Status Epilepticus Impairs Synaptic Plasticity in Rat Hippocampus and Is Followed by Changes in Expression of NMDA Receptors
T. Y. Postnikova1,2, O. E. Zubareva1,3, A. A. Kovalenko3, K. K. Kim1, L. G. Magazanik1,4, and A. V. Zaitsev1*
1Sechenov Institute of Evolutionary Physiology and Biochemistry, Russian Academy of Sciences, 194223 St. Petersburg, Russia; E-mail: aleksey_zaitsev@mail.ru2Peter the Great St. Petersburg Polytechnic University, 195251 St. Petersburg, Russia
3Institute of Experimental Medicine, 197376 St. Petersburg, Russia
4Saint Petersburg State University, 199034 St. Petersburg, Russia
* To whom correspondence should be addressed.
Received November 1, 2016; Revision received November 14, 2016
Cognitive deficits and memory loss are frequent in patients with temporal lobe epilepsy. Persistent changes in synaptic efficacy are considered as a cellular substrate underlying memory processes. Electrophysiological studies have shown that the properties of short-term and long-term synaptic plasticity in the cortex and hippocampus may undergo substantial changes after seizures. However, the neural mechanisms responsible for these changes are not clear. In this study, we investigated the properties of short-term and long-term synaptic plasticity in rat hippocampal slices 24 h after pentylenetetrazole (PTZ)-induced status epilepticus. We found that the induction of long-term potentiation (LTP) in CA1 pyramidal cells is reduced compared to the control, while short-term facilitation is increased. The experimental results do not support the hypothesis that status epilepticus leads to background potentiation of hippocampal synapses and further LTP induction becomes weaker due to occlusion, as the dependence of synaptic responses on the strength of input stimulation was not different in the control and experimental animals. The decrease in LTP can be caused by impairment of molecular mechanisms of neuronal plasticity, including those associated with NMDA receptors and/or changes in their subunit composition. Real-time PCR demonstrated significant increases in the expression of GluN1 and GluN2A subunits 3 h after PTZ-induced status epilepticus. The overexpression of obligate GluN1 subunit suggests an increase in the total number of NMDA receptors in the hippocampus. A 3-fold increase in the expression of the GluN2B subunit observed 24 h after PTZ-induced status epilepticus might be indicative of an increase in the proportion of GluN2B-containing NMDA receptors. Increased expression of the GluN2B subunit may be a cause for reducing the magnitude of LTP at hippocampal synapses after status epilepticus.
KEY WORDS: long-term synaptic potentiation (LTP), CA1, epilepsy, NMDA receptors, GluN2B, GluN2ADOI: 10.1134/S0006297917030063
Abbreviations: ACSF, artificial cerebrospinal fluid; CycA, cyclophilin A; fEPSP, field excitatory postsynaptic potentials; LTP, long-term synaptic potentiation; NMDA, N-methyl-D-aspartate; PTZ, pentylenetetrazole.
Epileptic seizures in patients with temporal lobe epilepsy often result
in disturbances of cognitive functions, especially memory [1]. Animal models have shown that even single episodes
of seizures can lead to such disorders [2, 3]. Synaptic plasticity is considered to be one of the
most important neural mechanisms of memory [4].
Electrophysiological studies show that the characteristics of short-
and long-term synaptic plasticity in the cortex and hippocampus can
change substantially after seizures [5-7]. However, the pattern and underlying mechanisms of
these changes are still generally unclear because of contradictory
results of experimental studies. According to one hypothesis, the
epileptic activity can lead to background NMDA (N-methyl-D-aspartate)
receptor-dependent long-term synaptic potentiation (LTP) of all
excitatory synapses [8]. In turn, this weakens or
even makes impossible LTP induction and intensifies long-term synaptic
depression (or depotentiation) during several hours or days after
epileptic activity. This hypothesis has been confirmed by experiments
with in vitro models [5, 9]; however, they do not always correspond to the data
from in vivo models showing both a decrease [7, 10, 11]
and an increase in LTP [6, 12,
13] depending on experimental conditions.
Therefore, it may be supposed that the changes in LTP generation after
seizures are determined also by impairment of the mechanisms of
plasticity induction, e.g. by changes in the level of expression and/or
subunit composition of NMDA receptors.
