REVIEW: Myosin Light Chain Kinase MYLK1: Anatomy, Interactions, Functions, and Regulation
A. Y. Khapchaev1,2* and V. P. Shirinsky1
1Russian Cardiology Research and Production Center, 121552 Moscow, Russia; E-mail: askerkhapcha@gmail.com2Lomonosov Moscow State University, Faculty of Fundamental Medicine, 119192, Moscow, Russia
* To whom correspondence should be addressed.
Received September 12, 2016
This review discusses and summarizes the results of molecular and cellular investigations of myosin light chain kinase (MLCK, MYLK1), the key regulator of cell motility. The structure and regulation of a complex mylk1 gene and the domain organization of its products is presented. The interactions of the mylk1 gene protein products with other proteins and posttranslational modifications of the mylk1 gene protein products are reviewed, which altogether might determine the role and place of MLCK in physiological and pathological reactions of cells and entire organisms. Translational potential of MLCK as a drug target is evaluated.
KEY WORDS: myosin light chain kinase, KRP/telokin, mylk1 gene organization and regulation, posttranslational modifications, protein–protein interactions, MLCK-dependent cellular reactions, MLCK in pathologyDOI: 10.1134/S000629791613006X
Abbreviations: CaM, calmodulin; Erk1/2, extracellular regulated kinase-1/2; IFNγ, interferon gamma; IL-1β, interleukin-1beta; KRP, kinase-related protein; MAP-kinase, mitogen-activated protein kinase; MLCK, myosin light chain kinase; MLCP, myosin light chain phosphatase; MYPT1, myosin phosphatase target subunit 1; PKA, cyclic AMP-dependent protein kinase, protein kinase A; PTM, posttranslational modification; RLC, regulatory light chain; ROCK, Rho-associated protein kinase; TNFα, tissue necrosis factor alpha; ZIPK, zipper-interacting protein kinase.
This review is intended to consolidate, analyze, and summarize molecular
and cell biology data on myosin light chain kinase (MLCK) including the
most recent data that have not been reviewed previously [1-10].
MLCK was discovered approximately 40 years ago as a skeletal and smooth muscle enzyme [11, 12]. Ever since, significant progress has been made. In vertebrates, at least three MLCK-encoding genes have been identified – mylk2 and mylk3 encode for tissue-specific skeletal muscle and cardiac MLCKs, respectively [13, 14]. In contrast to mylk2 and mylk3, mylk1 has a complex structure [15, 16], and multiple protein products of the mylk1 gene are expressed in most if not all cell types. This review deals with the mylk1 gene and its protein products – multiple MLCK isoforms and noncatalytic KRP/telokin protein.
Previously, phosphorylation of 20-kDa myosin regulatory light chains (RLC) was thought to be an exclusive MLCK function. Subsequent studies revealed that in some cases MLCK contributes to cellular responses by its scaffolding activity to recruit macromolecular complexes. A balance between MLCK catalytic and scaffolding activities, especially for high molecular weight MLCK isoform, is an urgent research issue that promises to elucidate roles of MLCK in cell (patho)physiology.
MLCK is a substrate for other protein kinases; moreover, MLCK is acetylated and methylated in vivo. Modern proteomic analysis methods have identified over 50 posttranslational modifications (PTM) of MLCK; many of these are found in vivo (http://www.phosphosite.org [17]). However, functional significance and the modifying enzymes are identified for only a few PTMs. Since PTMs often exert regulatory effects on a protein in vivo, it seems necessary to elucidate the roles of MLCK PTMs in the context of cellular functions. Here we provide a systematic review of the reported MLCK PTMs and suggest directions for further studies on this critical issue.
The involvement of MLCK in pathological processes is not the major theme of this review; nevertheless, tendencies in translation of fundamental MLCK studies to technologies for practical medicine are given in a separate section along with estimations of perspectives of MLCK as a molecular target for novel drug discovery efforts.
NOMENCLATURE AND DESIGNATIONS
Originally, MLCK has been isolated from skeletal muscle [12]. Next, a smooth muscle MLCK isoform was isolated [11], and a high molecular weight MLCK containing the whole smooth muscle MLCK sequence and a unique N-terminal extension was identified [18]. Moreover, the C-terminal fragment of smooth muscle MLCK is expressed as an independent non-kinase protein KRP/telokin (see below) [19]. These proteins (two MLCK isoforms and KRP/telokin) are encoded by a complex mylk gene initially described as a genetic locus [18]. In the literature, the designation mylk1 is often used to discriminate skeletal muscle and cardiac MLCKs. In this review, the numbering of exons is provided as outlined in a pioneering characterization of the mylk1 gene [15]; the numbering of amino acid residues corresponds to human high molecular weight MLCK, which contains 1914 amino acid residues. In the human genome, the mylk1 gene resides at 3qcen-q21 [16] and spans approximately 270 kb of DNA [20, 21]. Skeletal muscle MLCK (skMLCK) is encoded by the mylk2 gene [14]; mylk3 encodes a specific cardiac muscle MLCK (caMLCK) [13]. Moreover, a calmodulin (CaM)-independent cardiac MLCK isoform has recently been described [22].
In the literature, there are confusing designations for mylk1-derived proteins. Specifically, “non-muscle”, “heavy”, and “long” are synonyms of 210-220 kDa “high molecular weight” MLCK; for a 108-155 kDa low molecular weight MLCK, the terms “smooth muscle”, “light”, “small”, or “short” are applied. From our point of view, the terms “long MLCK” (L-MLCK) and “short MLCK” (S-MLCK) fit best to the current knowledge on the organization and tissue-specific expression of the MLCK isoforms encoded by the mylk1 gene. A 17-kDa non-kinase C-terminal fragment of MLCK [19] is known as KRP (Kinase-Related Protein) or telokin (telos + kinase). Considering that the term “KRP” is used also to designate Kinesin-Related Proteins, in this review we use the term “KRP/telokin”.
THE mylk1 GENE AND TRANSCRIPTIONAL CONTROL
Three mRNA classes (e.g. for avian species – 9.0, 5.5, and 2.7 kb) correspond to L-MLCK, S-MLCK, and KRP/telokin, respectively [15, 18, 23] (Fig. 1a). In mammals, the corresponding mRNA sizes differ slightly [24]. Each mRNA derives from an independent promoter. Thus, both S-MLCK and KRP/telokin promoters are located in introns of the mylk1 gene resulting in transcription of unique 5′-untranslated mRNA sequences from the corresponding introns, i.e. the starting exons utilized by either S-MLCK or KRP/telokin mRNAs contain unique 5′-upstream extensions not present in the larger transcripts [25-27].
Fig. 1. Schematic structure of the mylk1 gene and regulation of its multiple promoters. a) Three mRNA classes are transcribed from the mylk1 gene. They encode KRP/telokin, S-MLCK, and L-MLCK isoforms. KRP/telokin utilizes the larger exon 29A instead of exon 29 used by both S-MLCK and L-MLCK [15, 26]. S-MLCK utilizes a larger exon versus the exon 15 used by L-MLCK [25]. L-MLCK2 lacks exon 11, which is present in L-MLCK1 [28]. In more recent reports, exon 9 has been designated as exon 11 due to identification of additional 5′-untranslated exons [5]. Exon and intron sizes and distances here and below are not to scale. b) Regulation of KRP/telokin promoter. Position +1 corresponds to homologous human telokin mRNA start (chr3 reverse strand: 123,620,607). Nucleotide numbering refers to the human mylk1 sequence and is based on the reports describing KRP/telokin promoter in rabbit, mouse, and rat. Transcription activators and enhancers are coded by green range colors, whereas inhibitors and silencers are shown in red to purple range colors. Green arrows, stimulating activity; red lines with round cap, inhibitory action. c) Regulation of S-MLCK promoter. Homologous human mylk1 nucleotide numbering is based on extensive studies of mouse S-MLCK promoter. Position +1 corresponds to the homologous human S-MLCK mRNA start (chr3 reverse strand: 123,701,003). Dash line indicates that myocardin effect is much weaker compared to its effect on the KRP/telokin promoter. Foxf1 and TEF factors are shown bound to S-MLCK promoter based on their effect, although binding sites have not been defined. AR, androgen receptor. d) Regulation of L-MLCK promoter. Locations of homologous human cis-acting elements are indicated; two different +1 positions are indicated, which correspond to the transcription initiation sites driven by the two distinct promoters: human mRNA NM_053025 (chr3 reverse strand: 123,831,670) and NM_001321309 (chr3 reverse strand: 123,884,253). See text for details.
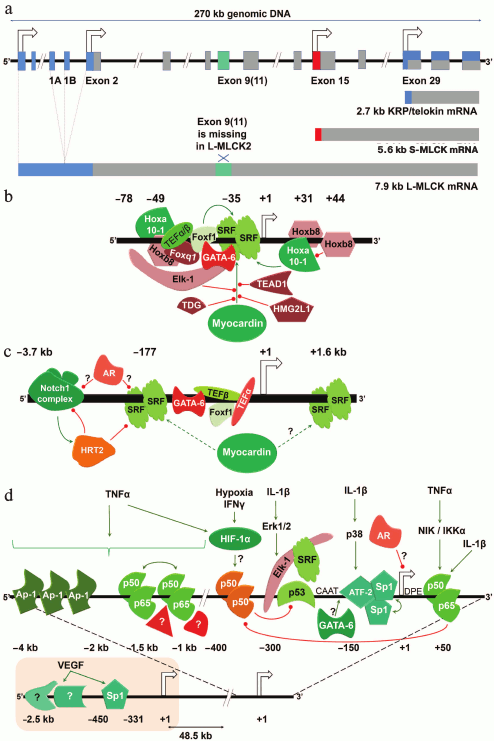
KRP/telokin promoter. The KRP/telokin promoter (Fig. 1b) is active only in smooth muscles of the adult organism [29]. In mice, a major driver of KRP/telokin expression is a short 30-bp sequence containing A/T-rich and CC(AT)6GG (CArG-box) cis-acting elements. The CArG-box binds serum response factor (SRF) [30] and is indispensable for KRP/telokin expression independently from S-MLCK expression [31]. Activation of the KRP/telokin promoter by SRF is enhanced by a cofactor, myocardin [32].
Additional transcription factors seem to play a secondary role [33, 34]. All identified KRP/telokin promoter regulators bind near the SRF binding site and either promote or inhibit the SRF/myocardin complex. Effects of these factors may be responsible for varying KRP/telokin expression levels in different smooth muscle types. The A/T-rich region attracts several enhancer factors like Hoxa10-1 [33], thyrotroph embryonic factor (TEF) α and β isoforms [34], and Foxf1 [35], as well as Foxq1 [36] and Hoxb8 [33] repressor factors. There are two additional Hoxb8 binding sites located downstream from the transcription initiation site; Hoxa10-1 competes for the one located more upstream. While Hoxa10-1 seems to be important for controlling the KRP/telokin expression in some smooth muscles, i.e. in uterus and intestine, silencing effects of Hoxa10-2 and Hoxb8 may be involved in the KRP/telokin promoter regulation in non-muscle tissues [33].
In the KRP/telokin promoter, adjacent to the CArG-box, there are binding sites for GATA-6 [37] and Elk-1 [38]. Both these factors compete with myocardin for SRF binding and exert a negative effect on the promoter activity. Interestingly, in the homologous region of human KRP/telokin promoter, there is a replacement in the conventional GATA-6 site (TATC to CATC). Effects of this replacement are unclear; however, a potential lack of GATA-6 effects in humans might be compensated by other repressor factors and/or might explain a trace KRP/telokin expression in the human heart [39]. While thymine DNA glycosylase (TDG) [40] and HMG2L1 [41] are competitive inhibitors of the SRF/myocardin complex, binding sites for these factors have not been identified within the KRP/telokin promoter. A negative influence of TEA Domain Transcription Factor-1 (TEAD1) on activity of the KRP/telokin promoter is expected based on the finding that several smooth muscle-specific genes including S-MLCK are inhibited by TEAD1, which interferes with the SRF/myocardin complex [42].
S-MLCK promoter. The S-MLCK promoter (Fig. 1c) has no conventional TATA-box and shares some common properties with the KRP/telokin promoter. A minimal S-MLCK promoter is located in a short (432 bp) intron, which separates exons 14 and 15. In the S-MLCK promoter, a CArG-box binds SRF, provides for the basal promoter activity, and is required for S-MLCK expression. The S-MLCK promoter is less dependent on myocardin compared to the KRP/telokin promoter [25, 32]. Interestingly, spontaneously hypertensive rats contain a 12-bp insertion adjacent to the CArG-box. This insertion promotes SRF binding to the CArG-box, i.e. positively regulates the S-MLCK promoter [43]. In mice, GATA-6 binds and suppresses the S-MLCK promoter [25]. It is plausible to suggest that in humans the effects of GATA-6 on the S-MLCK promoter might be compromised due to a replacement (TATC to CATC) found in the homologous GATA-6 binding site, like it is in the KRP/telokin promoter. While Foxf1 activates the S-MLCK promoter, Foxq1 has no effect [35]. Considering that in the KRP/telokin promoter Foxf1 and Foxq1 compete for a common cis-acting element, lack of the Foxq1 effect on the S-MLCK promoter activity may be explained by the action of unidentified transcription factors. The same explanation may be applied to the finding that the KRP/telokin promoter activators, TEFα and TEFβ, exert opposite effects on KRP/telokin activity: TEFα suppresses, but TEFβ activates S-MLCK expression in A10 smooth muscle cells [34].
In the first intron of the S-MLCK gene, there is an additional CArG-box that has no significant influence on S-MLCK expression in A10 smooth muscle cells and 10T1/2 fibroblasts [25]. However, deletion of an intronic region including this CArG-box selectively downregulated S-MLCK in smooth muscles of transgenic mice while it had no effect on either L-MLCK or KRP/telokin expression [44].
In mice, apart from regulation of the S-MLCK promoter activity by the SRF/myocardin complex, there is a conservative CTGGGAA cis-acting element responsible for the S-MLCK promoter activation by the Notch1 receptor complex. Decreased expression of S-MLCK in smooth muscles of transgenic Notch1-defective mice supports the involvement of the Notch1-signaling in S-MLCK expression [45]. The Notch1 receptor complex induces Hairy Related Transcription factor-2 (HRT2), which represses both myocardin- and Notch1-induced S-MLCK promoter activities. Notch1 dose escalation overwhelms the inhibitory effects of HRT2, indicating that the Notch1 effect on the S-MLCK promoter activity is autoregulatory and depends on intensity of the Notch1 signal [46].
L-MLCK promoter. The L-MLCK promoter (Fig. 1d) has no conventional TATA-box; however, a downstream promoter element (DPE) and other conventional regulatory cis-acting elements, a CAAT-box, and several E-boxes have been mapped. In epithelial cells, basal activity of the L-MLCK promoter is controlled independently by a p53 binding element [20] and two cooperative Sp1 binding sites [47]. In epithelial cells, inflammatory factors such as interferon gamma (IFNγ), tissue necrosis factor alpha (TNFα), or interleukin-1 beta (IL-1β) induce L-MLCK expression [20, 48] via NF-κB independently of p53 [20] or Sp1 [47]. It has been shown that a silencing effect of the NF-κB p50/p50 homodimer is antagonized by NF-κB p50/p65 heterodimer binding and activation of the L-MLCK promoter in response to stimulation with either TNFα [49] or IL-1β [50]. In epithelial cells, a TNFα-dependent pathway activates the p50/p65 heterodimer via NIK and IKKα protein kinases [51]. Additionally, IL-1β-dependent activation of Erk1/2 induces Elk-1, which directly interacts and activates the L-MLCK promoter [52], probably in cooperation with NF-κB p65. Simultaneously, IL-1β evokes p38 mitogen-activated protein (MAP)-kinase-dependent activation of Activating Transcription Factor-2 (ATF-2) [53]. Independent inhibition of either Elk-1 or ATF-2 blocks the activating effect of IL-1β, indicating that Elk-1 and ATF-2 may act cooperatively [52, 53]. More distant 5′-upstream regions of the L-MLCK promoter contain additional enhancer elements – two NF-κB binding sites and three Ap-1 binding sites. Similarly, there are experimental data consistent with unidentified silencing elements present in the same upstream region [47]. Complex multicomponent regulation of the L-MLCK promoter may explain both the lack of pharmacological NF-κB inhibition on the TNFα-dependent induction of L-MLCK in epithelial cells [48] as well as an attenuation of NF-κB and concurrent intensification of Ap-1 effects on induction of L-MLCK during epithelial cell differentiation [47]. Synergistic stimulation with TNFα and IFNγ significantly enhances L-MLCK induction compared to individual effects of these agents [47], confirming the complex pattern of L-MLCK expression regulation. Hypoxia Inducible Factor-1α (HIF-1α) and its antagonist, Factor Inhibiting HIF (FIH), may represent a convergence point for the IFNγ- and TNFα-activated pathways. Direct interaction of HIF-1α with the L-MLCK promoter has not been demonstrated, but HIF-1α activation either by hypoxia or in response to stimulation of epithelial cells with IFNγ or TNFα correlates with increased L-MLCK expression; moreover, specific inhibition of HIF-1α negatively affects L-MLCK expression [54, 55].
