MINI-REVIEW: Mitochondrial Genome and Longevity
R. A. Zinovkin1,2*, M. V. Skulachev1,2, and V. P. Skulachev1
1Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119991 Moscow, Russia; E-mail: roman.zinovkin@gmail.com2Lomonosov Moscow State University, Institute of Mitoengineering, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received August 14, 2016; Revision received September 7, 2016
The mitochondrial genome provides not only respiratory chain function, but it also ensures the impact of mitochondria on nearly all crucial metabolic processes. It is well known that mitochondria regulate aging and lifespan. However, until now there were no direct experimental data concerning the influence of various mitochondrial DNA variants on lifespan of animals with identical nuclear genome. In a recent paper of J. A. Enríquez and coworkers (Latorre-Pellicer, A., et al. (2016) Nature, 535, 561-565), it was shown that mice carrying nuclear DNA from one strain and mitochondrial DNA from another had longer median lifespan and retarded development of various aging traits. This review critically analyzes that paper and considers some aspects of the crosstalk between the nuclear and mitochondrial genomes. We also discuss new perspectives of gerontology in the light of the discovery made by Enríquez’s group.
KEY WORDS: mitochondria, mitochondrial DNA, nuclear genome, lifespan, agingDOI: 10.1134/S0006297916120014
Abbreviations: mtDNA, mitochondrial DNA; nDNA, nuclear DNA; ROS, reactive oxygen species.
Mitochondria are cellular organelles containing their own genome, like
cell nuclei. Mitochondrial DNA (mtDNA) of most mammalian species
contains genes necessary for the functioning of the respiratory chain:
ND1, 2, 3, 4, 4L, 5, and
6 of complex I; cytochrome b of complex III; COI,
COII, and COIII of complex IV; ATP6 and
ATP8 of complex V. In addition, mtDNA carries genes for 22 types
of tRNA, two ribosomal RNA, as well as a noncoding portion of about 1
kb involved in the regulation of mtDNA replication and its
transcription [1].
Mitochondrial genes are necessary for respiratory ATP synthesis, and therefore it would be expected that the mitochondrial genome should not be subject to frequent mutations. However, the mtDNA mutagenesis rate is very high compared to that of nuclear DNA (nDNA) [2]. This is probably due to the proximity of mtDNA (nucleoids) to sites of generation of reactive oxygen species (ROS), as well as a less complex mitochondrial reparation system. In the human population, there are several mtDNA variants (haplogroups) with different nucleotide sequences in both noncoding and coding regions, thus affecting amino acid sequence of the respiratory chain enzymes. It is assumed that human mtDNA haplogroups originated due to natural selection and the environment context [3].
Every nucleated cell of an organism contains two copies of nuclear genes and a few hundreds to thousands of mtDNA molecules. In a single cell, at the same time several variants of mtDNA may be found (so-called heteroplasmy). Previously, it was thought that the heteroplasmy is a relatively rare event, but the use of “deep” sequencing technology confirmed that a very low-level of heteroplasmic variance is present in all tested healthy individuals [4].
The majority of the thousand or more mitochondrial proteins are encoded in the nuclear genome. Therefore, the functioning of mitochondrial and nuclear genomes is closely related. Cellular information is transferred from the nucleus to the mitochondria and back (retrograde signaling). For a detailed study of the interaction between mitochondrial and nuclear genomes at the cellular level, so-called cybrids (from “cytoplasmic hybrids”) are used. At the organismal level, they use special conplastic strains derived from backcrossing of hybrid females F1 with the original strain for a number of generations (Fig. 1).
Fig. 1. Generation of the mouse strains used in the work of J. A. Enríquez and colleagues [5].
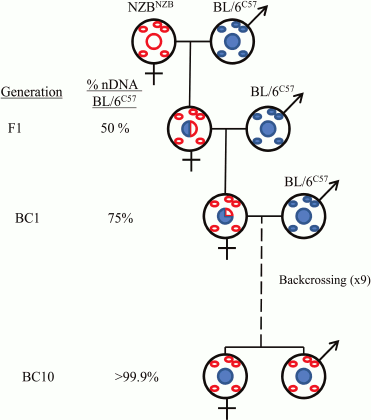
Evolution of the nuclear and mitochondrial genomes occurs simultaneously, thus ensuring maintenance of the functional activity of cells, tissues, and organs. This was first demonstrated in the elegant work of Kenyon and Moraes [6]. They obtained cybrid cells containing human nucleus and mitochondria of various primates. It was found that mitochondrial oxidative phosphorylation is maintained only with mitochondria from our closest relatives – chimpanzees and gorillas. The mitochondria from more evolutionarily distant orangutans and lemurs did not possess that ability. It is important to note that the incompatibility between mitochondrial and nuclear genomes contributes to the creation of interspecies reproductive barriers [7].