NMDA receptors are heteromeric complexes consisting of two obligate GluN1 subunits and two GluN2 or GluN3 subunits. Receptors containing GluN2A and GluN2B subunits are predominant in the cortex and hippocampus [14]. The functional properties of NMDA receptors directly depend on their subunit composition [15]. It is known that status epilepticus influences both the total number of NMDA receptors in synapses [16] and the ratio of NMDA receptors with different subunit compositions [17]. The sign of synaptic plasticity (potentiation or depression) often depends on the subunit composition of receptors [18, 19]; therefore, these changes in NMDA receptors are one of the potential mechanisms of plasticity impairment after seizures. Recently, a lithium-pilocarpine model of status epilepticus with 3-week-old rats used in our laboratory has shown a decrease in LTP lasting at least 1 week [10]. This may be accounted for by changes in the subunit composition of NMDA receptors.
The present work was aimed at investigating the changes in LTP after status epilepticus in the pentylenetetrazole seizure model with Wistar rats, and the changes in expression of genes encoding the subunits of NMDA receptors.
MATERIALS AND METHODS
Animals. The study was performed on 20-22-day-old Wistar rats (35-40 g) kept under standard conditions at room temperature with free access to water and food. All experiments were carried out in accordance with the Guidelines on the Treatment of Laboratory Animals effective at the Sechenov Institute of Evolutionary Physiology and Biochemistry, Russian Academy of Sciences, and these comply with Russian and international standards. A total of 26 animals were used in electrophysiological experiments and 28 animals were used for biochemical analysis.
Pentylenetetrazole model of status epilepticus. In experimental animals, status epilepticus was induced by intraperitoneal administration of pentylenetetrazole (PTZ, 70 mg/kg; Sigma, USA). The animals with generalized tonic–clonic seizure lasting no less than 15 min, i.e. with diagnosed status epilepticus, were taken for the experiment. The control rats were administered normal saline solution. Gene expression level was measured 3 and 24 h after the administration of PTZ or normal saline solution; synaptic plasticity was examined after 24 h.
Measurement of gene expression level of subunits of glutamate NMDA receptors. Total RNA from hippocampal cells was isolated by the method of acidic guanidine isocyanate–phenol–chloroform extraction with TRI reagent (Molecular Research Center, USA) according to the manufacturer’s protocol. Reverse transcription was performed using oligo dT primers (Medigen, Russia) and MMLV-reverse transcriptase (Promega, USA). The level of expression of the genes encoding the GluN1, GluN2A and GluN2B subunits of NMDA receptors was measured by real-time PCR with a CFX-96 REAL TIME C1000 Touch™ Thermal Cycler (Bio-Rad, USA). The reference gene was the cyclophilin A (peptidylprolyl isomerase A, CycA) gene with previously proven validity for application in experimental models [20]. The TaqMan technology was used. The sequences of PCR primers and probes (DNK-Sintez, Russia) are given in the table. Each sample was analyzed in two parallels. The relative gene production was calculated by the 2−ΔΔCt method [21].
Sequences of real-time PCR primers and probes
Preparation of hippocampal slices. The rats were decapitated 24 h after status epilepticus. The brain was quickly removed, the cerebellum, frontal lobes of the hemispheres, and a part of the dorsal surface of the brain were cut off. Horizontal 400-µm brain slices were obtained using a vibratome HM 650V (Microm International, Germany) in ice-cold (0°C) carbogen-aerated (95% O2/5% CO2) artificial cerebrospinal fluid (ACSF). ACSF contained: NaCl, 126 mM; KCl, 2.5 mM; NaH2PO4, 1.25 mM; MgSO4, 1 mM; CaCl2, 2 mM; NaHCO3, 24 mM; and glucose, 10 mM. The prepared slices were immersed in a chamber with aerated ACSF, which was placed into a temperature-controlled water bath (35°C) for 1 h; then the slices were stored at room temperature. After the incubation, the slices were transferred into the experimental chamber, where they were kept for 15-20 min prior to the electrophysiological study. In this chamber, a constant flow of oxygenated ACSF solution (no less than 5 ml/min) was maintained at room temperature. One to five slices from each rat were used in the experiment.