In endothelial cells, approximately 52 kb upstream from the regulatory elements described above, an alternative “minimal promoter” and transcription initiation site have been identified [21]. The discrepancy is probably due to alternative transcription of the L-MLCK gene in epithelial and endothelial cells resulting in mRNAs that differ in 5′-untranslated regions [21, 47]. Thus, in the shorter “epithelial” mRNA, exon 1A corresponds to exon 3 (mRNA NM_053025) reported in endothelial cells [21]. The latter study demonstrated that L-MLCK expression in human endothelial cells is sensitive to VEGF stimulation and depends on the binding of Sp1 transcription factor within –450 to –331 bp upstream of the most distant transcription initiation site.
Additionally, in A10 smooth muscle cells, GATA-6 enhances L-MLCK promoter activity in contrast to its silencing effects on both the S-MLCK and KRP/telokin promoters [25]. Finally, androgens have been implicated in regulation of the mylk1 gene. In prostate cancer cells, which express both L-MLCK and S-MLCK, androgens downregulated the expression of both MLCK isoforms [56].
Posttranscriptional control of mylk1 gene expression. MicroRNAs are short endogenous noncoding RNA species contributing to the control of many mRNAs. For endothelial cells, several microRNAs, including miR-374a, miR-374b, miR-1290, miR-520c-3p, and miR-155, have been shown to additively downregulate L-MLCK expression [57, 58]. These microRNAs are complementary to the 3′-untranslated region of L-MLCK mRNA, suggesting they might control the level of both S-MLCK and KRP/telokin mRNAs. Indeed, in cardiomyocytes of miR-1–/– mice, expression of smooth muscle myocardin and KRP/telokin is induced, but S-MLCK expression is not altered [59]. These different responses are probably due to a greater dependence of KRP/telokin expression on myocardin (see above), but the influence of additional regulatory factors cannot be excluded. In endothelial HUVEC cell line, oxidized low-density lipoprotein downregulated miR-1 and upregulated L-MLCK, indicating that in endothelial cells, miR-1, at least in part, may be responsible for the low expression level of L-MLCK [60]. In several cancer cell lines and in breast cancer patients, a decrease in miR-200c level correlates with increased L-MLCK expression and higher invasive behavior of the cancer cells. Moreover, in the 3′-untranslated region of L-MLCK mRNA, there are miR-200c binding sites [61] that may recruit miR-200c to downregulate L-MLCK mRNA and, probably, other mylk1-encoded transcripts.
Polymorphism of mylk1 gene transcripts. As noted above, the mylk1 gene encodes for three mRNA classes, for L-MLCK, S-MLCK, and KRP/telokin, respectively. Moreover, for L-MLCK, two distinct independent promoters and two alternative, probably tissue-specific, transcription initiation sites determine the length and alternative splicing of the 5′-untranslated exons [21, 47]. Splice variants of coding exons of L-MLCK and KRP/telokin mRNAs have been described. Alternative splicing of exons 1 and 2 of the KRP/telokin mRNA results in insertion of Glu29 in one of the KRP/telokin isoforms [39]. For human L-MLCK, several splice variants have been identified, but only two protein products are confirmed, the full-length L-MLCK1 and L-MLCK2 lacking residues 437-505 [28]. In addition, many SNPs have been reported in the human mylk1 gene, including those that result in amino acid replacements, e.g. Pro21His, Pro147Ser, Val261Ala, Ser1341Pro, Arg1450Gln, Ser1759Pro, and premature polypeptide chain termination (Arg1480X), or modulate the expression level [62-66].
KRP/telokin is characterized by significant heterogeneity. In tissue-purified avian KRP/telokin, three acetylated N-termini corresponding to Ala2, Met3, and Ser8 were found, with the latter form being predominant; moreover, one to six C-terminal glutamyl residues were missing [67]. Considering that the C-terminal region of KRP/telokin is involved in binding to the myosin rod, the C-terminal heterogeneity may result in KRP/telokin species with varying affinity to myosin and may modulate the ability of KRP/telokin to compete with S-MLCK for myosin binding (see below).
In the antisense strand of the mylk1 gene, there are two genes for noncoding RNAs [68]: MYLK-AS1, which comprises four exons, and a shorter MYLK-AS2, which comprises three exons. Interestingly, one of the MYLK-AS1 exons overlaps the coding sequence of the mylk1 gene, suggesting a role for MYLK-AS1 in posttranscriptional regulation of both S-MLCK and L-MLCK.
Tissue-specific expression of mylk1 gene-derived proteins. Tissue-specific expression of L-MLCK, S-MLCK, and KRP/telokin is controlled by independent regulation of the corresponding promoters as specified above and, apparently, additional still unidentified regulatory signals.
KRP/telokin is predominantly expressed in smooth muscle tissues with much higher levels found in phasic smooth muscles [15, 27, 69]. Trace amounts of KRP/telokin are present in human heart [39].
S-MLCK is expressed in most adult tissues [70]. In embryonic tissues, the S-MLCK levels are low and are upregulated after birth, in contrast to a decrease in L-MLCK expression [15, 71-73]. In many tissues and cell types, S-MLCK coexists with L-MLCK, e.g. this is a case for avian aorta, which was used as a source for the first-ever L-MLCK isolation [74, 75]. L-MLCK seems to be unique to neutrophils [76] and is a predominant form in endothelium, epithelium, monocytes, and T-cells [77-79]. Platelets contain S-MLCK and traces of L-MLCK [80].
Western blot analysis with anti-MLCK antibodies reveals that the L-MLCK levels are higher in non-muscle cell-enriched tissues with a relatively low content of smooth muscle cells (lung, liver, kidney, or spleen) [73, 75]. Upon cultivation of primary cells, L-MLCK expression increases and displaces S-MLCK either totally or partially [70, 71, 73]. As a rule, both MLCK isoforms coexist in cell lines, e.g. in A10, A7r5, and GbaSM-4 cells of smooth muscle origin [74, 81] or in non-muscle cell lines like HeLa, COS-7, or 3T3 [73, 82, 83]. Transformation to cancer cells is associated with upregulation of MLCK expression in some cell types [84, 85] and downregulation in others [61, 86]. Considering the complex regulation of the mylk1 gene, it is reasonable to suggest that a shifted balance of regulatory factors contributes to the developmental and tissue-specific differences in MLCK expression as well as its alterations in transformed cells and during primary cell cultivation.
MLCK DOMAIN STRUCTURE
Three proteins encoded by the mylk1 gene utilize the same open reading frame; the larger protein includes the whole sequence of the smaller. The structure of the largest protein, L-MLCK, is presented in Fig. 2; Met1 of S-MLCK corresponds to Met923 of L-MLCK; Met1 of KRP/telokin corresponds to Met1761 of L-MLCK. The biochemical structure of S-MLCK and issues concerning its enzymatic properties have been review [6, 10]. In this review, we specify the most noteworthy biochemical and structural properties of S-MLCK including the most recent data and take a closer look at the unique N-terminal tail of L-MLCK as well as at the identified regulatory mechanisms that modulate MLCK enzymatic and/or noncatalytic properties.
Fig. 2. Domain organization, protein–protein interactions, and posttranslational modifications of L-MLCK. The main structural elements of L-MLCK (see inset for legend) are presented in a linear mode. Selected modified amino acid residues with suggested physiological significance are superimposed with domain organization. Residue numbering is given for human L-MLCK1 (NM_053025). Responsible modifying enzymes are shown above the modified residues. Braces denote regions of L-MLCK involved in interaction with other proteins (listed below the braces). (?) – site(s) of interaction are not precisely mapped and/or lack solid evidence support. PKG, protein kinase G; PKC, protein kinase C; CaMKII, Ca-CaM-dependent kinase of type II. See text for details.
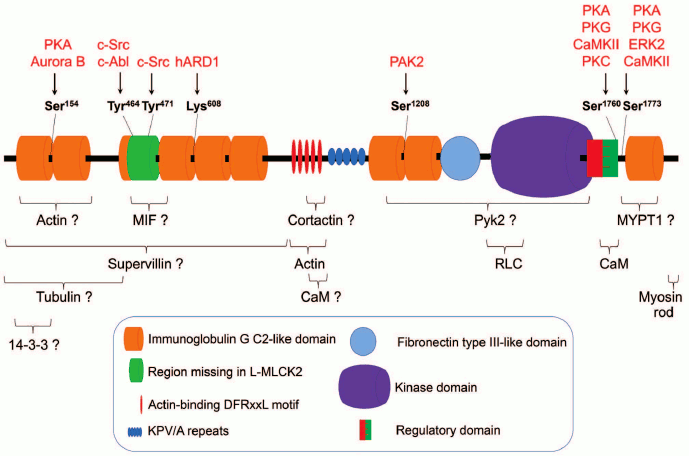
The catalytic core is located in the middle part of S-MLCK and demonstrates homology to the catalytic domains of many protein kinases. In the catalytic domain, there is a Gly-X-Gly-X-X-Gly-(X)14-Lys consensus Mg2+-ATP-binding sequence [23] that is constantly available for Mg2+-ATP [87]. A more N-terminally located acidic amino acid cluster plays an important role in myosin RLC binding [88]. RLC binds to Mg2+-ATP-bound MLCK [89]. Despite claims on the identification of additional MLCK substrates [76, 90, 91], myosin RLC is an unambiguous MLCK in vivo substrate with unquestionable physiological significance. In RLC, Arg16 plays a major role in substrate binding [92] and directs the MLCK catalytic core to Ser19 of RLC. Phosphorylation of two RLCs on a single myosin head proceeds in an ordered manner using different molecular mechanisms [93]. One of the reasons for this behavior may be a noncatalytic interaction of the MLCK C-terminal domain with myosin (see below). In vitro, at high MLCK concentrations, Thr18 of RLC is phosphorylated [94], but in vivo Thr18 is likely to be phosphorylated by alternative kinases, in particular, by Rho-associated protein kinase (ROCK) [95] or zipper-interacting protein kinase (ZIPK) [96]. Phosphorylation of Thr18 enhances the activating effect of Ser19 phosphorylation [97], but monophosphorylation of RLC at Thr18 correlates with smooth muscle relaxation [98], suggesting Thr18 monophosphorylation may not be sufficient for myosin II Mg2+-ATPase activation in non-muscle cells.
The regulatory segment of MLCK flanks the catalytic core at its C-terminus and consists of partially overlapping autoinhibitory and Ca2+/CaM-binding sites. Initially, a pseudosubstrate model was proposed based on similarity of the disposition of basic amino acids in the autoinhibitory region and near the RLC phosphorylation site [99]. However, the adjacent hydrophobic Tyr-Met-Ala cluster appeared as the most important for MLCK autoinhibition [100], indicating that the autoinhibitory mechanism is not restricted to imitation of the RLC sequence. Based on the MLCK autoinhibitory sequence, Lukas et al. developed the Peptide Inhibitor of Kinase (PIK) [101], which could penetrate through biological membranes [102, 103]. More recently, several proteolysis-resistant PIK analogs have been developed for use in in vitro and in vivo studies [102, 104, 105].
MLCK is a Ca2+/CaM-dependent enzyme. Even at basal intracellular Ca2+ concentrations, the C-terminal domain of CaM seems to be constantly bound to MLCK [106]. An increase in the Ca2+ level and subsequent Ca2+ binding at the N-terminal domain of CaM induces rearrangements in both CaM and MLCK and makes the MLCK active site available for RLC binding [107]. These data suggest that an increase in Ca2+ and release of the autoinhibitory segment are prerequisites for effective binding of RLC-imitating MLCK peptide inhibitors (e.g. peptide 11-19 [108]).
Finally, it should be noted that in vitro, the L-MLCK substrate specificity, activation by Ca2+/CaM, and enzymatic properties are identical to those of S-MLCK [74].
The myosin-binding site, which is distinct from the catalytic center, resides at the very C-terminus of the MLCK molecule and is common for L-MLCK, S-MLCK, and KRP/telokin. The acidic C-terminus of KRP/telokin plays a major role in KRP/telokin binding to myosin in vitro [109] to induce the myosin II unfolding and filament formation [110]. Moreover, the KRP/telokin C-terminus is responsible for competitive inhibition of RLC phosphorylation by MLCK [111] and alternative myosin II protein kinases [112].
IgG- and Fn-like domains. Adjacent to the N-terminal end of the catalytic domain is a fibronectin type III (Fn-like) domain of unknown function. Additionally, in S-MLCK, there are three immunoglobulin G (IgG) C2-type domains (IgG-like), each comprising approximately 100 amino acid residues. While two IgG-like domains reside N-terminally to the Fn-like domain, the third IgG-like domain resides at the C-terminus of the MLCK molecule. The C-terminal IgG-like domain with short N- and C-terminal flanking sequences is expressed as an independent KRP/telokin. In the unique N-terminal extension of L-MLCK, six additional IgG-like domains are found [18, 28]. Functional roles of these domains remain unclear, but they have been suggested to contribute to protein–protein interactions of L-MLCK directly or, by analogy with IgG-like domains of titin [113], they may unfold reversibly, thereby increasing the linear dimensions of L-MLCK and/or disclosing binding sites for L-MLCK protein partners (see below).
KPV/A repeats are found in mammalian MLCKs and reside more N-terminally to the first IgG-like domain of S-MLCK. These are 12-amino acid repeats containing conserved Lys-Pro-Val (KPV) and Lys-Pro-Ala (KPA) triplets. The number of KPV/A repeats is species-dependent and contributes to variation of the MLCK molecular mass among species. The physiological role of the KPV/A repeats remains unestablished.
The actin-binding domain of S-MLCK is located at the very N-terminus of the molecule and consists of three 28-amino acid repeats with internal DFRxxL motifs playing a leading role in interaction with actin [114]. Each DFRxxL motif is believed to bind to an individual actin monomer; thus, by three DFRxxL motifs, the S-MLCK N-terminal region can cross-link individual actin filaments in vitro [115]. The alternate binding/dissociation of individual DFRxxL motifs has been suggested to provide for migration of S-MLCK along actin filaments to expand the effective area of the kinase [116]. On an actin filament, sites of interaction with DFRxxL motifs are distinct from the binding sites for myosin, tropomyosin, caldesmon, and calponin [117]; thus, there are no steric obstructions for the lateral migration of S-MLCK. CaM might influence the association of MLCK with actin through the second CaM-binding site, which, according to in vitro studies, overlaps with the DFRxxL-containing region [118, 119]. Because of a relatively weak binding of DFRxxL motifs to actin, in non-muscle cells, which are less “saturated” with actomyosin structures, S-MLCK shows predominantly diffuse distribution and is readily washed out upon cell permeabilization [115]. In the unique N-terminus of L-MLCK, two additional DF/VRxxL motifs enhance the affinity of L-MLCK to actin [74]. However, an internal deletion or the five DFRxxL motifs did not induce L-MLCK dissociation from the cytoskeleton [115], indicating there is(are) an additional actin-binding site(s). Indeed, an actin-binding site structurally different from the DFRxxL-containing domain has been mapped to the two first IgG-like domains [120, 121].
PROTEIN–PROTEIN INTERACTIONS OF mylk1 GENE
PRODUCTS
Actin, myosin, and CaM are unquestionable MLCK partners with well-established physiological significance. However, experimental data indicate there are additional protein partners of MLCK (Fig. 2).
