Possible Mechanisms of Acquisition of Herpesvirus Virokines
E. A. Gorshkova1,2 and E. S. Shilov1,2*
1Lomonosov Moscow State University, Faculty of Biology, 119991 Moscow, Russia2Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, 119991 Moscow, Russia; E-mail: shilov_evgeny@inbox.ru
* To whom correspondence should be addressed.
Received June 17, 2016; Revision received July 29, 2016
The genomes of certain types of human and primate herpesviruses contain functional homologs of important host cytokines (IL-6, IL-17, and IL-10), or so-called virokines. Virokines can interact with immune cell receptors, transmit a signal to them, and thus switch the type of immune response that facilitates viral infection development. In this work, we have summarized possible ways of virokine origin and proposed an evolutionary scenario of virokine acquisition with involvement of retroviral coinfection of the host. This scenario is probably valid for vIL-6 of HHV-8 and MRV-5 viruses, vIL-17 of HVS virus, and vIL-10 of HHV-4, Bonobo-HV, RhLCV, and BaLCV viruses. The ability to acquire cytokine genes allows herpesviruses to implement unique strategies of avoiding the immune response and provides them an evolutionary advantage: more than 90% of the host population can be chronically infected with different herpesviruses. It is possible that the biological success of herpesviruses can be partially due to their cooperation with another group of viruses. This hypothesis emphasizes the importance of studies on the reciprocal influence of pathogens on their coinfection, as well as their impact on the host organism.
KEY WORDS: virokines, IL-6, IL-10, IL-17, herpesviruses, retroviruses, horizontal gene transferDOI: 10.1134/S0006297916110122
Abbreviations: BaLCV, baboon lymphocryptovirus; Bonobo-HV, bonobo herpesvirus; EBV, Epstein–Barr virus; HHV, human herpesvirus; HVS, herpesvirus saimiri; IFN, interferon; IL, interleukin; MRV, rhesus macaque rhadinovirus; RhCMV, rhesus macaque cytomegalovirus; RhLCV, rhesus macaque lymphocryptovirus; SaHV-2, Saimiriine herpesvirus 2; vIL, virokine, interleukin homolog.
Interaction between viruses and systems of host protection is crucial in
the evolution of both viruses and hosts. “Stealing” the
gene encoding a component of the host’s protection system is one
of the major strategies used by viruses for avoiding the host’s
immunity control [1]. During evolution, within the
viral genome a protein component of this gene can acquire new features,
for instance, it can become an inhibitor of the corresponding
protective system. Another strategy is also frequently realized when a
virus homolog retains the ability to transmit the signal to
non-infected cells involved in the antiviral immune response. In this
case, the virus uses the immune system cells and achieves a success in
retargeting the immune response and activating signaling pathways,
which promote its survival and replication. The term
“virokines” is applied to homologs of animal hosts whose
genes are present in the viral genomes [2]. These
proteins usually provide a selective advantage for their new owners,
the viruses, during their life cycle. Mechanisms of virus interaction
with cell are under active study, and the contribution of virokines to
the development of diseases are under active study, but their
evolutionary history, except high homology between the host and viral
genes, usually remains beyond the attention focus.
Herpesviruses (Herpesviridae family) were especially successful in borrowing genes of mammalian cytokines. The genome of herpesviruses is double-stranded DNA with length over 110 kb and contains about a hundred genes, the majority of which encode proteins that are necessary for avoiding the immune response, and frequently are “stolen” from the host [3]. This arsenal of immune regulators allows herpesviruses to exist for a long time in the host organism as a latent chronic infection. However, malignant transformation of the cells can be a side effect of the virus introduction into protective mechanisms.
The mechanism of borrowing genes use by herpesviruses needs separate consideration. The most obvious “know-how” of copying the host’s genes is recombination between the viral and host genomes during virus replication within the nucleus. A latent virus can also use nonhomologous recombination for repair of spontaneous double-stranded breaks: genomes of herpesviruses are found to contain many specific sequences detectable by the cellular repair systems [4]. In this case, the exon–intron structure of the “stolen” gene must be preserved and followed in the virus homologs. However, introns are present not in all genes borrowed from the host, thus only a quarter of genes of human herpesvirus 8 (HHV-8) contains introns [5]. It was reasonable to suppose that introns could be lost because of selection pressure, because there were also observed “transitional variants” of the structure, i.e. shortened introns or intron sets incomplete relatively to the cellular gene. However, another scenario also seems probable: the gene can enter the virus already in a spliced state without introns. This probability is indirectly confirmed by consideration of the gene regions on the border between the exon and intron. If the intron had been lost because of an occasional deletion, many insertions and deletions within the reading frame would have occurred in the beginning and ends of the virus gene regions homologous to the initial gene exons. These insertions and deletions would be caused by inaccurate removal of the intron at the splicing borders [6]. In reality, in the majority of cases of intron removal, the reading frame is neither elongated nor shortened; consequently, the DNA sequence of the gene really enters the viral genome in the spliced state due to reverse transcription. Herpesviruses themselves cannot realize reverse transcription, and endogenous or exogenous retrovirus acts as a mediator infecting the same cells as the herpesvirus.