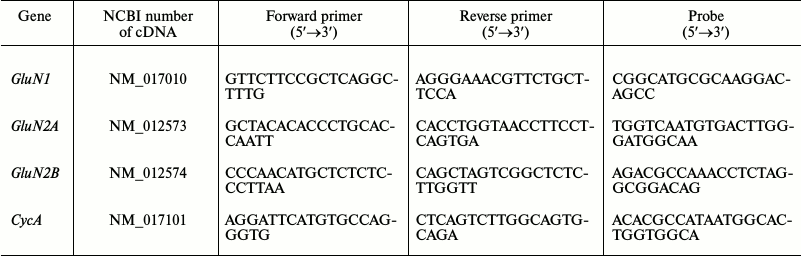
Electrophysiological experiments. The field excitatory postsynaptic potentials (fEPSP) were recorded in stratum radiatum of CA1 area with a glass microelectrode filled with ACSF (0.2-1.0 MΩ). A bipolar twisted stimulating electrode made of insulated nichrome wire (0.1 mm in diameter) was mounted in stratum radiatum, in the region of the Schaffer collateral pathway, between areas CA2 and CA1 of the rat hippocampus (Fig. 1a). The stimulation was performed with rectangular paired pulses (duration, 0.1 ms; interstimulus interval, 50 ms) every 20 s. The dependence of field response amplitude on stimulation strength was determined by increasing the current intensity from 25 to 400 µA at a pace of 25 µA. The stimulus intensity used in the experiment was chosen so that the amplitude of fEPSP would be 40-50% of the amplitude where the population spike appeared for the first time. The strength of stimulation was unvaried during the experiments, usually being 50-150 µA; the fEPSP amplitude was within the range of 0.5-1 mV. Short-term synaptic plasticity (facilitation) was investigated by comparing the ratios of fEPSP amplitudes during the paired stimulation.
Fig. 1. Positions of electrodes in the hippocampus and parameters of LTP induction. a) Scheme of the hippocampal slice showing positions of stimulating and recording electrodes; SC, Schaffer collaterals; b) parameters of theta-burst stimulation (TBS) for LTP induction.
The baseline fEPSPs were recorded for at least 30 min before LTP induction. LTP induction was started only if a stable amplitude of the baseline fEPSP had been recorded for 20 min. LTP was induced by theta-burst stimulation (five bursts of five 100-Hz pulses, with a 200-ms interval between bursts, applied five times every 10 s; Fig. 1b). The fEPSPs were recorded after induction protocol during 40 min.
Electrophysiological data were analyzed with the Clampfit 10.2 program (Axon Instruments, USA). For each fEPSP, the amplitude and the slope of the rising phase at a level of 20-80% of the peak amplitude were measured. The baseline fEPSP and the potentiated fEPSP (recorded 30-40 min after theta-burst stimulation) were averaged separately to measure LTP in a slice. Plasticity value was calculated as a ratio of the slope of the rising phase in the averaged potentiated and baseline fEPSP. The value of short-term synaptic facilitation was calculated as the amplitude ratio of the first to second fEPSP.
Statistical data processing. All data are presented as the mean with the standard error of the mean; the number of slices (for electrophysiological experiments) or the number of animals (for gene expression analysis) are given in brackets. The data were processed with SPSS Statistics 20 and OriginPro 8. The Kolmogorov–Smirnov test was used to examine normal distribution. The reliability of differences in two random samples was verified by Student’s t-test. The effect of the strength of stimulating current on the value of field responses in the control and experimental animals was tested by two-way analysis of variance.
RESULTS
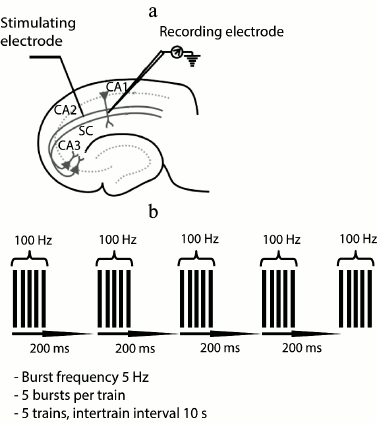
Status epilepticus reduces long-term synaptic potentiation in the hippocampus. We compared long-term synaptic potentiation in control and experimental rats 24 h after the PTZ-induced status epilepticus (Fig. 2). In the control animals, the theta-burst stimulation of Schaffer collaterals in hippocampal slices resulted in a marked LTP of fEPSP, and the slope of the rising phase of fEPSP increased 1.45 ± 0.05 times compared to the background level (n = 14). In the experimental rats, fEPSP increased only 1.29 ± 0.02 times (n = 10) with the same protocol, i.e. significantly less than in the control animals (t = 2.55; p < 0.05).