Tubulin, a structural component of microtubules, interacts with L-MLCK in vitro and colocalizes with L-MLCK in transfected CV-1 cells [122]. Via its interaction with tubulin, L-MLCK may provide an integration link between the actin and tubulin cytoskeleton structures.
Supervillin is a scaffold membrane-bound protein and interacts with actin, myosin, and several cytoskeletal proteins. The supervillin N-terminal fragment (residues 1-174) interacts with the L-MLCK unique N-terminal tail in vitro and colocalizes with activated myosin on the cell periphery [123]. It was suggested that an inhibitory effect of supervillin on cell spreading is brought about by myosin activation and myosin filament formation in the submembrane region of a cell. Inhibition of MLCK activity reverted the effects of supervillin.
Cortactin, an actin-binding cytoskeletal protein, interacts with L-MLCK in endothelial cells. Antibody against L-MLCK immunoprecipitated cortactin from endothelial cells; moreover, stimulation of endothelial cell tyrosine kinases increased the amount of precipitated cortactin [124]. Phosphorylation of either cortactin or the full-length L-MLCK (L-MLCK1) by c-Src tyrosine kinase enhanced the interaction of cortactin and L-MLCK1. Functionally, cortactin did not affect the activity of either MLCK isoform in vitro and, similarly, MLCK did not affect the actin-binding properties of cortactin but inhibited cortactin-stimulated activation of Arp2/3 [125]. While the cortactin SH3 domain is responsible for interaction with MLCK isoforms, putative cortactin-binding sites are located within the DFRxxL domain of MLCK [126] suggesting that the interaction might be regulated by Ca2+/CaM, which binds to the same MLCK region and may modulate MLCK interaction with actin. The unique N-terminal tail of L-MLCK might contain additional cortactin-binding sites. A L-MLCK-dependent increase in cortactin tyrosine phosphorylation in endothelial cells may be due to recruiting of non-receptor tyrosine kinases, in particular c-Src, via L-MLCK scaffolding activity [124] and/or due to L-MLCK-dependent conformational changes in cortactin that facilitate its interaction with c-Src. Involvement of L-MLCK in activation of tyrosine kinases was observed in neutrophils as well [76].
Pyk2, a FAK-family cytoplasmic tyrosine kinase, is recruited to β2-integrins and contributes to an adequate integrin activation and neutrophil adhesion. In murine L-MLCK–/– neutrophils, both Pyk2 activation and interaction with β2-integrins were impaired. Direct interaction of Pyk2 and L-MLCK is supported by coimmunoprecipitation studies and in vitro binding of purified Pyk2 and the recombinant MLCK catalytic domain. Interestingly, in neutrophils, the inhibition of L-MLCK enzymatic activity attenuated Pyk2 activation; moreover, the MLCK catalytic domain phosphorylated and activated Pyk2 in vitro. These data suggest both enzymatic and noncatalytic activities of MLCK contribute to Pyk2 activation [76].
MIF (macrophage migration inhibition factor) has been identified as an MLCK partner by the yeast two-hybrid assay system. Interaction between the two proteins was confirmed by coimmunoprecipitation of GST-tagged MLCK fragment from BPAEC cell lysates and indirect immunofluorescence data. Biochemical studies with truncated mutants show that the L-MLCK N-terminal 515 amino acids were sufficient for MIF binding and suggest that one MIF binding site on L-MLCK1 may reside between amino acids 437-505, a sequence that is spliced out in L-MLCK2. However, the functional significance of the MIF/MLCK complex is unclear, because MIF did not compete with c-Src-dependent L-MLCK phosphorylation within amino acids 415-500 and had no effect on the interaction of L-MLCK with actin or cortactin [127].
14-3-3 scaffold. In the L-MLCK regions flanking the first IgG-like domain, there are putative binding sites for 14-3-3 proteins. Interaction of L-MLCK with 14-3-3 proteins is supported by an enrichment of two 14-3-3 isoforms (24 and 27 kDa) in the immunoprecipitate of the FLAG-tagged L-MLCK fragment and ability of a 14-3-3 antagonist to abolish the attenuation of L-MLCK phosphorylation at Tyr464 [128]. Unfortunately, the putative interaction of L-MLCK with 14-3-3 is based only on indirect data and has not been confirmed by further studies.
Myosin light chain phosphatase (MLCP) is a dedicated MLCK antagonist and seems to be a protein partner of mylk1 gene-derived proteins. Based on copurification of MLCK and MLCP from smooth muscle, early reports suggested that MLCK and MLCP exist in a complex [129]. More recently, these indirect data gained support from several experimental findings in KRP/telokin–/– mice indicating a positive modulation of MLCP by KRP/telokin [31]. Because in a myosin II molecule, KRP/telokin shields RLC from activating protein kinases [111, 112] and KRP/telokin does not bind to phosphorylated myosin [130], it has been suggested that modulation of myosin dephosphorylation by KRP/telokin is due to its direct or indirect effects on MLCP [31]. Recent physiological and biochemical data are in favor of a hypothesis that direct interaction of KRP/telokin with regulatory MLCP subunit, MYPT1, abolishes the effect of inhibitory MYPT1 phosphorylation [131]. The interaction can probably be explained by some degree of similarity between KRP/telokin and a small MLCP subunit (M20) of unidentified function.
Using the yeast two-hybrid assay, coimmunoprecipitation, or peptide matrices, large-scale screening studies identified a score of putative MLCK protein partners; however, additional efforts are needed to confirm the interactions and identify the physiological significance of the confirmed ones.
Enzymes that use MLCK isoforms and/or KRP/telokin as substrates, e.g. c-Src tyrosine kinase [125], could be, with certain limitations, considered as MLCK/KRP protein partners. Because posttranslational modifications (PTM) play an important role in regulation of most if not all human proteins, below are summarized the most noteworthy PTMs that have been identified in the mylk1-derived proteins.
POSTTRANSLATIONAL MODIFICATIONS OF mylk1 GENE
PRODUCTS
According to the PhosphositePlus database (http://www.phosphosite.org) [17], there are over 10 acetylation and 40 phosphorylation sites in L-MLCK. In contrast, few methylation and ubiquitinylation sites have been identified. For the great majority of L-MLCK PTMs, the physiological significance remains unestablished, and there are few reports on this issue.
Acetylation-dependent modulation of mylk1 gene-encoded proteins. One of the earliest identified PTMs in KRP/telokin was a constitutive acetylation of N-termini of KRP/telokin isoforms that differed in the length of their N-terminal ends [67]. The Met1 residue of S-MLCK is acetylated as well [132]. These modifications seem to protect KRP/telokin and S-MLCK from degradation. Recently, hARD1 acetyltransferase was shown to modulate L-MLCK activity [133]. It was demonstrated that hARD1 binds within the L-MLCK region spanning the fourth and fifth IgG-like domains and acetylates the epsilon amino group of Lys608 (Fig. 2). In transfected cells, hARD1 downregulated both RLC phosphorylation and MLCK-dependent invasion and migration of HT1080 cancer cells. Moreover, the Lys608Arg mutation in L-MLCK completely abolished the effect of hARD1 on L-MLCK activity. Interestingly, the interaction of hARD1 with L-MLCK required a prior activation of L-MLCK by Ca2+/CaM, suggesting that there are complex interactions between L-MLCK domains that are located far from each other in the primary structure of the kinase.
Phosphorylation-dependent modulation of mylk1 gene-encoded proteins. Among the PTMs of the mylk1 gene-encoded proteins, phosphorylation-dependent modulation has been much more extensively studied. Proteomic techniques validated several in vivo phosphorylation sites of MLCK that partially matched the sites identified in earlier in vitro studies (Fig. 2). Among these is a functionally important MLCK phosphorylation in the CaM-binding domain (designated the phosphorylation site A) [134]. In site A, phosphorylation at Ser1760 by protein kinases A, G, C (PKA, PKG, and PKC, respectively), or Ca2+/CaM-dependent protein kinase II (CaMKII) results in inhibition of both CaM binding and RLC access to the MLCK catalytic center, which remains closed by the MLCK autoinhibitory region [134-136]. The p21-activated protein kinase-2 (PAK2) phosphorylates the adjacent Ser1759 and brings about the same inhibitory effect [137]. There is a commercial anti-(P)Ser1760 antibody that allows monitoring the level of Ser1760 phosphorylation and the corresponding level of MLCK inhibition, but in isolated studies, e.g. in [83], a positive reaction with this antibody is interpreted as being associated with MLCK activation. Conclusions drawn in reports of this kind should be considered with caution.
In L-MLCK, several phosphorylation sites have been identified that reside outside the catalytic domain but affect the activity of the enzyme. In the unique N-terminal tail of L-MLCK, c-Src-dependent phosphorylation at Tyr464 and Tyr471 in vitro increases the enzymatic activity of L-MLCK 2-3-fold [138]. These data indicate that L-MLCK may adopt a “folded” tertiary structure with its N- and C-terminal moieties coming into contact. This hypothesis is supported by the abovementioned effects of Lys608 acetylation on L-MLCK activity [133]. Unfortunately, no crystal structure is available for the full-length L-MLCK that would reflect the intramolecular interactions of distantly located L-MLCK domains.
Interestingly, both Tyr464 and Tyr471 are encoded by a single exon that is spliced out in L-MLCK2 [28]. Correspondingly, c-Src does not phosphorylate and regulate L-MLCK2. In vitro, c-Abl tyrosine kinase phosphorylates up to 10 tyrosine residues in L-MLCK1, including Tyr464, and it increases both L-MLCK1 catalytic activity and affinity to cortactin, as in the case with c-Src [139]. The same study reported autophosphorylation of L-MLCK at 19 serine and threonine residues. In this regard, it seems impossible to correlate the phosphorylation at a given residue with changes in L-MLCK catalytic activity. Moreover, phosphorylation of multiple residues may result from nonspecific modification due to excess of the protein kinase in in vitro experiments. This assumption is favored by the fact that only two out of nine c-Abl in vitro phosphorylation sites have been validated in cancer cells by mass-spectrometry (see http://www.phosphosite.org).
In chicken L-MLCK, Ser149Asp mutation (homologous to Ser154 in human L-MLCK and Ser149 in murine L-MLCK and identified as an in vivo phospho-site) attenuated the binding of the actin-binding N-terminal L-MLCK fragment to cytoskeletal structures in transfected HeLa cells [120]. In vitro, Ser149 in avian L-MLCK could be phosphorylated by PKA and Aurora B, which show similar substrate specificity. The latter kinase is active in mitosis and phosphorylates serine residues within the N-terminal tail of L-MLCK in mitotic HeLa cells [140]. In contrast, PKA may phosphorylate L-MLCK in interphase cells. Thus, PKA and Aurora B team up to represent a minimal set of protein kinases that can modulate actin-binding properties of L-MLCK in cells.
Most studies reporting L-MLCK phosphorylation in cells and tissues have not established links between phosphorylation at individual L-MLCK amino acid residues and alterations in cellular responses. Phosphorylation of L-MLCK at tyrosine residues has been demonstrated in endothelial cells following stimulation with diperoxyvanadate, a potent tyrosine kinase activator and tyrosine phosphatase inhibitor [124], and in fibroblasts following transformation with constitutively activated mutant epidermal growth factor receptor (v-ErbB) [141]. In both these studies, the authors reported alterations in the activation status of contractile proteins in cells. Phosphorylation of MLCK by MAP-kinases has been associated with MLCK activation, an increase in myosin RLC phosphorylation, and acceleration of cell migration [142]. Because MAP-kinase consensus sites are located outside the catalytic domain of MLCK, it seems likely that the MAP-kinase-dependent activation of MLCK required an interaction between distantly located MLCK domains, much as suggested for the phosphorylation of Tyr464 and Tyr471 and acetylation of Lys608 in the N-terminal tail of L-MLCK.
Both KRP/telokin and the corresponding domain within MLCK are multiply phosphorylated (see http://www.phosphosite.org). Within the unstructured N-terminus of KRP/telokin, eight of ten Ser/Thr residues are phosphorylated, suggesting that this region has an important regulatory role. However, there are no indications on the functional properties that are regulated by phosphorylation of the KRP/telokin domain except a report suggesting that PKA-dependent phosphorylation in this domain intensifies the inhibitory effect of the site A phosphorylation [136]. In KRP/telokin, PKA- and/or MAP-kinase-dependent phosphorylation of the homologous site does not alter the inhibitory effects of KRP/telokin on myosin phosphorylation and smooth muscle contraction [112]. In isolated phasic smooth muscle, KRP/telokin phosphorylation is increased during contraction and slightly varies during the subsequent relaxation [75]. Quantitative analysis of the phosphorylation dynamics at individual sites demonstrated that during smooth muscle contraction, phosphorylation at Ser13 of KRP/telokin increases from basal 20 to 100%; however, the level of KRP/telokin phosphorylation at Ser19 did not show significant changes during the contraction/relaxation cycle and amounted to about 25% [143]. According to the model suggested by A. Somlyo’s group, KRP/telokin activates MLCP and, in this way, contributes to smooth muscle relaxation. Moreover, the effect is augmented by PKG-dependent KRP/telokin phosphorylation at Ser13 [144, 145] because phospho-KRP/telokin binds to and activates MYPT1 by overriding the preexisting inhibitory phosphorylation of MYPT1 [131]. It should be noted, however, that direct interaction with MYPT1 in vitro was shown for the phospho-mimicking KRP/telokin mutant (Ser13Asp), whereas the behavior of KRP/telokin phosphorylated at Ser13 was not studied.
In summary, all proteins that derive from the mylk1 gene are extensively labeled by multiple PTMs. Isolated PTMs have been shown to regulate MLCK enzymatic activity, which affects the activation status of myosin II ATPase and, respectively, parameters of cell motility. For most PTMs that are found in MLCK and/or KRP/telokin, the functional significance remains unclear.
ROLE OF MLCK/KRP IN PHYSIOLOGICAL CELLULAR RESPONSES
The roles of MLCK in physiological processes are predominantly associated with myosin II activation, which drives a great variety of cellular responses. Thus, MLCK contributes to a variety of apparently unrelated processes, but all these depend on the motility apparatus of the cell. MLCK is a Ca2+/CaM-dependent myosin II activator and is activated by an increase in intracellular Ca2+ level that, in turn, is evoked by Ca2+-mobilizing agents like histamine, thrombin, bradykinin, bombesin, cholecystokinin, acetylcholine, catecholamines, fMet-Leu-Phe, oxidants, etc. Apart from MLCK protein kinase activity, interactions of MLCK with its protein partners (see section “Protein–Protein Interactions of mylk1 Gene Products”) may contribute to the MLCK-dependent functional responses of the cell.
Cell adhesion and migration. Adhesion and migration are fundamental features of substrate-dependent cells. MLCK is involved in both these cellular processes. Conjointly with focal adhesion kinase, protein kinase c-Src, and extracellular-regulated kinases (Erk), MLCK participates in both formation and disassembly of focal adhesions at the cell front and provides for onward migration [146]. Pharmacological inhibition of MLCK and CaM changes fibroblast-populated collagen lattice contraction, cell migration, focal adhesion formation, and wound contraction [147]. The Erk pathway regulates F9 parietal endoderm cell migration by affecting the formation of focal adhesions and lamellipodia via modulating MLCK activity [148].
In fibroblasts, MLCK-dependent focal adhesion formation proceeds at the cell periphery; in contrast, ROCK-dependent focal adhesions are located more centrally. Selective inhibition of these protein kinases results in disassembly of adhesions either in the center or at the periphery and alters the migration pattern of cells. MLCK-inhibited cells generated membrane protrusions all around the cell, turned more frequently, and migrated less effectively compared to ROCK-inhibited cells, which moved faster and straighter [149]. Other studies in different cell models support the conclusion regarding the spatial segregation of MLCK- and ROCK-dependent activation of myosin II [150-153].
