MINI-REVIEW: Uncouplers of Oxidation and Phosphorylation as Antiaging Compounds
D. A. Knorre and F. F. Severin*
Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119991 Moscow, Russia; E-mail: severin@belozersky.msu.ru* To whom correspondence should be addressed.
Received July 20, 2016; Revision received August 30, 2016
Food restriction causes a set of physiological changes that reduce the rate of aging. At the level of an organism, these changes are initiated by a hormonal response, which in turn activates certain intracellular signaling cascades. As a result, cells increase their antioxidant capacities and decrease the risk of cancerous transformation. A number of small molecule compounds activating these signaling cascades have been described. One could expect that direct pharmacological activation of the signaling can produce a stronger antiaging effect than that achieved by the indirect hormonal stimulation. Data from the literature point to the opposite. Possibly, a problem with pharmacological activators is that they cause generation of mitochondrial reactive oxygen species. Indeed, hyperpolarized mitochondria are known to induce oxidative stress. Such hyperpolarization could happen because of artificial activation of cellular response to caloric restriction in the absence of energy deficit. At the same time, energy deficit seems likely to be a natural consequence of the shortage of nutrients. Thus, there is a possibility that combining the pharmacological activators with compounds that decrease mitochondrial transmembrane potential, uncouplers, could be a powerful antiaging strategy.
KEY WORDS: aging, food restriction, caloric restriction, uncouplers, geroprotectors, mTOR, AMPKDOI: 10.1134/S0006297916120051
Abbreviations: AICAR, 5-aminoimidazole-4-carboxamide ribonucleoside; AMPK, AMP-dependent protein kinase; CR, caloric restriction; C12TPP, dodecyltriphenylphosphonium; FCCP, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone; GH, growth hormone (somatotropin); IGF1, insulin-like growth factor 1; ROS, reactive oxygen species.
Caloric restriction (CR) is the best-studied and most reliable way to
increase lifespan. CR affects most of experimental model organisms,
from unicellular ones to mammals (see [1] for
review). Signaling cascades responsible for the effects of CR were
studied in detail at the cellular level as well as at the levels of
tissues and the whole organisms. Increasing levels of AMP and
NAD+, which activates deacetylases, were shown to be the key
factors initiating these cascades at the cellular level [2-4]. Physical exercise [5] and certain stresses (see [6-8] for review) also display a geroprotective effect.
Interestingly, even such stresses as ionizing irradiation or cytotoxic
compound cisplatin can activate AMPK (see [9, 10] for review). Currently, there are several known
substances, mimetics of CR, capable of activating the processes
dependent on AMPK and NAD+ even in the absence of CR. Apart
from this, increasing the proton conductivity of mitochondrial inner
membrane by using the compounds called uncouplers displays CR-like
effects [11]. Uncouplers decrease the coupling
level of respiration and oxidative phosphorylation and, as a result,
part of the energy produced by the oxidation of nutrients dissipates as
heat instead of being used for ATP synthesis. Therefore, uncouplers
also can be considered as CR mimetics.
At the level of an organism, the signaling cascades induced by CR or its mimetics are regulated by hormones. The hormonal cascades are usually initiated by an endocrine tissue-CR sensor. This results in the initiation of a complex network of stimulatory and compensatory hormonal interactions leading to the activation of AMPK and the deacetylases in cells (see [1, 10] for review). Is CR the optimal way to activate AMPK- and NAD+-dependent processes that can be used for lifespan extension? In this review, we discuss potential advantages and side effects of activating animal CR-dependent lifespan-extending program using various CR mimetics and their combinations.
EFFECTS OF CR AND ITS MIMETICS AT THE CELLULAR LEVEL
Activation of AMP-dependent kinase (AMPK) and NAD+-dependent histone deacetylase Sirt1 are considered as key effects of CR at the cellular level. It also was shown that AMPK and Sirt1 stimulate each other (see [12] for review). There are a number of pharmacological compounds that activate these enzymes: resveratrol, metformin, 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR, natural AMP analog), and some others. Literature analysis shows that CR is likely to be a more reliable (with reported effects over a wider range of model organisms) and effective geroprotector than any of the pharmacological activators (see [13-15] for review).