Fig. 2. Induction of LTP is weaker in animals after PTZ-induced status epilepticus. a) Diagram showing the normalized slope of fEPSP in control and experimental animals before and after theta-burst stimulation (TS). Normalization was made by the averaged background fEPSP. The inset (above) shows the examples of fEPSP before induction (baseline response) and 40 min after theta-burst stimulation (LTP) in the hippocampal field CA1 of rats from the control group (control) and from the experimental group after PTZ-induced status epilepticus (PTZ). The arrow TS shows the moment of LTP induction. b) Diagram illustrating differences in LTP value between groups. The results were obtained from 14 control and 10 experimental slices. All data in this and the following figures are presented as mean ± standard error of mean; * p < 0.05.
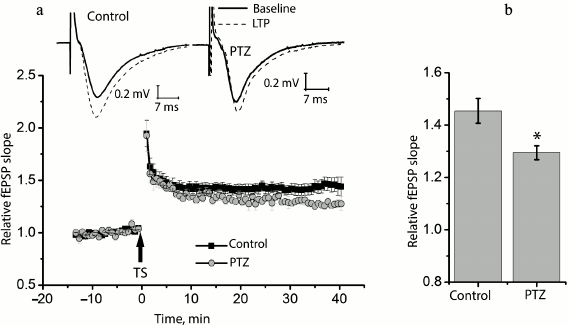
We checked whether there is a change in the paired-pulse ratio of amplitudes in potentiated field responses to determine the locus of induction of synaptic potentiation (Fig. 3). It is believed that the absence of changes is evidence in favor of postsynaptic locus of induction [22, 23]. In the control animals, the amplitude ratio did not change after response potentiation (baseline level, 1.36 ± 0.04; after potentiation, 1.35 ± 0.04; n = 14). It should be noted that the paired-pulse facilitation of responses in the experimental animals was initially higher (1.62 ± 0.10; n = 10); however, in this group, the theta-burst stimulation had no effect on paired-pulse facilitation either (1.63 ± 0.10). Thus, in both groups plasticity is induced on the postsynaptic membrane.
Fig. 3. The ratio of fEPSP amplitudes before and after the induction protocol. a) Examples of fEPSP responses before induction (baseline response) and 40 min after theta-burst stimulation (LTP) in hippocampal field CA1 of rats from the control group (control) and the experimental group after PTZ-induced status epilepticus (PTZ). b) Diagram showing that the amplitude ratio does not change after the induction protocol in both groups.
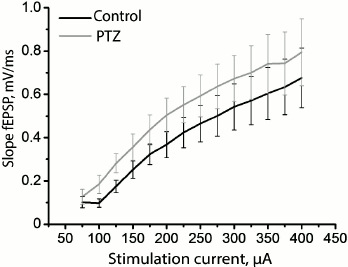
Since status epilepticus can cause potentiation of synaptic responses in the hippocampus, further potentiation of synapses will be impeded as a result of occlusion. Assuming that the hippocampal synapses in our model have been initially potentiated after status epilepticus, all other conditions being equal, the same force of stimulation will cause a response of greater amplitude. To test this hypothesis, we constructed a curve of dependence of the slope of the rising phase of fEPSP on the strength of the stimulating extracellular current (Fig. 4) and found that there are no reliable differences between the control and experimental animals (group factor F1.18 = 1.01; p = 0.33), while the runs of the two curves were not different either (group factor × current intensity factor F12.216 = 0.05; p = 0.999). Thus, the findings do not allow us to state that the synaptic responses of pyramidal neurons of the CA1 area to the stimulation of Schaffer collaterals are potentiated after status epilepticus.
