REVIEW: The Role of Cytokines in the Development of Atherosclerosis
A. R. Fatkhullina, I. O. Peshkova, and E. K. Koltsova*
Fox Chase Cancer Center, 333 Cottman Avenue, Philadelphia, PA, USA; E-mail: Ekaterina.Koltsova@fccc.edu* To whom correspondence should be addressed.
Received May 31, 2016; Revision received August 1, 2016
Atherosclerosis contributes to the development of many cardiovascular diseases, which remain the leading cause of death in developed countries. Atherosclerosis is a chronic inflammatory disease of large and medium-sized arteries. It is caused by dyslipidemia and mediated by both innate and adaptive immune responses. Inflammation is a key factor at all stages of atherosclerosis progression. Cells involved in pathogenesis of atherosclerosis were shown to be activated by soluble factors, cytokines, that strongly influence the disease development. Pro-inflammatory cytokines accelerate atherosclerosis progression, while anti-inflammatory cytokines ameliorate the disease. In this review, we discuss the latest findings on the role of cytokines in the development and progression of atherosclerosis.
KEY WORDS: cardiovascular diseases, atherosclerosis, inflammation, immune cells, adhesion molecules, cytokinesDOI: 10.1134/S0006297916110134
Abbreviations: ApoE, apolipoprotein E; CVDs, cardiovascular diseases; G-CSF, granulocyte colony-stimulating factor; ICAM-1, intercellular adhesion molecule 1; IFN-γ, interferon-γ; IL, interleukin; ILCs, innate lymphoid cells; LDLs, low-density lipoproteins; MCP-1 (CCL2), monocyte chemoattractant protein-1; NK cells, natural killer cells; oxLDLs, oxidized low-density lipoproteins; SMCs, smooth muscle cells; SOCS, suppressor of cytokine signaling; TGFβ, transforming growth factor beta; Th cells, T helper cells; TLR, toll-like receptor; TNF-α, tumor necrosis factor-alpha; Treg cells, regulatory T cells; VCAM-1, vascular adhesion molecule 1.
CARDIOVASCULAR DISEASES
Cardiovascular diseases (CVDs) remain the leading cause of death in industrially developed countries [1]. The number of CVD-related deaths has also been increasing in the developing world. Factors that drive CVD development include hyperlipidemia (lipid and carbohydrate metabolism disorder) caused by obesity or hereditary predisposition (family hyperlipidemia), arterial hypertension, diabetes mellitus, age, smoking, sedentary lifestyle, stress, or a combination of these factors. Atherosclerosis plays a predetermining role in the pathogenesis of the two most common CVDs – cardiac ischemia and cerebrovascular disease [2] (World Health Organization; http://www.who.int/mediacentre/factsheets/fs317/ru/). For many years, high blood pressure and elevated blood levels of cholesterol have been considered the major factors promoting atherosclerosis. However, recent studies have convincingly demonstrated that chronic inflammation also plays a key role in the pathogenesis of atherosclerosis [3].
ROLE OF INFLAMMATION AND IMMUNE CELLS IN ATHEROSCLEROSIS
The arterial wall is composed of three layers: the inner layer – intima, the intermediate layer – media, and the external layer – adventitia. Intima consists of a single layer of endothelial cells, thin basal membrane, and subendothelial layer of collagen fibers. Media is formed by smooth muscle cells (SMCs) and a network of elastin and collagen fibers. Adventitia is the outer layer and comprisized mostly of loose connective tissue. The atherosclerosis is characterized by the formation of atherosclerotic plaques in subendothelial layer, SMC proliferation, accumulation of activated immune cells, and thickening of adventitia at the site of plaque formation (figure). Various immune cells are normally present in the arterial wall; however, their number increases significantly during atherosclerosis progression (figure, panels (a) and (b)). Under normal conditions, immune cells migrate into the aortic wall and return to the circulation [4], thereby “patrolling” the tissue. At early stages of atherosclerosis, high concentration of low-density lipoproteins (LDLs) in the plasma, LDL accumulation in the aortic wall with subsequent LDL oxidation into oxLDLs, and high blood pressure activate endothelial cells, promote the expression of adhesion molecules, and facilitate the migration of monocytes into the aortic wall [5]. Monocytes differentiate into macrophages that engulf oxLDLs and convert into lipid-filled foam cells. Accumulation of modified LDLs by macrophages activates cytokine production by these cells. Cytokines promote the influx and activation of other inflammatory cells and mediate their retention in the plaque, leading to further accumulation of proinflammatory cells in the plaque and surrounding adventitia (figure, panel (b)).
a) Arterial cell wall is composed of three layers: intima (internal), media (intermediate), and adventitia (external). Healthy arterial wall is characterized by the presence of a small number of immune cells, mostly in the adventitia. b) Atherosclerosis progression is accompanied by the accumulation of various immune cells in the intima (atherosclerotic plaque) and adjacent adventitia. These immune cells produce cytokines that promote local inflammation in the vascular wall, resulting in the growth of atherosclerotic plaque and eventually its rupture
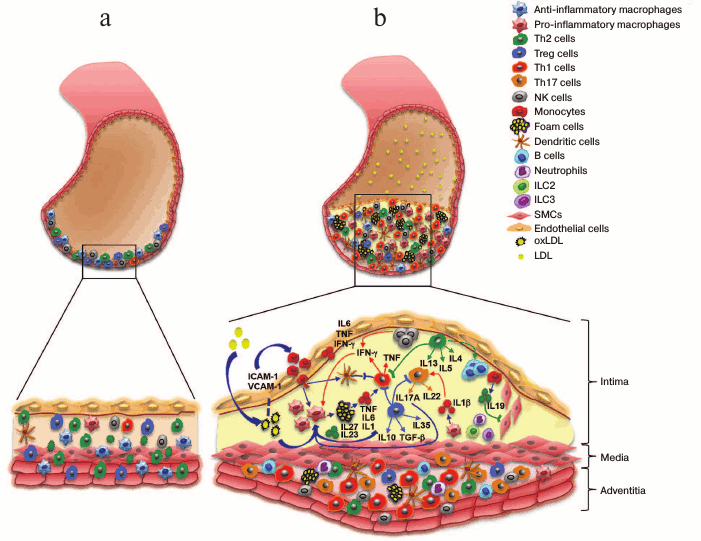
As in other tissues, the wall of a healthy artery contains resident macrophages that originate from the yolk sac during embryonic development [6]. Atherosclerosis is characterized by a high level of local proliferation of both resident and differentiated from monocytes macrophages [7-9].
Beside monocytes, other myeloid cells were shown to mediate inflammatory changes in the aortic wall [7, 10-12]. Recent studies have shown an important role of neutrophil extracellular traps (NETs), composed of extracellular DNA and neutrophil proteins, in the activation of interleukin (IL-1) production and inflammation in atherosclerosis [10].
Cells of adaptive immunity (T and B lymphocytes) also play an important role in the development of inflammation in the vessel wall [5]. Different types of T helper (Th) cells have been found in the aortic wall. The number of these cells, state of their activation, and the array of produced by them cytokines change along the disease progression. The role of Th cells in atherosclerosis will be discussed below. B lymphocytes (B1 and B2) are present in both healthy and atherosclerotic aortas. During the development of atherosclerosis, B1 cells perform protective functions by producing antibodies against various lipids, while B2 cells are pathogenic [5, 13].
Later stages of atherosclerosis are characterized by so-called unresolved inflammation that is maintained by various factors including increased levels of oxLDLs and high blood pressure [14].
The distinguishing feature of advanced atherosclerosis is progressive accumulation of foam cells in plaques. Foam cell are formed from macrophages because of excessive lipid accumulation by the latter, they cannot leave the plaque and eventually die, mostly via in situ necrosis leading to the formation of the necrotic nucleus. The necrotic nucleus destabilizes the compact structure of the plaque and causes its rapture leading to thrombus formation, which in turn can result in complete vessel blockage and cardiovascular complications, such as myocardial infarction and stroke [15].
The most commonly used animal models for studying atherosclerosis are mice with knockout of apolipoprotein E gene (Apoe–/–) or LDL receptor gene (Ldlr–/–). Mice lacking lipid-binding ApoE develop atherosclerosis spontaneously; the process is exacerbated by a high-lipid diet. In Ldlr–/– mice, atherosclerosis is induced by a high-lipid diet. In these mice, the absence of LDLR in non-hematopoietic cells, namely hepatocytes, is a prerequisite for the development of atherosclerosis. The Ldlr–/– model has certain advantages and allows bone marrow transplantation from other knockout models.