What are the differences in the actions of CR and the activators of AMPK/Sirt1? It is known that the activation of AMPK/Sirt1 stimulates transition from glycolysis to oxidative phosphorylation and lipolysis. The antioxidant systems are activated and the proliferative potential of cells is suppressed as well. These changes are believed to result in a general improvement of cellular physiology and reduce the risk of cancerous transformation (see [1] for review).
One could expect that under the conditions of CR the cells attempt to save energy. Many cellular changes indeed make bioenergetics more economical: CR decreases the rate of protein biosynthesis and activates autophagy [16]. It would be natural to presume that CR also raises the efficiency of mitochondrial energy production, i.e. that it increases the coupling of respiration and oxidative phosphorylation. The opposite appears to take place. It has been shown that mice under CR conditions accumulate proteins UCP2 and UCP3 in their muscle mitochondria [17, 18]. UCP3 also accumulates in rat muscles because of chronic exposure to AICAR [19]. UCP proteins (uncoupling proteins) catalyze an electrogenic process of transporting the dissociated forms of free fatty acids from the inner to the outer layers of mitochondrial inner membrane. In the outer layer the free fatty acids are protonated, and then in the neutral form return to the inner layer [20]. As this decreases the level of the transmembrane potential, the proteins of the UCP family act as natural uncouplers of respiration and oxidative phosphorylation (see [21, 22] for review). Indeed, during starvation there is simultaneous accumulation of UCP2 and UCP3, and a decrease in ADP/O ratio, the latter indicating a decrease in the efficiency of oxidative phosphorylation [18].
What is the physiological role of uncoupling activation upon CR? On one hand, CR induces mitochondrial biogenesis and respiration. On the other hand, it has been shown that mitochondrial hyperpolarization can induce a strong increase in ROS (reactive oxygen species) generation [23]. Probably, the increased expression of UCP is an “insurance” against the oxidative stress caused by mitochondrial hyperpolarization (see [24] for review).
A natural increase in ADP/ATP ratio due to nutrient limitation could provide another “insurance” against the CR-induced activation of mitochondrial respiratory activity. Such ratio increases, for example, in adipocytes under the conditions of CR (see [25] for review). On one hand, an increase in ADP level in cells induces an increase in AMP concentration, which leads to AMPK activation (see [10] for review). This is due to the activity of adenylate kinase, which catalyzes the following reaction: 2ADP ↔ ATP + AMP [26]. On the other hand, an increase in cellular ADP concentration can directly prevent mitochondrial hyperpolarization via activation of enzymes consuming the proton gradient: ATP-synthase and ATP/ADP antiporter. The latter exchanges one ATP molecule (charged 4–) from mitochondrial matrix for one ADP molecule (charged 3–) from mitochondrial intermembrane space (see [27] for review).
It is important to mention that no increase in ADP concentration in certain cases when CR-dependent molecular cascades are stimulated by chemicals should be expected. Indeed, the amounts of available glycolytic substrates may not change upon chemical stimulation. Therefore, one can expect that certain artificial ways of stimulation of CR-dependent signaling cascades (for example, by AICAR administration) to cause mitochondrial hyperpolarization-dependent increase in ROS levels. Obviously, this does not apply to the stimulation of CR-dependent cascades by the uncouplers – in such cases mitochondrial depolarization serves as an inducer of the cascade. This line of reasoning is illustrated by the figure arrows 1-5.
Scheme illustrating presumed effects of uncouplers on processes regulated by levels of nutrients in organisms
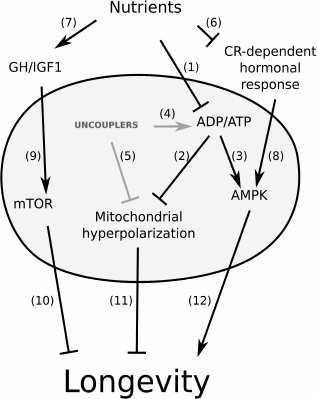
There is probably another advantage of using uncouplers as CR mimetics. As aforementioned, increase in NAD+ levels is one of the best-studied ways of geroprotection. There are many works showing that increasing NAD+ concentration via activation of its biosynthesis leads to lifespan increase in experimental animals (see [28, 29] for review). Importantly, in terms of lifespan increase in the sum of NAD+ and NADH concentrations is less relevant than the concentration of the oxidized form. At the same time, reduction of NAD+ to NADH takes place during cellular catabolic reactions (glycolysis and TCA cycle). Therefore, interfering with cell metabolism could be an efficient way of increasing NAD+ concentration. For example, a minor decrease in the transmembrane potential can strongly increase NAD+/NADH ratio without having any noticeable effect on ADP/ATP ratio. Theoretically, this can take place in cases when the activity of the respiratory chain is blocked by high transmembrane potential, and the rate of ATP synthesis is limited by the activities of ATP-synthase and ATP/ADP antiporter. The addition of the uncoupler (FCCP) at low concentration (100 nM) has been shown to increase ATP level in neurons due to a compensatory response to a temporal depolarization [30]. Earlier, it has been suggested that a slight decrease in the transmembrane potential can prevent the reaction of one-electron reduction of oxygen, which leads to ROS formation. At the same time, such a decrease may not affect the rate of ATP synthesis; thus, such uncoupling was called “mild” [24, 31, 32].