Fig. 4. Dynamics of changes in the slope of the rising phase of fEPSP in the control group (black curve) and in the animals after PTZ-induced status epilepticus (gray curve) in response to the increasing intensity of electrical stimulation of Schaffer collaterals.
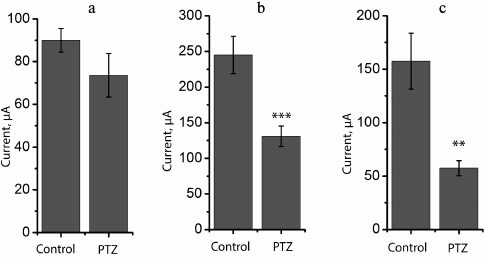
At the same time, we revealed a change in the shape of fEPSP response in most brain slices obtained from experimental animals. The typical fEPSP at different strengths of extracellular stimulation in the control and experimental animals are shown in Fig. 5. In brain slices of the experimental animals, population spikes started to appear in response to a relatively low current intensity (131 ± 14 µA, n = 17), while in the control animals they were recorded for the first time at a higher current intensity (245 ± 26 µA, n = 10, t = 4.21; p < 0.001; Figs. 5 and 6). Responses with several successive population spikes were recorded only in the brain slices of experimental animals (Fig. 5), which is indicative of neuronal hyperexcitability and impaired function of neural networks after status epilepticus.
Fig. 5. Change in shape of fEPSP in control rats and in two experimental rats (PTZ) after status epilepticus in response to increasing intensity of electrical stimulation of Schaffer collaterals. In the lower row, the same responses are shown with a vertical shift. The gray frame shows the formation of population spike in the slices.
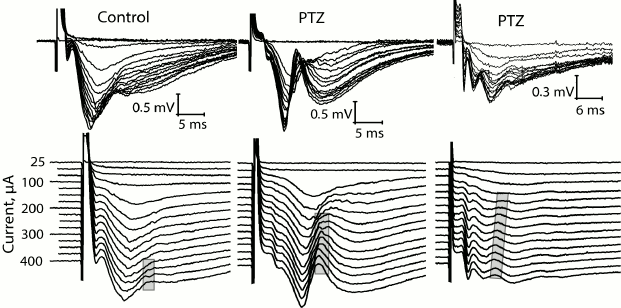
Fig. 6. Comparison of excitability of slices from animals of control (control) and experimental (PTZ) groups. Diagrams showing threshold value of extracellular current necessary for inducing fEPSP (a) and population spikes (b). Diagram (c) shows difference between these threshold currents; ** p < 0.01; *** p < 0.001.
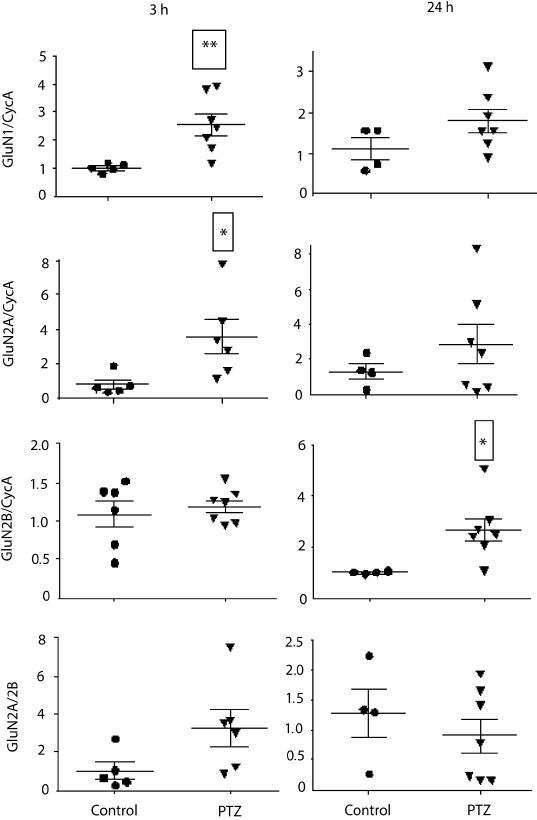
Variations in the level of expression of genes encoding NMDA receptor subunits. Since the change in LTP induction may be determined by changes in subunit composition of NMDA receptors [18, 19], we investigated the effect of PTZ-induced status epilepticus on the expression of NMDA receptor subunit mRNAs in the hippocampus by the method of real-time PCR (Fig. 7). The study was carried out 3 h after PTZ introduction, i.e. almost immediately after the termination of manifestations of status epilepticus, and in 24 h. In 3 h after the induction of convulsions, there was a considerable intensification of expression of the genes encoding subunits GluN1 (t = 3.9; p < 0.01) and GluN2A (t = 2.5; p < 0.05). The enhanced expression of obligate subunit GluN1 suggests an increase in the total number of NMDA receptors in the hippocampus. In 24 h, there was a 3-fold increase in the production of GluN2B mRNA (t = 2.7; p < 0.05), probably indicating a higher portion of GluN2B-containing NMDA receptors.