Since mtDNA is inherited exclusively from the mother, Nature herself constantly makes evolutionary experiments on the interaction of these genomes. In human and animal populations, there are many individuals with identical mtDNA, differing only in the nuclear genome. The study of these examples led to the understanding that certain variants of mtDNA are associated with the development of various diseases and aging. In extreme cases, the accumulation of mtDNA mutations results in the development of mitochondrial diseases [8]. However, the most pathogenic mtDNA variants are eliminated during oogenesis [1], thus ensuring species survival, otherwise the inevitable steady accumulation of mutations would lead to their extinction.
In 1998, it was first shown that the mtDNA genotype is one of the genetic factors determining human lifespan [9]. Since then, many other studies identified new correlations between different mtDNA haplogroups and lifespan (see review [10]). For example, haplogroup J was associated with longevity in Finns, but not in the southern Italians [11], thus indicating population specificity of the mitochondrial haplogroups. Moreover, analysis of different mtDNA haplogroups appeared to be insufficient for understanding of the whole picture. There was a need to consider the effect of all genetic variations in mtDNA. It was shown that mutations in subunits of complex I increased longevity, while the simultaneous presence of mutations in complexes I and III seemed to be harmful [12].
The direct effect of somatic mtDNA mutations on lifespan has been clearly demonstrated using “mutator” mice deficient in the proofreading activity of mitochondrial DNA polymerase γ [13]. These mice accumulated a large number of mutated mtDNA, prematurely grew old, and died. Importantly, wild-type mice containing mutated mtDNA from the mutator mice also had decreased lifespan compared to wild-type animals [14]. Thus, it was confirmed that mutations in mtDNA directly affect lifespan.
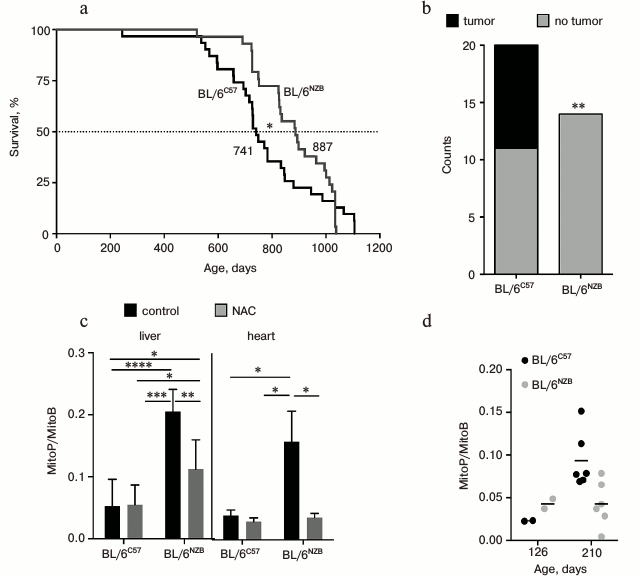
In a report recently published in Nature [5], J. A. Enríquez and coworkers investigated molecular, biochemical, and gerontological aspects of the two mouse strains differing only in the mitochondrial genome. They obtained the mouse strain BL/6NZB with the nuclear genome from C57BL/6 strain and mtDNA from NZB/OlaHsd strain. The original mouse strain C57BL/6 with the mitochondrial genome of C57BL/6 is referred to as BL/6C57. According to mtDNA, these strains differ from each other to about the same extent as the Eurasian and African haplogroups. It was found that the median lifespan of BL/6NZB mice was higher compared to BL/6C57 (Fig. 2a). After nine months, BL/6NZB mice gained less weight, had reduced rate of telomere shortening, and lower incidence of tumors at the time of death (Fig. 2b). Using transcriptomics, metabolomics, and proteomics studies, they revealed a number of differences between these two strains. For example, BL/6NZB conplastic mice had higher expression of lipid metabolism and lower expression of carbohydrate metabolism and inflammatory pathways.
Fig. 2. Functional differences between mice strains that differ in mtDNA (reproduced from [5] with permission from Nature). a) Survival curves (n = 31 per genotype, 19 males, 12 females). b) Tumor incidence in animals dying from natural causes (n = 14 BL/6NZB, n = 20 BL/6C57; ** p < 0.01, Fisher’s exact test). c) Relative amount of mitochondrial H2O2 ± N-acetylcysteine (NAC) in vivo in liver and heart tissues (12 weeks old; n = 6 per genotype). Ordinate axis: the ratio of the two mitochondria-directed cations MitoP/MitoB. Mitochondrial probe MitoB reacts with H2O2 to form MitoP. NAC, non-targeted antioxidant. d) Relative amount of H2O2 in liver mitochondria of young (126 days, n = 2 per genotype) and adult (210 days, n = 6 per genotype) mice of the two different strains. Ordinate as in Fig. 2c.