In other words, mild uncoupling is aimed at stimulating NAD+-dependent processes rather than at stimulation of AMPK. It is important to mention that hyperstimulation of AMPK can lead to cell death. It was shown that even a short-term depletion of cellular energy, which leads to a relatively small drop in ATP/ADP level, initiates programmed cell death [33]. This feature of AMPK found its use in anticancer therapy for killing tumor cells (see [16] for review). In particular, it has been shown that AMPK stimulates proapoptotic proteins Bax and Bim [34]. There are also other signaling routes of AMPK-dependent cell death, for example, activation of MEK1/ERK2 protein kinase module [35]. Apart from that, a general decrease in protein biosynthesis caused by activation of AMPK can also lead to cell death. The reason is that proapoptotic proteins tend to have longer half-lives than the antiapoptotic ones. Thus, deceleration of translation can cause apoptosis (see [36] and references within). Consistent with that, despite all the benefits of CR, its side effect is a decrease in the mass of muscle and bone (see [1] for review). Possibly, an increase in NAD+ concentration can also lead to cell death, for example, via Sirt1-dependent stimulation of AMPK (see [12] for review). Nevertheless, this way to kill a cell seems to be less straightforward than the one initiated by a direct stimulation of AMPK. This comes from the fact that AMP stimulates AMPK simultaneously in three ways: it activates allosteric changes, stimulates activating phosphorylation, and downregulates inhibitory dephosphorylation as well (see [10] for review). Sirt1 activates only one of the ways – phosphorylation (see [12] for review). In other words, stimulation of AMPK via Sirt1 appears to be milder than the one by AMP or its analogs. Therefore, mild uncoupling seems to be safer than administration of AMP analogs for lifespan interference because it does not tend to provoke cell death.
EFFECTS OF CR AND ITS MIMETICS AT LEVELS OF TISSUES AND
ORGANISMS
In multicellular organisms, in particular, in mammals, CR activates AMPK/Sirt1 in most tissues. This activation is usually mediated by hormones. It occurs as only a few cell types can “feel” the changes in the nutrients levels caused by a relatively small, “useful” (approximately 30%) CR. Such specialized cells secrete hormones that regulate AMPK/Sirt1 in the target cells, thus causing, in particular, a secondary CR-dependent hormonal response (see below).
What are the differences in the effects of CR and its mimetics at the level of an organism? Most works concerning this issue were performed on mammals. It has been shown that fat cells, adipocytes, activate the secretion of hormones, adipokines, in response to a decrease in concentrations of nutrients in blood. Activation of AMPK is considered as the key physiological intermediate of their hormonal response to CR (see [25] for review).
Endocrine response of hypothalamus is probably the best-studied secondary hormonal response triggered by adipokines. In many cases, the secondary response amplifies the primary one. Cellular reactions to the primary and secondary CR-induced hormonal stimuli were studied in much detail in muscle. It has been shown the adipokines leptin and adiponectin bind to their receptors at the cell surface and activate AMPK. Independently, leptin activates the secretion of the stress hormones that are produced by the hypothalamus. These hormones bind to adrenergic receptors of muscle cells and in this way also activate AMPK (see [10] for review).