Fig. 7. Relative levels of expression of NMDA receptor subunit mRNAs 3 and 24 h after administration of pentylenetetrazole (n = 6-7, circles) or normal saline solution (n = 4-5, triangles). Normalized by cyclophilin A; * p < 0.05; ** p < 0.01.
DISCUSSION
In this work, we found that induction of LTP in CA3–CA1 synapses of rat hippocampus decreases and short-term synaptic facilitation increases 24 h after PTZ-induced status epilepticus. An analogous decrease in LTP value was revealed after status epilepticus in the lithium-pilocarpine model [10]. It has been shown that short-term convulsions induced by kainate application potentiate synaptic transmission in the CA3 area of the hippocampus [24]. The potentiation of excitatory synapses between the CA3–CA1 and CA3–CA3 pyramidal cells in organotypic hippocampal culture was observed both after many hours [5] or a very short period of epileptiform activity (0.5-3 min) [9] induced by bicuculline application. This potentiation was NMDA receptor-dependent, and the high involvement of NMDA receptors in epileptic activity has been revealed in different models [17, 25]. The potentiation of synapses under seizure activity subsequently leads to the weakening of LTP production in these synapses as a result of occlusion [5, 9, 24]. However, the decrease in LTP value in our experiments cannot be accounted for by only the effect of occlusion due to synaptic potentiation during status epilepticus, because the dependence of the values of synaptic responses on the strength of input stimulation was not different in the control and experimental animals.
The weakening of LTP induction may be determined by the impairment of molecular mechanisms of induction caused by status epilepticus. It is known that PTZ-induced convulsions result in multiple disturbances in the hippocampus and the cortex, including morphological alterations, reduced cell proliferation, and the changes in many enzyme activities and in the functions of different metabolic pathways and receptors [26-29]. Alteration of the functional properties of NMDA receptors after status epilepticus may be one of the causes of impaired induction of plasticity. Status epilepticus may be accompanied by the rapid transportation of NMDA receptors from internal cellular compartments to synapses. For example, it has been shown that immunoreactivity to the GluN1 subunit in the area of postsynaptic density increases 3-fold during one hour after lithium-pilocarpine status epilepticus, and the amplitude of NMDA receptor-dependent miniature postsynaptic responses also increases [16].
The incorporation of already existing receptors into the postsynaptic membrane may be accompanied by changes in expression of the genes of different subunits and the synthesis of new NMDA receptors. Expression of the genes of NMDA receptor subunits has been investigated previously in several experimental epileptic models [17, 30, 31]. In this study, we analyzed the change in the level of expression of the genes encoding the GluN1, GluN2A, and GluN2B subunits in the rat hippocampus in 3 and 24 h after PTZ-induced status epilepticus. The enhanced expression of the GluN2A subunit gene one hour after PTZ introduction in rat hippocampus was shown by Ahmadirad et al. [30], which is in agreement with our results. According to our results, the production of the GluN2B mRNA is intensified 24 h after PTZ-induced convulsions. Previous data indicate increased share of GluN2B-containing NMDA receptors after status epilepticus [16, 32]. Similar results were obtained in rats with chronic epilepsy in a study of synapses formed by Schaffer collaterals [6]. Excessive expression of GluN1 and GluN2B subunits was demonstrated in primary hippocampal culture in vitro model 24 h after exposure to pilocarpine [17].