On the other hand, accumulating data suggest that noncatalytic properties of MLCK, e.g. interaction with actin and other protein partners, play an important role in migrating cells [76, 81, 154]. Indeed, as noted above, MLCK may act as a scaffold and/or integrate cytoskeletal structures [122-124]. In isolated model systems, e.g. in L-MLCK–/– neutrophils, an assay of neutrophil transmigration across endothelial monolayer revealed that an interaction between L-MLCK and c-Src and Pyk2 tyrosine kinases and subsequent recruitment of Pyk2 to β2-integrins was a sufficient MLCK-dependent event, while ROCK-dependent myosin activation assured motile activity [76]. Meanwhile, data from experiments with “substitution” of the MLCK catalytic activity by its actin-binding activity by overexpression of kinase-dead MLCK or its individual domains [81, 154] should be considered with caution. Ectopic overexpression of MLCK actin-binding domains would obviously stabilize the actin cytoskeleton even when the MLCK catalytic activity is lacking [122], and the motile component would be provided by alternative myosin-activating kinases like ROCK, ZIPK, citron kinase, etc.
Proliferation. MLCK takes part in the control of cell division. In proliferating rodent hepatocytes, inhibition of MLCK catalytic activity or downregulation of MLCK expression revealed a role for MLCK in the control of cell transition from late G1 to S phase involving Erk2-dependent p70S6 kinase activation [155]. Similarly, downregulation of MLCK expression or inhibition of MLCK catalytic activity slows the proliferation of various cancer cells [156-158]. In contrast, MLCK downregulation in GbaSM-4 smooth muscle cells increased the proliferation rate and shortened the doubling period of the population [159]. Effects of MLCK on mitosis seem to depend on the cytoskeleton status, activity of intracellular signaling pathways that affect MLCK activity, and the levels of MLCK protein partners. These parameters may vary significantly in normal versus tumor cells.
In mitotic HeLa cells, L-MLCK is concentrated in cortical cytoplasm during metaphase and in the cleavage furrow during anaphase and telophase [160]. Targeting of L-MLCK to the cleavage furrow required both the actin-binding DFRxxL motifs, which are located in the central part of the MLCK molecule, and additional sequences from the L-MLCK N-terminus. L-MLCK targeting to the cleavage furrow seems obvious, because the cleavage furrow is a typical actomyosin structure. Both L-MLCK catalytic and noncatalytic domains may contribute to cleavage furrow formation and contraction. Additionally, in the same study, the L-MLCK N-terminus disrupted normal spindle morphology during mitosis. The authors suggested the identification of a novel regulatory role of L-MLCK during mitosis, but there should be considered possible artefact consequences of overexpression of the L-MLCK N-terminus, which interacts with tubulin in vitro and microtubules in cells [122].
Endocytosis. Convincing experimental evidence has accumulated in support of active involvement of MLCK in endocytosis. Membrane internalization occurs for volume recovery in response to osmotic swelling. MLCK and myosin II localized with actin to swelling-induced membrane blebs just before retraction; inhibition of MLCK induced persistent blebbing and attenuated cell volume recovery. At the sites of membrane internalization, MLCK localized to dynamic actin-coated rings and patches, which contained also c-Src, cortactin, and dynamin; inhibition of either MLCK or c-Src altered the lifetimes of these actin-coated structures [161].
Endocytosis of synaptic vesicles recycles vesicle membranes to maintain synaptic transmission. The enzymatic activity of MLCK is essential in this process. MLCK inhibition or downregulation of MLCK expression slowed vesicle endocytosis and prevented depolarization-induced phosphorylation of RLC in rat hippocampal boutons. Similarly, inhibition of myosin II impaired endocytosis; moreover, the effect was shared by blockers of Ca2+-channels and CaM [162]. Earlier, a similar effect was observed at the calyx of Held synapse in rats [163]. The authors assumed that MLCK-dependent myosin phosphorylation and activation of synaptic vesicle endocytosis is a general neuronal process that facilitates recycling of vesicle membranes under enhanced neuronal activity.
Exocytosis. It is widely accepted that MLCK plays a role in exocytosis. In 1994, the essential role of MLCK in ATP-dependent initiation of Ca2+-induced release of catecholamines from adrenal chromaffin cells was reported [164]. Afterwards, involvement of MLCK in exocytosis/secretion was demonstrated in other cell types. In pancreatic β-cells, via activation of non-muscle myosin IIA, MLCK modulates the translocation of secretory granules and enhances insulin release [165]. In agonist-stimulated platelets, MLCK cooperates with ROCK to activate myosin II and induce platelet contraction and dense granule secretion [166].
In primary alveolar type II cells, L-MLCK and ROCK translocate to actin-coated plasma membrane-fused lamellar bodies and activate myosin II on actin coats. For efficient actin coat contraction, cofilin-1 and α-actinin are recruited to the surface of secretory granules to promote active extrusion of cargo [167]. While cofilin-1 regulates actin depolymerization on the contact surface of a lamellar granule with the plasma membrane to promote fusion, MLCK- and ROCK-dependent contraction of other parts of the actin coat probably promotes extrusion of cargo (surfactant components) via the pore.
The outlined scheme of molecular events during the late phases of exocytosis is in concert with experimental data and, thus, may be true regardless the exocytosis type – whether it is a release of catecholamines or insulin, externalization of receptors or glucose transporters, extrusion of Weibel–Palade or lamellar bodies, etc. In every instance, MLCK seems to activate myosin on the granules to be extruded, but an effect of scaffolding activity of MLCK cannot be excluded.
Regulation of mechanical properties of cells. Cells are characterized by a certain degree of stiffness, which under normoosmotic conditions depends presumably on the cytoskeleton and its remodeling activity. In fibroblasts overexpressing constitutively active S-MLCK, cytoskeletal stiffness was increased two-fold and balanced by microfilament depolymerization with cytochalasin D [168]. Recent study in L-MLCK–/– mice demonstrated a reduced flow-mediated NO-dependent dilation of small arteries that had L-MLCK-deficient endothelium. In wild-type mice, an inhibitor of MLCK produced a similar effect [169]. Thus, MLCK downregulation/inhibition impaired the sensitivity of endothelial NO-synthase to shear stress.
Using atomic force microscopy (AFM), Dudek et al. provided evidence supporting an important role of the c-Abl/cortactin/L-MLCK protein complex in both reorganization of cortical actin in endothelial cells and in a sphingosine-1-phosphate-induced 30% increase in cell stiffness [139]. This study did not show that L-MLCK directly regulates endothelial cell stiffness, but further studies supported this hypothesis. Indeed, AFM revealed that wild-type endothelial cells are stiffer compared to endothelial cells from L-MLCK–/– mice. Moreover, ROCK cooperates with L-MLCK to maintain endothelial cell stiffness [170, 171]. It is to be established whether noncatalytic domains of L-MLCK play a role in controlling cell stiffness. Potential cross-linking properties of L-MLCK, which are realized via distantly positioned actin-binding sites including those subject to phosphorylation-dependent modulation [120], may affect the cytoskeleton stiffness and the cell elastic modulus.
Epithelial and endothelial barriers. Epithelial and endothelial cells form tight semipermeable monolayers to allow passage of low molecular weight substances while preventing leakage of proteins [172]. The cytoplasmic domains of intercellular contact proteins interact with the actin cytoskeleton and meet with the actomyosin-induced drag force. Many natural stressors (histamine, thrombin, VEGF, TNFα, bacterial lipopolysaccharide, reactive oxygen species, excessive mechanical stress, etc.) evoke a disassembly of intercellular contacts in endothelium and epithelium and activate actomyosin-dependent contraction. In a number of situations, actomyosin activation in epithelium and endothelium is mediated by L-MLCK catalytic activity [1, 173]. Contraction drives the adjacent cells apart and hampers rapid contact recovery, which is achieved via cell spreading. Moreover, MLCK and myosin II accumulation in lamellipodia decreased lamellipodial lifetime and size, and stimulated retraction [153]. Thus, in endothelium and epithelium, L-MLCK is engaged in cell barrier destabilization and hyperpermeability.
On the other hand, in a complex with c-Abl tyrosine kinase and cortactin, L-MLCK is engaged in remodeling of the cortical actin in endothelium in response to sphingosine-1-phosphate, which is a platelet-derived barrier protective agent [139]. In this case, noncatalytic scaffolding and cross-linking activities of L-MLCK may play a role in the barrier protection, but focused studies are lacking. The L-MLCK–/– mice were less susceptible to a stress-induced endothelial and epithelial barrier dysfunction [102, 105, 174, 175]; conversely, overexpression of L-MLCK in murine endothelium enhanced its permeability to macromolecules [176].
Smooth muscle contraction. In contrast to S-MLCK, which initiates smooth muscle contraction via activation of myosin II, KRP/telokin seems to play an antagonistic role. Through the binding to myosin heads next to the RLC-binding site, KRP/telokin hinders RLC phosphorylation by myosin-activating protein kinases [112]. Additionally, KRP/telokin may be involved in MLCP activation. Intestinal smooth muscle of KRP/telokin–/– mice exhibited an approximately 30% decrease in MLCP activity accompanied by an increase in RLC phosphorylation [31]. Thus, in smooth muscle, independently of the molecular mechanism, KRP/telokin attenuates myosin activation and promotes relaxation. It remains to be established whether the KRP/telokin domain of MLCK plays a similar role.
ROLE OF MLCK IN PATHOLOGICAL PROCESSES
Endothelial/epithelial barrier dysfunction. Dysfunction of endothelial/epithelial barrier occurs in multiple pathological states in humans, including sepsis, myocardial infarction, stroke, wet macular degeneration, traumatic brain injury, and pancreatitis. Alterations in vascular permeability and lung and brain edema are complications of aggressive treatment for cancer, poisoning with pulmonotoxic substances, thermal burns of the body, tissue crashing, high altitude hypoxic exposure, and other conditions. Alterations of intestinal epithelium permeability are found in Crohn disease, celiac disease, autoimmune and inflammatory processes in the gut, infections, intestinal obstruction, and poisoning.
In mice, TNFα activates intestinal epithelial MLCK, which initiates caveolin-dependent endocytosis of occludin, the principal component of tight junctions in the gut epithelium [177]. Occludin endocytosis could be one of the mechanisms increasing intestinal epithelium permeability in response to various stress factors that increase TNFα levels in the organism.
A MLCK-dependent mechanism is apparently involved in the penetration of commensal bacteria in intestinal enterocytes in the presence of IFNγ. In such conditions, myosin II phosphorylation is increased in the terminal web, and so-called arches are formed. The fanning of brush border takes place, which is followed by transcytosis of bacterial cells. Introduction of an MLCK inhibitor into intestinal lumen or the use of anti-IFNγ antibodies blocks the penetration of bacteria into enterocytes. IFNγ-induced bacterial translocation is not observed in L-MLCK–/– mice, confirming the participation of MLCK in this process [178].
In several studies that used L-MLCK–/– mice and MLCK inhibitors (small organic molecules and/or cell-penetrating peptides), suppression of MLCK activity improved the integrity of the endothelial/epithelial barrier, reduced the transmigration of immune cells in tissues, and minimized clinical signs of pathology [1, 76, 104, 105, 174, 179-182].
Polymorphisms in the mylk1 gene are shown to associate with predisposition of various human populations (African Americans, African Caribbeans, Caucasians, Spanish descent families) to the development of acute lung injury, sepsis, and asthma [5]. These genetic variations may alter expression levels of MLCK in endothelium and epithelium of the lung and could be responsible for the increased content of this protein in the airway smooth muscles of asthmatic patients [183]. Several polymorphisms in the mylk1 gene are associated with aortic dissection and familial thoracic aortic aneurysm in humans [66].
Atherosclerosis. In ApoE–/–/L-MLCK–/– mice fed high fat diet, the size of aortic lesions is reduced as well as the content of lipids and macrophages in these lesions [77]. The authors link the alterations to the reduced permeability of aortic endothelium in these animals and confirm the suggestion by direct experiments. At the same time, they notice a decrease in c-Src phosphorylation in L-MLCK–/– endothelium following stimulation with thrombin [77] and relate the effect to noncatalytic activity of MLCK. Confirming the earlier report [76], they demonstrated a slower transmigration rate of L-MLCK-deficient monocytes through the endothelium and attributed the effect to the scaffold activity of L-MLCK.
Insulin resistance and diabetes. MLCK plays an important role in insulin signaling, the development of insulin resistance, and in diabetes. Inhibition of myosin II and MLCK decreased glucose-stimulated insulin secretion from pancreatic beta cells, which was associated with shortening of peripheral actin stress fibers, and reduced numbers of focal adhesions and insulin granules near the basal membrane [165]. Thus, MLCK catalytic activity is required for the modulation of insulin secretion by the pancreas.
Interaction of insulin with insulin receptors on the surface of skeletal muscle and adipose cells assumes the traverse of insulin from the bloodstream into tissue through the endothelial barrier. Transport of insulin across microvascular endothelium is accomplished by transcytosis using clathrin-coated vesicles [184]. It is known that myosin II participates in exocytosis of such vesicles [185], and MCLK may regulate myosin activity. Limited insulin granule exocytosis by endothelial cells due to alterations in the MLCK–myosin II system may therefore be a factor leading to insulin resistance. Decreased insulin concentration at the surface of the glucose-accumulating cells will result in hyperglycemia and reactive hyperinsulinemia.
Under physiological conditions, insulin stimulates the incorporation of GLUT4 glucose transporters in plasma membranes of adipocytes and skeletal myocytes. This process is a form of exocytosis; it depends on myosin IIA and myosin-activating protein kinases such as MLCK. Active (acto)myosin is required for the fusion of GLUT4-loaded vesicles with plasma membrane and for further maintenance of GLUT4 activity [83, 186, 187]. A decrease in efficiency of GLUT4 in adipocytes leads to hyperglycemia and the development of insulin resistance.
Persisting hyperglycemia typical for diabetes causes the accumulation of so-called advanced glycation end-products (AGE) such as albumin and hemoglobin that are non-enzymatically modified by glucose. AGE bind to their receptors (RAGE) on endothelium and induce its hyperpermeability. Because MLCK is involved in endothelial barrier dysfunction, inhibition of catalytic activity/expression of MLCK attenuates edemagenic effects of AGE and retards the development of diabetic vasculopathies [188].
Additionally, hyperglycemia and hyperlipidemia increase the expression of MLCK and the accumulation of phosphorylated (inhibited) MYPT1 in aortic endothelium of rats with streptozotocin-induced diabetes. These alterations favor the contractile phenotype and hyperpermeability of endothelium. These effects are largely reversed by melatonin administered to the animals [189]. In contrast, insulin restores the depressed expression of MLCK in the stomach and in the intestine of streptozotocin-diabetic rats. The decreased MLCK content in these organs is a possible cause of the delayed gastrointestinal passage of food typical for these animals [190].
Finally, a recent report states that MLCK concentration in the serum of patients with type 2 diabetes is significantly higher than in the serum of healthy donors. Thus, MLCK may soon become a novel biomarker of type 2 diabetes [191]. Perhaps MLCK immunoreactivity is increased in diabetic patients because of persistent damage of vascular endothelium due to oxidative stress. The latter condition might be augmented by endothelial L-MLCK through the modulation of cortactin–p47(phox) interaction, which is required for the assembly and activation of endothelial NADPH oxidase [192].
Cancer. Since MLCK is actively involved in physiological processes of cell adhesion, migration, and proliferation, alterations of its expression and activity regulation during oncogenic transformation of cells may be principal determinants of migratory and invasive potential of these cells, thereby affecting the dynamics of the disease and tactics of treatment. Studies show that MLCK is associated with various types of cancer, including breast, lung, prostate, colon cancer, etc. [5].
Decreasing MLCK content in normal breast epithelial cells leads to alterations in cell–cell interactions and activates invasive behavior of these cells. MLCK content in invasive breast cancer cells is lower than in noninvasive breast cancer cells, and low levels of MLCK correlate with poor prognosis of HER2-positive breast cancer patients [85]. At the same time, other reports demonstrate an opposite relationship between the content/activity of MLCK and the invasiveness of cancer cells [61, 193] that may reflect specific features of cells studied by various groups. Gene expression profiles in various cancer cells, even those derived from the same organ, may differ significantly; accordingly, cellular reactions may differ as well. Acetylation of Lys608 in L-MLCK by acetyltransferase hARD1 inactivates L-MLCK and leads to decreased myosin RLC phosphorylation in cancer cells and inhibition of cell migration and invasion [133].
MLCK is activated locally and activates actomyosin contraction in vascular endothelium, through which cancer cells invade the vasculature. Inhibition of myosin phosphorylation in endothelial cells attenuates transendothelial but not paracellular invasion of cancer cells [194].