ROLE OF CYTOKINES IN THE DEVELOPMENT OF ATHEROSCLEROSIS
Cytokines are protein mediators, which participate in many physiological processes and play a key role in inflammation. Cytokines are a very diverse group of molecules that includes over 100 secreted factors that could be subdivided into several classes: interleukins (ILs), tumor necrosis factors (TNFs), interferons (IFNs), transforming growth factors (TGFs), colony-stimulating factors (CSFs), and various chemokines. Cytokines are produced by T cells, monocytes, macrophages, and platelets, as well as by endothelial cells (ECs), SMCs, and adipocytes, in response to inflammation and other stimuli. An increased production of pro-inflammatory cytokines is related to disease progression and promotes atherosclerosis [16]. Cytokine-induced activation of ECs can cause endothelium dysfunction accompanied by upregulation of adhesion molecules and chemokines, which promotes migration of immune cells (monocytes, neutrophils, lymphocytes) into atherosclerosis site [17]. Cytokines also affect the function of SMCs by promoting their growth, proliferation, and migration [15]. At later stages of atherosclerosis, pro-inflammatory cytokines promote destabilization of atherosclerotic plaques, apoptosis of various cells, and matrix degradation, thereby accelerating plaque breakage and thrombus formation [14, 15].
For many years, cytokines produced by T helper cells were classified into two groups: cytokines produced by type I T helper cells (Th1) and cytokines produced by type II T helper cells (Th2). Recent studies showed the importance of type 17 T cells (Th17 cells) and regulatory T (Treg) cells in the pathogenesis of various immune disorders.
The major role of Th1 cytokines is activation of macrophages and T cells; cytokines produced by Th2 cells stimulate humoral response [18]. Th17 cells regulate infiltration and activation of myeloid cells in the inflammation locus [19]. Treg cells inhibit the activation of all types of T cells and suppress immune responses mediated by T cells [18] (table).
Role of cytokines in pathogenesis of atherosclerosis
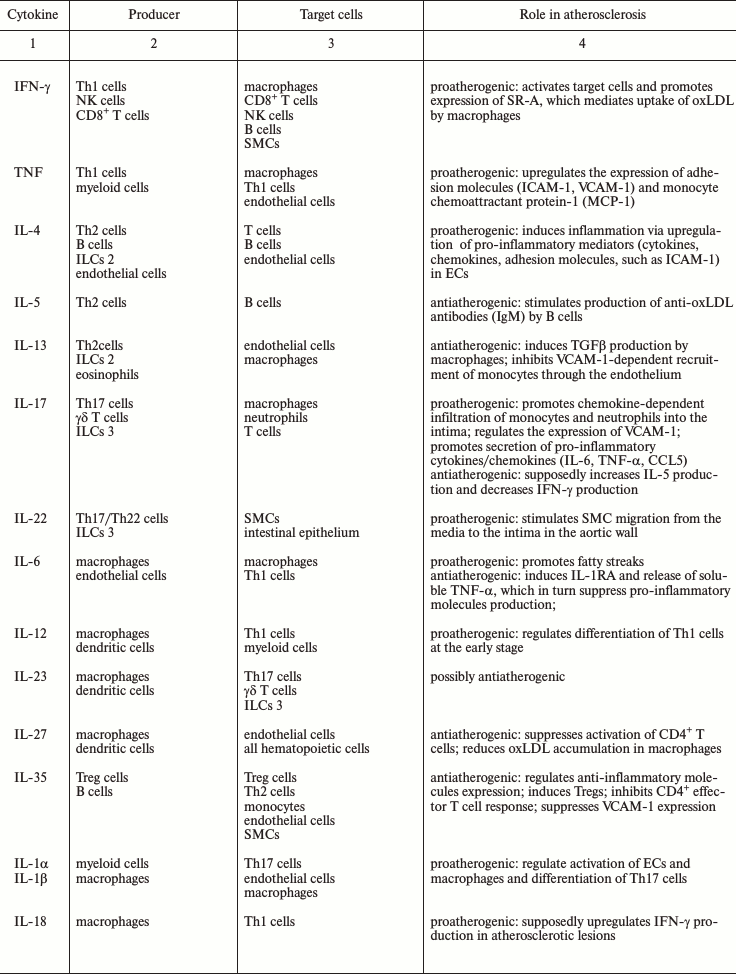

Type I cytokines. Type I cytokines are produced by Th1 cells (CD4+ T cells) and include interferon-gamma (IFN-γ) and tumor necrosis factor-α (TNF-α).
Interferon-gamma (IFN-γ). CVD patients exhibit increased blood levels of IFN-γ [20]. IFN-γ production is especially elevated in the atherosclerotic plaque, where IFN-γ is produced by Th1 cells (CD4+ cells), cytotoxic T cells (CD8+ cells), and natural killer (NK) cells [21].
IFN-γ has been found to act as a pathogenetic factor in atherosclerosis; it promotes inflammatory response by activating macrophages [22], T lymphocytes [23], NK cells, B cells, and vascular SMCs [24]. In particular, IFN-γ was shown to increase SR-A (scavenger receptor-A) expression on macrophages, thus, facilitating oxLDL accumulation and foam cell formation [25]. Genetic knocking-out of either IFN-γ receptor or IFN-γ considerably suppressed inflammation and increased collagen content in the plaque [26]. At the same time, administration of exogenous IFN-γ promoted the development of atherosclerosis [27]. Inhibition of IFN-γ-signaling by the administration of soluble mutant IFN-γ receptor (sIFN-γR) suppressed the inflammation and stabilized atherosclerotic plaques in Apoe–/– mice [28].
Tumor necrosis factor-α (TNF-α). TNF-α is a pro-inflammatory cytokine involved in cell homeostasis and immune response regulation [29]. TNF-α has been also found to play a key role in the development of atherosclerosis. It is produced by CD4+ T cells and myeloid cells in the aorta. Atherosclerosis progression always directly correlates with a local increase in TNF-α production in the atherosclerotic plaque and with TNF-α level in blood [30].
Experiments in mice with double knockout of the TNF-α (Tnf-α–/–) and ApoE (Apoe–/–) genes revealed significant reduction of plaque size in the aortic sinus of Tnf-α–/–Apoe–/– mice compared to control Apoe–/– group due to decreased expression of ICAM-1 and VCAM-1 adhesion molecules and monocyte chemotactic protein-1 (MCP-1) [31]. Note, that in rheumatoid arthritis patients predisposed to CVD, anti-TNF-α therapy decreased the occurrence of CVD events [32].
Therefore, experimental data obtained from animal models and the analysis of atherosclerosis in humans convincingly prove the pathogenic role of type I cytokines (IFN-γ and TNF-α).
Type II cytokines. Type II cytokines are produced by Th2 cells, innate lymphoid cells (ILCs), and eosinophils. The role of several Th2-like cytokines (IL-4, IL-5, and IL-13) has also been investigated in the pathogenesis of atherosclerosis. Early publications suggested that Th2 cells are main producers of type II cytokines. It has been demonstrated that IL-4 and IL-5 participate in atherosclerosis progression by regulating antibody production by B cells. Th2 cells have been considered strictly anti-inflammatory, since their action counteracts the functions of Th1 cells during the development of atherosclerosis and other vascular disorders. However, a number of studies have demonstrated a possible pathogenic role of cytokines produced by these cells.
Interleukin-4 (IL-4). IL-4 regulates differentiation of Th2 cells via STAT6. STAT6 activates GATA3 transcription factor, which promotes T cell differentiation into Th2 cells producing IL-4, IL-5, and IL-13. In atherosclerosis-resistant mouse models, production of cytokines is shifted toward type II, suggesting a protective role of these molecules [33]. However, in Ldlr–/– mice, IL-4 deficiency had virtually no effect on the progression of atherosclerosis [34]. Some studies convincingly demonstrated that IL-4, on the contrary, induces inflammation by acting on endothelial cells and increasing the expression of pro-inflammatory mediators, such as cytokines, chemokines, and adhesion molecules (ICAM-1) [35]. IL-4 was also found to induce apoptosis of endothelial cells via activation of caspase-3 signaling pathway, resulting in the endothelial cell dysfunction [36].
Interleukin-5 (IL-5)/Interleukin-13 (IL-13). Mouse models studies suggest an antiatherogenic role of IL-5 and IL-13. Indeed, Il-5–/–Ldlr–/– mice developed more severe atherosclerotic lesions than Ldlr–/– controls [37]. IL-5 was shown to stimulate the production of neutralizing antibodies (IgM) against oxLDLs, therefore contributing to the reduction of atherosclerotic plaque size [37].