The rate of respiration and ATP synthesis in liver mitochondria of newborn BL/6NZB mice was slightly lower than BL/6C57 mice. Subsequently, these variables equalized, then from 100-day-old onwards the rates in BL/6C57 mice decreased gradually, while in BL/6NZB they remained constant.
Interestingly, in young BL/6NZB mice (126 days) the liver and heart tissues had elevated mitochondrial ROS level measured by the mitochondrial probe MitoB [15] (Fig. 2c). On the contrary, in adult BL/6NZB animals (210 days) mitochondrial H2O2 level was lower than that of BL/6C57 (Fig. 2d). It is suggested that an adaptive response (hormesis) may take place, wherein an elevated level of ROS at the young age prevents its subsequent increase and thus prolongs lifespan. It should be noted that the method of mitochondrial H2O2 measurement using MitoB probe has been tested in just a few laboratories, so it is difficult to define its suitability. In addition to a number of high-tech procedures, oxidative damage of proteins, lipids, and nucleic acids in the animal tissues should be directly measured. Also, a straightforward experiment should be made: if the effect of hormesis indeed takes place, then antioxidants should decrease lifespan of BL/6NZB mice.
Continuing criticism of this work, it is necessary to note the following:
1) there are no data either for the control strain NZBC57 or even for the original NZB/OlaHsd strain. Therefore, it is not clear how to interpret the results concerning survival and metabolic activity of the animals;
2) the use of highly inbred strains may result in various artifacts due to the presence of homozygous alleles carrying potential harmful mutations. This obvious fact is confirmed by the authors themselves mentioning that C57BL/6 animals strain has a mutated gene coding for protein responsible for supercomplex III : IV formation. Consequently, the results should be confirmed in outbred animals;
3) the authors performed nDNA sequencing of the conplastic strain and claimed that it confirms identity of their nuclear genomes. Note, however, that backcrossing of F1 hybrid females with the males of the original strain may result in the persistence of some maternal alleles in the offspring due to natural selection preserving the effective interaction between the nucleus and mitochondria [16]. They showed that the nuclear genomes of BL/6NZB and BL/6C57 differ in only 197 positions (SNPs). One cannot exclude that some of the SNPs remained in the congenic strain BL/6NZB just because of the above-mentioned mechanism of natural selection;
4) it is known that inheritance of nuclear DNA methylation pattern from gametes is predominantly maternal [17]. The authors did not compare the methylation profiles of the animal strains. It is possible that the methylome of BL/6NZB strain kept some features of the original strain NZB/OlaHsd. Since methylation in the promoter region of genes determines their transcriptional activity, this would explain transcriptomic, proteomic, and other differences between the strains;
5) in addition to the nuclear epigenetics, it is also necessary to determine mtDNA methylation pattern of these strains since this parameter may also influence lifespan (see review [18]);
6) there is no respiratory control in the measurement of mitochondrial respiratory rate. Thus, it is impossible to estimate H+ leakage across the inner mitochondrial membrane. Such leak would explain steady ROS level in aging BL/6NZB animals;
7) theoretically, using the numerous “-omics” would help to understand exactly how molecular and biochemical processes are transformed in the animals with the foreign mitochondrial genome. However, despite the abundance of information, there is nearly nothing to add to the obvious fact that the difference between the derived strains is very high. Surely, this claim can be presented to the vast majority of the contemporary works applying systematic analysis of biological objects. However, it is possible that in the future some useful information will be obtained;
8) two mouse strains with mtDNA differing in 24 positions were used in this study. Perhaps it makes more sense to use a systematic approach to study the effect of individual mtDNA mutations on the mitochondrial function and aging. Probably soon we should expect to see animals carrying “optimized” mtDNA sequence to achieve maximum lifespan.
In the discussion of their work, the authors correctly assume that their findings also have practical implications. Recently, in the UK donation of mtDNA from a “third” parent was approved. This would potentially lead to the birth of children whose mitochondrial genome is poorly compatible with nuclear. Therefore, it is necessary to continue research in this area and clarify many aspects of the interaction of nuclear and mitochondrial genomes, as well as the role of mitochondrial ROS in the aging process. In this regard, special attention should be paid to Fig. 2d of Enríquez et al. [5]. Long-lived and slow aging BL/6NZB mouse strain at young age has a higher level of mitochondrial H2O2 than the original strain. However, this level is not increasing with age. As a result, the adult (210 days) long-lived mice have twice lower level of H2O2 than the short-lived strain. Exactly the same pattern described in the long-lived naked mole rat (lifespan >32 years compared to 3 years for the mouse) [19].
