Apoptotic Endonuclease EndoG Inhibits Telomerase Activity and Induces Malignant Transformation of Human CD4+ T Cells
D. A. Vasina1, D. D. Zhdanov1,2*, E. V. Orlova3, V. S. Orlova1, M. V. Pokrovskaya2, S. S. Aleksandrova2, and N. N. Sokolov2
1Peoples’ Friendship University of Russia, Ecological Faculty, 117198 Moscow, Russia; E-mail: zhdanovdd@mail.ru2Institute of Biomedical Chemistry, 119121 Moscow, Russia
3Institute of Theoretical and Experimental Biophysics, 142290 Pushchino, Moscow Region, Russia
* To whom correspondence should be addressed.
Received July 7, 2016; Revision received September 14, 2016
Telomerase activity is regulated by an alternative splicing of mRNA of the telomerase catalytic subunit hTERT (human telomerase reverse transcriptase). Increased expression of the inactive spliced hTERT results in inhibition of telomerase activity. Little is known about the mechanism of hTERT mRNA alternative splicing. This study was aimed at determining the effect of an apoptotic endonuclease G (EndoG) on alternative splicing of hTERT and telomerase activity in CD4+ human T lymphocytes. Overexpression of EndoG in CD4+ T cells downregulated the expression of the active full-length hTERT variant and upregulated the inactive alternatively spliced variant. Reduction of full-length hTERT levels caused downregulation of the telomerase activity, critical telomere shortening during cell division that converted cells into the replicative senescence state, activation of apoptosis, and finally cell death. Some cells survive and undergo a malignant transformation. Transformed cells feature increased telomerase activity and proliferative potential compared to the original CD4+ T cells. These cells have phenotype of T lymphoblastic leukemia cells and can form tumors and cause death in experimental mice.
KEY WORDS: EndoG, telomerase, hTERT, alternative splicing, malignant transformationDOI: 10.1134/S0006297917010035
Abbreviations: EndoG, endonuclease G; FITC, fluorescein isothiocyanate; GAPDH, glyceraldehyde-3-phosphatedehydrogenase; GFP, green fluorescent protein; hTERT, human telomerase reverse transcriptase; MFI, mean fluorescence intensity; TRAP, telomeric repeats amplification protocol; β-Gal, β-galactosidase.
Telomerase is a multi-protein complex synthesizing telomeric repeats
TTAGGG at the end of human chromosomes. The main components of
telomerase are hTR (human telomerase RNA), comprising RNA template for
synthesis of telomeres, and hTERT (human telomerase reverse
transcriptase) having reverse transcription activity that synthesizes
the telomeric repeats on the hTR template [1].
Telomerase is active in normal reproductive cells, stem cells,
activated lymphocytes, and most cancer cell types [2]; it maintains their high proliferative potential.
It is known that the enzyme activity is regulated by hTERT synthesis
levels [3] and by alternative splicing of its
mRNA.
The alternative splicing for hTERT mRNA has not been well studied. More than twenty splicing variants for hTERT are now known. However, only the full-length hTERT variant has catalytic activity [4]. Most of the splicing variants are expressed in minor quantities, and their function is not well understood. Two splicing variants represent the majority in total hTERT mRNA. Thirty-six-nucleotide deletion in exon 6 (α-variant) causes deletion of a part of a reverse transcriptase domain of the protein and leads to loss of catalytic activity. Deletion of 182 nucleotides from exons 7 and 8 (β-variant) results in a translational frameshift and appearance of a stop codon in exon 10. This is accompanied by synthesis of a truncated splicing variant of hTERT [5, 6], which is dominant-negative [7].
In normal somatic cells, telomerase is inactive, their telomeres are reduced by 40-200 bp every division cycle, and cells are only able to divide a limited number of times. Reducing the telomere to a critical length causes cell transition into so-called “replicative senescence” (M1 stage – mortality stage 1). During this stage, proliferation slows or stops, but the cells remain alive and metabolically active [8]. Change in the telomere structure at minimal critical length causes loss of main functions and triggers DNA damage response mechanisms. Thus, apoptosis is activated in the cells [9]. Nevertheless, some cells overcome this stage, for instance, in the presence of viral oncogenes causing loss of function of proteins p53 (protein 53) and Rb (Retinoblastoma) [10]. Telomeres of such cells keep reducing and completely lose their protective function. This leads to chromosomal instability accompanied by inhibition of cell proliferation, accumulation of genetic mutations, and cell death. This stage is referred to as “crisis” (M2 stage). A few cells that survive the M2 stage undergo malignant transformation. Apparently, as a result of numerous mutations, they trigger telomere elongation and become immortal [11]. Most cancer cells (about 90%) have telomerase activity [2]. Active telomerase in cancer cells does not fully restore the telomere length typical for normal cells (telomeres in cancer cells are significantly shorter than in normal cells), but maintains their size [12, 13].
It has been shown earlier that telomerase activity is dependent on activity of some apoptotic endonucleases [14, 15]. Apoptotic endonucleases are a group of enzymes that degrade DNA in cells at final apoptosis stages [16]. Endonuclease G (EndoG) is a site-specific endonuclease that cleaves DNA duplexes selectively at polyG regions. A trait of EndoG is its RNase activity [17]. EndoG causes cell aging and significantly decreases their replicative potential [18]. In this work, we focused on studying the mechanisms of connection between EndoG and hTERT alternative splicing in normal human T lymphocytes and on the effect of EndoG overexpression on cell proliferation.
MATERIALS AND METHODS
Blood sampling, selection and cell transfection. The research was approved by the ethics committee of the Institute of Biomedical Chemistry. Written consent was obtained from all donors participating in the research. Venous blood samples of four healthy donors were put into test tubes with anticoagulant K3EDTA (Greiner Bio-One, Austria). Mononuclear cells were obtained from blood samples using density-gradient centrifugation in Ficoll Lympholite-H (Cedarlane, Canada). The cell CD4+ fraction was obtained by the magnetic selection method using a CD4+ Isolation Kit, human (Miltenyi Biotec, Germany) according to the manufacturer’s manual.
Isolated CD4+ cells were plated at 5·105 cells/ml of medium. Cultural medium RPMI-1640 (Life Technologies, USA) was used with 10% FBS (fetal bovine serum; Gibco, USA), growth stimulators 100 U/ml IL-2 (R&D Systems, USA), 5 µg/ml anti-CD3 antibody (MedBioSpektr, Russia), 2 µg/ml anti-CD28 antibody (eBiosciences, USA), and antibiotics – 50 U/ml penicillin and 50 mg/ml streptomycin (Sigma, USA). The cells were cultivated in a CO2-incubator at 37°C, 5% CO2, and humidity 90%. Once in every three days of cultivation, the cells were re-plated at 5·105 cells/ml in the medium and re-stimulated with the complete medium. Cells were counted after staining with trypan blue on a Vi-cell XR Viability Cell Analyzer (Beckman Coulter, USA). Proliferating cells were photographed using a Leica DMI300 inverted microscope (Leica Biosystems, USA).
CD4+ T cells were transfected with one of the plasmids pEndoG-GFP, pDNase1-GFP, pDNaseX-GFP, or a control plasmid pGFP. Plasmids carrying endonuclease genes were synthesized on request by Clontech (USA) based on pGFP-N1 vector. Transfection was carried out using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s protocol. Transfection efficiency (typically 90-100%) was estimated by counting GFP-positive cells under a fluorescence microscope.
RNA extraction and real-time RT-PCR. Total cellular RNA was isolated using an RNeasy Mini Kit (Qiagen, USA) according to the manufacturer’s manual. Reverse transcription (RT) and real-time PCR were carried out according to the technique described by Basnakian and coauthors [19]. To do so, 5 µg of total RNA were used for RT in a 25 µl reaction mixture (Invitrogen) according to the manufacturer’s protocol. Platinum SYBR Green qPCR Supermix-UDG (Invitrogen) was used as a reaction mixture for real-time PCR according to the manufacturer’s manual. Primer structures (Syntol, Russia) are shown in Table 1. For amplification, a CFX96 Touch Real-Time PCR Detection System (Bio-Rad, USA) was used with two-temperature reaction (primer annealing/elongation). Amplicon content was determined by fluorescence at the end of the elongation cycle. Standard PCR efficiency curves were built using serial dilutions (1 : 40, 1 : 80, 1 : 160, and 1 : 320) of total cDNA. The data are represented as normalizations of levels of studied gene mRNAs to the 18S rRNA, which was used as a reference constitutively expressed gene.
Table 1. Primers used for real time
RT-PCR
Western blotting. Cells were disrupted in 1 ml of TBE (89 mM Tris, 89 mM H3BO3, 2 mM EDTA, pH 8.0) by ultrasonic treatment for 2 min at 50 W power using a Model 50 Sonic Dismembrator (Fisher Scientific, USA) and centrifuged for 10 min at 12,000g to remove cell debris. Total protein concentration in samples was measured with Bradford Protein Assay (Pierce, USA) using bovine serum albumin (BSA) to construct a calibration graph. Cell lysate (normalized to 50 µg of total protein) was dissolved in 50 mM Tris-HCl, pH 6.8, containing 1% SDS, 2 mM EDTA, 1% β-mercaptoethanol, and 7.5% glycerol, denatured by heating at 100°C for 10 min, and separated by electrophoresis in a gradient polyacrylamide gel [20] (at 100 V for 2 h) using NuPAGE Novex 4-12% Bis-Tris Protein Gels (Life Technologies, USA). The proteins were then transferred to a nitrocellulose membrane in the Novex transferring buffer (Invitrogen) at 40 V for 3 h. After the transfer, membranes were stained with Ponseau S (Sigma, USA) as described by Hofnagel and coauthors [21]. Then, the membranes were blocked in Blotting-Grade Blocker (Bio-Rad, USA) and incubated for 2 h with primary monoclonal antibodies anti-GAPDH, anti-hTERT (Abcam, USA) at 1 : 1000 dilution or polyclonal anti-EndoG (Millipore, USA) at 1 : 500 dilution. The primary antibodies were then washed out in phosphate buffered saline (PBS), pH 7.6, with 0.1% Tween-20, and the membranes were incubated with secondary antibody conjugated to horseradish peroxidase (Cell Signaling, USA). To visualize the bands, the SuperSignal chemiluminescent kit (Pierce Biotechnology, USA) was used with subsequent imaging with a ChemiDoc XRS imaging system (Bio-Rad). Protein content was quantified with densitometry in the GelAnalyzer 2010a program.
Determining telomerase activity. Telomerase activity was determined by the TRAP (Telomeric Repeat Amplification Protocol) method [2, 22]. Cells were lysed in buffer containing 10 mM Tris-HCl, pH 7.5, 1 mM MgCl2, 1 mM EGTA, 0.1 mM phenylmethanesulfonyl fluoride, 5 mM β-mercaptoethanol, 0.5% CHAPS, and 10% glycerol at the rate 1 µl of the buffer per 103 cells. Lysates were centrifuged (30 min, 4°C, 12,000g). Supernatant was collected and kept at –80°C. Primer elongation and further amplification were performed in 30 µl of reaction mixture containing 67 mM Tris-HCl, pH 8.8, 16.6 mM (NH4)2SO4, 0.01% Tween-20, 1.5 mM MgCl2, 1 mM EGTA, 0.25 mM each of the four dNTPs, 0.1 µg TS-primer (Telomerase Substrate primer; 5′-AATCCGTCGAGCAGAGTT-3′), and 2 µl of cell extract equivalent to 2000 cells diluted with lysis buffer, and incubated at 37°C for 25 min. After incubation, the mixture was heated for 10 min at 96°C to inactivate telomerase. Then, 0.1 µg of CX-primer (Copy Extended primer; 5′-CCCTTACCCTTACCCTTACCCTAA-3′) and 2.5 U of Taq polymerase were added to the mixture. The reaction mixture was subjected to thermal cycling as follows: for 2 min at 94°C, then 30 cycles (94°C – 30 s, 50°C – 30 s, 72°C – 40 s), and for 5 min at 72°C. Amplification products were separated by electrophoresis in 12% nondenaturing polyacrylamide gels in TBE buffer. A 10-µl sample was loaded into each well. The ChemiDoc XRS Imaging System was used for visualization of separated products after 30 min gel incubation in SYBR Green I (Invitrogen) stain solution. Telomerase activity was detected by densitometry of TRAP results in the GelAnalyzer 2010a program.
Determining absolute telomere length. To determine the absolute length of telomeres, a described method [23, 24] was applied. Genomic DNA was isolated using a PureLink Genomic DNA Mini Kit (Thermo Scientific, USA). Real-time PCR was carried out according to a technique suggested by Cawthon [24]. Telomere length in each sample was measured in triplicate. For further calculations, an average value was used when the coefficient of variation among three technical replicates did not exceed 2%. DNA of non-transfected CD4+ T cells was used as a control.
Immunophenotyping and identification of apoptosis and cell cycle. Peripheral blood from donors was lysed using as lysis buffer the Red Blood Cell Lysis buffer (Life Technologies, USA) according to the manufacturer’s protocol. Cell portions of 5·107 cells from the pellet or after malignant transformation were suspended in 200 µl of PBS (Gibco, USA). The cells were labeled with anti-CD45-Vio770 antibody and with each of CD3-FITC, CD4-FITC, CD8-FITC, CD14-FITC, CD16/56-FITC, CD2-FITC, CD5-FITC, CD7-FITC (all from Miltenyi Biotec, Germany) according to the manufacturer’s protocol. Cells were counted using flow cytometry in a FACSCalibur cytometer (Beckton Dickinson, USA). Marker expression was monitored by MFI (mean fluorescence intensity) during flow cytometry. To determine apoptosis, transfected CD4+ T cells were suspended in PBS and incubated with Annexin V-FITC and propidium iodide from an FITC Annexin V/Dead Cell Apoptosis kit (Invitrogen) according to the manufacturer’s instructions. Cell samples (5·104 cells) were analyzed using flow cytometry. For cell cycle determination, cells were fixed in 70% ethanol, treated with FxCycle PI/RNase Staining Solution (Thermo Scientific), according to the manufacturer’s protocol, and measured by flow cytometry. Cell cycle phases were determined by intensity of propidium iodide signal.
Measuring β-galactosidase activity. Activity of β-Gal in cells was measured using the Beta Galactosidase Detection Kit (Abcam, USA) according to the manufacturer’s protocol in a 96-well plates (Corning, USA). Fluorescence was measured using a Multiscan Go microplate spectrophotometer (Thermo Scientific, USA) at 490 nm (excitation) and 525 nm (emission) wavelengths. The activity was measured for 103 cells using a calibration curve constructed with commercial β-Gal (Abcam) in the 0.01-100 U/ml range.
Estimating cell tumorigenic potential in vivo. To estimate tumorigenic potential of transformed cells, 6-9-week-old male mice of Balb/c nude line (from the nursery for laboratory animals, Pushchino) were used. The mice were kept in a vivarium under conditions of absence of pathogenic microflora and natural illumination, fed with briquetted fodder, and having constant access to water. The mice were divided into two groups, 10 individuals in each group. The mice in the control group were administered freshly obtained human CD4+ T lymphocytes. Mice in the experimental group were administered malignantly transformed cells. The cells were suspended in RPMI-1640 medium at 1·106 cells in 200 µl and administered subdermally according to a standard method described by Pokrovsky and coauthors [25]. Lifespan of the mice and three-dimensional tumor size were estimated. After the experiment, the mice were euthanized by overdose of ether anesthesia.
Statistical analysis. The results were statistically analyzed using Student’s criterion with Statistica 9.0 software (StatSoft Inc., USA). Results are presented as mean value ± standard deviation. Numbers were considered statistically significant when p ≤ 0.05. The survival of the mice was statistically analyzed using the log-rank criterion and constructing Kaplan–Meier curves.
RESULTS
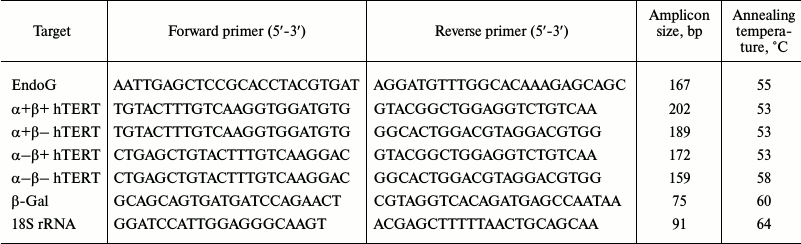
EndoG overexpression causes death of CD4+ T cells. To determine a possible role of EndoG in the proliferation of CD4+ T cells, lymphocytes from four donors were transformed with the plasmid, pEndoG-GFP, and a control plasmid, pGFP, cultivated in growth medium. Dead, living, and apoptotic cells were counted daily. In the group transfected with pEndoG-GFP, massive death was observed 24-28 days after transformation (Fig. 1). Therefore, on day 24, the number of apoptotic cells sharply increased to 13.31 ± 2.12%. On day 28, virtually all the cells were dead. Cells transfected with the control plasmid remained alive during 28 days of cultivation.
Fig. 1. EndoG overexpression causes death of CD4+ T cells during cultivation. Results of flow cytometry of CD4+ T cells transformed with (a) pEndoG-GFP or (b) pGFP and labeled with annexin V-FITC and propidium iodide. Percentage of viable cells (left lower quadrants), percentage of cells at the apoptosis stage (right lower quadrants), and percentage of dead cells (upper quadrants) are shown. One typical experiment out of four is shown. Histograms of viable, apoptotic, and dead CD4+ T cells transfected with (c) pEndoG-GFP or (d) pGFP (n = 4; * p ≤ 0.05 compared to cells transfected with pGFP).
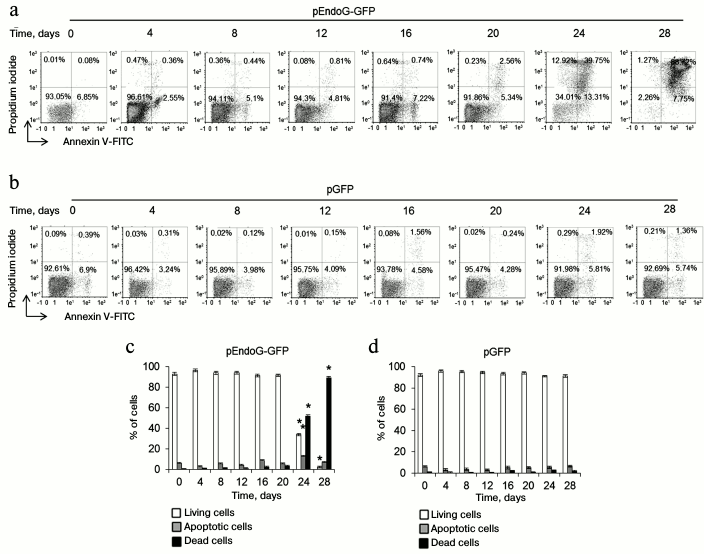
Overexpression of EndoG reduces expression of α+β+ variant of hTERT and decreases telomerase activity and telomere length. We studied changes in expression of hTERT splicing variants in the transfected CD4+ T cells during cultivation. Using real-time RT-PCR, we established that EndoG expression was constantly high during the cultivation period (Fig. 2a). Decrease in EndoG expression during long-term cultivation of the transfected cells may be explained by a large number of cell duplications and reduction of plasmid levels in the cells. Overexpression of EndoG was accompanied by reduction of α+β+ hTERT splicing variant expression (Fig. 2b) and increased expression of the α+β– variant (Fig. 2b). In the cells transfected with the control plasmid, expression of EndoG, α+β+, and α+β– hTERT splicing variants was not altered. Alteration of levels of the studied proteins in CD4+ T cells transfected with pEndoG-GFP compared to the pGFP-transfected cells was confirmed by Western blotting (Fig. 2, d-h).
Fig. 2. EndoG overexpression during cell cultivation causes expression of the full-size hTERT splicing variant, inhibition of telomerase activity, and reduction of telomeres. Expression levels of EndoG (a) and hTERT splicing variants (b, c) in CD4+ T cells transfected with pEndoG-GFP or pGFP. Gene expression levels are normalized to the expression of the reference 18S gene. Western blotting of the hTERT splicing variants and EndoG in CD4+ T cells transfected with pEndoG-GFP (d) or pGFP (e). Determining levels of EndoG (f) and hTERT splicing variants (g, h) in comparison with GAPDH. i) Gel electrophoresis of TRAP in transfected cells. j) Results of telomerase activity measured by the TRAP method. k) Absolute telomere length determined using real time PCR (n = 4; * p ≤ 0.05 compared to cells transfected with pGFP).
Replicative senescence occurs in the case of telomere reduction to the critical value. We followed changes in telomerase activity and telomere length in transfected CD4+ T cells during cultivation. Using the TRAP method, we found that the telomerase activity remained low in the pEndoG-GFP-transfected cells during the cultivation time (Fig. 2, i and j). Activity of the enzyme did not change in the control cells. Decrease in the telomerase activity in actively dividing cells should reduce telomere length. Using real time PCR, we found that telomere length was reduced in pEndoG-GFP-transfected cells by a factor of 6 in 24 days after transfection (Fig. 2k). During further cultivation, telomere length decreased insignificantly and remained low. In the pGFP-transfected cells, reduction of telomere length was also observed, but it was not as rapid as in the EndoG overexpressing cells.
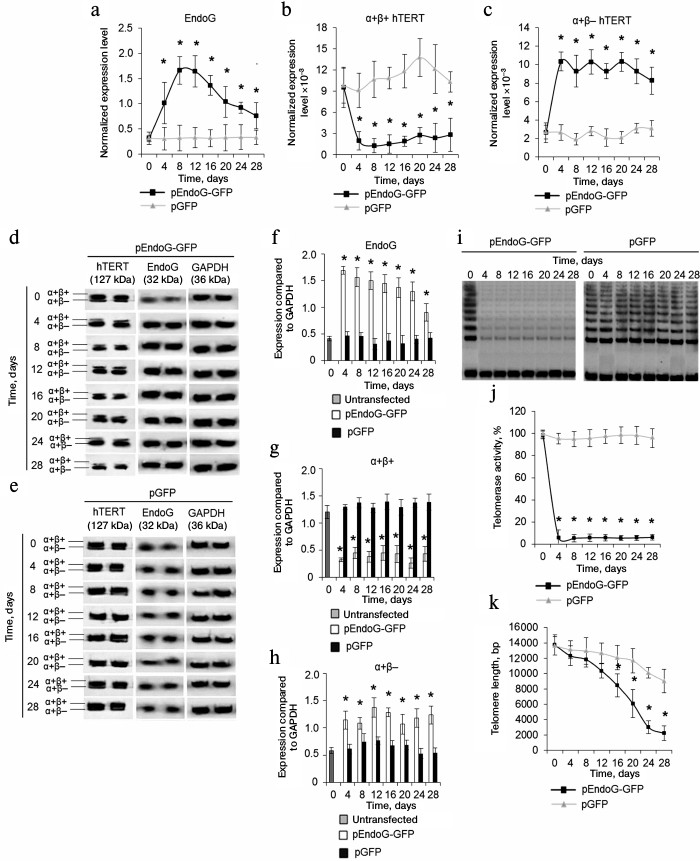
DNase 1 and DNase X cause death of CD4+ T cells and do not affect hTERT alternative splicing. As apoptotic endonuclease EndoG caused cell death and induced hTERT alternative splicing, it was of interest to study such activity for other apoptotic endonucleases with broad specificity. To do so, CD4+ T cells from four donors were transfected with the DNase 1 gene (pDNase 1-GFP plasmid) or DNase X gene (pDNase X-GFP) and cultivated in growth medium. The number of living, dead, and apoptotic cells was measured. Massive cell death was observed in the pDNase 1-GFP-transfected group already 24-48 h after transfection (Fig. 3, a and d). In the pDNase X-GFP-transfected group, cell death was observed 36-48 h after transfection (Fig. 3, b and e). Cells transfected with the control plasmid pGFP remained alive during 48 h (Fig. 3, c and f). Overexpression of both DNase 1 and DNase X did not alter expression of α+β+ and α+β– hTERT splicing variants (Fig. 3, g and h).
Fig. 3. Overexpression of DNase 1 and DNase X causes rapid CD4+ T cell death and does not affect expression of hTERT splicing variants. Flow cytometry results for CD4+ T cells transfected with pDNase 1-GFP (a), pDNase X-GFP (b), or pGFP (c) and labeled with the annexin V-FITC and propidium iodide. Percentages of living cells (left lower quadrants), apoptotic cells (right lower quadrants), and dead cells (upper quadrants) are shown. One of four typical experiments is shown. Histograms of living cells, apoptotic cells, and dead CD4+ T cells transfected with pDNase 1-GFP (d), pDNase X-GFP (e), or pGFP (f). Expression levels for α+β+ hTERT (g) and α+β– hTERT (h) in the transfected CD4+ T cells. The gene expression levels are normalized to the expression of the reference 18S gene (n = 4; * p ≤ 0.05 compared to the pGFP-transfected cells).
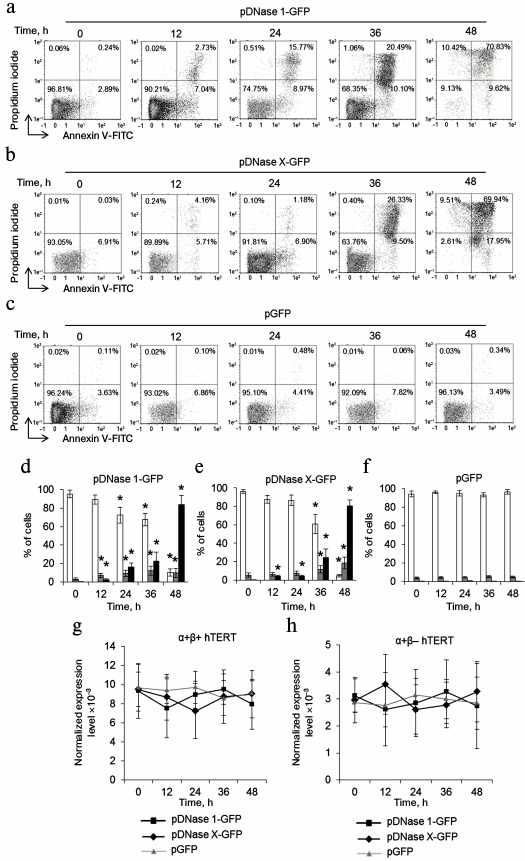
Telomere shortening provokes replicative senescence and inhibits cell cycle. It is known that inhibition of telomerase in actively proliferating cells causes replicative senescence. This is accompanied by G0/G1 cell cycle arrest [22, 26]. We measured the cell cycle in proliferating CD4+ T cells transfected with pEndoG-GFP and pGFP by flow cytometry using propidium iodide to label cell DNA. Upon transfection with pEndoG-GFP, we observed a significant decrease in the number of cells in the S and G2/M phases and reliable increase in the cell number in the G0/G1 phases 20 days after transfection (Fig. 4, a and c). However, the cells remained viable (Fig. 1, a and c). On day 24, we observed further decrease in the number of cells in G2/M phase and increase in number of apoptotic cells. Virtually all the cells entered apoptosis by day 28. The cell cycle was not altered in the cells transfected by the control plasmid (Fig. 4, b and d).
Fig. 4. Inhibition of the cell cycle during the transition to replicative senescence. Results of measuring cell cycle by flow cytometry; cell DNA was stained with propidium iodide. Percentage of CD4+ T cells transfected with pEndoG-GFP (a) or pGFP (b) that had entered apoptosis and G0/G1, S, and G2/M phases of the cell cycle. One typical experiment of four is shown. c, d) Histograms of transfected cell numbers in different cell cycle phases. e) Level of β-Gal expression in CD4+ T cells transfected with pEndoG-GFP or pGFP. Gene expression was normalized to the expression of the reference 18S gene. f) Enzymatic activity of β-Gal in the transfected cells. Activity of β-Gal is shown in units per 1000 cells. This was determined using a calibration curve (n = 4; * p ≤ 0.05 compared to pGFP-transfected cells).
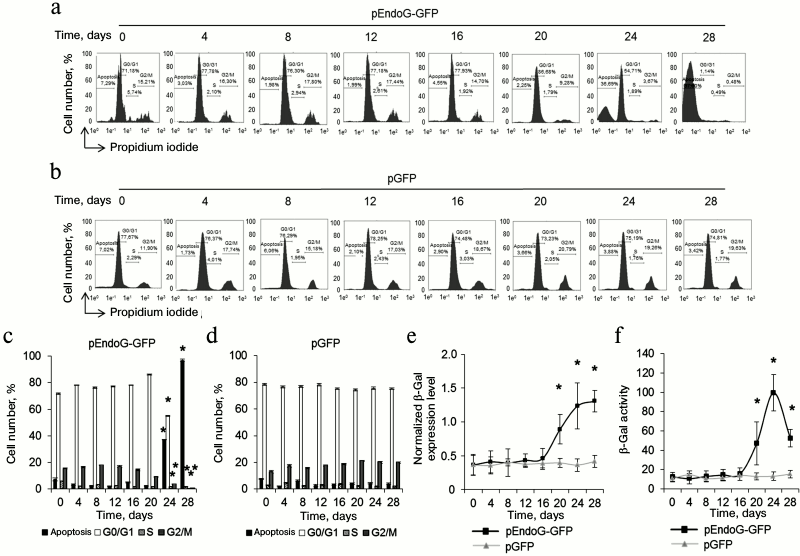
Increased expression and activity of β-Gal is a marker for transition of cells to replicative senescence. We examined expression and activity of this enzyme during cultivation of transfected cells. A reliable increase in β-Gal expression was observed in the cells transfected with pEndoG-GFP 20 days after transfection (Fig. 4e). Expression level remained stably high on days 24-28. Enzymatic activity of β-Gal also increased 20 days after transfection and remained high on day 24 (Fig. 4f). Reduction of enzymatic activity of β-Gal was observed on day 28, which is explained by massive cell death. Expression and activity of β-Gal remained unchanged in the cells transfected with the control plasmid. Activation of β-Gal on day 20 after transfection with the EndoG gene agrees with inhibition of S and G2/M phases of the cell cycle, indicating the transition of the cells to replicative senescence. Death of such cells by days 24-28 of cultivation was a further consequence of this state.
Dynamics of transfected CD4+ T cell proliferation. We studied the effect of EndoG overexpression on the absolute number of transfected CD4+ cells (Fig. 5a). In a population of pEndoG-GFP-transfected cells, exponential growth was observed from the beginning to day 16 of cultivation. Further cultivation led to cessation of cell proliferation within days 16-24, which agrees with telomere shortening to a critical value, cell transition to replicative senescence, and G0/G1 cell cycle arrest (Fig. 2k). On cultivation day 28, a sharp decrease in the number of living cells was observed, which corresponds to massive cell death due to apoptosis in the aging cells (Fig. 2). Massive cell death occurred until cultivation day 36; cell number was reduced to solitary living cells in the field of view. Cells remained alive, but were not able to divide. However, on cultivation day 62, massive unlimited cell growth was observed, which continued until cultivation day 67 and further. We propose that these cells have undergone a malignant transformation and have acquired unlimited proliferative potential. Similar dynamics was observed in the control population of the pGFP-transfected cells in the early cultivation stages. Exponential cell growth occurred until cultivation day 20. In general, cell number in the pGFP-transfected population was three orders of magnitude higher than in the pEndoG-GFP-transfected population. This indicates EndoG influence not only the telomerase complex system, but also other systems inhibiting cell proliferative activity. Cell growth stopped during days 28-32. Cells died massively on cultivation day 36. Cell death was complete by day 60. Living cells were not observed cells, and malignant transformation of cells did not occur.
Fig. 5. Development dynamics of transfected CD4+ T cells during cultivation (see description in text). a) Absolute cell number during cultivation (n = 4). b, c) Microphotographs of proliferating cells at ×200 magnification.
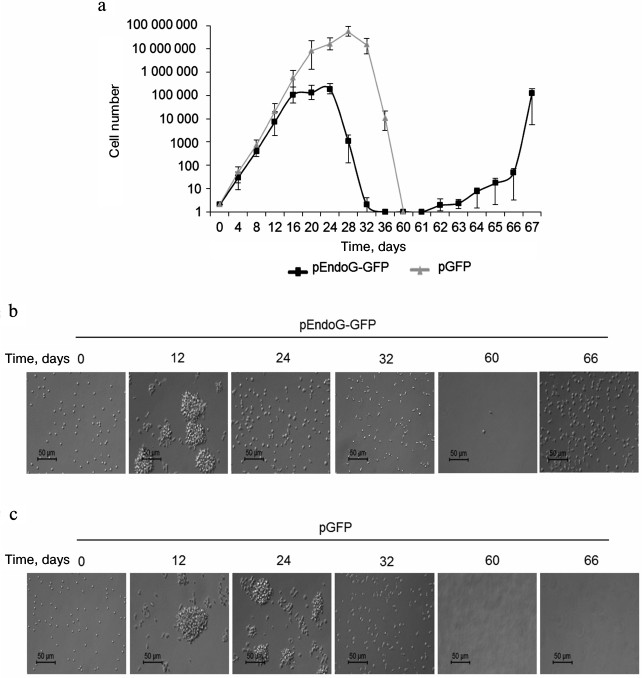
The CD4+ T cell population obtained by magnetic selection and subjected to transfection with pEndoG-GFP or pGFP comprises individual round cells 5-8 µm in size (Fig. 5, b and c, cultivation day 0). Proliferation of such cells occurred as proliferation clusters (cultivation day 12) representing cell aggregates 50-70 µm in size. Cells remaining in the replicative senescence state (cultivation day 24) were accompanied by proliferation cluster breakdown into individual cells. In parallel, increase in the individual cell size to 10-17 µm was observed. Cell shape in the replicative senescence state was not round. Development of apoptosis and massive cell death on day 32 was accompanied by appearance of a high number of cells with altered shape, and cell debris. After massive death in the population of pEndoG-GFP-transfected cells, only single cells remained alive (cultivation day 60). Living cells were not observed in the population of the control pGFP-transfected cells after massive cell death on cultivation day 60 and further. As a result of EndoG action, malignantly transformed cells did not proliferate (cultivation day 66) in proliferation clusters, and the shape of these cells was not round.
Malignantly transformed CD4+ T cells have increased telomerase activity of T cell lymphoma phenotype. We compared expression levels of EndoG and hTERT slicing variants in the original CD4+ T cells and in malignantly transformed cells. Using real time RT-PCR, we found that EndoG expression level in the transformed cells did not differ from the original cells (Fig. 6a). The expression level of the full size α+β+ hTERT splicing variant was reliably lower in the transformed cells (Fig. 6b), whereas expression of α+β– variant was increased (Fig. 6c). Changes in the expression of minor α–β+ and α–β– splicing variants were not observed (Fig. 6, d and e). Reliable change in the levels of the studied proteins in malignantly transformed cells was confirmed by Western blotting (Fig. 6, f and g). Using the TRAP method, we found that in the transformed cells telomerase activity was approximately twice as high as in the original CD4+ T cells (Fig. 6, h and i). Increased telomerase activity agrees with the increased proliferative activity of malignantly transformed cells and expression levels of α+β+ and α+β– splicing variants in these cells. Telomere length in the transformed cells was lower by a factor of 6 than in the original cells (Fig. 6j).
Fig. 6. Elevation of telomerase activity and phenotype of T cell lymphoma formed by malignantly transformed CD4+ T cells. Expression levels of EndoG (a) and hTERT splicing variants (b-e) in the original and malignantly transformed CD4+ T cells. Gene expression levels are normalized to the expression of the reference 18S gene. f) Western blotting of hTERT splicing variants and EndoG in the original and malignantly transformed CD4+ T cells. g) Results of determining quantities of EndoG and hTERT splicing variants compared to GAPDH. h) Gel electrophoresis of TRAP in the transformed cells. Lanes: 1) original cells; 2) transformed cells. i) Results of determining telomerase activity by the TRAP method. j) Absolute telomere length measured by real time PCR (n = 4; * p ≤ 0.05 compared to the original cells). Results of determining immune phenotype by flow cytometry in a population of the original cells (k) and the transformed cells (l). One typical experiment out of four is shown.
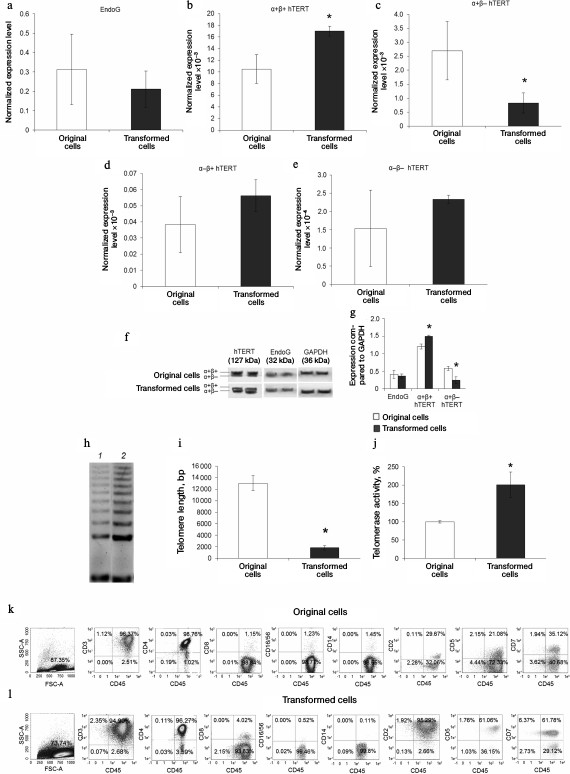
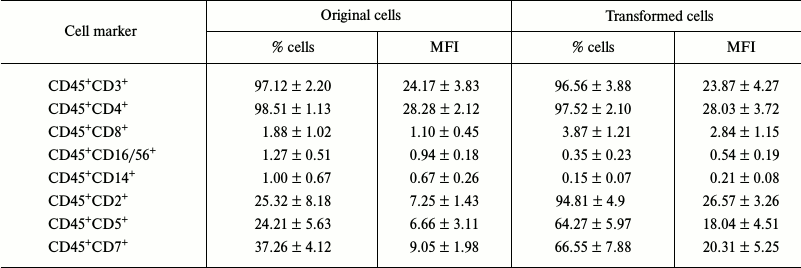
Studying the immune phenotype in a population of the transformed cells by flow cytometry revealed significantly increased number of cells expressing CD2, CD5, and CD7, which is typical in T cell lymphoma (Fig. 6, k and l, and Table 2). Immune phenotype of transformed cells can be described as CD45+CD3+CD4+CD2+CD5+CD7+. Increase in number of cells possessing such markers was accompanied by elevated expression of the molecules on every cell, which is indicated by increased MFI level (Table 2). Cytotoxic T lymphocytes (CD8+ marker), NK-cells (CD16/56+ markers), and macrophages (CD14+ marker) were absent from a population of the transformed cells. A small fraction (about 3%) of cells having CD8+ marker in a population of the transformed cells were cells that express both CD8+ and CD4+, which is also typical for T cell lymphoma.
Table 2. Expression levels of cell markers
(n = 4)
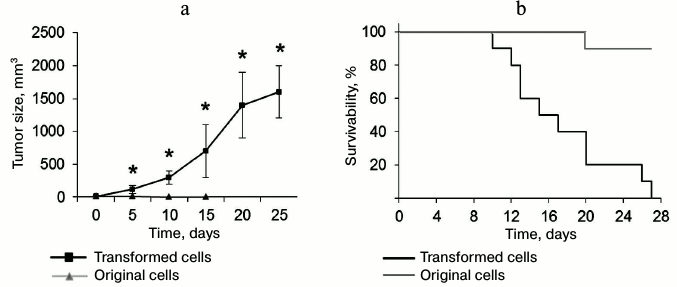
Transformed CD4+ T cells can form tumors and cause death in experimental mice. To estimate in vivo tumorigenic potential of the transformed CD4+ T cells, these cells and normal CD4+ T lymphocytes were administered to thymus-free Balb/c nude mice. Malignantly transformed CD4+ T lymphocytes formed rapidly developing tumor lumps (Fig. 7a). On day 5 after administration, tumor size was reliably different from that in the control group. By the end of the experiment, the tumor size was 1638 ± 421 mm3. In the control group mice (with normal CD4+ T lymphocytes) subcutaneous malignancies less than 11 mm3 in size were observed on the site of cell administration. At the same time, this malignancy gradually reduced: its size did not exceed 2 mm3 in 15 days, and in 20 days the cell administration area did not differ from the surrounding tissue. Along with the tumor development, we observed death in mice with introduced transformed cells (Fig. 7a). All the mice died in 27 days after cell administration, whereas in the control group only one mouse died on experiment day 20.
Fig. 7. Ability of transformed CD4+ T cells to form tumors in vivo and cause death in experimental mice. a) Size of tumor formed by the transformed and normal CD4+ T cells; * p ≤ 0.05 compared to the control group. b) Kaplan–Meier curves for survivability of mice after administration of transformed and normal CD4+ T cells.
DISCUSSION
Though alternative splicing of mRNA of the hTERT catalytic subunit of telomerase regulates telomerase activity in cells, little is known about this process. In our work, we investigated the ability of the apoptotic endonuclease EndoG for alternative splicing of hTERT mRNA in normal activated human CD4+ T lymphocytes. The resultants agree with the effect of EndoG on telomerase activity in CaCo-2 line tumor cells [27]. Alternative splicing of hTERT resulted in reduced telomerase activity and caused malignant transformation of normal CD4+ T cells. The following facts speak in favor of this. EndoG overexpression upon transfection of CD4+ T cells with pEndoG-GFP plasmid caused an increase in levels of inactive (α+β–) hTERT splicing variant and reduction in active (α+β+) variant levels. Reduced levels of active hTERT form upon EndoG overexpression accompanied by a reliable decrease in the telomerase activity. Prolonged cultivation of the transfected cells led to their massive death. These data do not agree with the data obtained by Listerman et al. [7]. They showed that overexpression of α+β– hTERT splicing variant can protect cells from apoptosis. Most probably, in our case cell fate was affected by EndoG overexpression as such, rather than by only the hTERT splicing and inhibition of telomerase activity that it causes.
Also, Listerman et al. did not monitor dynamics of telomere shortening in cells after transfection. As we mentioned above, EndoG can affect telomerase directly and reduce cell proliferative potential.
Cultivation of transfected cells with elevated EndoG expression led to shortening of their telomeres to a critical value and cell transition to replicative senescence, which is confirmed by elevated expression and activity of β-Gal and by G0/G1 cell cycle arrest.
Telomere shortening within 20-24 days of cultivation was accompanied with reduced expression of α+β+ hTERT variant (Fig. 2b) and decrease in telomerase activity (Fig. 2, i and j). Most probably, telomere length in the pEndoG-GFP-transfected cells was reduced to the critical value by cultivation day 24, and cells entered replicative senescence. These cells remained alive, but they lost their ability to divide. G0/G1 cell cycle arrest and decrease in number of cells in S and G2/M phases within 16-24 days of cultivation (Fig. 4) agrees with the inability of cells to divide. Massive cell death observed within 24-28 days of cultivation (Fig. 1) was probably caused by the development of apoptosis in the aging cells. Telomere shortening during cell division was also observed in cells transfected with the control plasmid pGFP. These data are consistent with the limited replicative potential of T lymphocytes [28].
Very low telomerase activity is detected in peripheral blood T lymphocytes [29]. Under in vitro conditions, telomerase activity significantly increases upon stimulation of cell proliferation with IL-2 and antibodies against CD3 and CD28 [30]. However, its activity is insufficient for unlimited proliferation of T cells [31]. In our case, exponential growth of pEndoG-GFP-transfected cells continued for 20 days of cultivation. Approximately 20 divisions occurred in the population during this time. This number of divisions is not sufficient for such rapid telomere shortening One may also explain a sharp decrease in telomere length in the pEndoG-GFP-transfected cells by degradation of telomeres by EndoG as such or other endonucleases upon their activation. This is the most probable explanation for rapid telomere shortening, as it is known that endonucleases can degrade G-quadruplex structures formed by telomeres [32-34]. Moreover, we have shown earlier that EndoG overexpression causes activation of other endonucleases [35]. Beyond any doubt, the direct effect of EndoG on telomere dynamics requires further investigation. Within 28 days of cultivation, telomere length reduced to approximately 2000 bp. Most probably, malignant transformation of the CD4+ T cells occurred when telomeres were reduced to these values. Such telomere size was preserved in transformed cells, while high telomerase activity provides high replicative potential of these cells. Studying the immune phenotype of the transformed cells revealed that these cells are leucocytes (as they express the leukocyte common antigen CD45), lymphocytes (as they express CD3), and T lymphocytes-helpers (as they express CD4). Malignantly transformed cells had immune phenotype of T cell lymphoma, for which simultaneous expression of CD2, CD5, and CD7 markers is typical [36]. Experiments on malignant transformation of the CD4+ T cells upon their transfection with the EndoG gene and death of cells transfected with the control plasmid were carried out in cells from four donors, which demonstrated reproducibility of the results. Malignancy of the transformed CD4+ T cells was confirmed in the in vivo experiment with thymus-free mice. These cells formed tumors and caused the death of the mice.
Overexpression of other apoptotic endonucleases with broad specificity, DNase 1 and DNase X, led to complete death of CD4+ T cells already 48 h after transfection and did not affect expression of hTERT splicing variants. The data indicate that EndoG has an intrinsic ability to trigger hTERT mRNA alternative splicing.
Telomerase activity is regulated by alternative splicing of its catalytic subunit hTERT. However, the alternative splicing mechanism is insufficiently studied. Results of this work describe several new phenomena. First, it was demonstrated that EndoG, but not DNase 1 or DNase X, can cause hTERT mRNA alternative splicing and inhibit telomerase activity. Second, it was demonstrated that prolonged cultivation of CD4+ T lymphocytes with overexpression of EndoG and inactive telomerase results in their malignant transformation. Apparently, the participation of EndoG in hTERT alternative splicing is a fundamental process in maintenance of cellular homeostasis and determines cell fate. Beyond any doubt, the described process requires further extensive study.
Acknowledgements
Authors are grateful to Dr. V. S. Pokrovsky for his help in in vivo experiments with thymus-free mice.
The study was financially supported by the Program of Fundamental Research in State Academies of Sciences for 2013-2020.
REFERENCES
1.Blackburn, E. H. (2000) Telomere states and cell
fates, Nature, 408, 53-56.
2.Kim, N. W., Piatyszek, M. A., Prowse, K. R.,
Harley, C. B., West, M. D., and Ho, P. L. (1994) Specific association
of human telomerase activity with immortal cells and cancer,
Science, 266, 2011-2015.
3.Meyerson, M., Counter, C. M., Eaton, E. N.,
Ellisen, L. W., Steiner, P., and Caddle, S. D. (1997) hEST2, the
putative human telomerase catalytic subunit gene, is up-regulated in
tumor cells and during immortalization, Cell, 90,
785-975.
4.Saeboe-Larssen, S., Fossberg, E., and Gaudernack,
G. (2006) Characterization of novel alternative splicing sites in human
telomerase reverse transcriptase (hTERT): analysis of expression and
mutual correlation in mRNA isoforms from normal and tumor tissues,
BMC Mol. Biol., 7, 26.
5.Ulaner, G. A., Hu, J. F., Vu, T. H., Oruganti, H.,
Giudice, L. C., and Hoffman, A. R. (2000) Regulation of telomerase by
alternate splicing of human telomerase reverse transcriptase (hTERT) in
normal and neoplastic ovary, endometrium and myometrium, Int. J.
Cancer, 85, 330-335.
6.Ulaner, G. A., Hu, J. F., Vu, T. H., Giudice, L.
C., and Hoffman, A. R. (1998) Telomerase activity in human development
is regulated by human telomerase reverse transcriptase (hTERT)
transcription and by alternate splicing of hTERT transcripts, Cancer
Res., 58, 4168-4172.
7.Listerman, I., Sun, J., Gazzaniga, F. S., Lukas, J.
L., and Blackburn, E. H. (2013) The major reverse
transcriptase-incompetent splice variant of the human telomerase
protein inhibits telomerase activity but protects from apoptosis,
Cancer Res., 73, 2817-2828.
8.Shay, J. W. (2003) Telomerase therapeutics:
telomeres recognized as a DNA damage signal: Commentary re: K. Kraemer
et al., Antisense-mediated hTERT inhibition specifically reduces the
growth of human bladder cancer cells, Clin. Cancer Res.,
9, 3521-3525.
9.Harley, C. B., Futcher, A. B., and Greider, C. W.
(1990) Telomeres shorten during ageing of human fibroblasts,
Nature, 345, 458-460.
10.Wright, W. E., Pereira-Smith, O. M., and Shay, J.
W. (1989) Reversible cellular senescence: implications for
immortalization of normal human diploid fibroblasts, Mol. Cell.
Biol., 9, 3088-3092.
11.Hanahan, D., and Weinberg, R. A. (2000) The
hallmarks of cancer, Cell, 100, 57-70.
12.Marian, C. O., Wright, W. E., and Shay, J. W.
(2010) The effects of telomerase inhibition on prostate
tumor-initiating cells, Int. J. Cancer, 127, 321-331.
13.Marian, C. O., Cho, S. K., McEllin, B. M., Maher,
E. A., Hatanpaa, K. J., and Madden, C. J. (2010) The telomerase
antagonist, imetelstat, efficiently targets glioblastoma
tumor-initiating cells leading to decreased proliferation and tumor
growth, Clin. Cancer Res., 16, 154-163.
14.Oulton, R., and Harrington, L. (2004) A human
telomerase-associated nuclease, Mol. Biol. Cell, 15,
3244-3256.
15.Lydeard, J. R., Jain, S., Yamaguchi, M., and
Haber, J. E. (2007) Break-induced replication and
telomerase-independent telomere maintenance require Pol32,
Nature, 448, 820-823.
16.Nagata, S., Nagase, H., Kawane, K., Mukae, N.,
and Fukuyama, H. (2003) Degradation of chromosomal DNA during
apoptosis, Cell Death Differ., 10, 108-116.
17.Ruiz-Carrillo, A., and Renaud, J. (1987)
Endonuclease G: a (dG)n X (dC)n-specific DNase from higher eukaryotes,
EMBO J., 6, 401-407.
18.Diener, T., Neuhaus, M., Koziel, R., Micutkova,
L., and Jansen-Durr, P. (2010) Role of endonuclease G in
senescence-associated cell death of human endothelial cells, Exp.
Gerontol., 45, 638-644.
19.Basnakian, A. G., Apostolov, E. O., Yin, X.,
Abiri, S. O., Stewart, A. G., and Singh, A. B. (2006) Endonuclease G
promotes cell death of non-invasive human breast cancer cells, Exp.
Cell Res., 312, 4139-4149.
20.Laemmli, U. K. (1970) Cleavage of structural
proteins during the assembly of the head of bacteriophage T4,
Nature, 227, 680-685.
21.Hofnagel, O., Luechtenborg, B., Stolle, K.,
Lorkowski, S., Eschert, H., and Plenz, G. (2004) Proinflammatory
cytokines regulate LOX-1 expression in vascular smooth muscle cells,
Arterioscler. Thromb. Vasc. Biol., 24, 1789-1795.
22.Kovalenko, N. A., Zhdanov, D. D., Bibikova,
M. V., and Gotovtseva, V. I. (2011) The influence of compound
aITEL1296 on telomerase activity and the growth of cancer cells,
Biomed. Khim., 57, 501-510.
23.O’Callaghan, N. J., and Fenech, M. (2011) A
quantitative PCR method for measuring absolute telomere length,
Biol. Proc. Online, 13, 3.
24.Cawthon, R. M. (2002) Telomere measurement by
quantitative PCR, Nucleic Acids Res., e47.
25.Pokrovsky, V. S., Treshalina, H. M., Lukasheva,
E. V., Sedakova, L. A., Medentzev, A. G., and Arinbasarova, A. Y.
(2013) Enzymatic properties and anticancer activity of L-lysine
α-oxidase from Trichoderma cf. aureoviride Rifai
BKMF-4268D, Anticancer Drugs, 24, 846-851.
26.Ruden, M., and Puri, N. (2013) Novel anticancer
therapeutics targeting telomerase, Cancer Treat. Rev.,
39, 444-456.
27.Zhdanov, D. D., Vasina, D. A., Orlova, V. S.,
Gotovtseva, V. Y., Bibikova, M. V., Pokrovsky, V. S., Pokrovskaya, M.
V., Aleksandrova, S. S., and Sokolov, N. N. (2016) Apoptotic
endonuclease EndoG induces alternative splicing of telomerase catalytic
subunit hTERT and death of tumor cells, Biomed. Chem.,
62, 239-250.
28.Effros, R. B., and Pawelec, G. (1997) Replicative
senescence of T cells: does the Hayflick limit lead to immune
exhaustion? Immunol. Today, 18, 450-454.
29.Counter, C. M., Gupta, J., Harley, C. B., Leber,
B., and Bacchetti, S. (1995) Telomerase activity in normal leukocytes
and in hematologic malignancies, Blood, 85,
2315-2320.
30.Moro-Garcia, M. A., Alonso-Arias, R., and
Lopez-Larrea, C. (2012) Molecular mechanisms involved in the aging of
the T-cell immune response, Curr. Genom., 13,
589-602.
31.Hodes, R. J., Hathcock, K. S., and Weng, N.
(2002) Telomeres in T and B cells, Nat. Rev. Immunol., 2,
699-706.
32.Read, M. A., Wood, A. A., Harrison, J. R., Gowan,
S. M., Kelland, L. R., and Dosanjh, H. S. (1999) Molecular modeling
studies on G-quadruplex complexes of telomerase inhibitors:
structure–activity relationships, J. Med. Chem.,
42, 4538-4546.
33.Zhou, Z., Du, Y., Zhang, L., and Dong, S. (2012)
A label-free, G-quadruplex DNAzyme-based fluorescent probe for
signal-amplified DNA detection and turn-on assay of endonuclease,
Biosens. Bioelectron., 34, 100-105.
34.Martadinata, H., Heddi, B., Lim, K. W., and Phan,
A. T. (2011) Structure of long human telomeric RNA (TERRA):
G-quadruplexes formed by four and eight UUAGGG repeats are stable
building blocks, Biochemistry, 50, 6455-6461.
35.Zhdanov, D. D., Fahmi, T., Wang, X., Apostolov,
E. O., Sokolov, N. N., and Javadov, S. (2015) Regulation of apoptotic
endonucleases by EndoG, DNA Cell Biol., 34, 316-326.
36.Sangle, N. A., Agarwal, A. M., Smock, K. J.,
Leavitt, M. O., Warnke, R., and Bahler, D. (2011) Diffuse large B-cell
lymphoma with aberrant expression of the T-cell antigens CD2 and CD7,
Appl. Immunohistochem. Mol. Morphol., 19, 579-583.