Possible Interventions to Modify Aging
Giacinto Libertini* and Nicola Ferrara
Federico II University, Department of Translational Medical Sciences, 80138 Naples, Italy; E-mail: giacinto.libertini@tin.it* To whom correspondence should be addressed.
Received May 25, 2016; Revision received July 25, 2016
The programmed aging paradigm interprets aging as a function favored by natural selection at a supra-individual level. This function is implemented, according to the telomere theory, through mechanisms that operate through the subtelomere–telomere–telomerase system. After reviewing some necessary technical and ethical reservations and providing a concise description of aging mechanisms, this work considers interventions that could lead to the control of some highly disabling characteristics of aging, such as Alzheimer’s and Parkinson’s syndromes and age-related macular degeneration, and afterwards to a full control of aging up to a condition equivalent to that of the species defined as “with negligible senescence”. The various steps needed for the development of such interventions are described along general lines.
KEY WORDS: aging, programmed aging paradigm, Alzheimer, Parkinson, telomere, subtelomereDOI: 10.1134/S0006297916120038
Aging, defined as “increasing mortality with increasing chronological age in populations in the wild” [1], also known as “actuarial senescence in the wild” [2], or “progressive loss of function accompanied by decreasing fertility and increasing mortality with advancing age” [3], is observed in the wild for many species [1, 4-6], our species included [7].
The theories that try to explain aging are many [8-10] and, in general, belong to one of only two very different general interpretations [11, 12], which for their important and opposite implications deserve the definition of paradigms in the meaning proposed by Kuhn [13].
The first, or “old”, paradigm includes various hypotheses that justify aging as due to inevitable damaging factors that are insufficiently contrasted by natural selection for various reasons. The second, or “new”, paradigm includes theories that explain aging as a specific function, or program, which is favored by natural selection, at the supra-individual level, under particular conditions [10, 12]. The new paradigm interprets aging as a particular type of phenoptosis [14] (“programmed death of an individual” [15]), a concept that includes many different phenomena, known for a long time [4], but not considered in their entirety and for their important implications until recent times [16].
In this work, the evidence in support of the new paradigm and against the old paradigm [10, 11] and a less concise description of aging physiology [17] is not expounded for brevity. This work tries to propose feasible interventions in the aging process that are consequent to the main concept of the new paradigm: indeed, if aging is a function, i.e. a physiological program that is genetically determined and regulated, and if aging mechanisms are sufficiently known, it should be possible to conceive possible modifications of this program that could hamper or even cancel age-related fitness decline. In contrast, this is not at all likely if, as proposed by the old paradigm, aging is an inevitable consequence of the cumulative effect of various damaging factors that act on many cellular and organismal processes [18].
Within the new paradigm, the theory that explains aging as a consequence of telomere shortening and the consequent: (i) progressive inhibition of cell functions and (ii) cell senescence (Fossel’s “cell senescence general model of aging” [17, 19, 20]) will be defined as “telomere theory” of aging and described in brief in the next section. Afterwards, some possible effective methods to modify aging will be proposed.
PREMISES
Technical reservations. The methods that subsequently will be expounded require in various steps the modifications of DNA segments by appropriate means. Currently, the technique that seems most feasible regarding both cost and feasibility, in comparison with other means such as ZFNs (zinc finger nucleases) and TALENs (transcriptional activator-like effector nucleases), is the so-called CRISPR-CAS9 (clustered regularly interspaced short palindromic repeat–CRISPR-associated nuclease 9) technique [21, 22].
Regarding this technique or about any other possible means, in particular for the possible necessity of inserting longer DNA segments, a critical element is the accuracy and reliability of DNA modifications associated with this method [23, 24]. Moreover, recent works have shown ways to greatly ameliorate the precision of the CRISPR-CAS9 technique [25, 26].
This criticality varies in a fundamental way according to the type of experiment:
– Level 1: For example, if we operate on yeast cells or on cultured cells or on animals, possible errors in DNA changes reduce the certainty and the accuracy of the results but do not involve consequences of pathological relevance;
– Level 2: If we act on human individuals no longer in the reproductive stage and suffering from diseases for which a cure is desperately sought, the imprecision of the changes could reduce the effectiveness of the results or even confer negative effects on the health of the affected subject as a result of unexpected DNA alterations, but will have no effect on genetic heritage;
– Level 3: In contrast, by operating on human subjects who may reproduce, the desired genetic modifications in germ line cells (in addition to the ethical reservations of the next point) could be accompanied by unexpected and undesired changes in other segments of DNA molecules that will be transmitted to subsequent generations.
These considerations imply that the degree of reliability of the possible techniques to modify DNA (i) is not critical for experiments of level 1; (ii) is critical and preliminary for experiments or treatments of level 2; (iii) is extremely critical and strongly preliminary for experiments or actions of level 3.
The observations outlined here, in very general terms, should always be considered for the possible actions discussed in this work. They will not be repeated but shall be considered as always implied in any proposal that will be formulated.
Ethical reservations. Any test or treatment or action on humans, or on living beings in general, requires an ethical evaluation; all the more for work on genetic information that will be transmitted to subsequent generations. For the proposals set out in this work, we need two kinds of ethical evaluations, quite different from each other.
The first type relates to the possible dangers to the health of those affected by the experiments or by the proposed actions. In general, for this type of hazard, it is essential first of all to conduct a series of experiments at the level of cultured cells and/or on other species. Only at a later stage will experiments on individuals suffering from serious and incurable diseases and not a condition to reproduce be admissible.
The second type of ethical evaluations is radically different. According to the prevailing conception, i.e. the old paradigm, aging results from the cumulative effect of various degenerative processes and so the care of one or more (or even all) aspects of aging is only the cure for various diseases. According to this paradigm, there is no ethical problem in procedures that contrast the aging process. Conversely, if the aging process is conceived of as a genetically determined and regulated phenomenon, an idea that is implicit to a detailed description of the mechanisms that carry it into effect and is a prerequisite for the proposals that afterwards will be here formulated, it is no longer possible to conceive aging as a sum of diseases or, on the whole, as a complex disease. By defining a disease as an alteration of a normal physiological process, it follows that aging may be “modified” but not “cured” because aging is a normal physiological process and not a disease. It would be different if aging, although normal, was equated to a medical condition due to its discomforts and impairments, but this different definition of disease should also be carefully evaluated in ethical terms.
Therefore, actions that modify aging require a particular ethical evaluation. Even more, possible actions that change the rates and manifestations of the aging process by gene modifications that will be transmitted to subsequent generations, as they are changes to human nature, need a very particular ethical evaluation that is beyond the scientific limits and falls within the ethical, religious, philosophical, and political fields. Consequently, the possibility of modifying aging, regardless of technical feasibility and of possible dangers associated with the procedures, needs assessments and decisions that are beyond mere scientific competence.
MECHANISMS OF AGING ACCORDING TO THE TELOMERE THEORY
Aging mechanisms in Saccharomyces cerevisiae. Saccharomyces cerevisiae (yeast), a unicellular species, shows some phenomena that may be somehow considered as aging, if it is precisely defined, as aforesaid, “increasing mortality with increasing chronological age in populations in the wild” [1].
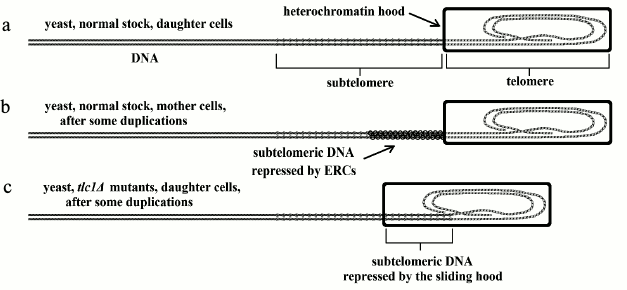
In fact, each yeast cell reproduces by division into two cells, “mother” and “daughter” cells, which are not perfectly equal. The daughter cell is identical to the parent cell, while the cells of the mother lineage can reproduce only a limited number of times (in 3 days, for about 25-35 duplications [27]). In proportion to the number of duplications, two phenomena are observed: (i) increasing metabolic alterations [28-32]; (ii) growing vulnerability to replicative senescence and apoptosis [28, 29, 31, 32]. These phenomena are a likely explanation of why some individuals die and others survive under particular conditions of stress. However, in the mother lineage, the death rate increases with an exponential dynamic as a function of the number of duplications [33]. This is similar to the age-related increase in mortality shown by many multicellular species [5, 6] and may be considered within the concept of aging, as underlined before.