MLCK as a molecular target for drug development. Due to the involvement of MLCK in major pathologies, many researchers consider this protein as a promising molecular target for the development of new drugs with wide application potential. Still, progress in this direction may encounter significant problems associated with potential negative influence of these compounds on physiological processes regulated by MLCK. In particular, inhibition of catalytic activity of L-MLCK in endothelial/epithelial cells could suppress the activity of smooth muscle S-MLCK and, possibly, other MLC kinases (skeletal, cardiac) in the absence of technologies for targeted drug delivery. Selective modulation of MLCK expression in a particular cell type will help understanding the tissue-specific regulation of the mylk1 gene multiple promoters. Focused manipulation of noncatalytic activities of MLCK will require detailed analysis of MLCK interaction with partner proteins and will critically depend on effective methods of drug targeting.
Concluding remarks. For several decades, MLCK has been attracting persistent interest, which highlights a widely appreciated biomedical role of the enzyme and indicates that many elusive issues are still to be discovered. These concern the organization and regulation of the mylk1 gene, structural and functional properties of the RNA and protein products that originate from the gene, and the role of these products in (patho)physiology of the cell and organism. We now know quite a lot about the differential regulation of the mylk1 gene, but there are only a few studies available concerning the links between the mylk1 gene polymorphisms and liability to diseases. Moreover, the list of mylk1 polymorphism-associated pathologies seems far from being complete.
Still, the initial steps have been made in identification of protein–protein interactions that may affect enzymatic/noncatalytic properties of L-MLCK, S-MLCK, and KRP/telokin. The unique N-terminus may target L-MLCK to specific intracellular domains and govern the participation of the kinase in various cellular reactions. Considering the confirmed involvement of L-MLCK in many physiological processes in cells and tissues, identification of L-MLCK protein partners and deciphering the functional role and regulation of these interactions becomes an urgent fundamental issue with promising perspectives in applied fields.
Posttranslational modifications are well known to affect protein structure and function, but for the mylk1 gene products, a regulatory role of PTMs is far from being completely elucidated; moreover, further questions are raised by reports that describe the relationships between particular PTMs and alterations of mylk1 protein features. In part, this is due to incomplete understanding of the L-MLCK structure and interactions with other proteins. Mapping of intramolecular interaction sites and sites of interaction with protein partners in the MLCK sequence is a clue to understanding the roles of particular PTMs. Conversely, localization of PTMs within the MLCK structure may reflect the sites essential for various interactions and regulation in vivo. Thus, solving a “PTM problem” is an actual issue for further studies of L-MLCK to expand our knowledge about the role of the enzyme in (patho)physiology of the cell. Progress in this direction depends mainly on gaining new insights into MLCK molecular anatomy and identification of partner binding sites.
A range of MLCK-dependent cellular and whole organism reactions has been outlined by studies utilizing molecular genetics and pharmacological methods for controlling either expression or activity of MLCK. This range, predictably, has included various aspects of cell motility. Later, alterations in cellular motility, which underlie cardiovascular, oncological, infectious, and other diseases, diabetes, and trauma, have been often associated with impairment in the “MLCK/partners” molecular system. These findings suggest MLCK as a promising molecular target for development of novel drugs. The primary steps have already been made in this direction. However, there are still many objective problems due to ubiquitous expression of MLCK and involvement of the enzyme in important physiological processes. In this regard, a critical issue for success is an understanding of the mechanisms that govern tissue-specific expression of the mylk1 gene, MLCK interactions and PTMs, as well as the development of targeted delivery of MLCK-selective agents (synthetic inhibitors, interfering peptides, molecular genetic constructs, etc.) in vivo. Future studies along these avenues of research should facilitate translation of the fundamental knowledge about MLCK into technologies and products urgently needed by clinical medicine.
Note added in proof
When this review was ready for publication, Gaceb et al. reported that circulating microvesicles from L-MLCK–/– mice exert a protective action against lipopolysaccharide-induced inflammatory effects both in vivo and in cultured aortic endothelial cells (Gaceb, A., Vergori, L., Martinez, M. C., and Andriantsitohaina, R. (2016) Activation of endothelial pro-resolving anti-inflammatory pathways by circulating microvesicles from non-muscular myosin light chain kinase-deficient mice, Front. Pharmacol., 7, 322; doi:10.3389/fphar.2016.00322).
Acknowledgements
This work was financially supported by the Russian Foundation for Basic Research (projects Nos. 16-04-01742 and 14-04-01813) and by the Russian Science Foundation (project No. 14-35-00026, section “Role of MLCK/KRP in Physiological Cellular Responses, subsection “Exocytosis”; section “Role of MLCK in Pathological Processes”, subsection “Insulin resistance and diabetes”).
REFERENCES
1.Cunningham, K. E., and Turner, J. R. (2012) Myosin
light chain kinase: pulling the strings of epithelial tight junction
function, Ann. N. Y. Acad. Sci., 1258, 34-42.
2.Herring, B. P., El-Mounayri, O., Gallagher, P. J.,
Yin, F., and Zhou, J. (2006) Regulation of myosin light chain kinase
and telokin expression in smooth muscle tissues, Am. J. Physiol.
Cell Physiol., 291, C817-827.
3.Kamm, K. E., and Stull, J. T. (2001) Dedicated
myosin light chain kinases with diverse cellular functions,
J. Biol. Chem., 276, 4527-4530.
4.Lukas, T. J., and Shirinsky, V. P. (2012) MYLK
(Myosin Light Chain Kinase), in Encyclopedia of Signaling
Molecules (Choi, S., ed.) Springer, New York, pp. 1160-1165.
5.Shen, K., Wang, T., and Garcia, J. G. (2012) MYLK
(myosin light chain kinase), Atlas Genet. Cytogenet. Oncol.
Haematol., 16, 901-908.
6.Gallagher, P. J., Herring, B. P., and Stull, J. T.
(1997) Myosin light chain kinases, J. Muscle Res. Cell Motil.,
18, 1-16.
7.Khapchaev, A. Y., Shirinsky, V. P., and Vorotnikov,
A. V. (2003) Structure, properties and regulation of the myosin light
chain kinase genetic locus protein products, Usp. Biol. Khim.,
43, 365-420.
8.Gao, Y., Ye, L. H., Kishi, H., Okagaki, T., Samizo,
K., Nakamura, A., and Kohama, K. (2001) Myosin light chain kinase as a
multifunctional regulatory protein of smooth muscle contraction,
IUBMB Life, 51, 337-344.
9.Takashima, S. (2009) Phosphorylation of myosin
regulatory light chain by myosin light chain kinase, and muscle
contraction, Circ. J., 73, 208-213.
10.Hong, F., Haldeman, B. D., Jackson, D., Carter,
M., Baker, J. E., and Cremo, C. R. (2011) Biochemistry of smooth muscle
myosin light chain kinase, Arch. Biochem. Biophys., 510,
135-146.
11.Dabrowska, R., Sherry, J. M., Aromatorio, D. K.,
and Hartshorne, D. J. (1978) Modulator protein as a component of the
myosin light chain kinase from chicken gizzard, Biochemistry,
17, 253-258.
12.Pires, E., Perry, S. V., and Thomas, M. A. (1974)
Myosin light-chain kinase, a new enzyme from striated muscle, FEBS
Lett., 41, 292-296.
13.Chan, J. Y., Takeda, M., Briggs, L. E., Graham,
M. L., Lu, J. T., Horikoshi, N., Weinberg, E. O., Aoki, H., Sato, N.,
Chien, K. R., and Kasahara, H. (2008) Identification of
cardiac-specific myosin light chain kinase, Circ. Res.,
102, 571-580.
14.Roush, C. L., Kennelly, P. J., Glaccum, M. B.,
Helfman, D. M., Scott, J. D., and Krebs, E. G. (1988) Isolation of the
cDNA encoding rat skeletal muscle myosin light chain kinase. Sequence
and tissue distribution, J. Biol. Chem., 263,
10510-10516.
15.Birukov, K. G., Schavocky, J. P., Shirinsky, V.
P., Chibalina, M. V., Van Eldik, L. J., and Watterson, D. M. (1998)
Organization of the genetic locus for chicken myosin light chain kinase
is complex: multiple proteins are encoded and exhibit differential
expression and localization, J. Cell. Biochem., 70,
402-413.
16.Potier, M. C., Chelot, E., Pekarsky, Y.,
Gardiner, K., Rossier, J., and Turnell, W. G. (1995) The human myosin
light chain kinase (MLCK) from hippocampus: cloning, sequencing,
expression, and localization to 3qcen-q21, Genomics, 29,
562-570.
17.Hornbeck, P. V., Zhang, B., Murray, B.,
Kornhauser, J. M., Latham, V., and Skrzypek, E. (2015) PhosphoSitePlus,
2014: mutations, PTMs and recalibrations, Nucleic Acids Res.,
43, D512-520.
18.Watterson, D. M., Collinge, M., Lukas, T. J., Van
Eldik, L. J., Birukov, K. G., Stepanova, O. V., and Shirinsky, V. P.
(1995) Multiple gene products are produced from a novel protein kinase
transcription region, FEBS Lett., 373, 217-220.
19.Ito, M., Dabrowska, R., Guerriero, V., Jr., and
Hartshorne, D. J. (1989) Identification in turkey gizzard of an acidic
protein related to the C-terminal portion of smooth muscle myosin light
chain kinase, J. Biol. Chem., 264, 13971-13974.
20.Ye, D., and Ma, T. Y. (2008) Cellular and
molecular mechanisms that mediate basal and tumour necrosis
factor-alpha-induced regulation of myosin light chain kinase gene
activity, J. Cell. Mol. Med., 12, 1331-1346.
21.Shimizu, Y., Camp, S. M., Sun, X., Zhou, T.,
Wang, T., and Garcia, J. G. (2015) Sp1-mediated nonmuscle myosin light
chain kinase expression and enhanced activity in vascular endothelial
growth factor-induced vascular permeability, Pulm. Circ.,
5, 707-715.
22.Chang, A. N., Mahajan, P., Knapp, S., Barton, H.,
Sweeney, H. L., Kamm, K. E., and Stull, J. T. (2016) Cardiac myosin
light chain is phosphorylated by Ca2+/calmodulin-dependent
and -independent kinase activities, Proc. Natl. Acad. Sci. USA,
113, E3824-3833.
23.Guerriero, V., Jr., Russo, M. A., Olson, N. J.,
Putkey, J. A., and Means, A. R. (1986) Domain organization of chicken
gizzard myosin light chain kinase deduced from a cloned cDNA,
Biochemistry, 25, 8372-8381.
24.Garcia, J. G., Lazar, V., Gilbert-McClain, L. I.,
Gallagher, P. J., and Verin, A. D. (1997) Myosin light chain kinase in
endothelium: molecular cloning and regulation, Am. J. Respir. Cell
Mol. Biol., 16, 489-494.
25.Yin, F., Hoggatt, A. M., Zhou, J., and Herring,
B. P. (2006) 130-kDa smooth muscle myosin light chain kinase is
transcribed from a CArG-dependent, internal promoter within the mouse
mylk gene, Am. J. Physiol. Cell Physiol., 290,
C1599-1609.
26.Collinge, M., Matrisian, P. E., Zimmer, W. E.,
Shattuck, R. L., Lukas, T. J., Van Eldik, L. J., and Watterson, D. M.
(1992) Structure and expression of a calcium-binding protein gene
contained within a calmodulin-regulated protein kinase gene, Mol.
Cell. Biol., 12, 2359-2371.
27.Gallagher, P. J., and Herring, B. P. (1991) The
carboxyl terminus of the smooth muscle myosin light chain kinase is
expressed as an independent protein, telokin, J. Biol. Chem.,
266, 23945-23952.
28.Lazar, V., and Garcia, J. G. (1999) A single
human myosin light chain kinase gene (MLCK; MYLK), Genomics,
57, 256-267.
29.Hoggatt, A. M., Simon, G. M., and Herring, B. P.
(2002) Cell-specific regulatory modules control expression of genes in
vascular and visceral smooth muscle tissues, Circ. Res.,
91, 1151-1159.
30.Herring, B. P., and Smith, A. F. (1997) Telokin
expression in A10 smooth muscle cells requires serum response factor,
Am. J. Physiol., 272, C1394-1404.
31.Khromov, A. S., Wang, H., Choudhury, N.,
McDuffie, M., Herring, B. P., Nakamoto, R., Owens, G. K., Somlyo, A.
P., and Somlyo, A. V. (2006) Smooth muscle of telokin-deficient mice
exhibits increased sensitivity to Ca2+ and decreased
cGMP-induced relaxation, Proc. Natl. Acad. Sci. USA, 103,
2440-2445.
32.Zhou, J., and Herring, B. P. (2005) Mechanisms
responsible for the promoter-specific effects of myocardin, J. Biol.
Chem., 280, 10861-10869.
33.El-Mounayri, O., Triplett, J. W., Yates, C. W.,
and Herring, B. P. (2005) Regulation of smooth muscle-specific gene
expression by homeodomain proteins, Hoxa10 and Hoxb8, J. Biol.
Chem., 280, 25854-25863.
34.Zhou, J., Hoggatt, A. M., and Herring, B. P.
(2004) Activation of the smooth muscle-specific telokin gene by
thyrotroph embryonic factor (TEF), J. Biol. Chem., 279,
15929-15937.
35.Hoggatt, A. M., Kim, J. R., Ustiyan, V., Ren, X.,
Kalin, T. V., Kalinichenko, V. V., and Herring, B. P. (2013) The
transcription factor Foxf1 binds to serum response factor and myocardin
to regulate gene transcription in visceral smooth muscle cells, J.
Biol. Chem., 288, 28477-28487.
36.Hoggatt, A. M., Kriegel, A. M., Smith, A. F., and
Herring, B. P. (2000) Hepatocyte nuclear factor-3 homologue 1 (HFH-1)
represses transcription of smooth muscle-specific genes, J. Biol.
Chem., 275, 31162-31170.
37.Yin, F., and Herring, B. P. (2005) GATA-6 can act
as a positive or negative regulator of smooth muscle-specific gene
expression, J. Biol. Chem., 280, 4745-4752.
38.Zhou, J., Hu, G., and Herring, B. P. (2005)
Smooth muscle-specific genes are differentially sensitive to inhibition
by Elk-1, Mol. Cell. Biol., 25, 9874-9885.
39.Watterson, D. M., Schavocky, J. P., Guo, L.,
Weiss, C., Chlenski, A., Shirinsky, V. P., Van Eldik, L. J., and
Haiech, J. (1999) Analysis of the kinase-related protein gene found at
human chromosome 3q21 in a multi-gene cluster: organization,
expression, alternative splicing, and polymorphic marker, J. Cell.
Biochem., 75, 481-491.
40.Zhou, J., Blue, E. K., Hu, G., and Herring, B. P.
(2008) Thymine DNA glycosylase represses myocardin-induced smooth
muscle cell differentiation by competing with serum response factor for
myocardin binding, J. Biol. Chem., 283, 35383-35392.
41.Zhou, J., Hu, G., and Wang, X. (2010) Repression
of smooth muscle differentiation by a novel high mobility group
box-containing protein, HMG2L1, J. Biol. Chem., 285,
23177-23185.
42.Liu, F., Wang, X., Hu, G., Wang, Y., and Zhou, J.
(2014) The transcription factor TEAD1 represses smooth muscle-specific
gene expression by abolishing myocardin function, J. Biol.
Chem., 289, 3308-3316.
43.Han, Y. J., Hu, W. Y., Chernaya, O., Antic, N.,
Gu, L., Gupta, M., Piano, M., and De Lanerolle, P. (2006) Increased
myosin light chain kinase expression in hypertension: regulation by
serum response factor via an insertion mutation in the promoter,
Mol. Biol. Cell, 17, 4039-4050.
44.Chen, M., Zhang, W., Lu, X., Hoggatt, A. M.,
Gunst, S. J., Kassab, G. S., Tune, J. D., and Herring, B. P. (2013)
Regulation of 130-kDa smooth muscle myosin light chain kinase
expression by an intronic CArG element, J. Biol. Chem.,
288, 34647-34657.
45.Basu, S., Srinivasan, D. K., Yang, K., Raina, H.,
Banerjee, S., Zhang, R., Fisher, S. A., and Proweller, A. (2013) Notch
transcriptional control of vascular smooth muscle regulatory gene
expression and function, J. Biol. Chem., 288,
11191-11202.
46.Basu, S., and Proweller, A. (2016) Autoregulatory
control of smooth muscle myosin light chain kinase promoter by notch
signaling, J. Biol. Chem., 291, 2988-2999.
47.Graham, W. V., Wang, F., Clayburgh, D. R., Cheng,
J. X., Yoon, B., Wang, Y., Lin, A., and Turner, J. R. (2006) Tumor
necrosis factor-induced long myosin light chain kinase transcription is
regulated by differentiation-dependent signaling events.
Characterization of the human long myosin light chain kinase promoter,
J. Biol. Chem., 281, 26205-26215.
48.Wang, F., Graham, W. V., Wang, Y., Witkowski, E.
D., Schwarz, B. T., and Turner, J. R. (2005) Interferon-gamma and tumor
necrosis factor-alpha synergize to induce intestinal epithelial barrier
dysfunction by up-regulating myosin light chain kinase expression,
Am. J. Pathol., 166, 409-419.
49.Ye, D., Ma, I., and Ma, T. Y. (2006) Molecular
mechanism of tumor necrosis factor-alpha modulation of intestinal
epithelial tight junction barrier, Am. J. Physiol. Gastrointest.
Liver Physiol., 290, G496-504.
50.Al-Sadi, R., Guo, S., Dokladny, K., Smith, M. A.,
Ye, D., Kaza, A., Watterson, D. M., and Ma, T. Y. (2012) Mechanism of
interleukin-1beta induced-increase in mouse intestinal permeability
in vivo, J. Interferon Cytokine Res., 32,
474-484.
51.Al-Sadi, R., Guo, S., Ye, D., Rawat, M., and Ma,
T. Y. (2016) TNF-alpha modulation of intestinal tight junction
permeability is mediated by NIK/IKK-alpha axis activation of the
canonical NF-kappaB pathway, Am. J. Pathol., 186,
1151-1165.
52.Al-Sadi, R., Ye, D., Said, H. M., and Ma, T. Y.
(2011) Cellular and molecular mechanism of interleukin-1beta modulation
of Caco-2 intestinal epithelial tight junction barrier, J. Cell.
Mol. Med., 15, 970-982.
53.Al-Sadi, R., Guo, S., Ye, D., Dokladny, K.,
Alhmoud, T., Ereifej, L., Said, H. M., and Ma, T. Y. (2013) Mechanism
of IL-1beta modulation of intestinal epithelial barrier involves p38
kinase and activating transcription factor-2 activation, J.
Immunol., 190, 6596-6606.
54.Cao, M., Wang, P., Sun, C., He, W., and Wang, F.
(2013) Amelioration of IFN-gamma and TNF-alpha-induced intestinal
epithelial barrier dysfunction by berberine via suppression of MLCK-MLC
phosphorylation signaling pathway, PLoS One, 8,
e61944.
55.Qi, H., Wang, P., Liu, C., Li, M., Wang, S.,
Huang, Y., and Wang, F. (2011) Involvement of HIF-1alpha in
MLCK-dependent endothelial barrier dysfunction in hypoxia, Cell.
Physiol. Biochem., 27, 251-262.
56.Leveille, N., Fournier, A., and Labrie, C. (2009)
Androgens down-regulate myosin light chain kinase in human prostate
cancer cells, J. Steroid Biochem. Mol. Biol., 114,
174-179.
57.Adyshev, D. M., Moldobaeva, N., Mapes, B.,
Elangovan, V., and Garcia, J. G. (2013) MicroRNA regulation of
nonmuscle myosin light chain kinase expression in human lung
endothelium, Am. J. Respir. Cell Mol. Biol., 49,
58-66.
58.Weber, M., Kim, S., Patterson, N., Rooney, K.,
and Searles, C. D. (2014) MiRNA-155 targets myosin light chain kinase
and modulates actin cytoskeleton organization in endothelial cells,
Am. J. Physiol. Heart Circ. Physiol., 306,
H1192-1203.
59.Heidersbach, A., Saxby, C., Carver-Moore, K.,
Huang, Y., Ang, Y. S., De Jong, P. J., Ivey, K. N., and Srivastava, D.
(2013) microRNA-1 regulates sarcomere formation and suppresses smooth
muscle gene expression in the mammalian heart, Elife, 2,
e01323.
60.Zhu, H. Q., Wang, F., Dong, L. Y., Zhou, Q., and
Wang, Y. (2014) MicroRNA1 modulates oxLDL-induced hyperlipidemia by
down-regulating MLCK and ERK/p38 MAPK pathway, Life Sci.,
107, 21-26.
61.Sundararajan, V., Gengenbacher, N., Stemmler, M.
P., Kleemann, J. A., Brabletz, T., and Brabletz, S. (2015) The
ZEB1/miR-200c feedback loop regulates invasion via actin interacting
proteins MYLK and TKS5, Oncotarget, 6, 27083-27096.
62.Gao, L., Grant, A., Halder, I., Brower, R.,
Sevransky, J., Maloney, J. P., Moss, M., Shanholtz, C., Yates, C. R.,
Meduri, G. U., Shriver, M. D., Ingersoll, R., Scott, A. F., Beaty, T.
H., Moitra, J., Ma, S. F., Ye, S. Q., Barnes, K. C., and Garcia, J. G.
(2006) Novel polymorphisms in the myosin light chain kinase gene confer
risk for acute lung injury, Am. J. Respir. Cell Mol. Biol.,
34, 487-495.
63.Gao, L., Grant, A. V., Rafaels, N.,
Stockton-Porter, M., Watkins, T., Gao, P., Chi, P., Munoz, M., Watson,
H., Dunston, G., Togias, A., Hansel, N., Sevransky, J., Maloney, J. P.,
Moss, M., Shanholtz, C., Brower, R., Garcia, J. G., Grigoryev, D. N.,
Cheadle, C., Beaty, T. H., Mathias, R. A., and Barnes, K. C. (2007)
Polymorphisms in the myosin light chain kinase gene that confer risk of
severe sepsis are associated with a lower risk of asthma, J. Allergy
Clin. Immunol., 119, 1111-1118.
64.Han, Y. J., Ma, S. F., Wade, M. S., Flores, C.,
and Garcia, J. G. (2012) An intronic MYLK variant associated with
inflammatory lung disease regulates promoter activity of the smooth
muscle myosin light chain kinase isoform, J. Mol. Med. (Berl.),
90, 299-308.
65.Lee, S. O., Cheong, H. S., Park, B. L., Bae, J.
S., Sim, W. C., Chun, J. Y., Isbat, M., Uh, S. T., Kim, Y. H., Jang, A.
S., Park, C. S., and Shin, H. D. (2009) MYLK polymorphism associated
with blood eosinophil level among asthmatic patients in a Korean
population, Mol. Cells, 27, 175-181.
66.Wang, L., Guo, D. C., Cao, J., Gong, L., Kamm, K.
E., Regalado, E., Li, L., Shete, S., He, W. Q., Zhu, M. S., Offermanns,
S., Gilchrist, D., Elefteriades, J., Stull, J. T., and Milewicz, D. M.
(2010) Mutations in myosin light chain kinase cause familial aortic
dissections, Am. J. Hum. Genet., 87, 701-707.
67.Rusconi, F., Potier, M. C., Le Caer, J. P.,
Schmitter, J. M., and Rossier, J. (1997) Characterization of the
chicken telokin heterogeneity by time-of-flight mass spectrometry,
Biochemistry, 36, 11021-11026.
68.Strausberg, R. L., Feingold, E. A., Grouse, L.
H., Derge, J. G., Klausner, R. D., Collins, F. S., Wagner, L., Shenmen,
C. M., Schuler, G. D., Altschul, S. F., Zeeberg, B., Buetow, K. H.,
Schaefer, C. F., Bhat, N. K., Hopkins, R. F., Jordan, H., Moore, T.,
Max, S. I., Wang, J., Hsieh, F., Diatchenko, L., Marusina, K., Farmer,
A. A., Rubin, G. M., Hong, L., Stapleton, M., Soares, M. B., Bonaldo,
M. F., Casavant, T. L., Scheetz, T. E., Brownstein, M. J., Usdin, T.
B., Toshiyuki, S., Carninci, P., Prange, C., Raha, S. S., Loquellano,
N. A., Peters, G. J., Abramson, R. D., Mullahy, S. J., Bosak, S. A.,
McEwan, P. J., McKernan, K. J., Malek, J. A., Gunaratne, P. H.,
Richards, S., Worley, K. C., Hale, S., Garcia, A. M., Gay, L. J.,
Hulyk, S. W., Villalon, D. K., Muzny, D. M., Sodergren, E. J., Lu, X.,
Gibbs, R. A., Fahey, J., Helton, E., Ketteman, M., Madan, A.,
Rodrigues, S., Sanchez, A., Whiting, M., Young, A. C., Shevchenko, Y.,
Bouffard, G. G., Blakesley, R. W., Touchman, J. W., Green, E. D.,
Dickson, M. C., Rodriguez, A. C., Grimwood, J., Schmutz, J., Myers, R.
M., Butterfield, Y. S., Krzywinski, M. I., Skalska, U., Smailus, D. E.,
Schnerch, A., Schein, J. E., Jones, S. J., and Marra, M. A. (2002)
Generation and initial analysis of more than 15,000 full-length human
and mouse cDNA sequences, Proc. Natl. Acad. Sci. USA, 99,
16899-16903.
69.Herring, B. P., and Smith, A. F. (1996) Telokin
expression is mediated by a smooth muscle cell-specific promoter,
Am. J. Physiol., 270, C1656-1665.
70.Herring, B. P., Dixon, S., and Gallagher, P. J.
(2000) Smooth muscle myosin light chain kinase expression in cardiac
and skeletal muscle, Am. J. Physiol. Cell Physiol., 279,
C1656-1664.
71.Gallagher, P. J., Garcia, J. G., and Herring, B.
P. (1995) Expression of a novel myosin light chain kinase in embryonic
tissues and cultured cells, J. Biol. Chem., 270,
29090-29095.
72.Fisher, S. A., and Ikebe, M. (1995) Developmental
and tissue distribution of expression of nonmuscle and smooth muscle
isoforms of myosin light chain kinase, Biochem. Biophys. Res.
Commun., 217, 696-703.
73.Blue, E. K., Goeckeler, Z. M., Jin, Y., Hou, L.,
Dixon, S. A., Herring, B. P., Wysolmerski, R. B., and Gallagher, P. J.
(2002) 220- and 130-kDa MLCKs have distinct tissue distributions and
intracellular localization patterns, Am. J. Physiol. Cell
Physiol., 282, C451-460.
74.Kudryashov, D. S., Chibalina, M. V., Birukov, K.
G., Lukas, T. J., Sellers, J. R., Van Eldik, L. J., Watterson, D. M.,
and Shirinsky, V. P. (1999) Unique sequence of a high molecular weight
myosin light chain kinase is involved in interaction with actin
cytoskeleton, FEBS Lett., 463, 67-71.
75.Krymsky, M. A., Kudryashov, D. S., Shirinsky, V.
P., Lukas, T. J., Watterson, D. M., and Vorotnikov, A. V. (2001)
Phosphorylation of kinase-related protein (telokin) in tonic and phasic
smooth muscles, J. Muscle Res. Cell Motil., 22,
425-437.
76.Xu, J., Gao, X. P., Ramchandran, R., Zhao, Y. Y.,
Vogel, S. M., and Malik, A. B. (2008) Nonmuscle myosin light-chain
kinase mediates neutrophil transmigration in sepsis-induced lung
inflammation by activating beta2 integrins, Nat. Immunol.,
9, 880-886.
77.Sun, C., Wu, M. H., and Yuan, S. Y. (2011)
Nonmuscle myosin light-chain kinase deficiency attenuates
atherosclerosis in apolipoprotein E-deficient mice via reduced
endothelial barrier dysfunction and monocyte migration,
Circulation, 124, 48-57.
78.Clayburgh, D. R., Rosen, S., Witkowski, E. D.,
Wang, F., Blair, S., Dudek, S., Garcia, J. G., Alverdy, J. C., and
Turner, J. R. (2004) A differentiation-dependent splice variant of
myosin light chain kinase, MLCK1, regulates epithelial tight junction
permeability, J. Biol. Chem., 279, 55506-55513.
79.Verin, A. D., Lazar, V., Torry, R. J., Labarrere,
C. A., Patterson, C. E., and Garcia, J. G. (1998) Expression of a novel
high molecular-weight myosin light chain kinase in endothelium, Am.
J. Respir. Cell Mol. Biol., 19, 758-766.
80.Hathaway, D. R., and Adelstein, R. S. (1979)
Human platelet myosin light chain kinase requires the calcium-binding
protein calmodulin for activity, Proc. Natl. Acad. Sci. USA,
76, 1653-1657.
81.Wang, H. H., Nakamura, A., Matsumoto, A.,
Yoshiyama, S., Qin, X., Ye, L. H., Xie, C., Zhang, Y., Gao, Y.,
Ishikawa, R., and Kohama, K. (2009) Nonkinase activity of MLCK in
elongated filopodia formation and chemotaxis of vascular smooth muscle
cells toward sphingosylphosphorylcholine, Am. J. Physiol. Heart
Circ. Physiol., 296, H1683-1693.
82.Poperechnaya, A., Varlamova, O., Lin, P. J.,
Stull, J. T., and Bresnick, A. R. (2000) Localization and activity of
myosin light chain kinase isoforms during the cell cycle, J. Cell
Biol., 151, 697-708.
83.Woody, S., Stall, R., Ramos, J., and Patel, Y. M.
(2013) Regulation of myosin light chain kinase during
insulin-stimulated glucose uptake in 3T3-L1 adipocytes, PLoS
One, 8, e77248.
84.Yu, H. J., Serebryannyy, L. A., Fry, M., Greene,
M., Chernaya, O., Hu, W. Y., Chew, T. L., Mahmud, N., Kadkol, S. S.,
Glover, S., Prins, G., Strakova, Z., and De Lanerolle, P. (2013) Tumor
stiffness is unrelated to myosin light chain phosphorylation in cancer
cells, PLoS One, 8, e79776.
85.Kim, D. Y., and Helfman, D. M. (2016) Loss of
MLCK leads to disruption of cell–cell adhesion and invasive
behavior of breast epithelial cells via increased expression of EGFR
and ERK/JNK signaling, Oncogene, 35, 4495-4508.
86.Minamiya, Y., Nakagawa, T., Saito, H., Matsuzaki,
I., Taguchi, K., Ito, M., and Ogawa, J. (2005) Increased expression of
myosin light chain kinase mRNA is related to metastasis in non-small
cell lung cancer, Tumour Biol., 26, 153-157.
87.Kennelly, P. J., Leng, J., and Marchand, P.
(1992) The MgATP-binding site on chicken gizzard myosin light chain
kinase remains open and functionally competent during the
calmodulin-dependent activation-inactivation cycle of the enzyme,
Biochemistry, 31, 5394-5399.
88.Herring, B. P., Gallagher, P. J., and Stull, J.
T. (1992) Substrate specificity of myosin light chain kinases, J.
Biol. Chem., 267, 25945-25950.
89.Sobieszek, A. (1991) Regulation of smooth-muscle
myosin-light-chain kinase. Steady-state kinetic studies of the reaction
mechanism, Eur. J. Biochem., 199, 735-743.
90.Sobieszek, A., Andruchov, O. Y., and Nieznanski,
K. (1997) Kinase-related protein (telokin) is phosphorylated by
smooth-muscle myosin light-chain kinase and modulates the kinase
activity, Biochem. J., 328 (Pt. 2), 425-430.
91.Davis, H. W., Crimmins, D. L., Thoma, R. S., and
Garcia, J. G. (1996) Phosphorylation of calmodulin in the first
calcium-binding pocket by myosin light chain kinase, Arch. Biochem.
Biophys., 332, 101-109.
92.Kemp, B. E., and Pearson, R. B. (1985) Spatial
requirements for location of basic residues in peptide substrates for
smooth muscle myosin light chain kinase, J. Biol. Chem.,
260, 3355-3359.