Previous studies addressing the role of IL-13 in atherosclerosis demonstrated that administartion of recombinant IL-13 stabilized the plaque due to increased content of collagen, decreased VCAM-1-dependent recruitment of monocytes and reduced accumulation of macrophages [38]. It is important to note that IL-13 deficiency accelerated atherosclerosis development in Ldlr–/– mice and does not affect cholesterol level in the blood [38]. Therefore, IL-13 displays protective properties in atherosclerosis and favorably modulates the morphology of the plaque.
IL-13 and IL-4 act through the same signaling pathway (IL-4Rα/IL-13Rα1 and STAT6) and, therefore, have similar functions, such as regulation of B cells, monocytes, dendritic cells, and fibroblasts [39-41]. However, some of their functions are differ. For example, IL-13 activates an alternative signal transduction via IL-13Rα2, a receptor that binds exclusively IL-13, and induces the production of transforming growth factor TGFβ in macrophages through the STAT-6-independent pathway, required for collagen biosynthesis in vivo [42]. Overall, the role of Th2 cytokines in atherosclerosis has not been fully elucidated, and, perhaps, is stage-dependent.
Th17-like cytokines. IL-17A belongs to the IL-17 cytokine family and is produced by Th17 cells, γδ T cells, and type 3 ILCs. Th17 lymphocytes synthesize IL-17 (or IL-17A), IL-17F, and IL-17C. Moreover, Th17 lymphocytes can also produce IL-21 and IL-22, which play an important role in the accumulation of macrophages and neutrophils as well as T cell activation [43]. IL-22 is involved in the regulation of the barrier function and microbiome activity in the intestine. Activation of the IL-17-producing cells (Th17 cells and type 3 ILCs) with subsequent production of Th17 cytokines depends on the RORγτ transcription factor and is regulated by IL-23, IL-6, and IL-1β produced by myeloid and epithelial cells [44, 45].
Interleukin-17A (IL-17A). Despite the fact that in the past few years the role of IL-17A in atherosclerosis has drawn considerable attention, the function of this cytokine still remains unclear [46]. Many studies described the presence and accumulation of IL-17A-producing cells in the aortic wall during atherosclerosis progression [47, 48]. Some reports suggested a protective role of this cytokine. Indeed, knockout of the IL-17A-encoding gene (Il17a–/–) in Apoe–/– mice accelerated the production of IFN-γ by CD4+ T cells in the spleen, thereby promoting formation of atherosclerotic plaques [49]. In addition, an increased content of macrophages and reduced SMC actin in the plaque fibrous cap in Il17a–/–Apoe–/– mice suggest a potential role of IL-17A, possibly, via IL-17A-dependent IFN-γ and IL-5 production at the early stages of the disease [50]. Mice lacking the suppressor of cytokine signaling 3 (SOCS3) gene in T cells developed less severe atherosclerosis, which correlated with an increased production of IL-17A and suggested indirect protective role of IL-17A [51]. However, the majority of studies revealed a proatherogenic role of this cytokine. Apoe–/– mice with genetic ablation of IL-17A or IL-17A receptor (IL-17RA) were characterized by ameliorated disease due to reduction of chemokine-dependent infiltration of monocytes and neutrophils to the aortic intima [52]. Blocking IL-17A by adenovirus-produced IL-17 receptor strongly suppressed plaque development in Apoe–/– mice [53] due to the reduction of pro-inflammatory molecules expression (IL-6 and granulocyte colony-stimulating factor, G-CSF) and macrophage accumulation in the aortas. Therefore, it has been suggested that IL-17A promotes atherosclerosis by regulating monocyte infiltration into the intima [53]. It is important to note that IL-17A neutralization in Apoe–/– mice decreased expression of VCAM-1 cell adhesion molecule, infiltration of immune cells into the aortic wall, and secretion of pro-inflammatory cytokines and chemokines (IL-6, TNF-α, CCL5), which altogether suppressed atherosclerosis development [54]. At the same time, administration of recombinant IL-17A promoted the formation of atherosclerotic plaques [55].
Interleukin-22 (IL-22). IL-22 is produced by activated T cells (Th17 cells) and ILCs. It is involved in tissue regeneration, metabolism regulation, and maintaining of bacterial homeostasis in the intestine [56]. The role of IL-22 in atherosclerosis is poorly investigated.
Recent studies demonstrated a reduction of atherosclerosis plaque burden in Il22–/–Apoe–/– mice compared to control Apoe–/– group. It was suggested that IL-22 activates migration of SMCs from the aortic media to the intima, and therefore promotes plaque development [57]. We found that IL-22 presumably acts as an antiatherogenic molecule, possibly, due to its capability to regulate barrier function and microbial activity in the intestine. Our experiments demonstrate that Ldlr–/– mice reconstituted with Il22–/– bone marrow displayed more rapid atherosclerosis progression than the control group (unpublished data).
Interleukin-6 (IL-6)/Interleukin-12 (IL-12) cytokine superfamily. Interleukin-6 (IL-6). Cytokines of this superfamily are dimeric molecules that signal via dimeric receptor complexes. gp130 receptor chain participates in the formation of some of the receptor complexes of this superfamily [58].
Interleukin-6 (IL-6) receptor is a heterodimer composed of IL-6R and gp130. Ligand binding activates STAT1 and STAT3 transcription factors [59]. IL-6 can play either a pro- or anti-inflammatory role in the pathogenesis of various autoimmune disorders. Thus, IL-6 can activate the expression of IL-1 receptor (IL-1RA) antagonist and release of soluble TNF-α receptor, which strongly suppresses IL-1 and TNF-α activities, respectively [60]. It was suggested that the role of IL-6 in atherosclerosis depends on the stage of disease and can be either pathogenic or protective [59, 61].
Earlier studies demonstrated that introduction of recombinant IL-6 results in a two-fold increase in the area of atherosclerotic lesions in Apoe–/– mice, suggesting the pro-inflammatory role of this cytokine [62]. At the same time, 24-week-old Apoe—/— mice lacking the IL-6 gene (Il6–/–Apoe–/–) were characterized by accelerated plaque formation associated with decreased collagen content, reduced IL-10 production, and reduced accumulation of inflammatory cells in the lesions [63]. However, in another study, 9-week-old Il6—/—Apoe—/— mice displayed no such differences when compared to the control group [64].
Recent studies revealed that IL-6 signals not only through the classical cell surface IL-6 receptor, but also through its soluble form (sIL-6R). The IL-6/sIL-6R complex binds directly to gp130 that is present on the surface of almost all cells in an organism and activates pro-inflammatory response. This process was named trans-signaling [65]. On the other hand, tissue regeneration and anti-inflammatory activity of this cytokine are mediated by the classical IL-6R signaling pathway. It was shown that administration of soluble gp130 (sgp130) that specifically inhibits the IL-6/sIL-6R complex but does not affect the classical IL-6R-dependent signaling pathway considerably suppressed atherosclerosis in Ldlr–/– mice [66].
Interleukin-12 (IL-12)/Interleukin-23 (IL-23). IL-12 is an important regulator of Th1 cells, whereas IL-23 controls differentiation and functions of Th17 cells and type 3 ILCs [67, 68]. IL-12 is a heterodimer composed of p35 and p40 subunits, while IL-23 is composed of p19 and p40 subunits. This complicates the interpretation of in vivo phenotypes of mice with the knockouts of individual subunits. The observed positive correlation between CVD and the levels of IL-12 and IL-23 in patients’ blood suggests proatherogenic function of these cytokines [69]. IL-12 has been shown to act as a proatherogenic molecule in animal models, since the size of atherosclerotic lesions in Il12–/–Apoe–/– mice was considerably smaller than in the control group [70]. In addition, the administration of recombinant IL-12 promoted atherosclerosis [71].
Although the role of IL-17A, a cytokine induced by IL-23, is well known, the effects of IL-23 itself in atherosclerosis still have to be investigated in genetically modified animal models. Our studies showed a protective role of this cytokine in atherosclerosis. Ldlr–/– mice transplanted with Il23–/– or Il23r–/– bone marrow had significantly larger atherosclerotic lesions (unpublished data).
Interleukin-27 (IL-27)/Interleukin-35 (IL-35). IL-27 is a heterodimer composed of p28 and Ebi3 subunits. The Ebi3 subunit is common for IL-27 and IL-35 cytokines [59]. IL-27 is an anti-inflammatory cytokine with a broad range of activities that affect multiple cell types [72]. IL-27 suppresses the activation of CD4+ T cells, since IL-27-receptor-deficient mice demonstrate increased accumulation and activation of Th1 and Th17 CD4+ T cells in the aorta and increased production of IL-17A and IL-17A-regulated chemokines (e.g. MCP-1) with subsequent accumulation of different types of myeloid cells [73]. IL-27 also inhibits lipid accumulation in macrophages, thereby suppressing formation of foam cells [74].