In yeast, DNA is linear, as for all eukaryotic cells, and not circular as for prokaryotes. In each replication, the enzyme DNA polymerase does not duplicate a small terminal part of the DNA molecule (the telomere) [34, 35]. The necessity of an enzyme able to restore telomere integrity was predicted in 1973 [36] and some years later the existence of this enzyme (telomerase) was demonstrated [37].
In yeast, telomerase is always perfectly active both in mother and daughter cells and there is no reduction in telomere length with each duplication [38-40].
In mother cells, the metabolic alterations and the vulnerability to replicative senescence and apoptosis, which increases after each duplication, are caused by a particular mechanism that is not related to telomere shortening, as in multicellular eukaryotes (see below). In yeast mother cells, proportionally to the number of replications, there is the accumulation of particular molecules, defined as extrachromosomal ribosomal DNA circles (ERCs) [41]; “several lines of evidence suggest that accumulation of ERCs is one determinant of life span” [30].
Specifically, a particular type of yeast mutant indirectly shows that the accumulation of ERCs interferes with the action of that part of the DNA molecule adjacent to the telomere, the subtelomere, and that in these mutants, interference is analogous to what happens in eukaryotic multicellular organisms, where subtelomere repression is a consequence of telomere shortening due to telomerase inactivity. In fact, tlc1Δ mutants, whose telomerase is inactive, show telomere shortening both in mother and daughter cells. However, the cells of the daughter cell lineage, although they show no ERC accumulation like normal strains, manifest all the alterations of mother lineage cells with an equal number of duplications. In particular, the overall expression of genes, defined as the transcriptome, is similar [30].
Experiments in yeast have allowed us to hypothesize that: “One model of telomere-gene expression linkage is an altered chromosomal structure (Ferguson et al., 1991), such as a heterochromatin “hood” that covers the telomere and a variable length of the subtelomeric chromosome (Fossel, 1996; Villeponteau, 1997; Wright et al., 1999). As the telomere shortens, the hood slides further down the chromosome (the heterochromatin hood remains invariant in size and simply moves with the shortening terminus)… the result is an alteration of transcription from portions of the chromosome immediately adjacent to the telomeric complex, usually causing transcriptional silencing, although the control is doubtless more complex than merely telomere effect through propinquity (Aparicio and Gottschling, 1994; Singer et al., 1998; Stevenson and Gottschling, 1999). These silenced genes may in turn modulate other, more distant genes (or set of genes). There is some direct evidence for such modulation in the subtelomere…” [19].
The gradual repression of subtelomeric DNA, caused by the accumulation of ERCs in normal yeast or by telomere shortening and heterochromatin hood sliding in yeast tlc1Δ mutants, has been known from some time and defined as the “telomere position effect” [42]. It has regulatory effects on gene expression in distant parts of the DNA molecule [43]. Perhaps, due to its effects, it is better to substitute the neutral expression “telomere position effect” with “gradual cell senescence”, which has a descriptive and interpretative value [10]. These concepts are illustrated in Fig. 1.
Regarding the passage from a normal state to replicative senescence and apoptosis, possible analogies with the cell senescence program (replicative senescence + on/off senescence) in multicellular eukaryotes have been proposed (see below).
Fig. 1. In yeast: a) normal stock, daughter lineage, the telomere is not shortened and the subtelomere is not repressed; b) normal stock, mother lineage, after each duplication, the telomere is not shortened but the subtelomere is progressively repressed by ERC accumulation; c) tlc1Δ mutants, daughter lineage, after each duplication, the telomere is shortened and the subtelomere is repressed by sliding of the heterochromatin hood and not by ERC accumulation.
AGING MECHANISMS IN MULTICELLULAR EUKARYOTES
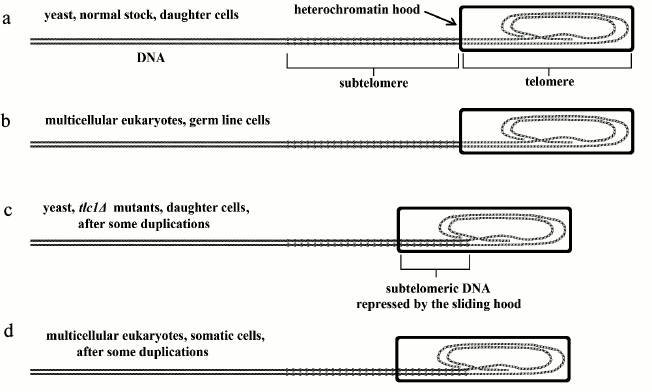
“Gradual cell senescence”. In vertebrates, in cells where telomerase is inactive or partially active, the telomere shortens with each duplication and the sliding of the heterochromatin hood progressively represses the subtelomere. In contrast, in cells where telomerase is perfectly active (e.g. germ line cells), the telomere does not shorten and the subtelomere is not repressed [19]. The first case is analogous to that of yeast tlc1Δ mutants, daughter lineage cells, while the second case is analogous to that of normal daughter lineage cells (Fig. 2).
Fig. 2. a, b) Analogy between normal yeast, daughter lineage, and germ line cells in vertebrates (or, in general, in multicellular eukaryotes); c, d) analogy between yeast tlc1Δ mutants, daughter lineage, and vertebrate somatic cells.
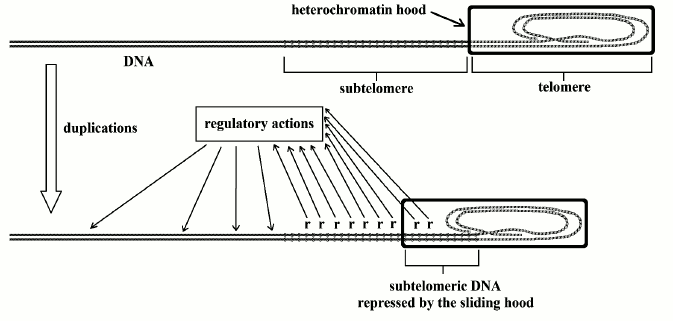
Regarding the alterations to subtelomere regulatory actions caused by the progressive sliding of the heterochromatin hood, it is possible to hypothesize that the subtelomere has a series of regulatory sequences (“r”), which carry on their actions on distant parts of the DNA, and, one after the other, are progressively covered and repressed by the heterochromatin hood (Fig. 3).
Fig. 3. The telomere shortens with each replication and the heterochromatin hood slides over the subtelomere, repressing an increasing portion of subtelomeric DNA, which probably means the repression of the hypothetical “r” sequences. This allows for alterations in gene expression in different and distant parts of the DNA molecule (modified and redrawn from Fig. 4 of [10]).
The hypothesis of the existence of “r” sequences is supported by the structure of the subtelomere, which has an “unusual structure: patchworks of blocks that are duplicated” [44]. Moreover, “a common feature associated with subtelomeric regions in different eukaryotes is the presence of long arrays of tandemly repeated satellite sequences” [45].
This “long arrays of tandemly repeated satellite sequences”, an “unusual structure” that characterizes the telomere and is apparently without meaning and useless, therefore would be a general regulator of cell functions progressively inhibited by the knob of telomere shortening (or, in yeast, of ERC accumulation) and so this could be the pivotal mechanism of aging.
According to this interpretation, the subtelomere might be defined as that part of DNA that is contiguous to the telomere and has general regulatory functions. One of its ends is easily defined by the beginning of telomere monotone sequence, while the other end is where repression is at the highest possible level before the obligatory triggering of the cell senescence mechanism caused by excessive telomere shortening.