Insulin and insulin-like hormones are the main antagonists of CR-induced hormonal response. Animals use the blood level of glucose as the key indicator of nutrient availability. This level is sensed first of all by β-cells, which secrete insulin in response to rising level of glucose in blood. The endocrine gut cells are another sensor – glucose induces their secretion of incretin hormones. One of the main functions of incretins is stimulation of insulin secretion by β-cells (see [37-40] for review). The secretory activity of β-cells and the specialized gut cells is regulated by their glucose receptors and thus does not directly depend on Sirt1/AMPK (see [41, 42] for review).
In terms of regulation of aging, the main antagonists of Sirt1/AMPK are insulin-like growth factor and growth hormone (IGF1/GH; see figure, arrows 7 and 9). Geroprotective effect of inactivation of this signaling path has been demonstrated for many organisms – from nematodes to mammals. In particular, mutations inactivating GH or its receptor in mice cause significant lifespan increase (see [43, 44] for review). At the same time, in many case therapy that includes GH administration also shows geroprotective effects [45-47]. We attempted to explain this contradiction by analyzing the integrative interactions of Sirt1/AMPK- and IGF1/GH-dependent cascades at a cellular level.
CELLS AS INTEGRATORS OF NUTRIENT-DEPENDENT HORMONAL
CASCADES
In cells, the protein kinase complex mTOR is the main antagonist of the signaling cascades initiated by Sirt1/AMPK. This complex is activated by insulin, IGF1/GH (see figure), and free amino acids. Activated mTOR stimulates protein biosynthesis, cell proliferation, and differentiation (see [48-50] for review). In addition, it has been shown that AMPK and mTOR inhibit each other (see [50, 51] for review). There are also data showing geroprotective effect of mTOR inhibition. In particular, the specific mTOR inhibitor rapamycin (see [52, 53] for review) and restrictions in the levels of certain amino acids in food (see [1] for review) were shown to extend the lifespan of laboratory mice.
What is the optimal balance of Sirt1/AMPK and mTOR activities from the point of view of geroprotection? Taking a closer look at their interactions in muscle tissue could help answer this question. Many studies show that while physical exercises delay aging in general, they are especially efficient in delaying muscle aging (see [54-56] for review). Physical exercise affects muscles in two ways. On one hand, the accelerated cycling of myosin (the main muscle motor protein) causes an increase in the rate ATP hydrolysis and, as a result, activation of AMPK. On the other hand, exercise activates mTOR. The activation occurs via three pathways: (i) mechanical stretching of Z-disks stimulates the intracellular activating cascade and (ii) the secretion of muscle growth factor (MGF, splice-variant of IGF1), which is an endocrine activator of mTOR. In addition, (iii) in response to muscle activity, the hypothalamus upregulates the secretion of GH, which also leads to the stimulation of muscle growth via mTOR (see [50] for review).
Therefore, studies on muscle tissue point to simultaneous activation of mTOR- and Sirt1/AMPK-dependent signaling cascades being an optimal metabolic geroprotector. Indeed, in such case, the cells profit from Sirt1/AMPK (stimulation of antioxidant systems, reliance on oxidative phosphorylation) and, at the same time, they are protected from the negative consequences of AMPK hyperactivation (a complete stop of the cell cycle and apoptosis). At first glance, the very absence of evidence pointing to the geroprotective effects of direct AMPK activators (e.g. AICAR) contradicts this logic. According to this line of reasoning, one can expect AICAR administration in the absence of CR to activate AMPK and, at the same time, not to suppress the GH/IGF1/mTOR module. Moreover, AICAR stimulates AMPK directly but not via a multistage hormonal cascade as in the case of CR. Therefore, the absence of CR combined with AICAR seems an even more robust geroprotector than CR. Why is it not so?
The reason might be that AICAR, unlike CR or physical exercise, neither causes an increase in ADP level nor accumulation of the uncoupling proteins, UCPs. This, in turn, can cause mitochondrial hyperpolarization and consequent oxidative stress [23]. It is important to mention that oxidative stress is considered one of the key factor driving the aging process [57]. Moreover, mitochondrial hyperactivation on its own can negatively affect the mitochondrial quality control mechanism and in this way accelerate aging [58]. If these presumptions are correct (they are illustrated by the figure, arrows 4 and 5), then the most efficient combination of treatments would be a direct stimulation of AMPK (for example, with AICAR), unlimited nutrition, and administration of the uncouplers.