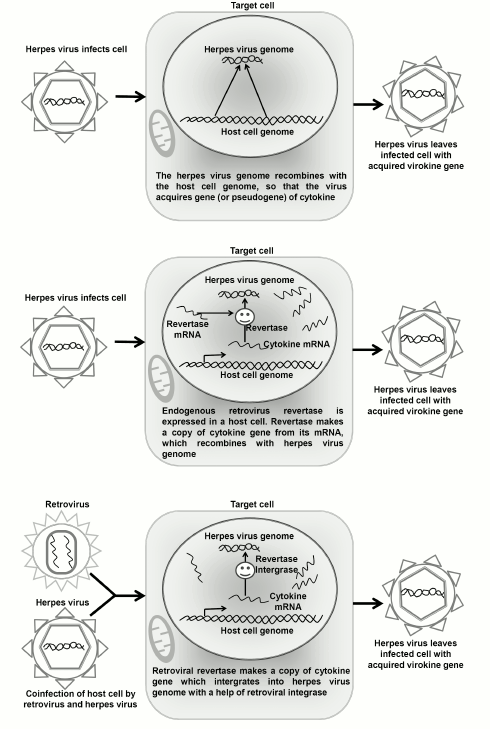
One has to understand that the initial gene of the cellular cytokine can enter the herpesvirus genome also directly, without involvement of retroelements, but under the influence of systems responsible for repair and nonhomologous recombination. In this case, the virokine gene will retain the initial exon–intron structure. For one-exon genes of virokines, we can suppose that a herpesvirus can borrow genes using several pathways (Fig. 1): (i) the activity of endogenous retroviruses in the germ line cells leads to origination in the host’s genome of processed pseudogenes, and after that the herpesvirus acquires a copy-pseudogene in the infected cells as a result of nonhomologous recombination with the host’s genome; (ii) an endogenous retrotransposon incorporates cDNA of the gene into the genome of a latent herpesvirus directly in the infected target cell; (iii) the reverse transcription and incorporation into the herpes viral genome are realized by a retrovirus co-infecting the target cell pre-infected with herpesvirus. In the present work, we compared these hypothetical scenarios of virokine acquisition by the known herpesvirus homologs IL-6, IL-17, and IL-10. For each virokine, we determined the systematic position of a host of the virus as the hypothetical donor of the virokine gene, performed a search for pseudogenes of the cytokines, and analyzed the probability of expression of endogenous and exogenous retroviruses in cells infected by herpesviruses to show the most probable scenario.
Fig. 1. Three probable scenarios of virokine gene acquisition by herpesvirus.
MATERIALS AND METHODS
Nucleotide and amino acid sequences of the virokines were taken from annotated genomes of herpesviruses (human herpesvirus 8, Saimiriine herpesvirus 2, and human herpesvirus 4), nucleotide and amino acid sequences of cytokines IL-6, IL-17, and IL-10 of primates (Homo sapiens, Pan troglodytes, Gorilla gorilla, Pongo abelii, Macaca mulatta, Papio anubis, Chlorocebus sabaeus, Callithrix jacchus, Saimiri boliviensis, Aotus nancymaae, Microcebus murinus, and Otolemur garnettii) and of rodents (Mus musculus and Rattus norvegicus) were taken from annotated genomes of the corresponding organisms. The list of identification numbers on the NCBI site for protein sequences of all organisms is presented in the table in the Supplement to this report on the site of the journal (http://protein.bio.msu.ru/biokhimiya) and Springer site (Link.springer.com). The percent of identical amino acids for the global alignment of sequences of virokines and of their cellular homologs were calculated using the EMBOSS Needle algorithm [7].
Multiple alignment of amino acid sequences of virokines and their homologs was performed using the MAFFT algorithm [8, 9]. Based on the multiple alignment, for each cytokine the most relevant evolutionary model was chosen, and then by the maximum likelihood method a phylogenetic tree was reconstructed based on this model. Stability of the tree topology was tested by bootstrapping (100 repeats). Portions of the amino acid sequences with incompletely covered alignments (“gaps”) were excluded from the analysis. Rodents were used as an outgroup for primates. The best evolutionary model was chosen and the phylogeny was reconstructed by the maximum likelihood method in the MEGA6 program [10]. Phylogenetic trees for every virokine were also constructed by Bayesian analysis with a relaxed molecular clock using the BEAST software package [11]. The trees were constructed using the same evolutionary models as for the reconstruction by the maximum likelihood method, and at least two launchings were performed of the algorithm with chain length of 10 million.
The search for sequences of pseudogenes of IL-6, IL-17, and IL-10 cytokines was performed in the human, rhesus macaque, and Saimiri genomes using BLAT instruments [12] and blastn at the NCBI site. The time of homolog divergence was assessed based on the divergence time of the taxons using the TimeTree service (http://timetree.org/) [13].
RESULTS
To determine the probable mechanism of virokine origin, we analyzed several genes of herpes virokines for the presence of introns and for the presence in the present-day host species genome of intron-free pseudogenes, and for the type of target cells and the presence of retroviruses co-infecting the same targets. These data and the supposed existence time and the systematic position of the hosts as of virokines sources are presented in the table (for brevity, only three from twelve IL-10 homologs are presented). We analyzed the present-day herpesvirus virokines from the standpoint of mechanisms of their acquisition and also of donor genomes and established that the most probable source of the viral IL-6 was a common ancestor of the dry-nosed monkeys (Haplorhini), of viral IL-17 — a common ancestor of the flat-nosed monkeys (Platyrrhini), and of viral IL-10 of HHV-4 – an ancestor of apes (Hominidae). Specific features of evolution and clinical significance of individual virokines are considered later in the corresponding subsections.
Characteristics of herpesvirus virokines
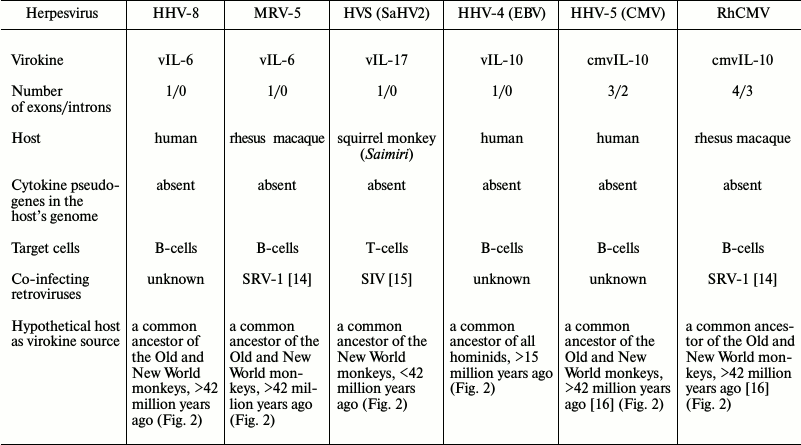
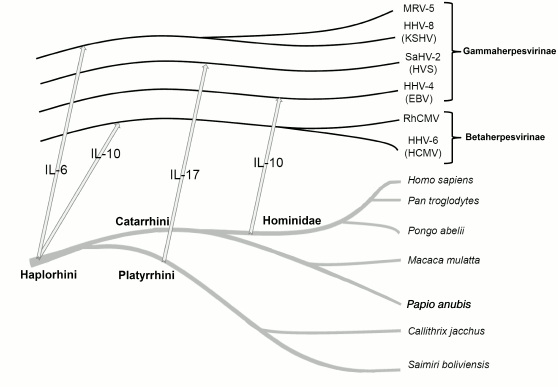
Fig. 2. Evolution of herpesviruses was collinear to evolution of their hosts and was accompanied by repeated transfer of virokine genes from the host to the virus. The arrows show the virokine gene transfer from host to herpesvirus.
Viral IL-6. Homologs of IL-6 are present in the genome of human herpesvirus 8 (HHV-8), which is also known as Kaposi’s sarcoma-associated herpesvirus, and in rhadinoviruses of some macaque species, especially of rhesus macaque (MRV-5). MRV-5 belongs to the so-called RV2-line of monkey rhadinoviruses that also includes rhadinovirus of the pig-tailed macaque (MneRV2), Japanese macaque (MfuRV2), and long-tailed macaque (MfaRV2). Together with herpesviruses of macaque retroperitoneal fibromatosis (RFHV) and with rhadinoviruses of other species of the Old World monkeys (chimpanzee, gorilla), HHV-8 forms the RV1-lineage, which is different from RV2 by the presence of additional genes. Viral IL-6 can be found in all human and macaque viruses belonging to the RV1- and RV2-lineages, but it is absent in rhadinoviruses of other monkey species [17]. Homologs of IL-6 of rhadinoviruses are free of introns. B-cells are the major reservoir of a latent HHV-8 infection with expressed early viral genes responsible for avoiding apoptosis and cytotoxic immunity and for inhibition of production of antiviral cytokines. The viral IL-6 acts in an autocrine manner, increasing cell viability and inducing expression of its IL-6 [18]. The virokine can suppress the cellular antiviral defense due to inhibition of IFNα production, but it should be noted that the viral IL-6 gene promoter contains an IFNα-dependent element, thus vIL-6 regulates the host’s IFNα through negative feedback [19]. The etiological role of vIL-6 has been established for such tumors as primary effusion lymphoma (PEL) and multicentric Castleman disease (MCD). The viral IL-6 can also cause development of syndrome of inflammatory cytokines that are associated with HHV-8 due to increasing production of the human IL-6, which leads to cytokine storm and systemic inflammation [20]. Rhadinovirus of rhesus macaque is a natural infectious agent – about 90% of animals of these species are shown to be seropositive for this virus in two American regional centers of studies on primates. However, oncological diseases similar to human PEL and MCD seldom appear in them; usually they develop on decrease in immune defense caused by simian immunodeficiency virus (SIV) [21]. Nevertheless, rhadinovirus infection during coinfection with SIV is the most relevant model of Kaposi’s sarcoma in AIDS-positive patients [22].
The amino acid sequence of vIL-6 (open reading frame K2 HHV-8) has only 25% identity with the sequence of human IL-6, but it has likeness on the level of the secondary and tertiary structure: the virokine has a motif containing four α-helices that is specific for all cytokines of this family. However, the vIL-6 virokine binding with cellular receptors has some specific features lacking in other proteins of the family [23]. As discriminated from the cellular IL-6, which binds with a specific high affinity IL-6R receptor and already within the complex interacts with gp130 molecule [24] transmitting into the cell the signal from the receptor through the molecular cascade JAK/STAT, the virokine can bind directly with gp130, producing tetrameric signaling complexes (2vIL-6–2gp130) [25]. However, for transmitting the signal, the virokine concentration has to be significantly higher than values characteristic for the cellular IL-6 [26].
The amino acid sequence of an IL-6 homolog from the rhesus macaque rhadinovirus has 36% identity with the host’s cytokine and 27% identity with K2 HHV-8; therefore, it can be supposed that the mechanism of interaction of this virokine with cellular receptors should be similar to that of the HHV-8 virokine. Phylogenetic analysis of the amino acid sequences of these two virokines, as well as of IL-6 from cytokines of primates, revealed (Fig. S1 of Supplement) that both cytokines are clustered into a separate branch located between the branch of the wet-nosed monkeys (lemurs and lorises) and the branch of dry-nosed monkeys (Haplorhini) (including flat-nosed (Platyrrhini) and narrow-nosed (Catarrhini) monkeys). According to literature data, the divergence of HHV-8 and MRV-5 viruses occurred ~38 million years ago [27], earlier than the divergence of human and rhesus macaque evolutionary branches (28 million years ago), but later than the divergence of flat-nosed and narrow-nosed monkeys (42 million years ago). Thus, virokines homologous to IL-6 belong to the most ancient herpesvirus virokines. We suppose that a common ancestor of HHV-8 and MRV-5 viruses received the gene of this cytokine from a common ancestor of dry-nosed monkeys who lived earlier than 42 million years ago, before the divergence of the branches of the Old and New World monkeys. The high divergence degree between two virokines suggests that the divergence of hosts was accompanied by divergence of viruses, which did not change the specificity, and evolution of the viruses was parallel to evolution of the hosts.
Viral IL-17. The IL-17 homolog has been found only in the genome of Saimiriine herpesvirus 2 (HVS or SaHV-2), which also belongs to rhadinoviruses and is free of introns. For squirrel saimiri, HVS is a natural infectious agent, whereas for other primates (tamarins, marmosets, macaques) this virus can cause malignant T-cell diseases. It has been shown that the HVS-C virus lineage is capable of transforming human T-cells [28]. The etiological role of the viral IL-17 has been established for human idiopathic pulmonary fibrosis [29].
The amino acid sequence of viral IL-17 (ORF13 of Saimiriine herpesvirus 2) has 74% identity with IL-17А of saimiri and 71% identity with human IL-17А, and the viral homolog can bind to the cellular receptor IL-17RA. Note that the IL-17 receptor of mouse IL-17RA was first identified and cloned just due to its binding with vIL-17 [30]. It was established later that in animals the receptor is usually present as an IL-17RA/IL17RC heterodimer and can interact with the homodimeric cellular cytokine IL-17A. The structure of the receptor complex has not been studied in detail, but the likeness of pictures of the tissue damage in chronic pulmonary diseases contributed by IL-17 and in idiopathic pulmonary fibrosis associated with the SaHV-2 infection indirectly confirms that the viral IL-17 not only binds a subunit of the receptor, but also transmits a proinflammatory signal similar to the cellular one [29, 31].
Analysis of six IL-17 homologs known in mammals revealed that the cellular IL-17A is the closest to the virokine, which is why we have taken for the phylogenetic analysis the sequences only of IL-17А genes of primates and rodents. On the phylogenetic tree, this virokine is clustered in the same branch with cytokines of flat-nosed monkeys (Fig. S2 of Supplement), but is significantly remote from them. Therefore, its acquisition can be considered a rather early event; it most probably occurred soon after the divergence of flat-nosed and narrow-nosed monkeys (42 million years ago).
Viral IL-10. Homologs of IL-10 have been found in more than twenty viruses belonging to three different families: 12 viruses of Herpesviridae, seven viruses of Poxviridae, and two viruses of Alloherpesviridae. Such wide distribution of IL-10 homologs among different families of viruses indicates a significant advantage acquired by a virus capable of producing immunosuppressive cytokine in the infected cells and evidences independent borrowing of the IL-10 gene during evolution, which could have occurred through different mechanisms. Thus, the best studied IL-10 homolog of human herpesvirus 4 (Epstein–Barr virus, HHV-4) and of the related species that also occur in other primates (bonobo herpesvirus (Bonobo-HV), rhesus macaque lymphocryptovirus (RhLCV), baboon lymphocryptovirus (BaLCV)) has over 90% identity with the host’s gene sequence and does not contain introns. However, homologs of IL-10 found in human and monkey cytomegaloviruses contain from one to three introns and encode proteins with amino acid sequences that have only 20-30% identity with the initial cytokine sequence. In addition, virokines with amino acid sequences possessing both high and low similarity degree with the cellular cytokines are capable of interacting with cellular receptors of IL-10R1 due to the conservative tertiary structure [32]. The phylogenetic tree for sequences of the typical intron-free virokine IL-10 of apes encoded by the BCRF1 locus of the HHV-4 virus indicates (Fig. S3 of Supplement) that the virokine gene donor was a member of Hominidae family and lived before their divergence began, i.e. ~15 million years ago.
DISCUSSION
Analysis of probable mechanisms of virokine acquisition. Evolution of herpesviruses closely followed evolution of their hosts and was accompanied by repeated transfers of virokine genes. Information about the most probable time of these transfers obtained in our work and taken from the literature is presented in Fig. 2.
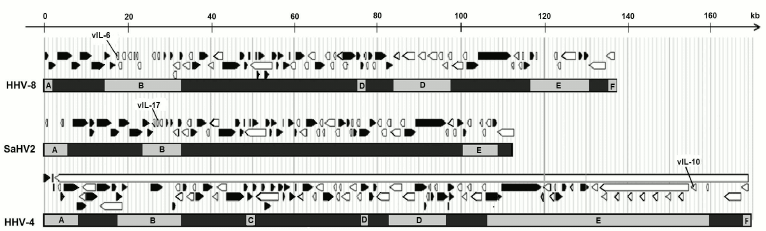
Among the hosts’ genes, not only genes of cytokines were acquired by herpesviruses. Thus, the HHV-8 virus has genes of thymidine kinase, thymidylate synthase, and dihydrofolate reductase, which are necessary for synthesis of pyrimidine nucleotides, ubiquitin ligases K3 and K5, etc. It is interesting that the genes of herpesviruses acquired by horizontal transfer are often linked between themselves: the above-mentioned genes of the herpesvirus IL-6, thymidylate synthase, and dihydrofolate reductase in the genome of HHV-8 are located within a small area with 4 kb size. The herpesvirus saimiri in the genome homologous region instead of the proinflammatory virokine IL-6 gene has the gene of another proinflammatory virokine IL-17, which indirectly suggests the existence of similar patterns of regulation that are required by viruses for successful use of proinflammatory cytokines. Genomes of certain herpesviruses possessing virokines are schematically presented in Fig. 3.
Fig. 3. Schematic imaging of genomes of herpesviruses carrying virokine genes. Light-gray rectangles with Latin letters A-F indicate divergent loci of herpesviruses, between them there are conservative genes specific for all herpesviruses. Above them, protein open reading frames are shown as arrows: black ones – from left to right, white ones — from right to left. In rhadinoviruses (HHV-8 and SaHV-2), virokines are located in the divergent locus B. The viral IL-10 is located in the divergent locus E of HHV-4 virus (Epstein–Barr virus, EBV). After data of Nicolas et al. [33] (with modifications).
We searched for pseudogenes of cytokines IL-6, IL-17, and IL-10 in the sequenced genome of present-day primates using BLAT and blastn algorithms, but did not find any homologous sequence with length above several tens of nucleotides. It seems that they were absent also in the genomes of extinct primates who were donors of the virokine genes. This favors the hypothesis that the genes IL-6, IL-17, and IL-10 “were stolen” by herpesviruses in target cells with involvement of retroviruses or endogenous retroviruses (the second and third mechanisms shown in Fig. 1). Retroviruses of primates that infect lymphoid tissue, first, T-cells (viruses of immunodeficiency and T-cell leukemia) and less frequently, В-cells (retrovirus 1 of primates), are widely distributed and well known. Expression of endogenous retroviral reverse transcriptase in lymphoid tissue is poorly studied, but because the majority of retrotransposons are located in the heterochromatin part of the genome, it can be expected that their activity in differentiated cells is a rare event associated with the entrance of retroelements into introns of the genes with a high level of tissue-specific expression. Thus, expression of some kinds of HERV-K retrotransposons is described in human peripheral blood lymphoid cells, but for the expression they need to be specifically activated [34]. Nevertheless, coinfection of the same host by retrovirus and herpesvirus is a regular event reproduced many times during evolution. Thus, it has been shown recently that genes of specific retroviral superantigens nonspecifically activating T-cell receptors can be transferred horizontally into the genome of herpesviruses beyond the context associated with the major histocompatibility complex (MHC) molecule of the peptide [35]. Such molecules nonspecifically activate T-cell immunity and thus prevent its targeting just the viral antigens, i.e. partially act like proinflammatory cytokines. The possibility of horizontal transfer of genes regulating immune reactions of the host between given groups of viruses is favorable for the hypothesis about arising of virokine genes in the course of coinfection with retroviruses. It is interesting that IL-6 promotes the polarization of T-lymphocytes to Th17 and suppresses the polarization to regulatory T-cells [36]. Thus, both types of proinflammatory herpesvirus virokines, vIL-6 and vIL-17, act through the same mechanism.
Having in mind the above-mentioned information, the horizontal transfer from host to virus in the target cell due to retroviral revertase activity seems probable for intron-free genes of lymphotropic herpesvirus virokines: vIL-6 of HHV-8 and MRV-5 viruses, vIL-17 of SaHV-2 virus, and vIL-10 of HHV-4, Bonobo-HV, RhLCV, and BaLCV viruses. For intronized homologs of herpesvirus vIL-10, the transfer into the viral genome is probable through the nonhomologous recombination system. However, the probability of such transfer of genes from host to a virus is high only in the scale of evolution, whereas it is extremely low for an individual case of herpesvirus infection. From this standpoint, it is interesting to attempt to assess experimentally in vitro the frequency of transfer of new host’s genes into the genome of a herpesvirus. In the easiest case, one can use a cell line concurrently coexpressing GFP (a marker gene) and revertase (analog of coinfection with retrovirus).
Advantages gained by a virus after acquisition of the cytokine gene are associated either with suppression of the immune response due to immunosuppressive IL-10, or with switching this response to Th17- axis, which is safe for the virus under the influence of proinflammatory cytokines. Since virokines significantly contribute to pathogenesis of some diseases associated with herpesviruses, it is clinically important to inhibit them, and this approach is already used. Thus, different inhibitors of the IL-6 signaling pathway are successfully used for treatment of the lymphoproliferative Castleman’s disease [37]. Although cellular receptors are capable to of discriminating virokines and true cytokines by their affinity that determines the difference in the effective working concentration [26], arising of an ideal system of cytokine signaling protected against cytokines seems to be impossible. Considering the higher rate of evolution of herpesvirus genes and the significance of virokines for their successful infection, from the “Red Queen hypothesis” standpoint, cytokines and their receptors will not be able “to break away” from virokines in the evolutionary race. However, significant differences in the primary sequences of virokines and their cellular prototypes allow us to expect that discriminating epitopes should exist, allowing the production of selective virokine-directed antibodies and cytolysis of virokine-expressing cells.
Acknowledgements
We are grateful to S. A. Nedospasov for fruitful discussion of the material and valuable remarks.
The work was supported by the Russian Science Foundation (project No. 14-25-00160).
REFERENCES
1.Kunin, E. V. (2014) Logic of Case. On the Nature
and Origin of Biological Evolution [in Russian], Tsentrolitograf,
Moscow.
2.Kotwal, G. J. (1999) Virokines: mediators of
virus-host interaction and future immunomodulators in medicine,
Arch. Immunol. Ther. Exp. (Warsz.), 47, 135-138.
3.Alcami, A. (2003) Viral mimicry of cytokines,
chemokines and their receptors, Nat. Rev. Immunol., 3,
36-50.
4.Brown, J. C. (2014) The role of DNA repair in
herpesvirus pathogenesis, Genomics, 104, 287-294.
5.Zheng, Z.-M. (2003) Split genes and their
expression in Kaposi’s sarcoma-associated herpesvirus, Rev.
Med. Virol., 13, 173-184.
6.Farlow, A., Meduri, E., and Schlotterer, C. (2011)
DNA double-strand break repair and the evolution of intron density,
Trends Genet., 27, 1-6.
7.Rice, P., Longden, I., and Bleasby, A. (2000)
EMBOSS: the European molecular biology open software suite, Trends
Genet., 16, 276-277.
8.Katoh, K., Misawa, K., Kuma, K. I., and Miyata, T.
(2002) MAFFT: a novel method for rapid multiple sequence alignment
based on fast Fourier transform, Nucleic Acids Res., 30,
3059-3066.
9.Katoh, K., Kuma, K., Toh, H., and Miyata, T. (2005)
MAFFT version 5: improvement in accuracy of multiple sequence
alignment, Nucleic Acids Res., 33, 511-518.
10.Tamura, K., Stecher, G., Peterson, D., Filipski,
A., and Kumar, S. (2013) MEGA6: Molecular Evolutionary Genetics
Analysis version 6.0, Mol. Biol. Evol., 30,
2725-2729.
11.Drummond, A. J., and Rambaut, A. (2007) BEAST:
Bayesian evolutionary analysis by sampling trees, BMC Evol.
Biol., 7, 1.
12.Kent, W. J. (2002) BLAT – the BLAST-like
alignment tool, Genome Res., 12, 656-664.
13.Hedges, S. B., Dudley, J., and Kumar, S. (2006)
TimeTree: a public knowledge base of divergence times among organisms,
Bioinformatics, 22, 2971-2972.
14.Maul, D. H., Zaiss, C. P., MacKenzie, M. R.,
Shiigi, S. M., Marx, P. A., and Gardner, M. B. (1988) Simian retrovirus
D serogroup 1 has a broad cellular tropism for lymphoid and nonlymphoid
cells, J. Virol., 62, 1768-1773.
15.Rogers, D. L., McClure, G. B., Ruiz, J. C., Abee,
C. R., and Vanchiere, J. A. (2015) Endemic viruses of squirrel monkeys
(Saimiri spp.), Comp. Med., 65, 232-240.
16.Ouyang, P., Rakus, K., van Beurden, S. J.,
Westphal, A. H., Davison, A. J., Gatherer, D., and Vanderplasschen, A.
F. (2014) IL-10 encoded by viruses: a remarkable example of independent
acquisition of a cellular gene by viruses and its subsequent evolution
in the viral genome, J. Gen. Virol., 95, 245-262.
17.Bruce, A. G., Thouless, M. E., Haines, A. S.,
Pallen, M. J., Grundhoff, A., and Rose, T. M. (2015) Complete genome
sequence of pig-tailed macaque rhadinovirus 2 and its evolutionary
relationship with rhesus macaque rhadinovirus and human herpesvirus
8/Kaposi’s sarcoma-associated herpesvirus, J. Virol.,
89, 3888-3909.
18.Sin, S. H., and Dittmer, D. P. (2012) Cytokine
homologs of human gammaherpesviruses, J. Interferon Cytokine
Res., 32, 53-59.
19.Chatterjee, M., Osborne, J., Bestetti, G., Chang,
Y., and Moore, P. S. (2002) Viral IL-6-induced cell proliferation and
immune evasion of interferon activity, Science, 298,
1432-1435.
20.Polizzotto, M. N., Uldrick, T. S., Wyvill, K. M.,
Aleman, K., Marshall, V., Wang, V., Whitby, D., Pittaluga, S., Jaffe,
E. S., Millo, C., Tosato, G., Little, R. F., Steinberg, S. M., Sereti,
I., and Yarchoan, R. (2016) Clinical features and outcomes of patients
with symptomatic Kaposi sarcoma herpesvirus (KSHV)-associated
inflammation: prospective characterization of KSHV inflammatory
cytokine syndrome (KICS), Clin. Infect. Dis., 62,
730-738.
21.Damania, B., and Desrosiers, R. C. (2001) Simian
homologs of human herpesvirus 8, Philos. Trans. R Soc. Lond. B:
Biol. Sci., 356, 535-543.
22.Estep, R. D., and Wong, S. W. (2013). Rhesus
macaque rhadinovirus-associated disease, Curr. Opin. Virol.,
3, 245-250.
23.Gorshkova, E. A., Nedospasov, S. A., and Shilov,
E. S. (2016) Evolutionary plasticity of the IL-6 family cytokines,
Mol. Biol., 50, in press.
24.Boulanger, M. J., Chow, D. C., Brevnova, E. E.,
and Garcia, K. C. (2003) Hexameric structure and assembly of the
interleukin-6/IL-6 α-receptor/gp130 complex, Science,
300, 2101-2104.
25.Chow, D. C., Brevnova, L., He, X. L., Martick, M.
M., Bankovich, A., and Garcia, K. C. (2002) A structural template for
gp130-cytokine signaling assemblies, Biochim. Biophys. Acta,
1592, 225-235.
26.Sakakibara, S., and Tosato, G. (2011) Viral
interleukin-6: role in Kaposi’s sarcoma-associated
herpesvirus-associated malignancies, J. Interferon Cytokine
Res., 31, 791-801.
27.McGeoch, D. J., Gatherer, D., and Dolan, A.
(2005) On phylogenetic relationships among major lineages of the
Gammaherpesvirinae, J. Gen. Virol., 86, 307-316.
28.Ensser, A., Thurau, M., Wittmann, S., and
Fickenscher, H. (2003) The genome of herpesvirus saimiri C488 which is
capable of transforming human T-cells, Virology, 314,
471-487.
29.Folcik, V. A., Garofalo, M., Coleman, J.,
Donegan, J. J., Rabbani, E., Suster, S., Nuovo, A., Magro, C. M., Di
Leva, G., and Nuovo, G. J. (2014) Idiopathic pulmonary fibrosis is
strongly associated with productive infection by herpesvirus saimiri,
Modern Pathol., 27, 851-862.
30.Yao, Z., Fanslow, W. C., Seldin, M. F., Rousseau,
A. M., Painter, S. L., Comeau, M. R., and Spriggs, M. K. (1995)
Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a
novel cytokine receptor, Immunity, 3, 811-821.
31.Gasse, P., Riteau, N., Vacher, R., Michel, M. L.,
Fautrel, A., Di Padova, F., Fick, L., Charron, S., Lagente, V., Eberl,
G., and Le Bert, M. (2011) IL-1 and IL-23 mediate early IL-17A
production in pulmonary inflammation leading to late fibrosis, PLoS
One, 6, e23185.
32.Ouyang, P., Rakus, K., Van Beurden, S. J.,
Westphal, A. H., Davison, A. J., Gatherer, D., and Vanderplasschen, A.
F. (2014) IL-10 encoded by viruses: a remarkable example of independent
acquisition of a cellular gene by viruses and its subsequent evolution
in the viral genome, J. Gen. Virol., 95, 245-262.
33.Nicholas, J., Zong, J. C., Alcendor, D. J.,
Ciufo, D. M., Poole, L. J., Sarisky, R. T., Chiou, C. J., Zhang, X.,
Wan, X., Guo, H. G., Reitz, M.S., and Hayward, G. S. (1998) Novel
organizational features, captured cellular genes, and strain
variability within the genome of KSHV/HHV8, J. Natl. Cancer Inst.
Monogr., 23, 79-88.
34.Brinzevich, D., Young, G. R., Sebra, R., Ayllon,
J., Maio, S. M., Deikus, G., Chen, B. K., Fernandez-Sesma, A., Simon,
V., and Mulder, L. C. (2014) HIV-1 interacts with HERV-K (HML-2)
envelopes derived from human primary lymphocytes, J. Virol.,
88, 6213-6223.
35.Aswad, A., and Katzourakis, A. (2015) Convergent
capture of retroviral superantigens by mammalian herpesviruses, Nat.
Commun., 6, 8299.
36.Kimura, A., and Kishimoto, T. (2010) IL-6:
regulator of Treg/Th17 balance, Eur. J. Immunol., 40,
1830-1835.
37.Rossi, J. F., Lu, Z. Y., Jourdan, M., and Klein,
B. (2015) Interleukin-6 as a therapeutic target, Clin. Cancer
Res., 6, 1248-1257.
Supplementary Table, Figures S1, S2 and S3 (PDF)