93.Persechini, A., and Hartshorne, D. J. (1983)
Ordered phosphorylation of the two 20,000 molecular weight light chains
of smooth muscle myosin, Biochemistry, 22, 470-476.
94.Ikebe, M., Hartshorne, D. J., and Elzinga, M.
(1986) Identification, phosphorylation, and dephosphorylation of a
second site for myosin light chain kinase on the 20,000-dalton light
chain of smooth muscle myosin, J. Biol. Chem., 261,
36-39.
95.Amano, M., Ito, M., Kimura, K., Fukata, Y.,
Chihara, K., Nakano, T., Matsuura, Y., and Kaibuchi, K. (1996)
Phosphorylation and activation of myosin by Rho-associated kinase
(Rho-kinase), J. Biol. Chem., 271, 20246-20249.
96.Murata-Hori, M., Suizu, F., Iwasaki, T., Kikuchi,
A., and Hosoya, H. (1999) ZIP kinase identified as a novel myosin
regulatory light chain kinase in HeLa cells, FEBS Lett.,
451, 81-84.
97.Uehara, R., Hosoya, H., and Mabuchi, I. (2008)
In vivo phosphorylation of regulatory light chain of myosin II
in sea urchin eggs and its role in controlling myosin localization and
function during cytokinesis, Cell Motil. Cytoskelet., 65,
100-115.
98.Puetz, S., Schroeter, M. M., Piechura, H.,
Reimann, L., Hunger, M. S., Lubomirov, L. T., Metzler, D., Warscheid,
B., and Pfitzer, G. (2012) New insights into myosin phosphorylation
during cyclic nucleotide-mediated smooth muscle relaxation, J.
Muscle Res. Cell Motil., 33, 471-483.
99.Pearson, R. B., Wettenhall, R. E., Means, A. R.,
Hartshorne, D. J., and Kemp, B. E. (1988) Autoregulation of enzymes by
pseudosubstrate prototopes: myosin light chain kinase, Science,
241, 970-973.
100.Tanaka, M., Ikebe, R., Matsuura, M., and Ikebe,
M. (1995) Pseudosubstrate sequence may not be critical for
autoinhibition of smooth muscle myosin light chain kinase, EMBO
J., 14, 2839-2846.
101.Lukas, T. J., Mirzoeva, S., Slomczynska, U.,
and Watterson, D. M. (1999) Identification of novel classes of protein
kinase inhibitors using combinatorial peptide chemistry based on
functional genomics knowledge, J. Med. Chem., 42,
910-919.
102.Clayburgh, D. R., Barrett, T. A., Tang, Y.,
Meddings, J. B., Van Eldik, L. J., Watterson, D. M., Clarke, L. L.,
Mrsny, R. J., and Turner, J. R. (2005) Epithelial myosin light chain
kinase-dependent barrier dysfunction mediates T cell activation-induced
diarrhea in vivo, J. Clin. Invest., 115,
2702-2715.
103.Owens, S. E., Graham, W. V., Siccardi, D.,
Turner, J. R., and Mrsny, R. J. (2005) A strategy to identify stable
membrane-permeant peptide inhibitors of myosin light chain kinase,
Pharm. Res., 22, 703-709.
104.Khapchaev, A. Y., Kazakova, O. A., Samsonov, M.
V., Sidorova, M. V., Bushuev, V. N., Vilitkevich, E. L., Az’muko,
A. A., Molokoedov, A. S., Bespalova, Z. D., and Shirinsky, V. P. (2016)
Design of peptidase-resistant peptide inhibitors of myosin light chain
kinase, J. Peptide Sci., in press.
105.Mirzapoiazova, T., Moitra, J., Moreno-Vinasco,
L., Sammani, S., Turner, J. R., Chiang, E. T., Evenoski, C., Wang, T.,
Singleton, P. A., Huang, Y., Lussier, Y. A., Watterson, D. M., Dudek,
S. M., and Garcia, J. G. (2011) Non-muscle myosin light chain kinase
isoform is a viable molecular target in acute inflammatory lung injury,
Am. J. Respir. Cell Mol. Biol., 44, 40-52.
106.Johnson, J. D., Snyder, C., Walsh, M., and
Flynn, M. (1996) Effects of myosin light chain kinase and peptides on
Ca2+ exchange with the N- and C-terminal Ca2+
binding sites of calmodulin, J. Biol. Chem., 271,
761-767.
107.Krueger, J. K., Zhi, G., Stull, J. T., and
Trewhella, J. (1998) Neutron-scattering studies reveal further details
of the Ca2+/calmodulin-dependent activation mechanism of
myosin light chain kinase, Biochemistry, 37,
13997-14004.
108.Pearson, R. B., Misconi, L. Y., and Kemp, B. E.
(1986) Smooth muscle myosin kinase requires residues on the
COOH-terminal side of the phosphorylation site. Peptide inhibitors,
J. Biol. Chem., 261, 25-27.
109.Silver, D. L., Vorotnikov, A. V., Watterson, D.
M., Shirinsky, V. P., and Sellers, J. R. (1997) Sites of interaction
between kinase-related protein and smooth muscle myosin, J. Biol.
Chem., 272, 25353-25359.
110.Shirinsky, V. P., Vorotnikov, A. V., Birukov,
K. G., Nanaev, A. K., Collinge, M., Lukas, T. J., Sellers, J. R., and
Watterson, D. M. (1993) A kinase-related protein stabilizes
unphosphorylated smooth muscle myosin minifilaments in the presence of
ATP, J. Biol. Chem., 268, 16578-16583.
111.Nieznanski, K., and Sobieszek, A. (1997)
Telokin (kinase-related protein) modulates the oligomeric state of
smooth-muscle myosin light-chain kinase and its interaction with myosin
filaments, Biochem. J., 322 (Pt. 1), 65-71.
112.Shcherbakova, O. V., Serebryanaya, D. V.,
Postnikov, A. B., Schroeter, M. M., Zittrich, S., Noegel, A. A.,
Shirinsky, V. P., Vorotnikov, A. V., and Pfitzer, G. (2010)
Kinase-related protein/telokin inhibits Ca2+-independent
contraction in Triton-skinned guinea pig taenia coli, Biochem.
J., 429, 291-302.
113.Minajeva, A., Neagoe, C., Kulke, M., and Linke,
W. A. (2002) Titin-based contribution to shortening velocity of rabbit
skeletal myofibrils, J. Physiol., 540, 177-188.
114.Smith, L., Su, X., Lin, P., Zhi, G., and Stull,
J. T. (1999) Identification of a novel actin binding motif in smooth
muscle myosin light chain kinase, J. Biol. Chem., 274,
29433-29438.
115.Smith, L., Parizi-Robinson, M., Zhu, M. S.,
Zhi, G., Fukui, R., Kamm, K. E., and Stull, J. T. (2002) Properties of
long myosin light chain kinase binding to F-actin in vitro and
in vivo, J. Biol. Chem., 277, 35597-35604.
116.Hong, F., Brizendine, R. K., Carter, M. S.,
Alcala, D. B., Brown, A. E., Chattin, A. M., Haldeman, B. D., Walsh, M.
P., Facemyer, K. C., Baker, J. E., and Cremo, C. R. (2015) Diffusion of
myosin light chain kinase on actin: a mechanism to enhance myosin
phosphorylation rates in smooth muscle, J. Gen. Physiol.,
146, 267-280.
117.Hatch, V., Zhi, G., Smith, L., Stull, J. T.,
Craig, R., and Lehman, W. (2001) Myosin light chain kinase binding to a
unique site on F-actin revealed by three-dimensional image
reconstruction, J. Cell Biol., 154, 611-617.
118.Ye, L. H., Hayakawa, K., Kishi, H., Imamura,
M., Nakamura, A., Okagaki, T., Takagi, T., Iwata, A., Tanaka, T., and
Kohama, K. (1997) The structure and function of the actin-binding
domain of myosin light chain kinase of smooth muscle, J. Biol.
Chem., 272, 32182-32189.
119.Lin, P., Luby-Phelps, K., and Stull, J. T.
(1997) Binding of myosin light chain kinase to cellular actin-myosin
filaments, J. Biol. Chem., 272, 7412-7420.
120.Vilitkevich, E. L., Khapchaev, A. Y.,
Kudryashov, D. S., Nikashin, A. V., Schavocky, J. P., Lukas, T. J.,
Watterson, D. M., and Shirinsky, V. P. (2015) Phosphorylation regulates
interaction of 210-kDa myosin light chain kinase N-terminal domain with
actin cytoskeleton, Biochemistry (Moscow), 80,
1288-1297.
121.Yang, C. X., Chen, H. Q., Chen, C., Yu, W. P.,
Zhang, W. C., Peng, Y. J., He, W. Q., Wei, D. M., Gao, X., and Zhu, M.
S. (2006) Microfilament-binding properties of N-terminal extension of
the isoform of smooth muscle long myosin light chain kinase, Cell
Res., 16, 367-376.
122.Kudryashov, D. S., Stepanova, O. V.,
Vilitkevich, E. L., Nikonenko, T. A., Nadezhdina, E. S., Shanina, N.
A., Lukas, T. J., Van Eldik, L. J., Watterson, D. M., and Shirinsky, V.
P. (2004) Myosin light chain kinase (210 kDa) is a potential
cytoskeleton integrator through its unique N-terminal domain, Exp.
Cell Res., 298, 407-417.
123.Takizawa, N., Ikebe, R., Ikebe, M., and Luna,
E. J. (2007) Supervillin slows cell spreading by facilitating myosin II
activation at the cell periphery, J. Cell Sci., 120,
3792-3803.
124.Garcia, J. G., Verin, A. D., Schaphorst, K.,
Siddiqui, R., Patterson, C. E., Csortos, C., and Natarajan, V. (1999)
Regulation of endothelial cell myosin light chain kinase by Rho,
cortactin, and p60(src), Am. J. Physiol., 276,
L989-998.
125.Dudek, S. M., Birukov, K. G., Zhan, X., and
Garcia, J. G. (2002) Novel interaction of cortactin with endothelial
cell myosin light chain kinase, Biochem. Biophys. Res. Commun.,
298, 511-519.
126.Dudek, S. M., Jacobson, J. R., Chiang, E. T.,
Birukov, K. G., Wang, P., Zhan, X., and Garcia, J. G. (2004) Pulmonary
endothelial cell barrier enhancement by sphingosine 1-phosphate: roles
for cortactin and myosin light chain kinase, J. Biol. Chem.,
279, 24692-24700.
127.Wadgaonkar, R., Dudek, S. M., Zaiman, A. L.,
Linz-McGillem, L., Verin, A. D., Nurmukhambetova, S., Romer, L. H., and
Garcia, J. G. (2005) Intracellular interaction of myosin light chain
kinase with macrophage migration inhibition factor (MIF) in
endothelium, J. Cell. Biochem., 95, 849-858.
128.Shen, K., Ramirez, B., Mapes, B., Shen, G. R.,
Gokhale, V., Brown, M. E., Santarsiero, B., Ishii, Y., Dudek, S. M.,
Wang, T., and Garcia, J. G. (2015) Structure-function analysis of the
non-muscle myosin light chain kinase (nmMLCK) isoform by NMR
spectroscopy and molecular modeling: influence of MYLK variants,
PLoS One, 10, e0130515.
129.Sobieszek, A., Borkowski, J., and Babiychuk, V.
S. (1997) Purification and characterization of a smooth muscle myosin
light chain kinase–phosphatase complex, J. Biol. Chem.,
272, 7034-7041.
130.Komatsu, S., Miyazaki, K., Tuft, R. A., and
Ikebe, M. (2002) Translocation of telokin by cGMP signaling in smooth
muscle cells, Am. J. Physiol. Cell Physiol., 283,
C752-761.
131.Khromov, A. S., Momotani, K., Jin, L.,
Artamonov, M. V., Shannon, J., Eto, M., and Somlyo, A. V. (2012)
Molecular mechanism of telokin-mediated disinhibition of myosin light
chain phosphatase and cAMP/cGMP-induced relaxation of gastrointestinal
smooth muscle, J. Biol. Chem., 287, 20975-20985.
132.Faux, M. C., Mitchelhill, K. I., Katsis, F.,
Wettenhall, R. E., and Kemp, B. E. (1993) Chicken smooth muscle myosin
light chain kinase is acetylated on its NH2-terminal
methionine, Mol. Cell. Biochem., 127-128, 81-91.
133.Shin, D. H., Chun, Y. S., Lee, K. H., Shin, H.
W., and Park, J. W. (2009) Arrest defective-1 controls tumor cell
behavior by acetylating myosin light chain kinase, PLoS One,
4, e7451.
134.Conti, M. A., and Adelstein, R. S. (1981) The
relationship between calmodulin binding and phosphorylation of smooth
muscle myosin kinase by the catalytic subunit of 3′:5′
cAMP-dependent protein kinase, J. Biol. Chem., 256,
3178-3181.
135.Lukas, T. J., Mirzoeva, S., and Watterson, D.
M. (1998) Calmodulin-regulated protein kinases, in Calmodulin and
Signal Transduction (Van Eldik, L. J., and Watterson, D. M., eds.)
Academic Press, New York, pp. 66-168.
136.Nishikawa, M., Shirakawa, S., and Adelstein, R.
S. (1985) Phosphorylation of smooth muscle myosin light chain kinase by
protein kinase C. Comparative study of the phosphorylated sites, J.
Biol. Chem., 260, 8978-8983.
137.Goeckeler, Z. M., Masaracchia, R. A., Zeng, Q.,
Chew, T. L., Gallagher, P., and Wysolmerski, R. B. (2000)
Phosphorylation of myosin light chain kinase by p21-activated kinase
PAK2, J. Biol. Chem., 275, 18366-18374.
138.Birukov, K. G., Csortos, C., Marzilli, L.,
Dudek, S., Ma, S. F., Bresnick, A. R., Verin, A. D., Cotter, R. J., and
Garcia, J. G. (2001) Differential regulation of alternatively spliced
endothelial cell myosin light chain kinase isoforms by p60(Src), J.
Biol. Chem., 276, 8567-8573.
139.Dudek, S. M., Chiang, E. T., Camp, S. M., Guo,
Y., Zhao, J., Brown, M. E., Singleton, P. A., Wang, L., Desai, A.,
Arce, F. T., Lal, R., Van Eyk, J. E., Imam, S. Z., and Garcia, J. G.
(2010) Abl tyrosine kinase phosphorylates nonmuscle myosin light chain
kinase to regulate endothelial barrier function, Mol. Biol.
Cell, 21, 4042-4056.
140.Dulyaninova, N. G., and Bresnick, A. R. (2004)
The long myosin light chain kinase is differentially phosphorylated
during interphase and mitosis, Exp. Cell Res., 299,
303-314.
141.McManus, M. J., Boerner, J. L., Danielsen, A.
J., Wang, Z., Matsumura, F., and Maihle, N. J. (2000) An oncogenic
epidermal growth factor receptor signals via a p21-activated
kinase–caldesmon–myosin phosphotyrosine complex, J.
Biol. Chem., 275, 35328-35334.
142.Klemke, R. L., Cai, S., Giannini, A. L.,
Gallagher, P. J., De Lanerolle, P., and Cheresh, D. A. (1997)
Regulation of cell motility by mitogen-activated protein kinase, J.
Cell Biol., 137, 481-492.
143.Khapchaev, A. Y., Krymsky, M. A., Sidorova, M.
V., Bespalova Zh, D., Wang, C. L., Shirinsky, V. P., and Vorotnikov, A.
V. (2004) Novel phosphospecific antibodies for monitoring
phosphorylation of proteins encoded by the myosin light chain kinase
genetic locus, Biochemistry (Moscow), 69, 789-798.
144.MacDonald, J. A., Walker, L. A., Nakamoto, R.
K., Gorenne, I., Somlyo, A. V., Somlyo, A. P., and Haystead, T. A.
(2000) Phosphorylation of telokin by cyclic nucleotide kinases and the
identification of in vivo phosphorylation sites in smooth
muscle, FEBS Lett., 479, 83-88.
145.Wu, X., Haystead, T. A., Nakamoto, R. K.,
Somlyo, A. V., and Somlyo, A. P. (1998) Acceleration of myosin light
chain dephosphorylation and relaxation of smooth muscle by telokin.
Synergism with cyclic nucleotide-activated kinase, J. Biol.
Chem., 273, 11362-11369.
146.Webb, D. J., Donais, K., Whitmore, L. A.,
Thomas, S. M., Turner, C. E., Parsons, J. T., and Horwitz, A. F. (2004)
FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion
disassembly, Nat. Cell Biol., 6, 154-161.
147.Levinson, H., Moyer, K. E., Saggers, G. C., and
Ehrlich, H. P. (2004) Calmodulin-myosin light chain kinase inhibition
changes fibroblast-populated collagen lattice contraction, cell
migration, focal adhesion formation, and wound contraction, Wound
Repair Regen., 12, 505-511.
148.Hong, T., and Grabel, L. B. (2006) Migration of
F9 parietal endoderm cells is regulated by the ERK pathway, J. Cell.
Biochem., 97, 1339-1349.
149.Totsukawa, G., Wu, Y., Sasaki, Y., Hartshorne,
D. J., Yamakita, Y., Yamashiro, S., and Matsumura, F. (2004) Distinct
roles of MLCK and ROCK in the regulation of membrane protrusions and
focal adhesion dynamics during cell migration of fibroblasts, J.
Cell Biol., 164, 427-439.
150.Katoh, K., Kano, Y., and Ookawara, S. (2007)
Rho-kinase dependent organization of stress fibers and focal adhesions
in cultured fibroblasts, Genes Cells, 12, 623-638.
151.Smith, A., Bracke, M., Leitinger, B., Porter,
J. C., and Hogg, N. (2003) LFA-1-induced T cell migration on ICAM-1
involves regulation of MLCK-mediated attachment and ROCK-dependent
detachment, J. Cell Sci., 116, 3123-3133.
152.Yokomori, H., Yoshimura, K., Nagai, T.,
Fujimaki, K., Nomura, M., Hibi, T., Ishii, H., and Oda, M. (2004)
Sinusoidal endothelial fenestrae organization regulated by myosin light
chain kinase and Rho-kinase in cultured rat sinusoidal endothelial
cells, Hepatol. Res., 30, 169-174.
153.Lou, S. S., Diz-Munoz, A., Weiner, O. D.,
Fletcher, D. A., and Theriot, J. A. (2015) Myosin light chain kinase
regulates cell polarization independently of membrane tension or Rho
kinase, J. Cell Biol., 209, 275-288.
154.Chen, C., Tao, T., Wen, C., He, W. Q., Qiao, Y.
N., Gao, Y. Q., Chen, X., Wang, P., Chen, C. P., Zhao, W., Chen, H. Q.,
Ye, A. P., Peng, Y. J., and Zhu, M. S. (2014) Myosin light chain kinase
(MLCK) regulates cell migration in a myosin regulatory light chain
phosphorylation-independent mechanism, J. Biol. Chem.,
289, 28478-28488.
155.Bessard, A., Coutant, A., Rescan, C., Ezan, F.,
Fremin, C., Courselaud, B., Ilyin, G., and Baffet, G. (2006) An
MLCK-dependent window in late G1 controls S phase entry of
proliferating rodent hepatocytes via ERK-p70S6K pathway,
Hepatology, 44, 152-163.
156.Barkan, D., Kleinman, H., Simmons, J. L.,
Asmussen, H., Kamaraju, A. K., Hoenorhoff, M. J., Liu, Z. Y., Costes,
S. V., Cho, E. H., Lockett, S., Khanna, C., Chambers, A. F., and Green,
J. E. (2008) Inhibition of metastatic outgrowth from single dormant
tumor cells by targeting the cytoskeleton, Cancer Res.,
68, 6241-6250.
157.Zhou, X., Liu, Y., You, J., Zhang, H., Zhang,
X., and Ye, L. (2008) Myosin light-chain kinase contributes to the
proliferation and migration of breast cancer cells through cross-talk
with activated ERK1/2, Cancer Lett., 270, 312-327.
158.Zou, D. B., Wei, X., Hu, R. L., Yang, X. P.,
Zuo, L., Zhang, S. M., Zhu, H. Q., Zhou, Q., Gui, S. Y., and Wang, Y.
(2015) Melatonin inhibits the migration of colon cancer RKO cells by
down-regulating myosin light chain kinase expression through cross-talk
with p38 MAPK, Asian Pac. J. Cancer Prev., 16,
5835-5842.
159.Wang, H. H., Nakamura, A., Yoshiyama, S.,
Ishikawa, R., Cai, N., Ye, L. H., Takano-Ohmuro, H., and Kohama, K.
(2012) Down-regulation of myosin light chain kinase expression in
vascular smooth muscle cells accelerates cell proliferation:
requirement of its actin-binding domain for reversion to normal rates,
J. Pharmacol. Sci., 119, 91-96.
160.Dulyaninova, N. G., Patskovsky, Y. V., and
Bresnick, A. R. (2004) The N-terminus of the long MLCK induces a
disruption in normal spindle morphology and metaphase arrest, J.
Cell Sci., 117, 1481-1493.
161.Barfod, E. T., Moore, A. L., Van de Graaf, B.
G., and Lidofsky, S. D. (2011) Myosin light chain kinase and Src
control membrane dynamics in volume recovery from cell swelling,
Mol. Biol. Cell, 22, 634-650.
162.Li, L., Wu, X., Yue, H. Y., Zhu, Y. C., and Xu,
J. (2016) Myosin light chain kinase facilitates endocytosis of synaptic
vesicles at hippocampal boutons, J. Neurochem., 138,
60-73.
163.Yue, H. Y., and Xu, J. (2014) Myosin light
chain kinase accelerates vesicle endocytosis at the calyx of Held
synapse, J. Neurosci., 34, 295-304.
164.Kumakura, K., Sasaki, K., Sakurai, T.,
Ohara-Imaizumi, M., Misonou, H., Nakamura, S., Matsuda, Y., and
Nonomura, Y. (1994) Essential role of myosin light chain kinase in the
mechanism for MgATP-dependent priming of exocytosis in adrenal
chromaffin cells, J. Neurosci., 14, 7695-7703.
165.Arous, C., Rondas, D., and Halban, P. A. (2013)
Non-muscle myosin IIA is involved in focal adhesion and actin
remodelling controlling glucose-stimulated insulin secretion,
Diabetologia, 56, 792-802.
166.Getz, T. M., Dangelmaier, C. A., Jin, J.,
Daniel, J. L., and Kunapuli, S. P. (2010) Differential phosphorylation
of myosin light chain (Thr)18 and (Ser)19 and functional implications
in platelets, J. Thromb. Haemost., 8, 2283-2293.
167.Miklavc, P., Ehinger, K., Sultan, A., Felder,
T., Paul, P., Gottschalk, K. E., and Frick, M. (2015) Actin
depolymerisation and crosslinking join forces with myosin II to
contract actin coats on fused secretory vesicles, J. Cell Sci.,
128, 1193-1203.
168.Cai, S., Pestic-Dragovich, L., O’Donnell,
M. E., Wang, N., Ingber, D., Elson, E., and De Lanerolle, P. (1998)
Regulation of cytoskeletal mechanics and cell growth by myosin light
chain phosphorylation, Am. J. Physiol., 275,
C1349-1356.
169.Ohlmann, P., Tesse, A., Loichot, C., Ralay
Ranaivo, H., Roul, G., Philippe, C., Watterson, D. M., Haiech, J., and
Andriantsitohaina, R. (2005) Deletion of MLCK210 induces subtle changes
in vascular reactivity but does not affect cardiac function, Am. J.
Physiol. Heart Circ. Physiol., 289, H2342-2349.
170.Samsonov, M. V., Khalisov, M. M., Khapchaev, A.
Y., Penniyaynen, V. A., Ankudinov, A. V., Krylov, B. V., and Shirinsky,
V. P. (2016) The role of 210 kDa myosin light chain kinase and
RhoA-activated protein kinase in control of microvascular endothelial
cell stiffness, in Materials of Int. Symp. “Biological
Motility”, Pushchino, Russia, pp. 208-209.
171.Shirinsky, V. P., Kazakova, O. A., Samsonov, M.
V., Khalisov, M. M., Khapchaev, A. Y., Penniyaynen, V. A., Ankudinov,
A. V., and Krylov, B. V. (2016) Spatiotemporal activity profiling of
key myosin regulators in endothelial cells with regard to control of
cell stiffness and barrier dysfunction, in Experimental and
Computational Biomedicine: Russ. Conf. with Int. Participation in
Memory of Prof. Vladimir S. Markhasin, Ekaterinburg, p. 53.
172.Shirinsky, V. P. (2011) Molecular physiology of
endothelium and mechanisms of vascular permeability, Usp. Fiziol.
Nauk, 42, 18-32.
173.Mehta, D., and Malik, A. B. (2006) Signaling
mechanisms regulating endothelial permeability, Physiol. Rev.,
86, 279-367.
174.Wainwright, M. S., Rossi, J., Schavocky, J.,
Crawford, S., Steinhorn, D., Velentza, A. V., Zasadzki, M., Shirinsky,
V., Jia, Y., Haiech, J., Van Eldik, L. J., and Watterson, D. M. (2003)
Protein kinase involved in lung injury susceptibility: evidence from
enzyme isoform genetic knockout and in vivo inhibitor treatment,
Proc. Natl. Acad. Sci. USA, 100, 6233-6238.
175.Yu, Y., Lv, N., Lu, Z., Zheng, Y. Y., Zhang, W.
C., Chen, C., Peng, Y. J., He, W. Q., Meng, F. Q., Zhu, M. S., and
Chen, H. Q. (2012) Deletion of myosin light chain kinase in endothelial
cells has a minor effect on the lipopolysaccharide-induced increase in
microvascular endothelium permeability in mice, FEBS J.,
279, 1485-1494.
176.Moitra, J., Evenoski, C., Sammani, S.,
Wadgaonkar, R., Turner, J. R., Ma, S. F., and Garcia, J. G. (2008) A
transgenic mouse with vascular endothelial over-expression of the
non-muscle myosin light chain kinase-2 isoform is susceptible to
inflammatory lung injury: role of sexual dimorphism and age, Transl.
Res., 151, 141-153.
177.Marchiando, A. M., Shen, L., Graham, W. V.,
Weber, C. R., Schwarz, B. T., Austin, J. R., 2nd, Raleigh, D. R., Guan,
Y., Watson, A. J., Montrose, M. H., and Turner, J. R. (2010)
Caveolin-1-dependent occludin endocytosis is required for TNF-induced
tight junction regulation in vivo, J. Cell Biol.,
189, 111-126.
178.Wu, L. L., Peng, W. H., Kuo, W. T., Huang, C.
Y., Ni, Y. H., Lu, K. S., Turner, J. R., and Yu, L. C. (2014) Commensal
bacterial endocytosis in epithelial cells is dependent on myosin light
chain kinase-activated brush border fanning by interferon-gamma, Am.
J. Pathol., 184, 2260-2274.
179.Fazal, F., Bijli, K. M., Murrill, M., Leonard,
A., Minhajuddin, M., Anwar, K. N., Finkelstein, J. N., Watterson, D.
M., and Rahman, A. (2013) Critical role of non-muscle myosin light
chain kinase in thrombin-induced endothelial cell inflammation and lung
PMN infiltration, PLoS One, 8, e59965.
180.Tan, J., Wang, Y., Xia, Y., Zhang, N., Sun, X.,
Yu, T., and Lin, L. (2014) Melatonin protects the esophageal epithelial
barrier by suppressing the transcription, expression and activity of
myosin light chain kinase through ERK1/2 signal transduction, Cell.
Physiol. Biochem., 34, 2117-2127.
181.Reynoso, R., Perrin, R. M., Breslin, J. W.,
Daines, D. A., Watson, K. D., Watterson, D. M., Wu, M. H., and Yuan, S.
(2007) A role for long chain myosin light chain kinase (MLCK-210) in
microvascular hyperpermeability during severe burns, Shock,
28, 589-595.
182.Marchenko, A. V., Sidorova, M. V., Sekridova,
A. V., Bushuev, V. N., Lakomkin, V. L., Orlova, Ts. R., Stepanova, O.
V., Kapel’ko, V. I., Watterson, D. M., Van Eldik, L. J.,
Bespalova, Zh. D., and Shirinskii, V. P. (2009) Penetrating peptide
inhibitor of the myosin light chain kinase suppresses hyperpermeability
of vascular endothelium, Ross. Fiziol. Zh. im. I. M. Sechenova,
95, 507-515.
183.Stephens, N. L., Cheng, Z. Q., and Fust, A.
(2007) Sensitized airway smooth muscle plasticity and hyperreactivity:
a review, Can. J. Physiol. Pharmacol., 85, 679-685.
184.Azizi, P. M., Zyla, R. E., Guan, S., Wang, C.,
Liu, J., Bolz, S. S., Heit, B., Klip, A., and Lee, W. L. (2015)
Clathrin-dependent entry and vesicle-mediated exocytosis define insulin
transcytosis across microvascular endothelial cells, Mol. Biol.
Cell, 26, 740-750.
185.Jerdeva, G. V., Wu, K., Yarber, F. A., Rhodes,
C. J., Kalman, D., Schechter, J. E., and Hamm-Alvarez, S. F. (2005)
Actin and non-muscle myosin II facilitate apical exocytosis of tear
proteins in rabbit lacrimal acinar epithelial cells, J. Cell
Sci., 118, 4797-4812.
186.Choi, Y. O., Ryu, H. J., Kim, H. R., Song, Y.
S., Kim, C., Lee, W., Choe, H., Leem, C. H., and Jang, Y. J. (2006)
Implication of phosphorylation of the myosin II regulatory light chain
in insulin-stimulated GLUT4 translocation in 3T3-F442A adipocytes,
Exp. Mol. Med., 38, 180-189.
187.Fulcher, F. K., Smith, B. T., Russ, M., and
Patel, Y. M. (2008) Dual role for myosin II in GLUT4-mediated glucose
uptake in 3T3-L1 adipocytes, Exp. Cell Res., 314,
3264-3274.
188.Wu, F., Guo, X., Xu, J., Wang, W., Li, B.,
Huang, Q., Su, L., and Xu, Q. (2016) Role of myosin light chain and
myosin light chain kinase in advanced glycation end product-induced
endothelial hyperpermeability in vitro and in vivo,
Diab. Vasc. Dis. Res., 13, 137-144.
189.Tang, S. T., Su, H., Zhang, Q., Tang, H. Q.,
Wang, C. J., Zhou, Q., Wei, W., Zhu, H. Q., and Wang, Y. (2016)
Melatonin attenuates aortic endothelial permeability and
arteriosclerosis in streptozotocin-induced diabetic rats: possible role
of MLCK- and MLCP-dependent MLC phosphorylation, J. Cardiovasc.
Pharmacol. Ther., 21, 82-92.
190.Hu, W., and Feng, P. (2012) Myosin light chain
kinase is involved in the mechanism of gastrointestinal dysfunction in
diabetic rats, Dig. Dis. Sci., 57, 1197-1202.
191.Di, Y., Pan, W., and Li, X. (2015) Serum myosin
light chain kinase in type 2 diabetes mellitus: a cross-sectional
study, Ann. Clin. Lab. Sci., 45, 54-57.
192.Usatyuk, P. V., Singleton, P. A., Pendyala, S.,
Kalari, S. K., He, D., Gorshkova, I. A., Camp, S. M., Moitra, J.,
Dudek, S. M., Garcia, J. G., and Natarajan, V. (2012) Novel role for
non-muscle myosin light chain kinase (MLCK) in hyperoxia-induced
recruitment of cytoskeletal proteins, NADPH oxidase activation, and
reactive oxygen species generation in lung endothelium, J. Biol.
Chem., 287, 9360-9375.
193.Kaneko, K., Satoh, K., Masamune, A., Satoh, A.,
and Shimosegawa, T. (2002) Myosin light chain kinase inhibitors can
block invasion and adhesion of human pancreatic cancer cell lines,
Pancreas, 24, 34-41.
194.Khuon, S., Liang, L., Dettman, R. W., Sporn, P.
H., Wysolmerski, R. B., and Chew, T. L. (2010) Myosin light chain
kinase mediates transcellular intravasation of breast cancer cells
through the underlying endothelial cells: a three-dimensional FRET
study, J. Cell Sci., 123, 431-440.