According to our data [20, 21] mitochondria-targeted antioxidant SkQ1 prolongs median (not maximum) lifespan of mice, and this effect is even more pronounced than in the case of mitochondria replacement in the experiments of Enríquez and coworkers (we – 100%, Enríquez et al. – 16%). At the same time, SkQ1, like mitochondria replacement, retards the traits of aging such as kyphosis, graying, hair loss, and age-related weight increase due to fat deposition. It is important to note that both SkQ1 and mtDNA replacement reduce the level of mitochondrial ROS in adulthood, which likely provides an increase in the animals’ lifespan.
Acknowledgements
The work was supported by the Russian Science Foundation (project No. 14-24-00107).
REFERENCES
1.Wallace, D. C., Lott, M. T., and Procaccio, V.
(2013) Mitochondrial medicine: the mitochondrial biology and genetics
of metabolic and degenerative diseases, cancer, and aging, in Emery
and Rimoin’s Essential Medical Genetics (Rimoin, D. L.,
Pyeritz, R. E., and Korf, B. R., eds.) Churchill Livingstone,
Philadelphia.
2.Brown, W. M., George, M., Jr., and Wilson, A. C.
(1979) Rapid evolution of animal mitochondrial DNA, Proc. Natl.
Acad. Sci. USA, 76, 1967-1971.
3.Wallace, D. C. (2015) Mitochondrial DNA variation
in human radiation and disease, Cell, 163, 33-38.
4.Payne, B. A., Wilson, I. J., Yu-Wai-Man, P.,
Coxhead, J., Deehan, D., Horvath, R., Taylor, R. W., Samuels, D. C.,
Santibanez-Koref, M., and Chinnery, P. F. (2013) Universal heteroplasmy
of human mitochondrial DNA, Hum. Mol. Genet., 22,
384-390.
5.Latorre-Pellicer, A., Moreno-Loshuertos, R.,
Lechuga-Vieco, A. V., Sanchez-Cabo, F., Torroja, C., Acin-Perez, R.,
Calvo, E., Aix, E., Gonzalez-Guerra, A., Logan, A., Bernad-Miana, M.
L., Romanos, E., Cruz, R., Cogliati, S., Sobrino, B., Carracedo, A.,
Perez-Martos, A., Fernandez-Silva, P., Ruiz-Cabello, J., Murphy, M. P.,
Flores, I., Vazquez, J., and Enriquez, J. A. (2016) Mitochondrial and
nuclear DNA matching shapes metabolism and healthy ageing,
Nature, 535, 561-565.
6.Kenyon, L., and Moraes, C. T. (1997) Expanding the
functional human mitochondrial DNA database by the establishment of
primate xenomitochondrial cybrids, Proc. Natl. Acad. Sci. USA,
94, 9131-9135.
7.Ma, H., Gutierrez, N. M., Morey, R., Van Dyken, C.,
Kang, E., Hayama, T., Lee, Y., Li, Y., Tippner-Hedges, R., and Wolf, D.
P. (2016) Incompatibility between nuclear and mitochondrial genomes
contributes to an interspecies reproductive barrier, Cell
Metab., 24, 283-294.
8.Wallace, D. C. (1999) Mitochondrial diseases in man
and mouse, Science, 283, 1482-1488.
9.Tanaka, M., Gong, J. S., Zhang, J., Yoneda, M., and
Yagi, K. (1998) Mitochondrial genotype associated with longevity,
Lancet, 351, 185-186.
10.Sevini, F., Giuliani, C., Vianello, D.,
Giampieri, E., Santoro, A., Biondi, F., Garagnani, P., Passarino, G.,
Luiselli, D., Capri, M., Franceschi, C., and Salvioli, S. (2014) mtDNA
mutations in human aging and longevity: controversies and new
perspectives opened by high-throughput technologies, Exp.
Gerontol., 56, 234-244.
11.Dato, S., Passarino, G., Rose, G., Altomare, K.,
Bellizzi, D., Mari, V., Feraco, E., Franceschi, C., and De Benedictis,
G. (2004) Association of the mitochondrial DNA haplogroup J with
longevity is population specific, Eur. J. Hum. Genet.,
12, 1080-1082.
12.Raule, N., Sevini, F., Li, S., Barbieri, A.,
Tallaro, F., Lomartire, L., Vianello, D., Montesanto, A., Moilanen, J.
S., Bezrukov, V., Blanche, H., Hervonen, A., Christensen, K., Deiana,
L., Gonos, E. S., Kirkwood, T. B., Kristensen, P., Leon, A., Pelicci,
P. G., Poulain, M., Rea, I. M., Remacle, J., Robine, J. M., Schreiber,
S., Sikora, E., Eline Slagboom, P., Spazzafumo, L., Antonietta Stazi,
M., Toussaint, O., Vaupel, J. W., Rose, G., Majamaa, K., Perola, M.,
Johnson, T. E., Bolund, L., Yang, H., Passarino, G., and Franceschi, C.
(2014) The co-occurrence of mtDNA mutations on different oxidative
phosphorylation subunits, not detected by haplogroup analysis, affects
human longevity and is population specific, Aging Cell,
13, 401-407.
13.Trifunovic, A., Wredenberg, A., Falkenberg, M.,
Spelbrink, J. N., Rovio, A. T., Bruder, C. E., Bohlooly, Y. M., Gidlof,
S., Oldfors, A., Wibom, R., Tornell, J., Jacobs, H. T., and Larsson, N.
G. (2004) Premature ageing in mice expressing defective mitochondrial
DNA polymerase, Nature, 429, 417-423.
14.Ross, J. M., Coppotelli, G., Hoffer, B. J., and
Olson, L. (2014) Maternally transmitted mitochondrial DNA mutations can
reduce lifespan, Sci. Rep., 4, 6569-6571.
15.Cocheme, H. M., Logan, A., Prime, T. A.,
Abakumova, I., Quin, C., McQuaker, S. J., Patel, J. V., Fearnley, I.
M., James, A. M., Porteous, C. M., Smith, R. A., Hartley, R. C.,
Partridge, L., and Murphy, M. P. (2012) Using the mitochondria-targeted
ratiometric mass spectrometry probe MitoB to measure
H2O2 in living Drosophila, Nat.
Protoc., 7, 946-958.
16.Gregorova, S., Divina, P., Storchova, R.,
Trachtulec, Z., Fotopulosova, V., Svenson, K. L., Donahue, L. R.,
Paigen, B., and Forejt, J. (2008) Mouse consomic strains: exploiting
genetic divergence between Mus m. musculus and Mus m.
domesticus subspecies, Genome Res., 18, 509-515.
17.Muers, M. (2011) Methylation from mother, Nat.
Rev. Genet., 12, 62011.
18.Zinovkina, L. A., and Zinovkin, R. A. (2015) DNA
methylation, mitochondria, and programmed aging, Biochemistry
(Moscow), 80, 1571-1577.
19.Edrey, Y. H., Casper, D., Huchon, D., Mele, J.,
Gelfond, J. A., Kristan, D. M., Nevo, E., and Buffenstein, R. (2012)
Sustained high levels of neuregulin-1 in the longest-lived rodents; a
key determinant of rodent longevity, Aging Cell, 11,
213-222.
20.Skulachev, M. V., Antonenko, Y. N., Anisimov, V.
N., Chernyak, B. V., Cherepanov, D. A., Chistyakov, V. A., Egorov, M.
V., Kolosova, N. G., Korshunova, G. A., Lyamzaev, K. G., Plotnikov, E.
Y., Roginsky, V. A., Savchenko, A. Y., Severina, I. I., Severin, F. F.,
Shkurat, T. P., Tashlitsky, V. N., Shidlovsky, K. M., Vyssokikh, M. Y.,
Zamyatnin, A. A., Zorov, D. B., and Skulachev, V. P. (2011)
Mitochondrial-targeted plastoquinone derivatives. Effect on senescence
and acute age-related pathologies, Curr. Drug Targets,
12, 800-826.
21.Skulachev, V. P., Anisimov, V. N., Antonenko, Y.
N., Bakeeva, L. E., Chernyak, B. V., Erichev, V. P., Filenko, O. F.,
Kalinina, N. I., Kapelko, V. I., Kolosova, N. G., Kopnin, B. P.,
Korshunova, G. A., Lichinitser, M. R., Obukhova, L. A., Pasyukova, E.
G., Pisarenko, O. I., Roginsky, V. A., Ruuge, E. K., Senin, I. I.,
Severina, I. I., Skulachev, M. V., Spivak, I. M., Tashlitsky, V. N.,
Tkachuk, V. A., Vyssokikh, M. Y., Yaguzhinsky, L. S., and Zorov, D. B.
(2009) An attempt to prevent senescence: a mitochondrial approach,
Biochim. Biophys. Acta, 1787, 437-461.