It is well known that the subunit composition of NMDA receptors is directly associated with their functional activity [15, 33]. Some works have shown that the predominance of NMDA receptors of particular subunit composition may influence the sign of long-term synaptic plasticity. It has been shown that the GluN2A- and GluN2B-containing NMDA receptors play a key role in the induction of long-term potentiation and long-term depression, respectively [18, 34]. At the same time, the fundamentally important factor of regulation of long-term potentiation and depression thresholds is the GluN2A and GluN2B subunit production ratio [35].
The expression of GluN2B-containing NMDA receptors can increase both in synaptic and extrasynaptic areas of neurons; however, Frasca et al. revealed that this subtype of NMDA receptors can be redistributed among synaptic and extrasynaptic areas in favor of extrasynaptic localization [36]. Recent studies have shown that synaptic and extrasynaptic NMDA receptors have different effects on long-term synaptic plasticity: synaptic NMDA receptors contribute to LTP induction, while extrasynaptic receptors contribute to depression [37]. Thus, the enhanced expression of the GluN2B mRNA and, probably, the preferential incorporation of GluN2B-containing NMDA receptors into extrasynaptic areas may a cause of LTP decrease in hippocampal synapses after status epilepticus.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (projects Nos. 14-04-00413, 15-04-02951, 16-04-00998) and by the Program I.19 of the Presidium of the Russian Academy of Sciences.
REFERENCES
1.Giovagnoli, A. R., and Avanzini, G. (1999) Learning
and memory impairment in patients with temporal lobe epilepsy: relation
to the presence, type, and location of brain lesion, Epilepsia,
40, 904-911.
2.Aniol, V. A., Ivanova-Dyatlova, A. Y., Keren, O.,
Guekht, A. B., Sarne, Y., and Gulyaeva, N. V. (2013) A single
pentylenetetrazole-induced clonic-tonic seizure episode is accompanied
by a slowly developing cognitive decline in rats, Epilepsy
Behav., 26, 196-202.
3.Kalemenev, S. V., Zubareva, O. E., Frolova, E. V.,
Sizov, V. V., Lavrentyeva, V. V., Lukomskaya, N. Y., Kim, K., Zaitsev,
A. V., and Magazanik, L. G. (2015) Impairment of exploratory behavior
and spatial memory in adolescent rats in lithium–pilocarpine
model of temporal lobe epilepsy, Dokl. Biol. Sci., 463,
175-177.
4.Kandel, E. R. (2004) The molecular biology of
memory storage: a dialog between genes and synapses, Biosci.
Rep., 24, 475-522.
5.Abegg, M. H., Savic, N., Ehrengruber, M. U.,
McKinney, R. A., and Gahwiler, B. H. (2004) Epileptiform activity in
rat hippocampus strengthens excitatory synapses, J. Physiol.,
554, 439-448.
6.Muller, L., Tokay, T., Porath, K., Kohling, R., and
Kirschstein, T. (2013) Enhanced NMDA receptor-dependent LTP in the
epileptic CA1 area via upregulation of NR2B, Neurobiol. Dis.,
54, 183-193.
7.Zhou, J. L., Shatskikh, T. N., Liu, X., and Holmes,
G. L. (2007) Impaired single cell firing and long-term potentiation
parallels memory impairment following recurrent seizures, Eur. J.
Neurosci., 25, 3667-3677.
8.Meador, K. J. (2007) The basic science of memory as
it applies to epilepsy, Epilepsia, 48, 23-25.
9.Debanne, D., Thompson, S. M., and Gahwiler, B. H.
(2006) A brief period of epileptiform activity strengthens excitatory
synapses in the rat hippocampus in vitro, Epilepsia,
47, 247-256.
10.Kryukov, K. A., Kim, K. K., Magazanik, L. G., and
Zaitsev, A. V. (2016) Status epilepticus alters hippocampal long-term
synaptic potentiation in a rat lithium-pilocarpine model,
Neuroreport, 27, 1191-1195.
11.Suarez, L. M., Cid, E., Gal, B., Inostroza, M.,
Brotons-Mas, J. R., Gomez-Dominguez, D., de la Prida, L. M., and Solis,
J. M. (2012) Systemic injection of kainic acid differently affects LTP
magnitude depending on its epileptogenic efficiency, PLoS One,
7, e48128.
12.Guli, X., Tokay, T., Kirschstein, T., and
Kohling, R. (2016) