and speck formation, Nat. Commun., 5,
3209.
37.Chavarria-Smith, J., and Vance, R. E. (2015) The
NLRP1 inflammasomes, Immunol. Rev., 265, 22-34.
38.Agostini, L., Martinon, F., Burns, K., McDermott,
M. F., Hawkins, P. N., and Tschopp, J. (2004) NALP3 forms an
IL-1beta-processing inflammasome with increased activity in
Muckle–Wells autoinflammatory disorder, Immunity,
20, 319-325.
39.Kagan, J. C. (2014) Common mechanisms activate
plant guard receptors and TLR4, Trends Immunol., 35,
454-456.
40.Py, B. F., Kim, M. S., Vakifahmetoglu-Norberg,
H., and Yuan, J. (2013) Deubiquitination of NLRP3 by BRCC3 critically
regulates inflammasome activity, Mol. Cell, 49,
331-338.
41.Schmid-Burgk, J. L., Chauhan, D., Schmidt, T.,
Ebert, T. S., Reinhardt, J., Endl, E., and Hornung, V. (2016) A
genome-wide CRISPR (Clustered Regularly Interspaced Short Palindromic
Repeats) screen identifies NEK7 as an essential component of NLRP3
inflammasome activation, J. Biol. Chem., 291,
103-109.
42.Shi, H., Wang, Y., Li, X., Zhan, X., Tang, M.,
Fina, M., Su, L., Pratt, D., Bu, C. H., Hildebrand, S., Lyon, S.,
Scott, L., Quan, J., Sun, Q., Russell, J., Arnett, S., Jurek, P., Chen,
D., Kravchenko, V. V., Mathison, J. C., Moresco, E. M., Monson, N. L.,
Ulevitch, R. J., and Beutler, B. (2016) NLRP3 activation and mitosis
are mutually exclusive events coordinated by NEK7, a new inflammasome
component, Nat. Immunol., 17, 250-258.
43.Fernandes-Alnemri, T., Kang, S., Anderson, C.,
Sagara, J., Fitzgerald, K. A., and Alnemri, E. S. (2013) Cutting edge:
TLR signaling licenses IRAK1 for rapid activation of the NLRP3
inflammasome, J. Immunol., 191, 3995-3999.
44.Kayagaki, N., Wong, M. T., Stowe, I. B., Ramani,
S. R., Gonzalez, L. C., Akashi-Takamura, S., Miyake, K., Zhang, J.,
Lee, W. P., Muszynski, A., Forsberg, L. S., Carlson, R. W., and Dixit,
V. M. (2013) Noncanonical inflammasome activation by intracellular LPS
independent of TLR4, Science, 341, 1246-1249.
45.Shi, J., Zhao, Y., Wang, Y., Gao, W., Ding, J.,
Li, P., Hu, L., and Shao, F. (2014) Inflammatory caspases are innate
immune receptors for intracellular LPS, Nature, 514,
187-192.
46.Lamkanfi, M., and Dixit, V. M. (2012)
Inflammasomes and their roles in health and disease, Annu. Rev.
Cell. Dev. Biol., 28, 137-161.
47.Miao, E. A., Mao, D. P., Yudkovsky, N., Bonneau,
R., Lorang, C. G., Warren, S. E., Leaf, I. A., and Aderem, A. (2010)
Innate immune detection of the type III secretion apparatus through the
NLRC4 inflammasome, Proc. Natl. Acad. Sci. USA, 107,
3076-3080.
48.Matusiak, M., Van Opdenbosch, N., Vande Walle,
L., Sirard, J. C., Kanneganti, T. D., and Lamkanfi, M. (2015)
Flagellin-induced NLRC4 phosphorylation primes the inflammasome for
activation by NAIP5, Proc. Natl. Acad. Sci. USA, 112,
1541-1546.
49.Zhang, L., Chen, S., Ruan, J., Wu, J., Tong, A.
B., Yin, Q., Li, Y., David, L., Lu, A., Wang, W. L., Marks, C., Ouyang,
Q., Zhang, X., Mao, Y., and Wu, H. (2015) Cryo-EM structure of the
activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization,
Science, 350, 404-409.
50.Elinav, E., Strowig, T., Kau, A. L., Henao-Mejia,
J., Thaiss, C. A., Booth, C. J., Peaper, D. R., Bertin, J., Eisenbarth,
S. C., Gordon, J. I., and Flavell, R. A. (2011) NLRP6 inflammasome
regulates colonic microbial ecology and risk for colitis, Cell,
145, 745-757.
51.Wang, P., Zhu, S., Yang, L., Cui, S., Pan, W.,
Jackson, R., Zheng, Y., Rongvaux, A., Sun, Q., Yang, G., Gao, S., Lin,
R., You, F., Flavell, R., and Fikrig, E. (2015) Nlrp6 regulates
intestinal antiviral innate immunity, Science, 350,
826-830.
52.Vladimer, G. I., Weng, D., Paquette, S. W.,
Vanaja, S. K., Rathinam, V. A., Aune, M. H., Conlon, J. E., Burbage, J.
J., Proulx, M. K., Liu, Q., Reed, G., Mecsas, J. C., Iwakura, Y.,
Bertin, J., Goguen, J. D., Fitzgerald, K. A., and Lien, E. (2012) The
NLRP12 inflammasome recognizes Yersinia pestis, Immunity,
37, 96-107.
53.Khare, S., Dorfleutner, A., Bryan, N. B., Yun,
C., Radian, A. D., de Almeida, L., Rojanasakul, Y., and Stehlik, C.
(2012) An NLRP7-containing inflammasome mediates recognition of
microbial lipopeptides in human macrophages, Immunity,
36, 464-476.
54.Unterholzner, L., Keating, S. E., Baran, M.,
Horan, K. A., Jensen, S. B., Sharma, S., Sirois, C. M., Jin, T., Latz,
E., Xiao, T. S., Fitzgerald, K. A., Paludan, S. R., and Bowie, A. G.
(2010) IFI16 is an innate immune sensor for intracellular DNA, Nat.
Immunol., 11, 997-1004.
55.Man, S. M., and Kanneganti, T. D. (2015)
Regulation of inflammasome activation, Immunol. Rev.,
265, 6-21.
56.Strowig, T., Henao-Mejia, J., Elinav, E., and
Flavell, R. (2012) Inflammasomes in health and disease, Nature,
481, 278-286.
57.Finlay, B. B., and McFadden, G. (2006)
Anti-immunology: evasion of the host immune system by bacterial and
viral pathogens, Cell, 124, 767-782.
58.Vanden Berghe, T., Linkermann, A.,
Jouan-Lanhouet, S., Walczak, H., and Vandenabeele, P. (2014) Regulated
necrosis: the expanding network of non-apoptotic cell death pathways,
Nat. Rev. Mol. Cell Biol., 15, 135-147.
59.Kaufmann, S. H. E., and Andersen, P. (2011)
Immune intervention strategies against tuberculosis, in The Immune
Response to Infection (Kaufmann, S. H. E., Rouse, B. T., and Sacks,
D. L., eds.) ASM Press, Washington, D. C., pp. 571-586.
60.Cunha, L. D., and Zamboni, D. S. (2013)
Subversion of inflammasome activation and pyroptosis by pathogenic
bacteria, Front. Cell. Infect. Microbiol., 3, 76.
61.Lamkanfi, M., and Dixit, V. M. (2011) Modulation
of inflammasome pathways by bacterial and viral pathogens, J.
Immunol., 187, 597-602.
62.Lupfer, C. R., and Kanneganti, T. D. (2012) The
role of inflammasome modulation in virulence, Virulence,
3, 262-270.
63.Schotte, P., Denecker, G., Van Den Broeke, A.,
Vandenabeele, P., Cornelis, G. R., and Beyaert, R. (2004) Targeting
Rac1 by the Yersinia effector protein YopE inhibits
caspase-1-mediated maturation and release of interleukin-1beta, J.
Biol. Chem., 279, 25134-25142.
64.Bustelo, X. R., Sauzeau, V., and Berenjeno, I. M.
(2007) GTP-binding proteins of the Rho/Rac family: regulation,
effectors and functions in vivo, BioEssays, 29,
356-370.
65.Brodsky, I. E., Palm, N. W., Sadanand, S.,
Ryndak, M. B., Sutterwala, F. S., Flavell, R. A., Bliska, J. B., and
Medzhitov, R. (2010) A Yersinia effector protein promotes
virulence by preventing inflammasome recognition of the type III
secretion system, Cell Host Microbe, 7, 376-387.
66.LaRock, C. N., and Cookson, B. T. (2012) The
Yersinia virulence effector YopM binds caspase-1 to arrest
inflammasome assembly and processing, Cell Host Microbe,
12, 799-805.
67.Hofling, S., Grabowski, B., Norkowski, S.,
Schmidt, M. A., and Ruter, C. (2015) Current activities of the
Yersinia effector protein YopM, Int. J. Med. Microbiol.,
305, 424-432.
68.Zheng, Y., Lilo, S., Mena, P., and Bliska, J. B.
(2012) YopJ-induced caspase-1 activation in Yersinia-infected
macrophages: independent of apoptosis, linked to necrosis, dispensable
for innate host defense, PLoS One, 7, e36019.
69.Tseneva, G. Ya., Solodovnikova, N. Yu., and
Voskresenskaya, E. A. (2002) Molecular aspects of Yersinia
virulence, Klin. Mikrobiol. Antimikrob. Khimioter.,
4, 248-256.
70.Sutterwala, F. S., Mijares, L. A., Li, L., Ogura,
Y., Kazmierczak, B. I., and Flavell, R. A. (2007) Immune recognition of
Pseudomonas aeruginosa mediated by the IPAF/NLRC4
inflammasome, J. Exp. Med., 204, 3235-3245.
71.Galle, M., Schotte, P., Haegman, M., Wullaert,
A., Yang, H. J., Jin, S., and Beyaert, R. (2008) The Pseudomonas
aeruginosa type III secretion system plays a dual role in the
regulation of caspase-1 mediated IL-1beta maturation, J. Cell. Mol.
Med., 12, 1767-1776.
72.Diacovich, L., and Gorvel, J. P. (2010) Bacterial
manipulation of innate immunity to promote infection, Nat. Rev.
Microbiol., 8, 117-128.
73.Liu, M., Haenssler, E., Uehara, T., Losick, V.
P., Park, J. T., and Isberg, R. R. (2012) The Legionella
pneumophila EnhC protein interferes with immunostimulatory
muramyl peptide production to evade innate immunity, Cell
Host Microbe, 12, 166-176.
74.Skoble, J., Portnoy, D. A., and Welch, M. D.
(2000) Three regions within ActA promote Arp2/3 complex-mediated actin
nucleation and Listeria monocytogenes motility, J. Cell
Biol., 150, 527-538.
75.Brodsky, I. E., and Medzhitov, R. (2009)
Targeting of immune signalling networks by bacterial pathogens, Nat.
Cell Biol., 11, 521-526.
76.Johannessen, M., Askarian, F., Sangvik, M., and
Sollid, J. E. (2013) Bacterial interference with canonical NFkappaB
signalling, Microbiology, 159 (Pt. 10), 2001-2013.
77.Broz, P., and Monack, D. M. (2011) Molecular
mechanisms of inflammasome activation during microbial infections,
Immunol. Rev., 243, 174-190.
78.Castro-Eguiluz, D., Pelayo, R., Rosales-Garcia,
V., Rosales-Reyes, R., Alpuche-Aranda, C., and Ortiz-Navarrete, V.
(2009) B cell precursors are targets for Salmonella infection,
Microb. Pathog., 47, 52-56.
79.Perez-Lopez, A., Rosales-Reyes, R.,
Alpuche-Aranda, C. M., and Ortiz-Navarrete, V. (2013) Salmonella
downregulates Nod-like receptor family CARD domain containing protein 4
expression to promote its survival in B-cells by preventing
inflammasome activation and cell death, J. Immunol., 190,
1201-1209.
80.Shimada, T., Park, B. G., Wolf, A. J., Brikos,
C., Goodridge, H. S., Becker, C. A., Reyes, C. N., Miao, E. A., Aderem,
A., Gotz, F., Liu, G. Y., and Underhill, D. M. (2010) Staphylococcus
aureus evades lysozyme-based peptidoglycan digestion that links
phagocytosis, inflammasome activation, and IL-1beta secretion, Cell
Host Microbe, 7, 38-49.
81.Abdelaziz, D. H., Gavrilin, M. A., Akhter, A.,
Caution, K., Kotrange, S., Khweek, A. A., Abdulrahman, B. A., Grandhi,
J., Hassan, Z. A., Marsh, C., Wewers, M. D., and Amer, A. O. (2011)
Apoptosis-associated speck-like protein (ASC) controls Legionella
pneumophila infection in human monocytes, J. Biol. Chem.,
286, 3203-3208.
82.Creasey, E. A., and Isberg, R. R. (2012) The
protein SdhA maintains the integrity of the
Legionella-containing vacuole, Proc. Natl. Acad. Sci.
USA, 109, 3481-3486.
83.Ulland, T. K., Buchan, B. W., Ketterer, M. R.,
Fernandes-Alnemri, T., Meyerholz, D. K., Apicella, M. A., Alnemri, E.
S., Jones, B. D., Nauseef, W. M., and Sutterwala, F. S. (2010) Cutting
edge: mutation of Francisella tularensis mviN leads to increased
macrophage absent in melanoma 2 inflammasome activation and a loss of
virulence, J. Immunol., 185, 2670-2674.
84.Witzenrath, M., Pache, F., Lorenz, D., Koppe, U.,
Gutbier, B., Tabeling, C., Reppe, K., Meixenberger, K., Dorhoi, A., Ma,
J., Holmes, A., Trendelenburg, G., Heimesaat, M. M., Bereswill, S., van
der Linden, M., Tschopp, J., Mitchell, T. J., Suttorp, N., and Opitz,
B. (2011) The NLRP3 inflammasome is differentially activated by
pneumolysin variants and contributes to host defense in pneumococcal
pneumonia, J. Immunol., 187, 434-440.
85.Master, S. S., Rampini, S. K., Davis, A. S.,
Keller, C., Ehlers, S., Springer, B., Timmins, G. S., Sander, P., and
Deretic, V. (2008) Mycobacterium tuberculosis prevents
inflammasome activation, Cell Host Microbe, 3,
224-232.
86.Jacobs, S. R., and Damania, B. (2012) NLRs,
inflammasomes, and viral infection, J. Leukoc. Biol., 92,
469-477.
87.Best, S. M. (2008) Viral subversion of apoptotic
enzymes: escape from death row, Annu. Rev. Microbiol.,
62, 171-192.
88.Zhou, Q., Krebs, J. F., Snipas, S. J., Price, A.,
Alnemri, E. S., Tomaselli, K. J., and Salvesen, G. S. (1998)
Interaction of the baculovirus anti-apoptotic protein p35 with
caspases. Specificity, kinetics, and characterization of the
caspase/p35 complex, Biochemistry, 37, 10757-10765.
89.Ray, C. A., Black, R. A., Kronheim, S. R.,
Greenstreet, T. A., Sleath, P. R., Salvesen, G. S., and Pickup, D. J.
(1992) Viral inhibition of inflammation: cowpox virus encodes an
inhibitor of the interleukin-1 beta converting enzyme, Cell,
69, 597-604.
90.Komiyama, T., Ray, C. A., Pickup, D. J., Howard,
A. D., Thornberry, N. A., Peterson, E. P., and Salvesen, G. (1994)
Inhibition of interleukin-1 beta converting enzyme by the cowpox virus
serpin CrmA. An example of cross-class inhibition, J. Biol.
Chem., 269, 19331-19337.
91.MacNeill, A. L., Moldawer, L. L., and Moyer, R.
W. (2009) The role of the cowpox virus crmA gene during
intratracheal and intradermal infection of C57BL/6 mice,
Virology, 384, 151-160.
92.Turner, S. J., Silke, J., Kenshole, B., and Ruby,
J. (2000) Characterization of the ectromelia virus serpin, SPI-2, J.
Gen. Virol., 81 (Pt. 10), 2425-2430.
93.Sawada, M., Kawayama, T., Imaoka, H., Sakazaki,
Y., Oda, H., Takenaka, S., Kaku, Y., Azuma, K., Tajiri, M., Edakuni,
N., Okamoto, M., Kato, S., and Hoshino, T. (2013) IL-18 induces airway
hyperresponsiveness and pulmonary inflammation via CD4+
T-cell and IL-13, PLoS One, 8, e54623.
94.Smith, V. P., Bryant, N. A., and Alcami, A.
(2000) Ectromelia, vaccinia and cowpox viruses encode secreted
interleukin-18-binding proteins, J. Gen. Virol., 81, (Pt.
5), 1223-1230.
95.Kettle, S., Alcami, A., Khanna, A., Ehret, R.,
Jassoy, C., and Smith, G. L. (1997) Vaccinia virus serpin B13R (SPI-2)
inhibits interleukin-1beta-converting enzyme and protects
virus-infected cells from TNF- and Fas-mediated apoptosis, but does not
prevent IL-1beta-induced fever, J. Gen. Virol., 78,
677-685.
96.Gregory, S. M., Davis, B. K., West, J. A.,
Taxman, D. J., Matsuzawa, S.-I., Reed, J. C., Ting, J. P. Y., and
Damania, B. (2011) Discovery of a viral NLR homolog that inhibits the
inflammasome, Science, 331, 330-334.
97.Komune, N., Ichinohe, T., Ito, M., and Yanagi, Y.
(2011) Measles virus V protein inhibits NLRP3 inflammasome-mediated
interleukin-1beta secretion, J. Virol., 85,
13019-13026.
98.Moriyama, M., Chen, I. Y., Kawaguchi, A.,
Koshiba, T., Nagata, K., Takeyama, H., Hasegawa, H., and Ichinohe, T.
(2016) The RNA- and TRIM25-binding domains of influenza virus NS1
protein are essential for suppression of NLRP3 inflammasome-mediated
interleukin-1beta secretion, J. Virol., 90,
4105-4114.
99.Johnston, J. B., Barrett, J. W., Nazarian, S. H.,
Goodwin, M., Ricciuto, D., Wang, G., and McFadden, G. (2005) A
poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit
host inflammatory and apoptotic responses to infection,
Immunity, 23, 587-598.
100.Lu, A., Li, Y., Schmidt, F. I., Yin, Q., Chen,
S., Fu, T. M., Tong, A. B., Ploegh, H. L., Mao, Y., and Wu, H. (2016)
Molecular basis of caspase-1 polymerization and its inhibition by a new
capping mechanism, Nat. Struct. Mol. Biol., 23,
416-425.
101.Taylor, A. L., and Llewelyn, M. J. (2010)
Superantigen-induced proliferation of human
CD4+CD25– T-cells is followed by a switch
to a functional regulatory phenotype, J. Immunol., 185,
6591-6598.
102.Garib, F. Yu., and Rizopulu, A. P. (2015)
T-regulatory cells as part of strategy of immune evasion by pathogens,
Biochemistry (Moscow), 80, 1141-1159.
103.Belkaid, Y., and Tarbell, K. (2009) Regulatory
T-cells in the control of host–microorganism interactions,
Annu. Rev. Immunol., 27, 551-589.
104.Dorfleutner, A., Talbott, S. J., Bryan, N. B.,
Funya, K. N., Rellick, S. L., Reed, J. C., Shi, X., Rojanasakul, Y.,
Flynn, D. C., and Stehlik, C. (2007) A Shope fibroma virus PYRIN-only
protein modulates the host immune response, Virus Genes,
35, 685-694.
105.Matusiak, M., Van Opdenbosch, N., and Lamkanfi,
M. (2015) CARD- and pyrin-only proteins regulating inflammasome
activation and immunity, Immunol. Rev., 265, 217-230.