IL-35 is a heterodimer composed of p35 and Ebi3 subunits. This anti-inflammatory cytokine is produced by Tregs [75]. IL-35 regulates the expression of anti-inflammatory cytokines, facilitates the development of Tregs, inhibits CD4+ T cell response, suppresses the progression of inflammatory and autoimmune disorders [76]. Ebi3 and p35 subunits were found in atherosclerotic aorta [77], and deletion of the Ebi3 subunit gene promotes the disease in atherosclerosis-prone mouse models [74]. Recent studies showed that IL-35 inhibits lipopolysaccharide (LPS)-induced acute inflammation in vascular wall by suppressing the expression of VCAM-1 by endothelial cells due to inactivation of the mitogen activated protein kinase (MAPK) signaling pathway [78]. Therefore, IL-27 and IL-35 display pronounced antiatherogenic properties and might be used as agents for anti-atherosclerosis therapy in the future.
Interleukin-1 (IL-1) cytokine family. The IL-1 family includes 11 proteins, such as IL-1α, IL-1β, IL-1 receptor antagonist (IL-1RA), IL-18, IL-33 (ligand of the membrane-bound ST2L receptor), and other less investigated cytokines [79].
Interleukin-1 (IL-1). IL-1α and IL-1β are pro-inflammatory cytokines produced by myeloid cells. Secretion of IL-1 family cytokines and expression of their receptors are increased in atherosclerotic aortas [80]. IL-1β is an essential factor of Th17 cell differentiation [81] that can exacerbate inflammation in the vascular wall. Experiments in mouse models confirmed proatherogenic properties of IL-1α and IL-1β that are involved into the upregulation of adhesion molecules expression by endothelial cells as well as macrophage activation [80, 82]. At the same time, IL-1RA displays endogenous anti-inflammatory properties since IL-1RA is a potent inhibitor of IL-1 signaling pathways. The production of IL-1β in atherosclerosis depends on the activation of the NLRP3 inflammasome caused by lysosomal distruction by accumulated in macrophages cholesterol crystals. The receptor complex composed of CD36, TLR4 and TLR6 is required for binding and internalization of modified lipoproteins (oxLDLs) as well as for the activation of NLRP3 and subsequent IL-1β production. Knockout of any component of the CD36/TLR4/TLR6 complex considerably decreases the production of IL-1β active form and reduces atherosclerotic plaque burden [83]. The production of IL-1α is stimulated by fatty acids via NLRP3-independent pathway [84]. IL-1α, IL-1β, or IL-1R deficiency strongly reduces atherosclerosis progression [80, 84, 85]. Recombinant IL-1RA (or IL-1RA-based drug Anakinra) suppresses inflammation in atherosclerosis, whereas IL-1RA deficincy significantly exacerbates the disease [86]. The administartion of recombinant IL-1RA into Apoe–/– mice [87], or IL-1RA overexpression in Ldlr–/– [88] or Apoe–/– mice, notably suppress plaque burden [89]. On the contrary, IL-1RA knockout C57BL/6J mice fed a high-fat diet tended to accumulate foam cells in the aortic wall and, thus, had accelerated disease [88].
Interleukin-18 (IL-18). IL-18 is a pro-inflammatory cytokine; its expression is elevated in atherosclerotic plaques [90]. IL-18 production is also elevated in patients with myocardium infarction and diabetes mellitus [91]. IL-18 administration in Apoe–/– mice accelerates atherosclerosis [92], whereas overexpression of the IL-18-binding protein, an endogenous inhibitor of IL-18, strongly supresses the disease [93]. It was suggested that the proatherogenic effect of IL-18 is mediated by IFN-γ, since atherosclerosis progression is diminished in IFN-γ-deficient Apoe–/– mice [92]. The administration of recombinant IL-18 into Apoe–/– mice upregulates IFN-γ production in the lesions and promotes the disease progression [94].
Interleukin-33 (IL-33). IL-33 exhibits strong immunomodulatory properties [95]. It regulates the production of Th2 cytokines (IL-4, IL-5, and IL-13) by Th2 cells, type 2 ILCs, and eosinophils. The administration of recombinant IL-33 upregulates IL-4, IL-5 and IL-13 production as well as immunoglobulins A, E, and G1, while suppresses IFN-γ, thereby stimulating the protective response and suppressing atherosclerosis development [96]. The administration of soluble IL-33 receptor (ST2L) strongly reduces atherosclerosis in mouse models [96]. Moreover, IL-33 is a potent inhibitor of oxLDL uptake and foam cell formation [97].
Interleukin-10 (IL-10) cytokine family. The IL-10 family includes IL-10, IL-28A, IL-28B, IL-29, and so-called IL-20 subfamily [98] composed of IL-19, IL-20, IL-22, IL-24, and IL-26 [99]. These cytokines stimulate various protective immune mechanisms and are essential for maintanence of tissue homeostasis [99].
Interleukin-10 (IL-10). IL-10 plays a key role in the regulation of innate and adaptive immune responses by suppressing the activation of Th1 cells and macrophages and activating antibody production by B cells [100]. IL-10 is produced by myeloid and Treg cells. Experiments in mouse models showed that genetic inactivation of IL-10 accelerates atherosclerosis due to increased infiltration of inflammatory cells and production of pro-inflammatory cytokines in atherosclerotic lesions [101-103]. Therefore, IL-10 is a key pro-inflammatory cytokine in pathogenesis of atherosclerosis.
Interleukin-20 (IL-20) subfamily. Cytokines of the IL-20 subfamily are produced by both non-immune and immune cells, including myeloid cells and lymphocytes [98]. IL-19, IL-20, and IL-24 were shown to mediate signaling cascades by binding to the β-subunit of the IL-20 receptor (IL-20Rβ), whereas IL-22 and IL-26 bind to the IL-10Rβ receptor.
Interleukin-19 (IL-19)/Interleukin-20 (IL-20). IL-19 belongs to the IL-20 subfamily of the IL-19 family of cytokines. IL-19 acts through the receptor complex composed of IL-20R1 and IL-20R2 subunits [98]. IL-19 is produced mostly by monocytes, endothelial cells, fibroblasts, and CD8+ T cells. IL-19 regulates the development of Th2-dependent immune responses, controls the function of SMCs, and reduces hyperplasia of the intima in vasclular wall inflammation [104]. Recent studies demonstrated that IL-19 deficiency causes vascular SMCs (VSMC) activation and pro-inflammatory molecules production, including IL-1β, TNF-α, and MCP-1. Beside VSMC activation, IL-19 also controls endothelial cells activation, since elevated adhesion molecules espression was found in Il19–/– atherosclerosis-prone mice [105]. Taken together these data suggest that IL-19 is a potent suppressor of atherosclerosis development, which controls VSMC migration, proliferation and pro-inflammatory molecules expression [106].
The role of IL-20 is not completely understood. This cytokine is produced mostly by epithelial cells and adipocytes [90]. Both IL-20 and its receptor IL-20R1/IL-20R2 can be detected in human atherosclerotic plaques. In mouse models, the administration of recombinant IL-20 exacerbates the disease in Apoe–/– mice [107], suggesting a pathogenic role of this cytokine.
Transforming growth factor TGFβ. Three isoforms of TGFβ have been described – TGFβ1, TGFβ2, and TGFβ3. All isoforms have been implicated into the regulation of various biological processes by engaging three types of cell surface receptors known as types I, II, and III. All subsets of cells in our body, including epithelial, endothelial, hematopoietic, and connective tissues, express TGFβ and its receptor [108]. TGFβ regulates cell proliferation and differentiation and therefore is critical for embryonic development. It is essential for supporting normal structure of blood vessel wall [109]. TGFβ also plays an important role in the regulation of immune cells, since it inhibits proliferation, activation, and differentiation of Th1 and Th2 cells. It is also required for differentiation of Tregs [16]. In atherosclerosis, TGFβ plays anti-inflammatory and antiatherogenic role [110]. Neutralization [111] or genetic ablation [112] of TGFβ promotes the development of atherosclerosis in Apoe–/– mice and facilitates the recruitment of pro-inflammatory macrophages and T cells into the site of inflammation. At the same time, TGFβ was shown to decrease collagen content in the aorta. Therefore, TGFβ is a key antiatherogenic cytokine required for Tregs differentiation, which in turn suppresses T cells.
Inflammation plays an important role at all stages of atherosclerosis development – from attracting immune cells and atherosclerotic plaque formation to its rupture. Chronic inflammation in the aortic wall is caused by dyslipidemia, innate and adaptive immune responses and is mediated by various pro-inflammatory cytokines. The balance between pro- and anti-inflammatory cytokines is the major factor that determines the stability of atherosclerotic plaque.