“Replicative senescence”, “on/off senescence”, “cell senescence program”. A simplistic hypothesis could be that the cell becomes unable to duplicate (replicative senescence) only when telomere shortening reaches a critical value. However, the growth potential of a cell culture shows not an abrupt collapse of duplication capacities, i.e. contemporary replicative senescence for all cells after a certain number of duplications, but a progressive decrease related to telomere length reduction [46, 47]. A brilliant explanation for this phenomenon was proposed by Blackburn [48].
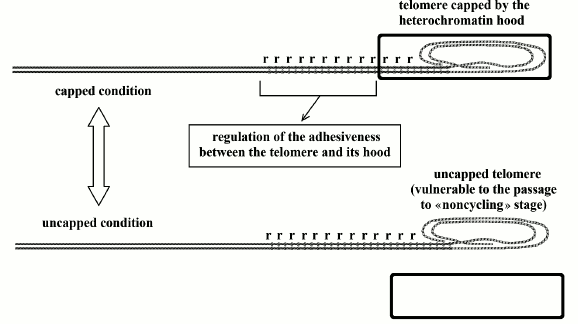
A protein hood (likely the same aforementioned heterochromatin hood [10]) caps the telomere, and there is a continuous oscillation between two telomere states: “uncapped” and “capped”. The first state, whose duration is related to telomere shortening, is vulnerable to the passage to replicative senescence (“noncycling” stage), while the other state is resistant to replicative senescence. Even with activated telomerase and telomeres at the maximum length, with each division there is a short period of uncapped state and a small percentage of cells shows replicative senescence [48].
As the percentage of time in which the telomere is uncapped and vulnerable is related to the progressive subtelomere repression, and as this repression is related to the gradual reduction of the relative telomere length (see before) and not to the initial length of the telomere (see next section), it is proper to assume that the oscillation between capped and uncapped states is somehow regulated by subtelomeric repetitive sequences, which might be the same before defined as “r”. In Fig. 4, these sequences are indicated with the letter “r”.
Fig. 4. Telomeres oscillate between the “capped” and “uncapped” conditions. The probability of uncapped telomeres increases in proportion to subtelomere repression of hypothetical repetitive sequences (“r”) that regulate the degree of adhesion between the heterochromatin hood and the telomere. In the uncapped state, the non-protected telomere is a free end of the DNA molecule and is susceptible to end-to-end joining that blocks cell replication [48].
Stem cells, which, unlike germ line cells, show telomerase activity that only partially preserves telomere length [49], are therefore limited in their replacement capacities of the elements eliminated by cell turnover [19].
The cellular phenomenon described by Blackburn as the shift to a “noncycling state”, i.e. replicative senescence, in fact is part of so-called “cell senescence”, well-defined as a “fundamental cellular program” [50]. It is characterized both by replicative senescence and by the altered expression of many genes in a way that jeopardizes cell functions, cellular secretions included – and thus also the extracellular matrix and those cells physiologically dependent on the altered cells or simply nearby – up to the maximum degree that may be caused by gradual senescence. As the transition to the altered physiological conditions determined by cell senescence, and the reverse passage – by the activation of telomerase [51-55] – towards a condition in which all cellular functions are intact, is a bidirectional on/off transformation, this type of cell alteration has been defined as “on/off senescence” [10].
Absence of a relationship between longevity and initial telomere length or telomerase activity. In the comparison between species, an easy prediction, however falsified by the evidence, is a direct relation between longevity and initial telomere length (i.e. that in the germ cell) and, similarly, between longevity and the degree of telomerase activity. However, many facts disprove these predictions:
a) hamsters and mice have longer telomeres than our species but age more precociously [56];
b) among rodents, there is no relationship between telomerase activity and maximum lifespan [57];
c) mice show a baseline activity of telomerase in most somatic cells [58], but have limited longevity;
d) two Mus strains, the first with a telomere length of 20 kb and the other of only 10 kb, show equal life spans and the same timing patterns of cell senescence [19];
e) analogously, cloned animals derived from somatic cells, which have shortened telomeres, show the same senescence timing of donor animals [19];
f) in telomerase knockout (mTR–/–) mice, which have genetically inactivated telomerase, only after four [59] to six [60] generations, when telomeres are very shortened, it is possible to see, at least in artificially protected laboratory conditions, that fertility and viability are jeopardized. However, in organs with high cell turnover, the alterations are found in early generations [59, 61], so, fitness would likely be reduced under natural conditions.
These phenomena, which are apparently inexplicable if you want to assign an exclusive role in longevity determination to telomere length, on the contrary are completely explicable if it is assumed that in the germ cell, in a phase – defined as the “reset” phase – before the first cell replication, the heterochromatin hood is formed with a size proportional to telomere length. In this reset phase, regarding the longevity, the absolute “telomere length is irrelevant” [19], provided of course that telomere length is not less than a critical size [19]. Afterwards, in proportion to the number of duplications and so to the progressive shortening of the telomere (if it is not elongated by telomerase), there is progressive sliding of the heterochromatin hood, and thus proportional repression of the subtelomere, which causes the manifestations of gradual senescence and increases the probability of cell senescence, i.e. on/off senescence plus replicative senescence.
For this model, it is necessary that: (i) the heterochromatin hood must have a fixed length in all the cells of the organism; (ii) the telomere elongation by telomerase up to the initial length must somehow have the hood length as a limiting factor. It is certainly necessary to study the mechanisms underlying these phenomena, which must be phylogenetically very old.
In fact, as described above, in yeast, with an evolutionary history that diverged from that of our species at least at the beginning of Cambrian era, i.e. about 600 million years ago [62]: (i) in the normal stocks, at each duplication, each cell restores telomere length on the basis of the heterochromatin hood size, which is then kept fixed despite the succession of generations; (ii) in the daughter cells of tlc1Δ mutants, where telomerase is inactive, the telomere shortens with each division, but the hood remains at a fixed length and represses the subtelomere by sliding on it.
All this indicates that, as regards longevity, the critical element is not the absolute initial length of the telomere, but the progressive inhibition of subtelomeric DNA caused by telomere shortening [10, 19]. The aforementioned phenomena (a-f) are illustrated and interpreted in Fig. 5 for phenomena a, b, and c; in Fig. 6 for d and e; and in Fig. 7 for f.
Fig. 5. In case 1, there are shorter subtelomeres and longer telomeres than in case 2. After a certain number of duplications, by assuming that telomeres are equally shortened at each generation in the two cases, in case 1 the shorter subtelomeres are more impaired by the sliding of the heterochromatin hood, which should lead to earlier aging. If we assume for case 1 also a greater mean telomerase activity, which contrasts telomere shortening, this may be insufficient to compensate for greater subtelomeric repression. This model is an easy explanation for phenomena (a), (b), and (c).
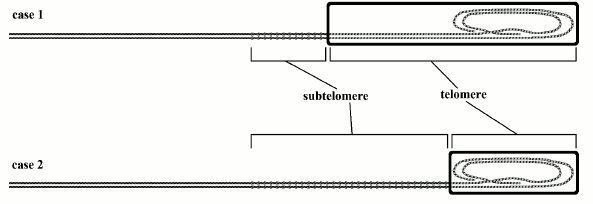
Fig. 6. Case 1: Mus strain with 20 kb telomeres; case 2: Mus strain with 20 kb telomeres (or case 1: donor animals; case 2: cloned animals). In case 1, cells have longer telomeres and heterochromatin hoods than in case 2 cells, but the longevity is the same: the progressive gradual senescence and the increasing probability of cell senescence activation are not a function of telomere absolute initial length but of progressive subtelomere repression, caused by relative telomere shortening. This model explains phenomena (d) and (e).