CONCLUSION: UNCOUPLING AS A GEROPROTECTOR
What level of uncoupling is most suitable for the purposes of geroprotection? As mentioned, a small decrease in proton resistance of the strongly energized mitochondrial membranes can induce a significant decrease in ROS production and an increase in NAD+/NADH ratio without affecting ATP concentration. According to our line of reasoning, such level of uncoupling combined with AMPK activation is sufficient for efficient interference with the aging process. Theoretically, one could consider a higher level of uncoupling leading to a strong depolarization of the membranes and, as a consequence, a significant increase in ADP/ATP ratio. Apparently, such treatment could lead to a lethal deenergization of cells [33]. Therefore, a relatively weak level of uncoupling seems to be preferential.
Which uncouplers should be used? Probably, the anionic compound dinitrophenol is the best-studied uncoupler in terms of its effects on mammalian physiology. In particular, it has been used on humans as a weight loss treatment. However, it was reported that its use was accompanied by a set of negative side effects [59, 60].
Recently, we reported uncoupling activity of a unique type of chemical compounds – lipophilic penetrating cations. Most of the studies on such compounds were performed on dodecyltriphenylphosphonium, C12TPP. A potential advantage of using such compounds is that their mitochondrial accumulation is proportional to the level of the transmembrane potential. For this reason, penetrating cations affect highly polarized mitochondria to greater extent than mitochondria with relatively low potential levels [61, 62]. In other words, they cause self-limiting (mild) uncoupling.
Two independent research groups ([63] and M. Lovat, personal communication) have shown that C12TPP administration positively affects mice that were kept on high fat diet: it reduces weight gain and decreases glucose level in the blood.
Finally, it is necessary to mention that despite the fact that the artificial activators of Sirt1/AMPK display lower extent or absence of geroprotective effect, these compounds are widely used as treatments against diabetes and obesity. Possibly, if combined with mild, C12TPP-like uncouplers, the antiaging effect of such compounds can be even higher that of CR.
Acknowledgements
The work was supported by the Russian Science Foundation (project No. 14-14-00181).
REFERENCES
1.Spindler, S. R. (2010) Caloric restriction: from
soup to nuts, Ageing Res. Rev., 9, 324-353.
2.Ruiz, R., Pedrez-Villegas, E. M., and Manuel
Carrion, A. (2016) AMPK function in aging process, Curr. Drug
Targets, 17, 932-941.
3.Burkewitz, K., Zhang, Y., and Mair, W. B. (2014)
AMPK at the nexus of energetics and aging, Cell Metab.,
20, 10-25.
4.Verdin, E. (2015) NAD+ in aging,
metabolism, and neurodegeneration, Science, 350,
1208-1213.
5.Gremeaux, V., Gayda, M., Lepers, R., Sosner, P.,
Juneau, M., and Nigam, A. (2012) Exercise and longevity,
Maturitas, 73, 312-317.
6.Lagisz, M., Hector, K. L., and Nakagawa, S. (2013)
Life extension after heat shock exposure: assessing meta-analytic
evidence for hormesis, Ageing Res. Rev., 12, 653-660.
7.Munkacsy, E., and Rea, S. L. (2014) The paradox of
mitochondrial dysfunction and extended longevity, Exp.
Gerontol., 56, 221-233.
8.Ristow, M., and Zarse, K. (2010) How increased
oxidative stress promotes longevity and metabolic health: the concept
of mitochondrial hormesis (mitohormesis), Exp. Gerontol.,
45, 410-418.
9.Sanli, T., Steinberg, G. R., Singh, G., and
Tsakiridis, T. (2014) AMP-activated protein kinase (AMPK) beyond
metabolism: a novel genomic stress sensor participating in the DNA
damage response pathway, Cancer Biol. Ther., 15,
156-169.
10.Kahn, B. B., Alquier, T., Carling, D., and
Hardie, D. G. (2005) AMP-activated protein kinase: ancient energy gauge
provides clues to modern understanding of metabolism, Cell
Metab., 1, 15-25.
11.Caldeira da Silva, C. C., Cerqueira, F. M.,
Barbosa, L. F., Medeiros, M. H. G., and Kowaltowski, A. J. (2008) Mild
mitochondrial uncoupling in mice affects energy metabolism, redox
balance and longevity, Aging Cell, 7, 552-560.
12.Ruderman, N. B., Xu, X. J., Nelson, L., Cacicedo,
J. M., Saha, A. K., Lan, F., and Ido, Y. (2010) AMPK and SIRT1: a
long-standing partnership? Am. J. Physiol., 298,
137-163.
13.Ingram, D. K., and Roth, G. S. (2015) Calorie
restriction mimetics: can you have your cake and eat it, too? Ageing
Res. Rev., 20, 46-62.
14.Mercken, E. M., Carboneau, B. A., Krzysik-Walker,
S. M., and De Cabo, R. (2012) Of mice and men: the benefits of caloric
restriction, exercise, and mimetics, Ageing Res. Rev.,
11, 390-398.
15.Testa, G., Biasi, F., Poli, G., and Chiarpotto,
E. (2014) Calorie restriction and dietary restriction mimetics: a
strategy for improving healthy aging and longevity, Curr. Pharm.
Design., 20, 2950-2977.
16.Green, D. R., Galluzzi, L., and Kroemer, G.
(2014) Cell biology. Metabolic control of cell death, Science,
345, 1250256.
17.Seifert, E. L., Bezaire, V., Estey, C., and
Harper, M.-E. (2008) Essential role for uncoupling protein-3 in
mitochondrial adaptation to fasting but not in fatty acid oxidation or
fatty acid anion export, J. Biol. Chem., 283,
25124-25131.
18.Wang, C.-M., Almsherqi, Z. A., McLachlan, C. S.,
Matthews, S., Ramachandran, M., Tay, S. K. H., and Deng, Y. (2011)
Acute starvation in C57BL/6J mice increases myocardial UCP2 and UCP3
protein expression levels and decreases mitochondrial bio-energetic
function, Stress, 14, 66-72.
19.Suwa, M., Nakano, H., and Kumagai, S. (2003)
Effects of chronic AICAR treatment on fiber composition, enzyme
activity, UCP3, and PGC-1 in rat muscles, J. Appl. Physiol.,
95, 960-968.
20.Fedorenko, A., Lishko, P. V., and Kirichok, Y.
(2012) Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat
mitochondria, Cell, 151, 400-413.
21.Sluse, F. E., Jarmuszkiewicz, W., Navet, R.,
Douette, P., Mathy, G., and Sluse-Goffart, C. M. (2006) Mitochondrial
UCPs: new insights into regulation and impact, Biochim. Biophys.
Acta, 1757, 480-485.
22.Garlid, K. D., Jaburek, M., and Jezek, P. (2001)
Mechanism of uncoupling protein action, Biochem. Soc. Trans.,
29, 803-806.
23.Korshunov, S. S., Skulachev, V. P., and Starkov,
A. A. (1997) High protonic potential actuates a mechanism of production
of reactive oxygen species in mitochondria, FEBS Lett.,
416, 15-18.
24.Skulachev, V. P. (1996) Role of uncoupled and
non-coupled oxidations in maintenance of safely low levels of oxygen
and its one-electron reductants, Quart. Rev. Biophys.,
29, 169-202.
25.Daval, M., Foufelle, F., and Ferre, P. (2006)
Functions of AMP-activated protein kinase in adipose tissue, J.
Physiol., 574, 55-62.
26.Atkinson, D. E. (1968) The energy charge of the
adenylate pool as a regulatory parameter. Interaction with feedback
modifiers, Biochemistry, 7, 4030-4034.
27.Klingenberg, M. (2008) The ADP and ATP transport
in mitochondria and its carrier, Biochim. Biophys. Acta,
1778, 1978-2021.
28.Massudi, H., Grant, R., Guillemin, G. J., and
Braidy, N. (2012) NAD+ metabolism and oxidative stress: the
golden nucleotide on a crown of thorns, Redox Rep., 17,
28-46.
29.Hubbard, B. P., and Sinclair, D. A. (2014) Small
molecule SIRT1 activators for the treatment of aging and age-related
diseases, Trends Pharmacol. Sci., 35, 146-154.
30.Weisova, P., Anilkumar, U., Ryan, C., Concannon,
C. G., Prehn, J. H. M., and Ward, M. W. (2012) “Mild
mitochondrial uncoupling” induced protection against neuronal
excitotoxicity requires AMPK activity, Biochim. Biophys. Acta,
1817, 744-753.
31.Zorov, D. B. (1996) Mitochondrial damage as a
source of diseases and aging: a strategy of how to fight these,
Biochim. Biophys. Acta, 1275, 10-15.
32.Starkov, A. A. (1997) “Mild”
uncoupling of mitochondria, Biosci. Rep., 17,
273-279.
33.Izyumov, D. S., Avetisyan, A. V., Pletjushkina,
O. Y., Sakharov, D. V., Wirtz, K. W., Chernyak, B. V., and Skulachev,
V. P. (2004) “Wages of Fear”: transient threefold decrease
in intracellular ATP level imposes apoptosis, Biochim. Biophys.
Acta, 1658, 141-147.
34.Bodur, C., Karakas, B., Timucin, A. C., Tezil,
T., and Basaga, H. (2015) AMP-activated protein kinase couples
3-bromopyruvate-induced energy depletion to apoptosis via activation of
FoxO3a and upregulation of proapoptotic Bcl-2 proteins, Mol.
Carcinog., doi: 10.1002/mc.22411.
35.Shin, S., Buel, G. R., Wolgamott, L., Plas, D.
R., Asara, J. M., Blenis, J., and Yoon, S. O. (2015) ERK2 mediates
metabolic stress response to regulate cell fate, Mol. Cell,
59, 382-398.
36.Meynet, O., Zunino, B., Happo, L., Pradelli, L.
A., Chiche, J., Jacquin, M. A., Mondragon, L., Tanti, J. F., Taillan,
B., Garnier, G., Reverso-Meinietti, J., Mounier, N., Michiels, J. F.,
Michalak. E. M., Carles, M., Scott, C. L., and Ricci, J. E. (2013)
Caloric restriction modulates Mcl-1 expression and sensitizes lymphomas
to BH3 mimetic in mice, Blood, 122, 2402-2411.
37.Tasyurek, H. M., Altunbas, H. A., Balci, M. K.,
and Sanlioglu, S. (2014) Incretins: their physiology and application in
the treatment of diabetes mellitus, Diab. Metab. Res. Rev.,
30, 354-371.
38.Tian, L., and Jin, T. (2016) The incretin hormone
GLP-1 and mechanisms underlying its secretion, J. Diabetes, http://dx.doi.org/10.1111/1753-0407.12439.
39.Reimann, F., and Gribble, F. M. (2016) Mechanisms
underlying glucose-dependent insulinotropic polypeptide and
glucagon-like peptide-1 secretion, J. Diab. Invest., 7
(Suppl. 1), 13-19.
40.Svendsen, B., and Holst, J. J. (2016) Regulation
of gut hormone secretion. Studies using isolated perfused intestines,
Peptides, 77, 47-53.
41.Rorsman, P., and Braun, M. (2013) Regulation of
insulin secretion in human pancreatic islets, Annu. Rev.
Physiol., 75, 155-179.
42.Pais, R., Gribble, F. M., and Reimann, F. (2016)
Stimulation of incretin secreting cells, Ther. Adv. Endocrinol.
Metab., 7, 24-42.
43.Brown-Borg, H. M., and Bartke, A. (2012) GH and
IGF1: roles in energy metabolism of long-living GH mutant mice, J.
Gerontol. Ser. A Biol. Sci. Med. Sci., 67, 652-660.
44.Bartke, A., Sun, L. Y., and Longo, V. (2013)
Somatotropic signaling: trade-offs between growth, reproductive
development, and longevity, Physiol. Rev., 93,
571-598.
45.Esteban, S., Garau, C., Aparicio, S., Moranta,
D., Barcelo, P., Ramis, M., Tresguerres, J. A., and Rial, R. (2010)
Improving effects of long-term growth hormone treatment on
monoaminergic neurotransmission and related behavioral tests in aged
rats, Rejuv. Res., 13, 707-716.
46.Zouboulis, C. C., and Makrantonaki, E. (2012)
Hormonal therapy of intrinsic aging, Rejuv. Res., 15,
302-312.
47.Paredes, S. D., Forman, K. A., Garcia, C., Vara,
E., Escames, G., and Tresguerres, J. A. F. (2014) Protective actions of
melatonin and growth hormone on the aged cardiovascular system,
Horm. Mol. Biol. Clin. Invest., 18, 79-88.
48.Bartke, A., List, E. O., and Kopchick, J. J.
(2016) The somatotropic axis and aging: benefits of endocrine defects,
Growth Horm. Res., 27, 41-45.
49.Cheng, Z., Tseng, Y., and White, M. F. (2010)
Insulin signaling meets mitochondria in metabolism, Trends
Endocrinol. Metab., 21, 589-598.
50.Sharples, A. P., Hughes, D. C., Deane, C. S.,
Saini, A., Selman, C., and Stewart, C. E. (2015) Longevity and skeletal
muscle mass: the role of IGF signaling, the sirtuins, dietary
restriction and protein intake, Aging Cell, 14,
511-523.
51.Hindupur, S. K., Gonzalez, A., and Hall, M. N.
(2015) The opposing actions of target of rapamycin and AMP-activated
protein kinase in cell growth control, Cold Spring Harb. Perspect.
Biol., 7, a019141.
52.Blagosklonny, M. V. (2012) Prospective treatment
of age-related diseases by slowing down aging, Am. J. Pathol.,
181, 1142-1146.
53.Xu, S., Cai, Y., and Wei, Y. (2014) mTOR
signaling from cellular senescence to organismal aging, Aging
Dis., 5, 263-273.
54.McLeod, M., Breen, L., Hamilton, D. L., and
Philp, A. (2016) Live strong and prosper: the importance of skeletal
muscle strength for healthy ageing, Biogerontology, 17,
497-510.
55.Russell, A. P., Foletta, V. C., Snow, R. J., and
Wadley, G. D. (2014) Skeletal muscle mitochondria: a major player in
exercise, health and disease, Biochim. Biophys. Acta,
1840, 1276-1284.
56.Vina, J., Sanchis-Gomar, F., Martinez-Bello, V.,
and Gomez-Cabrera, M. C. (2012) Exercise acts as a drug; the
pharmacological benefits of exercise, Br. J. Pharmacol.,
167, 1-12.
57.Harman, D. (1956) Aging: a theory based on free
radical and radiation chemistry, J. Gerontol., 11,
298-300.
58.Knorre, D. A., and Severin, F. F. (2012)
Longevity and mitochondrial membrane potential, Biochemistry
(Moscow), 77, 793-794.
59.Kamour, A., George, N., Gwynnette, D., Cooper,
G., Lupton, D., Eddleston, M., Thompson, J. P., Vale, J. A.,
Thanacoody, H. K., Hill, S., and Thomas, S. H. (2015) Increasing
frequency of severe clinical toxicity after use of 2,4-dinitrophenol in
the UK: a report from the National Poisons Information Service,
Emerg. Med. J., 32, 383-386.
60.Grundlingh, J., Dargan, P. I., El-Zanfaly, M.,
and Wood, D. M. (2011) 2,4-dinitrophenol (DNP): a weight loss agent
with significant acute toxicity and risk of death, J. Med.
Toxicol., 7, 205-212.
61.Severin, F. F., Severina, I. I., Antonenko, Y.
N., Rokitskaya, T. I., Cherepanov, D. A., Mokhova, E. N., Vyssokikh, M.
Y., Pustovidko, A. V., Markova, O. V., Yaguzhinsky, L. S., Korshunova,
G. A., Sumbatyan, N. V., Skulachev, M. V., and Skulachev, V. P. (2010)
Penetrating cation/fatty acid anion pair as a mitochondria-targeted
protonophore, Proc. Natl. Acad. Sci. USA, 107,
663-668.
62.Antonenko, Y. N., Avetisyan, A. V., Cherepanov,
D. A., Knorre, D. A., Korshunova, G. A., Markova, O. V., Ojovan, S. M.,
Perevoshchikova, I. V., Pustovidko, A. V., Rokitskaya, T. I., Severina,
I. I., Simonyan, R. A., Smirnova, E. A., Sobko, A. A., Sumbatyan, N.
V., Severin, F. F., and Skulachev, V. P. (2011) Derivatives of
rhodamine 19 as mild mitochondria-targeted cationic uncouplers, J.
Biol. Chem., 286, 17831-17840.
63.Kalinovich, A. V., Mattsson, C. L., Mohamed R.
Youssef, M. R., Petrovic, N., Ost, M., Skulachev, V. P., and Shabalina,
I. G. (2016) Mitochondria-targeted dodecyltriphenylphosphonium (C12TPP)
combats high-fat 2 diet-induced obesity in mice, Int. J.
Obesity, accepted manuscript.