Experimental data obtained from mouse models of atherosclerosis has shown that inhibition of pro-inflammatory cytokines suppresses atherosclerosis development and progression. For example, anti-TNF-α therapy decreases the risk of CVD in patients with rheumatoid arthritis. At the same time, all attempts to use clinically anti-inflammatory TGFβ and IL-10 have failed, in part due to incomplete understanding of the function of these cytokines in atherosclerosis. Therefore, discovering and studying functions of new atheroprotective cytokines can significantly contribute to the development of new approaches to anti-atherosclerosis therapy. One of the potential strategies in the development of new treatment methods could be a suppression of inflammatory immune response by shifting the balance toward anti-inflammatory mediators to achieve the stabilization of atherosclerotic plaque.
Acknowledgements
This study was financially supported by the NIH/NCI P30 Cancer Grant (FCCC), AHA SDG 13SDG14490059 and NIH/NCI R21 CA202396 grants.
REFERENCES
1.Pagidipati, N. J., and Gaziano, T. A. (2013)
Estimating deaths from cardiovascular disease: a review of global
methodologies of mortality measurement, Circulation, 127,
749-756.
2.Dahlof, B. (2010) Cardiovascular disease risk
factors: epidemiology and risk assessment, Am. J. Cardiol.,
105, 3A-9A.
3.Nagornev, V. A., and Ketlinsky, S. A. (2009)
Humoral and cell immunity against atherosclerosis: the possibility of
vaccine development, Med. Akad. Zh., 9, 2-15.
4.Galkina, E., Kadl, A., Sanders, J., Varughese, D.,
Sarembock, I. J., and Ley, K. (2006) Lymphocyte recruitment into the
aortic wall before and during development of atherosclerosis is
partially L-selectin dependent, J. Exp. Med., 203,
1273-1282.
5.Galkina, E., and Ley, K. (2009) Immune and
inflammatory mechanisms of atherosclerosis, Annu. Rev. Immunol.,
27, 165-197.
6.Perdiguero, G. E., Klapproth, K., Schulz, C.,
Busch, K., Azzoni, E. L., Crozet, L., Garner, H., Trouillet, C., de
Bruijn, M. F., Geissmann, F., and Rodewald, H. R. (2015)
Tissue-resident macrophages originate from yolk-sac-derived
erythro-myeloid progenitors, Nature, 518, 547-551.
7.Swirski, F. K. (2014) Monocyte recruitment and
macrophage proliferation in atherosclerosis, Kardiol. Pol.,
72, 311-314.
8.Ensan, S., Li, A., Besla, R., Degousee, N. J.,
Cosme, J., Roufaiel, M., Shikatani, E. A., El-Maklizi, M., Williams, J.
W., Robins, L., Li, C., Lewis, B., Yun, T. J., Lee, J. S., Wieghofer,
P., Khattar, R., Farrokhi, K., Byrne, J., Ouzounian, M., Zavitz, C. C.,
Levy, G. A., Bauer, C. M., Libby, P., Husain, M., Swirski, F. K.,
Cheong, C., Prinz, M., Hilgendorf, I., Randolph, G. J., Epelman, S.,
Gramolini, A. O., Cybulsky, M. I., Rubin, B. B., and Robbins, C. S.
(2016) Self-renewing resident arterial macrophages arise from embryonic
CX3CR1(+) precursors and circulating monocytes immediately after birth,
Nat. Immunol., 17, 159-168.
9.Ye, Y. X., Calcagno, C., Binderup, T., Courties,
G., Keliher, E. J., Wojtkiewicz, G. R., Iwamoto, Y., Tang, J.,
Perez-Medina, C., Mani, V., Ishino, S., Johnbeck, C. B., Knigge, U.,
Fayad, Z. A., Libby, P., Weissleder, R., Tawakol, A., Dubey, S.,
Belanger, A. P., Di Carli, M. F., Swirski, F. K., Kjaer, A., Mulder, W.
J., and Nahrendorf, M. (2015) Imaging macrophage and hematopoietic
progenitor proliferation in atherosclerosis, Circ. Res.,
117, 835-845.
10.Warnatsch, A., Ioannou, M., Wang, Q., and
Papayannopoulos, V. (2015) Inflammation. Neutrophil extracellular traps
license macrophages for cytokine production in atherosclerosis,
Science, 349, 316-320.
11.Koltsova, E. K., Hedrick, C. C., and Ley, K.
(2013) Myeloid cells in atherosclerosis: a delicate balance of
anti-inflammatory and proinflammatory mechanisms, Curr. Opin.
Lipidol., 24, 371-380.
12.Doring, Y., Drechsler, M., Soehnlein, O., and
Weber, C. (2015) Neutrophils in atherosclerosis: from mice to man,
Arterioscler. Thromb. Vasc. Biol., 35, 288-295.
13.Binder, C. J., Shaw, P. X., Chang, M. K.,
Boullier, A., Hartvigsen, K., Horkko, S., Miller, Y. I., Woelkers, D.
A., Corr, M., and Witztum, J. L. (2005) The role of natural antibodies
in atherogenesis, J. Lipid Res., 46, 1353-1363.
14.Tabas, I., Garcia-Cardena, G., and Owens, G. K.
(2015) Recent insights into the cellular biology of atherosclerosis,
J. Cell Biol., 209, 13-22.
15.Hansson, G. K., Libby, P., and Tabas, I. (2015)
Inflammation and plaque vulnerability, J. Intern. Med.,
278, 483-493.
16.Ait-Oufella, H., Taleb, S., Mallat, Z., and
Tedgui, A. (2011) Recent advances on the role of cytokines in
atherosclerosis, Arterioscler. Thromb. Vasc. Biol., 31,
969-979.
17.Szmitko, P. E., Wang, C. H., Weisel, R. D., De
Almeida, J. R., Anderson, T. J., and Verma, S. (2003) New markers of
inflammation and endothelial cell activation: Part I,
Circulation, 108, 1917-1923.
18.Mallat, Z., Taleb, S., Ait-Oufella, H., and
Tedgui, A. (2009) The role of adaptive T cell immunity in
atherosclerosis, J. Lipid Res., 50, 364-369.
19.Taleb, S., Tedgui, A., and Mallat, Z. (2015)
IL-17 and Th17 cells in atherosclerosis: subtle and contextual roles,
Arterioscler. Thromb. Vasc. Biol., 35, 258-264.
20.Ranjbaran, H., Sokol, S. I., Gallo, A., Eid, R.
E., Iakimov, A. O., D’Alessio, A., Kapoor, J. R., Akhtar, S.,
Howes, C. J., Aslan, M., Pfau, S., Pober, J. S., and Tellides, G.
(2007) An inflammatory pathway of IFN-gamma production in coronary
atherosclerosis, J. Immunol., 178, 592-604.
21.Young, J. L., Libby, P., and Schonbeck, U. (2002)
Cytokines in the pathogenesis of atherosclerosis, Thromb.
Haemost., 88, 554-567.
22.Koltsova, E. K., Garcia, Z., Chodaczek, G.,
Landau, M., McArdle, S., Scott, S. R., von Vietinghoff, S., Galkina,
E., Miller, Y. I., Acton, S. T., and Ley, K. (2012) Dynamic T cell-APC
interactions sustain chronic inflammation in atherosclerosis, J.
Clin. Invest., 122, 3114-3126.
23.Whitman, S. C., Ravisankar, P., and Daugherty, A.
(2002) IFN-gamma deficiency exerts gender-specific effects on
atherogenesis in apolipoprotein E–/– mice, J.
Interferon Cytokine Res., 22, 661-670.
24.Harvey, E. J., and Ramji, D. P. (2005)
Interferon-gamma and atherosclerosis: pro- or anti-atherogenic,
Cardiovasc. Res., 67, 11-20.
25.Wuttge, D. M., Zhou, X., Sheikine, Y., Wagsater,
D., Stemme, V., Hedin, U., Stemme, S., Hansson, G. K., and Sirsjo, A.
(2004) CXCL16/SR-PSOX is an interferon-gamma-regulated chemokine and
scavenger receptor expressed in atherosclerotic lesions,
Arterioscler. Thromb. Vasc. Biol., 24, 750-755.
26.Gupta, S., Pablo, A. M., Jiang, X., Wang, N.,
Tall, A. R., and Schindler, C. (1997) IFN-gamma potentiates
atherosclerosis in ApoE knock-out mice, J. Clin. Invest.,
99, 2752-2761.
27.Whitman, S. C., Ravisankar, P., Elam, H., and
Daugherty, A. (2000) Exogenous interferon-gamma enhances
atherosclerosis in apolipoprotein E–/– mice,
Am. J. Pathol., 157, 1819-1824.