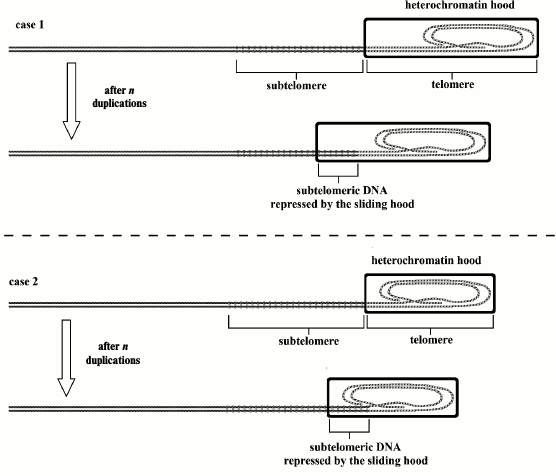
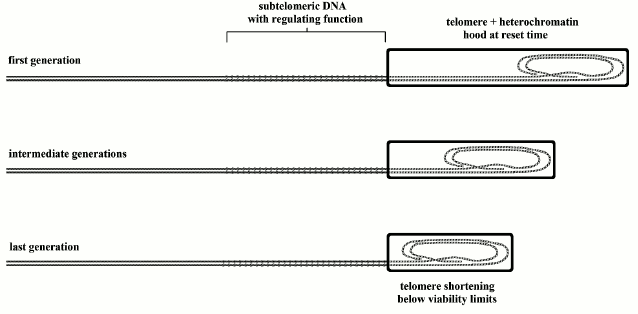
Fig. 7. In mice with telomerase genetically inactivated, the length of the heterochromatin hood, modeled on the telomere in the reset phase, and that of the telomere in the first generation are equal to that of normal stocks. As telomerase is inactive, in each subsequent generation, the initial length becomes shorter, up to fourth to sixth generation when the telomere shortens below viability limits. With each generation, the subtelomere length is unvaried, because it is not influenced by telomerase. Within each generation, with each cell duplication, the telomere shortens and the subtelomere is progressively inhibited by sliding of the heterochromatin hood. Therefore, as shown in the previous figure, this genetic repression is a function of relative telomere shortening and not a function of the absolute initial telomere length [19]. This explains phenomenon (f) (modified and redrawn from Fig. 6 of [10]).
Consequences of gradual senescence and cell senescence. The effects of gradual senescence and cell senescence (on/off senescence + replicative senescence) on the organism as a whole, considered under natural conditions, are the likely roots of all aging manifestations, as already proposed and explained elsewhere [17-19].
In short, we have an “atrophic syndrome” of all tissues and organs, which is the consequence of: (i) an age-related increase in the percentage of cells with altered functions, to varying degrees, due to gradual senescence and on/off senescence; (ii) an age-related decline in the speed and completeness of cell turnover due to the progressive increase in the percentage of stem and somatic cells in replicative senescence; (iii) perennial cells, i.e. cells without turnover (e.g. almost all neurons), are compromised and die by the phenomena (i)-(ii) in satellite cells (glial cells), which show turnover and are essential for their functionality, causing problems that are considered distinct diseases (Alzheimer’s disease, Parkinson’s disease, age-related macular degeneration, etc. [63]); (iv) the consequent (to phenomena (i)-(iii)) reduction in functional cells in a tissue or organ, partially replaced by nonfunctional cells, and the related decline in tissue or organ functionality [17, 18].
The related alterations reduce fitness, i.e. the ability to survive under natural conditions. In contrast, under protected conditions, even a remarkable fitness reduction can be compatible with survival, but, at older ages, the progressive worsening of the above-mentioned tissue/organ alterations becomes lethal even under artificial conditions of greatest protection, which are non-existent in the wild [18, 64].
Distinction between aging and diseases whose frequency and severity increases with age. It is essential to make a clear distinction between the physiological process of aging and several diseases that are virtually absent under natural conditions [18, 64] and often increase in frequency and severity over the years (e.g. hypertension, type 2 diabetes, vascular diseases, various types of cancer, etc. [18]).
These diseases, as a rule, are due to changes in the ecological niche (eating habits, lifestyles, pollutants in the environment, etc.) to which the species is not adapted and which are therefore harmful [18]. Some of these alterations, here defined as “risk factors”, are countered, at least in part, by medications or other measures (“protective factors”).
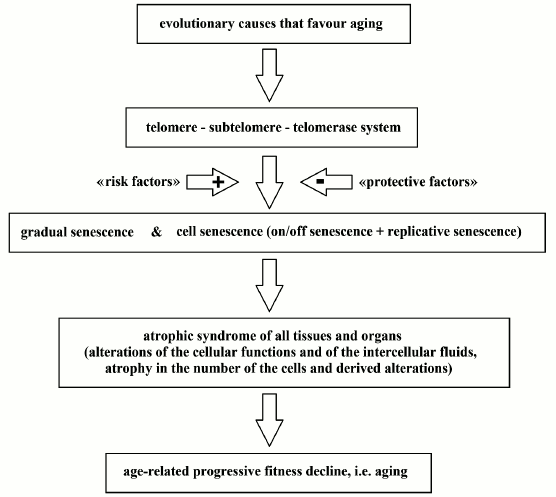
The distinction between the aforesaid diseases and the aging process is that these problems are, at least partially, preventable or curable by the appropriate restoration of the correct ecological niche or by opportune drugs, while, on the contrary, aging, in principle – being a physiological phenomenon – is not contrasted by preventive, protective, or curative actions. Lifestyles and environmental conditions that are ideally perfect to preserve health allow the achievement of physiological, i.e. normal, aging but cannot block or reverse aging [18]. However, “risk factors” may cause an acceleration of the aging process; “protective factors” may contrast this acceleration and give the false impression of contrasting aging while they only counteract pathologically altered aging. A specific example is the gradual weakening of endothelial function, which is a major cause of vascular diseases and can be measured by counting endothelial progenitor cells (EPCs) [65, 66]. EPC number, which is a predictor of cardiovascular disease as reliable as the Framingham score [67], decreases over the years and is also reduced by risk factors. A healthy lifestyle or the action of various drugs can restore the normal EPC number for the age but do not block the reduction related to age [65]. These concepts are summarized in Fig. 8.
Fig. 8. A scheme of the aging process. Evolutionary causes are not discussed in this work.
Surely, it is possible to propose alternative interpretations of the aging process that might be compatible with the programmed aging paradigm (e.g. Olovnikov’s hypothetical “chronomeres” and “printomeres” as pivotal parts of aging mechanisms [68, 69]), but we think that our explanation is more consistent with evidence.
POSSIBLE INTERVENTIONS
The aim of this work is to search for feasible methods to slacken, block, or reverse the normal mechanisms of aging. It must be stressed that many common diseases caused by alterations in the ecological niche, or by other factors: (i) are not part of the normal process of aging, even if their frequency and seriousness may often be age-related; (ii) can be prevented or treated with appropriate measures; and (iii) are not part of this goal.
For our species, in individuals who are in the best physical condition and free from any disease that could endanger their fitness, there is a progressive age-related fitness decline, which starts from about 30 years: “No one would consider a man in his thirties senile, yet, according to athletic records and life tables, senescent is rampant during this decade” [70]. In fact, this decline may somehow be quantified by observing the world speed records for each age group (Fig. 9).
Fig. 9. Quantification of age-related fitness decline based on data from modern populations, namely on speed records for various distances in individuals younger than 35 (world records; source: http://en.wikipedia.org/wiki/World_records_in_athletics), and in other age groups (source: http://www.world-masters-athletics.org/records_output/rec_list_outdoor_m.php). From the beginning of adulthood to an age of about 30-35 years, we observe the best performance, i.e. the greatest fitness, and this is the period with the lowest mortality observed under natural conditions [7].
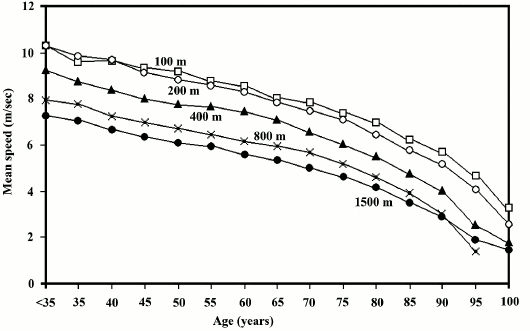
Methods to reverse aging should transform the curve of fitness decline into a straight horizontal line, i.e. a fitness or mortality rate that is constant at any age. The search for these methods will be based exclusively on the interpretation of the aging mechanisms outlined above.
Method 1: telomerase activation. The evaluation of aging mechanisms and the experiments carried out so far suggest immediately that the simplest and best way to arrest aging is telomerase activation to bring telomere length to the initial one, defined by the size of the heterochromatin hood.
Well-known and already mentioned experiments on cultured cells have shown since 1998 that telomerase activation can completely reverse the features of cell senescence, namely to totally rejuvenate cells with the markers of cell senescence [51-55].
At the tissue level, in vitro-aged fibroblasts with “substantial alterations in gene expression” were treated with telomerase and then “assessed by incorporation into reconstituted human skin”, which showed no biological difference with skin obtained from young fibroblasts [71].
At the organismal level, in aged mice with artificially blocked telomerase, telomerase reactivation shows a marked reversal of degenerative manifestations, even for the nervous system [72]. Moreover, in one- and two-year-old normal mice, telomerase expression, induced by adeno-associated viruses carrying the mouse telomerase reverse transcriptase, delays aging and increases longevity (increase in median lifespan of 24 and 13%, respectively) without increasing cancer risk [73].
The use of telomerase reactivation to control aging is, however, wrongly hampered by a formidable non-technical difficulty originating in the assumptions of the old paradigm, which is still the dominant thesis although its validity as a scientific theory is strongly opposed by evidence and theoretical arguments [12]. In fact, according to the old paradigm, genetically determined and regulated mechanisms that progressively impair fitness simply cannot exist. Therefore, the subtelomere–telomere–telomerase system, concisely described in the previous section and supported by strong evidence, for the old paradigm, cannot have aging as an evolutionary motivation but must absolutely have another reason that justifies its existence. The only motivation that has so far been proposed to justify these mechanisms is the old one that interprets them as a general defense against cancer [74, 75]. Replicative senescence would constitute an effective obstacle to cancer proliferation. Thus, aging would be just a side effect of this defense, in a sort of terrible evolutionary trade-off between the problems of aging and the need to defend the organism from cancer [76], in excellent compatibility with the ideas maintained by some old traditional evolutionary hypotheses about the evolutionary causes of aging (antagonistic pleiotropy theory [70, 77]; disposable soma theory [78, 79]).
However, several strong arguments are against the aforesaid explanation, as in part already explained elsewhere [18, 64]: a) there are animal species without any age-related fitness decline (“animals with negligible senescence” [4]) and old individuals of such species (e.g. rainbow trout and lobster) have under natural conditions the same telomerase activity shown by young individuals [80, 81], but there is no age-related increased vulnerability to cancer, as their constant mortality at any age shows; b) gradual senescence and on/off senescence cause a progressive weakening of immune system efficiency [19], and this increases vulnerability to cancer and so cancer incidence [82]; c) shortened telomeres cause DNA instability, and this increases the probabilities of cancer onset [83-85]; d) in eukaryotic unicellular species such as yeast, replicative senescence and apoptosis, not caused by shortened telomeres but by ERC accumulation, are well-documented [27, 33, 86], but cannot be a defense against cancer, as it is impossible in unicellular species; e) gradual senescence, i.e. the existence of critical regulatory sequences in subtelomeric DNA that are gradually repressed because of telomere shortening, is an implausible defense against neoplastic cell proliferation; f) in mice, the selective elimination of senescent cells (p16Ink4a+ cells) contrasts several age-dependent changes, delays the progression of malignant diseases, and increases lifespan [87], and this is against the possibility that senescent cells are a defense against cancer; g) in humans studied in the wild: (i) the survivors at ages 60 and 70 were approximately 30 and 20%, respectively; (ii) cancer cases were not reported and only in a few older individuals (>70 years) there was the possibility that death was caused by cancer. In the same population, the age-related increase in mortality, i.e. aging, was evident starting from the thirties. The hypothesis that the mechanisms underlying this increment of mortality could be a defense against a rare disease which shows its deadly effects at later ages is clearly illogical [64]; h) telomerase activation, which is a common feature in cancer, as it is subsequent to and does not precede cancer onset, must be considered a cancer aggravating phenomenon and not a cause of it [19].
It must be stressed that with the rejection of the aforesaid explanation for cell aging mechanisms as a defense against cancer, the old paradigm loses a valuable last trench and becomes even more untenable. This explains historically and psychologically, but does not justify scientifically, the tenacious defense of this explanation, which is a heavy unjustified brake against the use of telomerase to counter aging.
However, in overcoming this difficulty, the main way to slow aging is clearly telomerase activation and the subsequent restoration of the telomere to its initial length [88].
One possibility is the use of drugs that are capable of such action. Certain substances, called astragalosides, have shown some action in reactivating telomerase [89, 90], but are remarkably expensive and their effect is limited [88].
However, the technique that appears to be more effective and more feasible in a short time is telomerase activation by telomerase reverse transcriptase (TRT) introduced into the organism using an adenovirus as a vector, as in an experiment carried out with significant positive results in mice and already mentioned in this work [73].
This must necessarily be accomplished through several phases:
1) new experiments on animals to study: (i) the best techniques to introduce TRT into the organism with the highest standards of efficiency and security; (ii) further details on the results, also as a function of the age of the individual when the technique is applied;
2) first experiments on elderly human subjects suffering from diseases that are seriously debilitating and are part of aging process, such as Alzheimer’s disease, Parkinson’s disease, age-related macular degeneration, etc. (excluding precocious cases likely due to genetic defects or to harmful lifestyles), and possibly in individuals not in a condition to reproduce, to avoid possible transmission of an altered genome;
3) experiments on subjects with less disabling diseases, or even on individuals with reproductive capacity if the possibility of genome alteration has been excluded;
4) experiments for the treatment of subjects suffering from other diseases that may be considered a consequence of aging process acceleration;
5) experiments for the treatment of healthy elderly individuals, i.e. simply to rejuvenate them.
Method 1 would allow for the rejuvenation of the organism with the limit that any irreversible changes (for example, macro-structural alterations, reduction in stem cell number) would not be correctable. Therefore, it should be applied early enough (e.g. before the age of 40) and repeated relatively frequently (e.g. 10 years). Clearly these are reasonable assumptions that require due confirmation.
Method 2: subtelomere modification. It is possible to envisage a different method based on genetic modifications to subtelomeres such as to increase the age at which fitness decline begins to manifest (i.e. about 30 years in our species [70]). For example, if it were possible to increase this age from 30 to 60 and if, in conjunction with this method, we apply method 1 at the age of 60 and then every 30 years, we would lengthen the period of biological youth without any treatment and minimize possible irreversible changes at the age when method 1 is applied.
Now, let us see the method that is proposed here.
Some abbreviations are necessary for a more concise exposition:
– The telomere is composed of a monotonous repetition of a motif (TTAGGG in vertebrates), which will be indicated with “<m>”. So, if the motif is repeated n times, the telomere will be described as “<m>n”;
– That part of the subtelomere which is next to the telomere and may be defined as the telomere–subtelomere junction; it will be indicated with “<J>”;
– All the remaining part of the subtelomere will be indicated with “<ST>”. According to these abbreviations, the terminal part of a chromosome will be indicated with: <ST><J><m>n;
– The abbreviation “<NS>” means a neutral sequence, i.e. a sequence that has no negative or positive action on the remaining part of the DNA molecule or on any genetic or cellular function;
– The abbreviations “<c1>” and “<c2>” indicate two sequences with distinct marker codes without any identical sequence in the genome.
It is necessary to specify, inter alia, that:
– The study of the <J> sequence is preliminary to the development of the method. It is essential that the <J> sequence is, or is delimited, such that there is no identical sequence in the genome;
– Likewise, the definition of one or more neutral sequences (<NS>) will be preliminary.
The objective of method 2 is to achieve the insertion of one or more sequences <NS> between <ST> and <J>, namely to get the following structure of the terminal part of the chromosome: <ST><J><m>n → <ST><NS>y<J><m>n, where y is an integer arbitrarily chosen for convenience.
To achieve this result, by using a technique such as CRISPR-CAS9, the following procedure might be feasible:
1) substitution of <J> with the sequence <c2><NS><c1>:
<ST><J><m>n → <ST><c2><NS><c1><m>n;
2) substitution of <c1> with <J>:
→ <ST><c2><NS><J><m>n;
3) substitution of <c2> with the sequence <c1><NS>:
→ <ST><c1><NS>2<J><m>n;
4) substitution of <c1> with the sequence <c2><NS>:
→ <ST><c2><NS>3<J><m>n;
5) repetition of the steps 3 and 4 until <NS> is repeated y times:
→ <ST>
6) removal of
→
<ST><NS>y<J><m>n,
which is the required result.
We should note that each sequence that marks where an operation must be
performed (e.g. <J> in (1)) is always destroyed by the action, so
that it cannot be executed more than once.
This change would have effect both on the individual on which it is
performed, and for individuals of the following generations if it has
been accomplished in germ cells.
From a theoretical point of view, it would be useless to lengthen the
subtelomere beyond the maximum length for which the subtelomere can be
inhibited by telomere shortening before the telomere reaches a critical
minimum length and cell senescence is obligatorily triggered.
This method has an important advantage. As it takes place on the
subtelomere and not on the telomere, experiments would be easily
feasible in a S. cerevisiae animal model that “ages”
because of subtelomere inhibition not caused by telomere shortening but
by progressive ERC accumulation. As the optimal result, in a strain of
S. cerevisiae modified by this method, the maternal lineage
cells should stop their replications after several duplications greater
than the 25-35 limit found by Jazwinski [27].
After acquiring a good mastery of the techniques to be employed, the
method should be applied on a multicellular animal model with short
longevity (e.g. mice) and afterwards on animals whose longevity and
physiology are closer to our species (e.g. pigs).
As for method 1, with the reservations expressed in the Introduction (in
particular for the following step (c)), the subsequent steps may be
conceived with the accompanying application (possibly repeated) of
method 1: a) application on human subjects who are unable to procreate
and suffer from serious diseases such as Alzheimer’s disease,
Parkinson’s disease, age-related macular degeneration, etc.; the
possible success of this approach, particularly for diseases like
Alzheimer’s and Parkinson’s, would also allow a decisive
discrimination between their interpretation as coherent and critical
parts of the aging mechanisms [63] and other
interpretations related to biochemical events and functional disorders
of the same diseases (e.g.: [91-94]); b) application on elderly subjects who are
unable to procreate but are healthy; c) modification of the germ cells
of healthy individuals.
Method 3: telomere elongation without the use of telomerase. By
operating after the reset phase, telomerase can lengthen the telomere
to the maximum length allowed by the heterochromatin hood. By using an
alternative method on germ cells, it is possible to envisage telomere
elongation up to the desired length. In both cases, since the
heterochromatin hood is modeled in the reset phase according to
telomere length, this does not affect longevity. However, if the
subtelomere has been modified by method 2, this would allow more
telomere shortening before the telomere length reaches a critical
level. In short, the combined use of method 2 and method 3 would lead
to a longer period before reaching the stage at which fitness begins to
decline and there is a need to resort to method 1 for restoring
fitness.
So, if the goal is to achieve telomere elongation by adding y repeats of
the sequence <m>f (where y and f are integers
arbitrarily chosen according to convenience), namely:
<ST><J><m>n →
<ST><J><m>n+yf, the following
procedure might be feasible:
1) substitution of <J> with the sequence
<c1><m>f<c2>:
<ST><J><m>n →
<ST><c1><m>f<c2><m>n;
2) substitution of <c1> with <J>:
→
<ST><J><m>f<c2><m>n;
3) substitution of <c2> with the sequence
<m>f<c1>:
→
<ST><J><m>2f<c1><m>n;
4) substitution of <c1> with the sequence
<m>f<c2>:
→
<ST><J><m>3f<c2><m>n;
5) repetition of the steps 3 and 4 until the sequence
<m>f is repeated y times:
→ <ST><J><m>yf
6) removal of
→ <ST><J><m>n+yf, which is the
required result.
Figure 10 illustrates by way of example the three
methods outlined here.
Clearly, what is proposed in the last section might be considered
exceedingly speculative. However, the purpose of this work is precisely
to speculate what would be possible to perform to slacken or block the
aging process, starting from the necessary condition of the
truthfulness of the new paradigm – not discussed here – and
of what is known about the subtelomere–telomere–telomerase
system as a result of a considerable amount of evidence.
In no way will this be accepted by those who reject a priori the
new paradigm, but the same are requested to consider what is proposed
here by accepting the new paradigm only as a working hypothesis, i.e.
the revolutionary idea that aging is a physiological phenomenon
determined and regulated by genes and therefore in principle
modifiable.
1.Libertini, G. (1988) An adaptive theory of the
increasing mortality with increasing chronological age in populations
in the wild, J. Theor. Biol., 132, 145-162.
REFERENCES
2.Holmes, D. J., and Austad, S. N. (1995) Birds as
animal models for the comparative biology of aging: a prospectus, J.
Gerontol. A Biol. Sci., 50, B59-B66.
3.Kirkwood, T. B. L., and Austad, S. N. (2000) Why do
we age? Nature, 408, 233-238.
4.Finch, C. E. (1990) Longevity, Senescence, and
the Genome, The University of Chicago Press, Chicago.
5.Ricklefs, R. E. (1998) Evolutionary theories of
aging: confirmation of a fundamental prediction, with implications for
the genetic basis and evolution of life span, Am. Nat.,
152, 24-44.
6.Nussey, D. H., Froy, H., Lemaitre, J. F., Gaillard,
J. M., and Austad, S. N. (2013) Senescence in natural populations of
animals: widespread evidence and its implications for bio-gerontology,
Ageing Res. Rev., 12, 214-225.
7.Hill, K., and Hurtado, A. M. (1996) Ache Life
History, Aldine De Gruyter, New York.
8.Comfort, A. (1979) The Biology of
Senescence, Elsevier North Holland, New York.
9.Medvedev, Z. A. (1990) An attempt at a rational
classification of theories of ageing, Biol. Rev. Camb. Philos.
Soc., 65, 375-398.
10.Libertini, G. (2015) Phylogeny of aging and
related phenoptotic phenomena, Biochemistry (Moscow), 80,
1529-1546.
11.Libertini, G. (2008) Empirical evidence for
various evolutionary hypotheses on species demonstrating increasing
mortality with increasing chronological age in the wild, Sci. World
J., 8, 182-193.
12.Libertini, G. (2015) Non-programmed versus
programmed aging paradigm, Curr. Aging Sci., 8,
56-68.
13.Kuhn, T. S. (1962) The Structure of Scientific
Revolutions, The University of Chicago Press, Chicago.
14.Skulachev, V. P. (1997) Aging is a specific
biological function rather than the result of a disorder in complex
living systems: biochemical evidence in support of Weismann’s
hypothesis, Biochemistry (Moscow), 62, 1191-1195.
15.Skulachev, V. P. (1999) Phenoptosis: programmed
death of an organism, Biochemistry (Moscow), 64,
1418-1426.
16.Libertini, G. (2012) Classification of
phenoptotic phenomena, Biochemistry (Moscow), 77,
707-715.
17.Libertini, G. (2014) The programmed aging
paradigm: how we get old, Biochemistry (Moscow), 79,
1004-1016.
18.Libertini, G. (2009) Prospects of a longer life
span beyond the beneficial effects of a healthy lifestyle, in
Handbook on Longevity: Genetics, Diet & Disease
(Bentely, J. V., and Keller, M. A., eds.) Nova Science Publishers Inc.,
New York, pp. 35-96.
19.Fossel, M. B. (2004) Cells, Aging and
Human Disease, Oxford University Press, New York.
20.Libertini, G. (2009) The role of
telomere–telomerase system in age-related fitness decline, a
tameable process, in Telomeres: Function, Shortening and
Lengthening (Mancini, L., ed.) Nova Science Publishers Inc., New
York, pp. 77-132.
21.Harrison, M. M., Jenkins B. V.,
O’Connor-Giles, K. M., and Wildonger, J. (2014) A CRISPR view of
development, Genes Dev., 28, 1859-1872.
22.Zhang, F., Wen, Y., and Guo, X. (2014)
CRISPR/Cas9 for genome editing: progress, implications and challenges,
Hum. Mol. Genet., 23, R40-46.
23.Chandrasegaran, S., and Carroll, D. (2016)
Origins of programmable nucleases for genome engineering, J. Mol.
Biol., 428, 963-989.
24.O’Geen, H., Yu, A. S., and Segal, D. J.
(2015) How specific is CRISPR/Cas9 really? Curr. Opin. Chem.
Biol., 29, 72-78.
25.Kleinstiver, B. P., Pattanayak, V., Prew, M. S.,
Tsai, S. Q., Nguyen, N. T., Zheng, Z., and Joung, J. K. (2016)
High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide
off-target effects, Nature, 529, 490-495.
26.Slaymaker, I. M., Gao, L., Zetsche, B., Scott, D.
A., Yan, W. X., and Zhang, F. (2016) Rationally engineered Cas9
nucleases with improved specificity, Science, 351,
84-88.
27.Jazwinski, S. M. (1993) The genetics of aging in
the yeast Saccharomyces cerevisiae, Genetica, 91,
35-51.
28.Laun, P., Pichova, A., Madeo, F., Fuchs, J.,
Ellinger, A., Kohlwein, S., Dawes, I., Frohlich, K. U., and
Breitenbach, M. (2001) Aged mother cells of Saccharomyces cerevisiae
show markers of oxidative stress and apoptosis, Mol. Microbiol.,
39, 1166-1173.
29.Herker, E., Jungwirth, H., Lehmann, K. A.,
Maldener, C., Frohlich, K. U., Wissing, S., Buttner, S., Fehr, M.,
Sigrist, S., and Madeo, F. (2004) Chronological aging leads to
apoptosis in yeast, J. Cell Biol., 164, 501-507.
30.Lesur, I., and Campbell, J. L. (2004) The
transcriptome of prematurely aging yeast cells is similar to that of
telomerase-deficient cells, Mol. Biol. Cell, 15,
1297-1312.
31.Buttner, S., Eisenberg, T., Herker, E.,
Carmona-Gutierrez, D., Kroemer, G., and Madeo, F. (2006) Why yeast
cells can undergo apoptosis: death in times of peace, love, and war,
J. Cell Biol., 175, 521-525.
32.Fabrizio, P., and Longo, V. D. (2008)
Chronological aging-induced apoptosis in yeast, Biochim. Biophys.
Acta, 1783, 1280-1285.
33.Laun, P., Bruschi, C. V., Dickinson, J. R.,
Rinnerthaler, M., Heeren, G., Schwimbersky, R., Rid, R., and
Breitenbach, M. (2007) Yeast mother cell-specific ageing, genetic
(in)stability, and the somatic mutation theory of ageing, Nucleic
Acids Res., 35, 7514-7526.
34.Olovnikov, A. M. (1971) Principle of marginotomy
in template synthesis of polynucleotides, Doklady Biochem.,
201, 394-397.
35.Watson, J. D. (1972) Origin of concatemeric T7
DNA, Nat. New Biol., 239, 197-201.
36.Olovnikov, A. M. (1973) A theory of marginotomy:
The incomplete copying of template margin in enzyme synthesis of
polynucleotides and biological significance of the problem, J.
Theor. Biol., 41, 181-190.
37.Greider, C. W., and Blackburn, E. H. (1985)
Identification of a specific telomere terminal transferase activity in
Tetrahymena extracts, Cell, 43, 405-413.
38.D’Mello, N. P., and Jazwinski, S. M. (1991)
Telomere length constancy during aging of Saccharomyces
cerevisiae, J. Bacteriol., 173, 6709-6713.
39.Smeal, T., Claus, J., Kennedy, B., Cole, F., and
Guarente, L. (1996) Loss of transcriptional silencing causes sterility
in old mother cells of Saccharomyces cerevisiae, Cell,
84, 633-642.
40.Maringele, L., and Lydall, D. (2004) Telomerase-
and recombination-independent immortalization of budding yeast,
Genes Dev., 18, 2663-2675.
41.Sinclair, D. A., and Guarente, L. (1997)
Extrachromosomal rDNA circles – a cause of aging in yeast,
Cell, 91, 1033-1042.
42.Gottschling, D. E., Aparicio, O. M., Billington,
B. L., and Zakian, V. A. (1990) Position effect at S. cerevisiae
telomeres: reversible repression of Pol II transcription, Cell,
63, 751-762.
43.Robin, J. D., Ludlow, A. T., Batten, K.,
Magdinier, F., Stadler, G., Wagner, K. R., Shay, J. W., and Wright, W.
E. (2014) Telomere position effect: regulation of gene expression with
progressive telomere shortening over long distances, Genes Dev.,
28, 2464-2476.
44.Mefford, H. C., and Trask, B. J. (2002) The
complex structure and dynamic evolution of human subtelomeres, Nat.
Rev. Genet., 3, 91-102.
45.Torres, G. A., Gong, Z., Iovene, M., Hirsch, C.
D., Buell, C. R., Bryan, G. J., Novak, P., Macas, J., and Jiang, J.
(2011) Organization and evolution of subtelomeric satellite repeats in
the potato genome, G3 (Bethesda), 1, 85-92.
46.Ponten, J., Stein, W. D., and Shall, S. (1983) A
quantitative analysis of the aging of human glial cells in culture,
J. Cell Physiol., 117, 342-352.
47.Jones, R. B., Whitney, R. G., and Smith, J. R.
(1985) Intramitotic variation in proliferative potential: stochastic
events in cellular aging, Mech. Ageing Dev., 29,
143-149.
48.Blackburn, E. H. (2000) Telomere states and cell
fates, Nature, 408, 53-56.
49.Holt, S. E., Shay, J. W., and Wright, W. E.
(1996) Refining the telomere–telomerase hypothesis of aging and
cancer, Nature Biotechnol., 14, 836-839.
50.Ben-Porath, I., and Weinberg, R. A. (2005) The
signals and pathways activating cellular senescence, Int. J.
Biochem. Cell Biol., 37, 961-976.
51.Bodnar, A. G., Ouellette, M., Frolkis, M., Holt,
S. E., Chiu, C., Morin, G. B., Harley, C. B., Shay, J. W.,
Lichtsteiner, S., and Wright, W. E. (1998) Extension of life-span by
introduction of telomerase into normal human cells, Science,
279, 349-352.
52.Counter, C. M., Hahn, W. C., Wei, W., Caddle, S.
D., Beijersbergen, R. L., Lansdorp, P. M., Sedivy, J. M., and Weinberg,
R. A. (1998) Dissociation among in vitro telomerase activity,
telomere maintenance, and cellular immortalization, Proc. Natl.
Acad. Sci. USA, 95, 14723-14728.
53.Vaziri, H. (1998) Extension of life span in
normal human cells by telomerase activation: a revolution in cultural
senescence, J. Anti-Aging Med., 1, 125-130.
54.Vaziri, H., and Benchimol, S. (1998)
Reconstitution of telomerase activity in human normal cells leads to
elongation of telomeres and extended replicative life span, Curr.
Biol., 8, 279-282.
55.De Lange, T., and Jacks, T. (1999) For better or
worse? Telomerase inhibition and cancer, Cell, 98,
273-275.
56.Slijepcevic, P., and Hande, M. P. (1999) Chinese
hamster telomeres are comparable in size to mouse telomeres,
Cytogenet. Cell Genet., 85, 196-199.
57.Gorbunova, V., Bozzella, M. J., and Seluanov, A.
(2008) Rodents for comparative aging studies: from mice to beavers,
Age (Dordrecht), 30, 111-119.
58.Prowse, K. R., and Greider, C. W. (1995)
Developmental and tissue-specific regulation of mouse telomerase and
telomere length, Proc. Natl. Acad. Sci. USA, 92,
4818-4822.
59.Herrera, E., Samper, E., Martin-Caballero, J.,
Flores, J. M., Lee, H. W., and Blasco, M. A. (1999) Disease states
associated with telomerase deficiency appear earlier in mice with short
telomeres, EMBO J., 18, 2950-2960.
60.Blasco, M. A., Lee, H. W., Hande, M. P., Samper,
E., Lansdorp, P. M., DePinho, R. A., and Greider, C. W. (1997) Telomere
shortening and tumor formation by mouse cells lacking telomerase RNA,
Cell, 91, 25-34.
61.Lee, H. W., Blasco, M. A., Gottlieb, G. J.,
Horner, J. W., 2nd, Greider, C. W., and DePinho, R. A. (1998) Essential
role of mouse telomerase in highly proliferative organs, Nature,
392, 569-574.
62.Minkoff, E. C. (1983) Evolutionary
Biology, Addison-Wesley, Reading, Massachusetts (USA).
63.Libertini, G., and Ferrara, N. (2016) Aging of
perennial cells and organ parts according to the programmed aging
paradigm, Age (Dordrecht), 38, 1-13.
64.Libertini, G. (2013) Evidence for aging theories
from the study of a hunter-gatherer people (Ache of Paraguay),
Biochemistry (Moscow), 78, 1023-1032.
65.Hill, J. M., Zalos, G., Halcox, J. P., Schenke,
W. H., Waclawiw, M. A., Quyyumi, A. A., and Finkel, T. (2003)
Circulating endothelial progenitor cells, vascular function, and
cardiovascular risk, N. Engl. J. Med., 348,
593-600.
66.Werner, N., Kosiol, S., Schiegl, T., Ahlers, P.,
Walenta, K., Link, A., Bohm, M., and Nickenig, G. (2005) Circulating
endothelial progenitor cells and cardiovascular outcomes, N. Engl.
J. Med., 353, 999-1007.
67.Wilson, P. W., Castelli, W. P., and Kannel, W. B.
(1987) Coronary risk prediction in adults (the Framingham Heart Study),
Am. J. Cardiol., 59, 91G-94G.
68.Olovnikov, A. M. (2003) The redusome hypothesis
of aging and the control of biological time during individual
development, Biochemistry (Moscow), 68, 2-33.
69.Olovnikov, A. M. (2015) Chronographic theory of
development, aging, and origin of cancer: role of chronomeres and
printomeres, Curr. Aging Sci., 8, 76-88.
70.Williams, G. C. (1957) Pleiotropy, natural
selection and the evolution of senescence, Evolution, 11,
398-411.
71.Funk, W. D., Wang, C. K., Shelton, D. N., Harley,
C. B., Pagon, G. D., and Hoeffler, W. K. (2000) Telomerase expression
restores dermal integrity to in vitro-aged fibroblasts in a
reconstituted skin model, Exp. Cell Res., 258,
270-278.
72.Jaskelioff, M., Muller, F. L., Paik, J. H.,
Thomas, E., Jiang, S., Adams, A. C., Sahin, E., Kost-Alimova, M.,
Protopopov, A., Cadinanos, J., Horner, J. W., Maratos-Flier, E., and
Depinho, R. A. (2011) Telomerase reactivation reverses tissue
degeneration in aged telomerase-deficient mice, Nature,
469, 102-106.
73.Bernardes de Jesus, B., Vera, E., Schneeberger,
K., Tejera, A. M., Ayuso, E., Bosch, F., and Blasco, M. A. (2012)
Telomerase gene therapy in adult and old mice delays aging and
increases longevity without increasing cancer, EMBO Mol. Med.,
4, 691-704.
74.Campisi, J. (1997) The biology of replicative
senescence, Eur. J. Cancer, 33, 703-709.
75.Wright, W. E., and Shay, J. W. (2005) Telomere
biology in aging and cancer, J. Am. Geriatr. Soc., 53,
S292-S294.
76.Campisi, J. (2000) Cancer, aging and cellular
senescence, In vivo, 14, 183-188.
77.Rose, M. R. (1991) Evolutionary Biology of
Aging, Oxford University Press, New York.
78.Kirkwood, T. B. L. (1977) Evolution of ageing,
Nature, 270, 301-304.
79.Kirkwood, T. B. L., and Holliday, R. (1979) The
evolution of ageing and longevity, Proc. R. Soc. Lond. B Biol.
Sci., 205, 531-546.
80.Klapper, W., Heidorn, K., Kuhne, K., Parwaresch,
R., and Krupp, G. (1998) Telomerase activity in “immortal”
fish, FEBS Lett., 434, 409-412.
81.Klapper, W., Kuhne, K., Singh, K. K., Heidorn,
K., Parwaresch, R., and Krupp, G. (1998). Longevity of lobsters is
linked to ubiquitous telomerase expression, FEBS Lett.,
439, 143-146.
82.Rosen, P. (1985) Aging of the immune system,
Med. Hypotheses, 18, 157-161.
83.DePinho, R. A. (2000) The age of cancer,
Nature, 408, 248-254.
84.Artandi, S. E. (2002) Telomere shortening and
cell fates in mouse models of neoplasia, Trends Mol. Med.,
8, 44-47.
85.Artandi, S. E., and DePinho, R. A. (2010)
Telomeres and telomerase in cancer, Carcinogenesis, 31,
9-18.
86.Fabrizio, P., and Longo, V. D. (2007) The
chronological life span of Saccharomyces cerevisiae, Methods Mol.
Biol., 371, 89-95.
87.Baker, D. J., Childs, B. G., Durik, M., Wijers,
M. E., Sieben, C. J., Zhong, J., Saltness, R. A., Jeganathan, K. B.,
Verzosa, G. C., Pezeshki, A., Khazaie, K., Miller, J. D., and Van
Deursen, J. M. (2016) Naturally occurring p16(Ink4a)-positive cells
shorten healthy lifespan, Nature, 530, 184-189.
88.Fossel, M. B. (2015) The Telomerase
Revolution, BenBella Books, Dallas.
89.Harley, C. B., Liu, W., Blasco, M., Vera, E.,
Andrews, W. H., Briggs, L. A., and Raffaele, J. M. (2011) A natural
product telomerase activator as part of a health maintenance program,
Rejuv. Res., 14, 45-56.
90.Harley, C. B., Liu, W., Flom, P. L., and
Raffaele, J. M. (2013) A natural product telomerase activator as part
of a health maintenance program: metabolic and cardiovascular response,
Rejuv. Res., 16, 386-395.
91.Femminella, G. D., Leosco, D., Ferrara, N., and
Rengo, G. (2016) Adrenergic drugs blockers or enhancers for cognitive
decline? What to choose for Alzheimer’s disease patients? CNS
Neurol. Disord. Drug Targets, Epub ahead of print.
92.Pagano, G., Ferrara, N., Brooks, D. J., and
Pavese, N. (2016) Age at onset and Parkinson disease phenotype,
Neurology, 86, 1400-1407.
93.Pagano, G., Rengo, G., Pasqualetti, G.,
Femminella, G. D., Monzani, F., Ferrara, N., and Tagliati, M. (2015)
Cholinesterase inhibitors for Parkinson’s disease: a systematic
review and meta-analysis, J. Neurol. Neurosurg. Psychiatry,
86, 767-773.
94.Femminella, G. D., Rengo, G., Komici, K.,
Iacotucci, P., Petraglia, L., Pagano, G., De Lucia, C., Canonico, V.,
Bonaduce, D., Leosco, D., and Ferrara, N. (2014) Autonomic dysfunction
in Alzheimer’s disease: tools for assessment and review of the
literature, J. Alzheimer’s Dis., 42, 369-377.