28.Koga, M., Kai, H., Yasukawa, Yamamoto, T., Kawai,
Y., Kato, S., Kusaba, K., Kai, M., Egashira, K., Kataoka, Y., and
Imaizumi, T. (2007) Inhibition of progression and stabilization of
plaques by postnatal interferon-gamma function blocking in
ApoE-knockout mice, Circ. Res., 101, 348-356.
29.Kalliolias, G. D., and Ivashkiv, L. B. (2016) TNF
biology, pathogenic mechanisms and emerging therapeutic strategies,
Nat. Rev. Rheumatol., 12, 49-62.
30.Canault, M., Peiretti, F., Poggi, M., Mueller,
C., Kopp, F., Bonardo, B., Bastelica, D., Nicolay, A., Alessi, M. C.,
and Nalbone, G. (2008) Progression of atherosclerosis in ApoE-deficient
mice that express distinct molecular forms of TNF-alpha, J.
Pathol., 214, 574-583.
31.Ohta, H., Wada, H., Niwa, T., Kirii, H., Iwamoto,
N., Fujii, H., Saito, K., Sekikawa, K., and Seishima, M. (2005)
Disruption of tumor necrosis factor-alpha gene diminishes the
development of atherosclerosis in ApoE-deficient mice,
Atherosclerosis, 180, 11-17.
32.Jacobsson, L. T., Turesson, C., Gulfe, A.,
Kapetanovic, M. C., Petersson, I. F., Saxne, T., and Geborek, P. (2005)
Treatment with tumor necrosis factor blockers is associated with a
lower incidence of first cardiovascular events in patients with
rheumatoid arthritis, J. Rheumatol., 32, 1213-1218.
33.Huber, S. A., Sakkinen, P., David, C., Newell, M.
K., and Tracy, R. P. (2001) T helper-cell phenotype regulates
atherosclerosis in mice under conditions of mild hypercholesterolemia,
Circulation, 103, 2610-2616.
34.King, V. L., Cassis, L. A., and Daugherty, A.
(2007) Interleukin-4 does not influence development of
hypercholesterolemia or angiotensin II-induced atherosclerotic lesions
in mice, Am. J. Pathol., 171, 2040-2047.
35.Thornhill, M. H., Kyan-Aung, U., and Haskard, D.
O. (1990) IL-4 increases human endothelial cell adhesiveness for
T-cells but not for neutrophils, J. Immunol., 144,
3060-3065.
36.Lee, Y. W., Kuhn, H., Hennig, B., and Toborek, M.
(2000) IL-4 induces apoptosis of endothelial cells through the
caspase-3-dependent pathway, FEBS Lett., 485,
122-126.
37.Binder, C. J., Hartvigsen, K., Chang, M. K.,
Miller, M., Broide, D., Palinski, W., Curtiss, M., Corr, L. K., and
Witztum, J. L. (2004) IL-5 links adaptive and natural immunity specific
for epitopes of oxidized LDL and protects from atherosclerosis, J.
Clin. Invest., 114, 427-437.
38.Cardilo-Reis, L., Gruber, S., Schreier, S. M.,
Drechsler, M., Papac-Milicevic, N., Weber, C., Wagner, O., Stangl, H.,
Soehnlein, O., and Binder, C. J. (2012) Interleukin-13 protects from
atherosclerosis and modulates plaque composition by skewing the
macrophage phenotype, EMBO Mol. Med., 4, 1072-1086.
39.Chomarat, P., and Banchereau, J. (1998)
Interleukin-4 and interleukin-13: their similarities and discrepancies,
Int. Rev. Immunol., 17, 1-52.
40.Kuperman, D. A., and Schleimer, R. P. (2008)
Interleukin-4, interleukin-13, signal transducer and activator of
transcription factor 6, and allergic asthma, Curr. Mol. Med.,
8, 384-392.
41.Tedgui, A., and Mallat, Z. (2006) Cytokines in
atherosclerosis: pathogenic and regulatory pathways, Physiol.
Rev., 86, 515-581.
42.Fichtner-Feigl, S., Strober, W., Kawakami, K.,
Puri, R. K., and Kitani, A. (2006) IL-13 signaling through the
IL-13alpha2 receptor is involved in induction of TGF-beta1 production
and fibrosis, Nat. Med., 12, 99-106.
43.Korn, T., Bettelli, E., Oukka, M., and Kuchroo,
V. K. (2009) IL-17 and Th17 Cells, Annu. Rev. Immunol.,
27, 485-517.
44.Ivanov, I. I., McKenzie, B. S., Zhou, L.,
Tadokoro, C. E., Lepelley, A., Lafaille, J. J., Cua, D. J., and
Littman, D. R. (2006) The orphan nuclear receptor RORgammat directs the
differentiation program of proinflammatory IL-17+ T helper
cells, Cell, 126, 1121-1133.
45.Patel, D. D., and Kuchroo, V. K. (2015) Th17 cell
pathway in human immunity: lessons from genetics and therapeutic
interventions, Immunity, 43, 1040-1051.
46.Taleb, S., Tedgui, A., and Mallat, Z. (2010)
Interleukin-17: friend or foe in atherosclerosis, Curr. Opin.
Lipidol., 21, 404-408.
47.Xie, J. J., Wang, J., Tang, T. T., Chen, J., Gao,
X. L., Yuan, J., Zhou, Z. H., Liao, M. Y., Yao, R., Yu, X., Wang, D.,
Cheng, Y., Liao, Y. H., and Cheng, X. (2010) The Th17/Treg functional
imbalance during atherogenesis in ApoE–/– mice,
Cytokine, 49, 185-193.
48.Ma, T., Gao, Q., Zhu, F., Guo, C., Wang, Q., Gao,
F., and Zhang, L. (2013) Th17 cells and IL-17 are involved in the
disruption of vulnerable plaques triggered by short-term combination
stimulation in apolipoprotein E-knockout mice, Cell. Mol.
Immunol., 10, 338-348.
49.Madhur, M. S., Funt, S. A., Li, L., Vinh, A.,
Chen, W., Lob, H. E., Iwakura, Y., Blinder, Y., Rahman, A., Quyyumi, A.
A., and Harrison, D. G. (2011) Role of interleukin 17 in inflammation,
atherosclerosis, and vascular function in apolipoprotein E-deficient
mice, Arterioscler. Thromb. Vasc. Biol., 31,
1565-1572.
50.Danzaki, K., Matsui, Y., Ikesue, M., Ohta, D.,
Ito, K., Kanayama, M., Kurotaki, D., Morimoto, J., Iwakura, Y., Yagita,
H., Tsutsui, H., and Uede, T. (2012) Interleukin-17A deficiency
accelerates unstable atherosclerotic plaque formation in apolipoprotein
E-deficient mice, Arterioscler. Thromb. Vasc. Biol., 32,
273-280.
51.Taleb, S., Romain, M., Ramkhelawon, B.,
Uyttenhove, C., Pasterkamp, G., Herbin, O., Esposito, B., Perez, N.,
Yasukawa, H., Van Snick, J., Yoshimura, A., Tedgui, A., and Mallat, Z.
(2009) Loss of SOCS3 expression in T cells reveals a regulatory role
for interleukin-17 in atherosclerosis, J. Exp. Med., 206,
2067-2077.
52.Butcher, M. J., Gjurich, B. N., Phillips, T., and
Galkina, E. V. (2012) The IL-17A/IL-17RA axis plays a proatherogenic
role via the regulation of aortic myeloid cell recruitment, Circ.
Res., 110, 675-687.
53.Smith, E., Prasad, K. M., Butcher, M., Dobrian,
A., Kolls, J. K., Ley, K., and Galkina, E. (2010) Blockade of
interleukin-17A results in reduced atherosclerosis in apolipoprotein
E-deficient mice, Circulation, 121, 1746-1755.
54.Erbel, C., Chen, L., Bea, F., Wangler, S., Celik,
S., Lasitschka, F., Wang, Y., Bockler, D., Katus, H. A., and Dengler,
T. J. (2009) Inhibition of IL-17A attenuates atherosclerotic lesion
development in apoE-deficient mice, J. Immunol., 183,
8167-8175.
55.Gao, Q., Jiang, Y., Ma, T., Zhu, F., Gao, F.,
Zhang, P., Guo, C., Wang, Q., Wang, X., Ma, C., Zhang, Y., Chen, W.,
and Zhang, L. (2010) A critical function of Th17 proinflammatory cells
in the development of atherosclerotic plaque in mice, J.
Immunol., 185, 5820-5827.
56.Wang, X., Ota, N., Manzanillo, P., Kates, L.,
Zavala-Solorio, J., Eidenschenk, C., Zhang, J., Lesch, J., Lee, W. P.,
Ross, J., Diehl, L., Van Bruggen, N., Kolumam, G., and Ouyang, W.
(2014) Interleukin-22 alleviates metabolic disorders and restores
mucosal immunity in diabetes, Nature, 514, 237-341.
57.Rattik, S., Hultman, K., Rauch, U., Soderberg,
I., Sundius, L., Ljungcrantz, I., Hultgardh-Nilsson, A., Wigren, M.,
Bjorkbacka, H., Fredrikson, G. N., and Nilsson, J. (2015) IL-22 affects
smooth muscle cell phenotype and plaque formation in apolipoprotein E
knockout mice, Atherosclerosis, 242, 506-514.
58.Jones, L. L., and Vignali, D. A. (2011) Molecular
interactions within the IL-6/IL-12 cytokine/receptor superfamily,
Immunol. Res., 51, 5-14.
59.Garbers, C., Hermanns, H. M., Schaper, F.,
Muller-Newen, G., Grotzinger, J., Rose-John, S., and Scheller, J.
(2012) Plasticity and cross-talk of interleukin 6-type cytokines,
Cytokine Growth Factor Rev., 23, 85-97.
60.Xing, Z., Gauldie, J., Cox, G., Baumann, H.,
Jordana, M., Lei, X. F., and Achong, M. K. (1998) IL-6 is an
antiinflammatory cytokine required for controlling local or systemic
acute inflammatory responses, J. Clin. Invest., 101,
311-320.
61.Fontes, J. A., Rose, N. R., and Cihakova, D.
(2015) The varying faces of IL-6: from cardiac protection to cardiac
failure, Cytokine, 74, 62-68.
62.Huber, S. A., Sakkinen, P., Conze, D., Hardin,
N., and Tracy, R. (1999) Interleukin-6 exacerbates early
atherosclerosis in mice, Arterioscler. Thromb. Vasc. Biol.,
19, 2364-2367.
63.Schieffer, B., Selle, T., Hilfiker, A.,
Hilfiker-Kleiner, D., Grote, K., Tietge, U. J., Trautwein, C.,
Luchtefeld, M., Schmittkamp, C., Heeneman, S., Daemen, M. J., and
Drexler, H. (2004) Impact of interleukin-6 on plaque development and
morphology in experimental atherosclerosis, Circulation,
110, 3493-3500.
64.Elhage, R., Clamens, S., Besnard, S., Mallat, Z.,
Tedgui, A., Arnal, J., Maret, A., and Bayard, F. (2001) Involvement of
interleukin-6 in atherosclerosis but not in the prevention of fatty
streak formation by 17beta-estradiol in apolipoprotein E-deficient
mice, Atherosclerosis, 156, 315-320.
65.Rose-John, S. (2012) IL-6 trans-signaling via the
soluble IL-6 receptor: importance for the pro-inflammatory activities
of IL-6, Int. J. Biol. Sci., 8, 1237-1247.
66.Schuett, H., Oestreich, R., Waetzig, G. H.,
Annema, W., Luchtefeld, M., Hillmer, A., Bavendiek, U., von Felden, J.,
Divchev, D., Kempf, T., Wollert, K. C., Seegert, D., Rose-John, S.,
Tietge, U. J., Schieffer, B., and Grote, K. (2012) Transsignaling of
interleukin-6 crucially contributes to atherosclerosis in mice,
Arterioscler. Thromb. Vasc. Biol., 32, 281-290.
67.Teng, M. W., Bowman, E. P., McElwee, J. J.,
Smyth, M. J., Casanova, J. L., Cooper, A. M., and Cua, D. J. (2015)
IL-12 and IL-23 cytokines: from discovery to targeted therapies for
immune-mediated inflammatory diseases, Nat. Med., 21,
719-729.
68.Diefenbach, A., Colonna, M., and Koyasu, S.
(2014) Development, differentiation, and diversity of innate lymphoid
cells, Immunity, 41, 354-365.
69.Abbas, A., Gregersen, I., Holm, S., Daissormont,
I., Bjerkeli, V., Krohg-Sorensen, K., Skagen, K. R., Dahl, T. B.,
Russell, D., Almas, T., Bundgaard, D., Alteheld, L. H., Rashidi, A.,
Dahl, C. P., Michelsen, A. E., Biessen, E. A., Aukrust, P., Halvorsen,
B., and Skjelland, M. (2015) Interleukin 23 levels are increased in
carotid atherosclerosis: possible role for the interleukin
23/interleukin 17 axis, Stroke, 46, 793-799.
70.Davenport, P., and Tipping, P. G. (2003) The role
of interleukin-4 and interleukin-12 in the progression of
atherosclerosis in apolipoprotein E-deficient mice, Am. J.
Pathol., 163, 1117-1125.
71.Lee, T. S., Yen, H. C., Pan, C. C., and Chau, L.
Y. (1999) The role of interleukin 12 in the development of
atherosclerosis in ApoE-deficient mice, Arterioscler. Thromb. Vasc.
Biol., 19, 734-742.
72.Yoshida, H., and Hunter, C. A. (2015) The
immunobiology of interleukin-27, Annu. Rev. Immunol., 33,
417-443.
73.Koltsova, E. K., Kim, G., Lloyd, K. M., Saris, C.
J., Von Vietinghoff, S., Kronenberg, M., and Ley, K. (2012) IL-27
receptor limits atherosclerosis in Ldlr–/– mice,
Circ. Res., 111, 1274-1285.
74.Hirase, T., Hara, H., Miyazaki, Y., Ide, N.,
Nishimoto-Hazuku, A., Fujimoto, H., Saris, C. J., Yoshida, H., and
Node, K. (2013) Interleukin 27 inhibits atherosclerosis via
immunoregulation of macrophages in mice, Am. J. Physiol. Heart Circ.
Physiol., 305, 420-429.
75.Collison, L. W., Workman, C. J., Kuo, T. T.,
Boyd, K., Wang, Y., Vignali, K. M., Cross, R., Sehy, D., Blumberg, R.
S., and Vignali, D. A. (2007) The inhibitory cytokine IL-35 contributes
to regulatory T-cell function, Nature, 450, 566-569.
76.Collison, L. W., Delgoffe, G. M., Guy, C. S.,
Vignali, K. M., Chaturvedi, V., Fairweather, D., Satoskar, A. R.,
Garcia, K. C., Hunter, C. A., Drake, C. G., Murray, P. J., and Vignali,
D. A. (2012) The composition and signaling of the IL-35 receptor are
unconventional, Nat. Immunol., 13, 290-299.
77.Kempe, S., Heinz, P., Kokai, E., Devergne, O.,
Marx, N., and Wirth, T. (2009) Epstein–Barr virus-induced gene-3
is expressed in human atheroma plaques, Am. J. Pathol.,
175, 440-447.
78.Sha, X., Meng, S., Li, X., Xi, H., Maddaloni, M.,
Pascual, D. W., Shan, H., Jiang, X., Wang, H., and Yang, X. F. (2015)
Interleukin-35 inhibits endothelial cell activation by suppressing
MAPK-AP-1 pathway, J. Biol. Chem., 290, 19307-19318.
79.Dinarello, C. A. (2009) Immunological and
inflammatory functions of the interleukin-1 family, Annu. Rev.
Immunol., 27, 519-550.
80.Kirii, H., Niwa, T., Yamada, Y., Wada, H., Saito,
K., Iwakura, Y., Asano, M., Moriwaki, H., and Seishima, M. (2003) Lack
of interleukin-1beta decreases the severity of atherosclerosis in
ApoE-deficient mice, Arterioscler. Thromb. Vasc. Biol.,
23, 656-660.
81.Mills, K. H. (2008) Induction, function and
regulation of IL-17-producing T cells, Eur. J. Immunol.,
38, 2636-2649.
82.Clarke, M. C., Talib, S., Figg, N. L., and
Bennett, M. R. (2010) Vascular smooth muscle cell apoptosis induces
interleukin-1-directed inflammation: effects of hyperlipidemia-mediated
inhibition of phagocytosis, Circ. Res., 106, 363-372.
83.Sheedy, F. J., Grebe, A., Rayner, K. J.,
Kalantari, P., Ramkhelawon, B., Carpenter, S. B., Becker, C. E.,
Ediriweera, H. N., Mullick, A. E., Golenbock, D. T., Stuart, L. M.,
Latz, E., Fitzgerald, K. A., and Moore, K. J. (2013) CD36 coordinates
NLRP3 inflammasome activation by facilitating intracellular nucleation
of soluble ligands into particulate ligands in sterile inflammation,
Nat. Immunol., 14, 812-820.
84.Freigang, S., Ampenberger, F., Weiss, A.,
Kanneganti, T. D., Iwakura, Y., Hersberger, M., and Kopf, M. (2013)
Fatty acid-induced mitochondrial uncoupling elicits
inflammasome-independent IL-1alpha and sterile vascular inflammation in
atherosclerosis, Nat. Immunol., 14, 1045-1053.
85.Kamari, Y., Shaish, A., Shemesh, S., Vax, E.,
Grosskopf, I., Dotan, S., White, M., Voronov, E., Dinarello, C. A.,
Apte, R. N., and Harats, D. (2011) Reduced atherosclerosis and
inflammatory cytokines in apolipoprotein-E-deficient mice lacking bone
marrow-derived interleukin-1alpha, Biochem. Biophys. Res.
Commun., 405, 197-203.
86.Isoda, K., Sawada, S., Ishigami, N., Matsuki, T.,
Miyazaki, K., Kusuhara, M., Iwakura, Y., and Ohsuzu, F. (2004) Lack of
interleukin-1 receptor antagonist modulates plaque composition in
apolipoprotein E-deficient mice, Arterioscler. Thromb. Vasc.
Biol., 24, 1068-1073.
87.Elhage, R., Maret, A., Pieraggi, M. T., Thiers,
J. C., Arnal, J. F., and Bayard, F. (1998) Differential effects of
interleukin-1 receptor antagonist and tumor necrosis factor binding
protein on fatty-streak formation in apolipoprotein E-deficient mice,
Circulation, 97, 242-244.
88.Devlin, C. M., Kuriakose, G., Hirsch, E., and
Tabas, I. (2002) Genetic alterations of IL-1 receptor antagonist in
mice affect plasma cholesterol level and foam cell lesion size,
Proc. Natl. Acad. Sci. USA, 99, 6280-6285.
89.Merhi-Soussi, F., Kwak, B. R., Magne, D.,
Chadjichristos, C., Berti, M., Pelli, G., James, R. W., Mach, F., and
Gabay, C. (2005) Interleukin-1 plays a major role in vascular
inflammation and atherosclerosis in male apolipoprotein E-knockout
mice, Cardiovasc. Res., 66, 583-593.
90.Mallat, Z., Corbaz, A., Scoazec, A., Besnard, S.,
Leseche, G., Chvatchko, Y., and Tedgui, A. (2001) Expression of
interleukin-18 in human atherosclerotic plaques and relation to plaque
instability, Circulation, 104, 1598-1603.
91.Troseid, M., Seljeflot, I., and Arnesen, H.
(2010) The role of interleukin-18 in the metabolic syndrome,
Cardiovasc. Diabetol., 9, 11.
92.Whitman, S. C., Ravisankar, P., and Daugherty, A.
(2002) Interleukin-18 enhances atherosclerosis in apolipoprotein
E–/– mice through release of interferon-gamma,
Circ. Res., 90, E34-38.
93.Mallat, Z., Corbaz, A., Scoazec, A., Graber, P.,
Alouani, S., Esposito, B., Humbert, Y., Chvatchko, Y., and Tedgui, A.
(2001) Interleukin-18/interleukin-18 binding protein signaling
modulates atherosclerotic lesion development and stability, Circ.
Res., 89, E41-45.
94.Tenger, C., Sundborger, A., Jawien, J., and Zhou,
X. (2005) IL-18 accelerates atherosclerosis accompanied by elevation of
IFN-gamma and CXCL16 expression independently of T-cells,
Arterioscler. Thromb. Vasc. Biol., 25, 791-796.
95.Schmitz, J., Owyang, A., Oldham, E., Song, Y.,
Murphy, E., McClanahan, T. K., Zurawski, G., Moshrefi, M., Qin, J., Li,
X., Gorman, D. M., Bazan, J. F., and Kastelein, R. A. (2005) IL-33, an
interleukin-1-like cytokine that signals via the IL-1 receptor-related
protein ST2 and induces T-helper type 2-associated cytokines,
Immunity, 23, 479-490.
96.Miller, A. M., Xu, D., Asquith, D. L., Denby, L.,
Li, Y., Sattar, N., Baker, A. H., McInnes, I. B., and Liew, F. Y.
(2008) IL-33 reduces the development of atherosclerosis, J. Exp.
Med., 205, 339-346.
97.McLaren, J. E., Michael, D. R., Salter, R. C.,
Ashlin, T. G., Calder, C. J., Miller, A. M., Liew, F. Y., and Ramji, D.
P. (2010) IL-33 reduces macrophage foam cell formation, J.
Immunol., 185, 1222-1229.
98.Rutz, S., Wang, X., and Ouyang, W. (2014) The
IL-20 subfamily of cytokines – from host defence to tissue
homeostasis, Nat. Rev. Immunol., 14, 783-795.
99.Ouyang, W., Rutz, S., Crellin, N. K., Valdez, P.
A., and Hymowitz, S. G. (2011) Regulation and functions of the IL-10
family of cytokines in inflammation and disease, Annu. Rev.
Immunol., 29, 71-109.
100.Moore, K. W., De Waal Malefyt, R., Coffman, R.
L., and O’Garra, A. (2001) Interleukin-10 and the interleukin-10
receptor, Annu. Rev. Immunol., 19, 683-765.
101.Mallat, Z., Besnard, S., Duriez, M., Deleuze,
V., Emmanuel, F., Bureau, M. F., Soubrier, F., Esposito, B., Duez, H.,
Fievet, C., Staels, B., Duverger, N., Scherman, D., and Tedgui, A.
(1999) Protective role of interleukin-10 in atherosclerosis, Circ.
Res., 85, 17-24.
102.Pinderski Oslund, L. J., Hedrick, C. C.,
Olvera, T., Hagenbaugh, A., Territo, M., Berliner, J. A., and Fyfe, A.
I. (1999) Interleukin-10 blocks atherosclerotic events in vitro
and in vivo, Arterioscler. Thromb. Vasc. Biol.,
19, 2847-2853.
103.Caligiuri, G., Rudling, M., Ollivier, V.,
Jacob, M. P., Michel, J. B., Hansson, G. K., and Nicoletti, A. (2003)
Interleukin-10 deficiency increases atherosclerosis, thrombosis, and
low-density lipoproteins in apolipoprotein E knockout mice, Mol.
Med., 9, 10-17.
104.Tian, Y., Sommerville, L. J., Cuneo, A.,
Kelemen, S. E., and Autieri, M. V. (2008) Expression and suppressive
effects of interleukin-19 on vascular smooth muscle cell
pathophysiology and development of intimal hyperplasia, Am. J.
Pathol., 173, 901-909.
105.Ellison, S., Gabunia, K., Richards, J. M.,
Kelemen, S. E., England, R. N., Rudic, D., Azuma, Y. T., Munroy, M. A.,
Eguchi, S., and Autieri, M. V. (2014) IL-19 reduces ligation-mediated
neointimal hyperplasia by reducing vascular smooth muscle cell
activation, Am. J. Pathol., 184, 2134-2143.
106.Gabunia, K., Jain, S., England, R. N., and
Autieri, M. V. (2011) Anti-inflammatory cytokine interleukin-19
inhibits smooth muscle cell migration and activation of cytoskeletal
regulators of VSMC motility, Am. J. Physiol. Cell Physiol.,
300, C896-906.
107.Chen, W. Y., Cheng, B. C., Jiang, M. J., Hsieh,
M. Y., and Chang, M. S. (2006) IL-20 is expressed in atherosclerosis
plaques and promotes atherosclerosis in apolipoprotein E-deficient
mice, Arterioscler. Thromb. Vasc. Biol., 26,
2090-2095.
108.Blobe, G. C., Schiemann, W. P., and Lodish, H.
F. (2000) Role of transforming growth factor beta in human disease,
N. Engl. J. Med., 342, 1350-1358.
109.Pepper, M. S. (1997) Transforming growth
factor-beta: vasculogenesis, angiogenesis, and vessel wall integrity,
Cytokine Growth Factor Rev., 8, 21-43.
110.Lutgens, E., and Daemen, M. J. (2001)
Transforming growth factor-beta: a local or systemic mediator of plaque
stability, Circ. Res., 89, 853-855.
111.Mallat, Z., Gojova, A., Marchiol-Fournigault,
C., Esposito, B., Kamate, C., Merval, R., Fradelizi, D., and Tedgui, A.
(2001) Inhibition of transforming growth factor-beta signaling
accelerates atherosclerosis and induces an unstable plaque phenotype in
mice, Circ. Res., 89, 930-934.
112.Grainger, D. J., Mosedale, D. E., Metcalfe, J.
C., and Bottinger, E. P. (2000) Dietary fat and reduced levels of
TGFbeta1 act synergistically to promote activation of the vascular
endothelium and formation of lipid lesions, J. Cell Sci.,
113, 2355-2361.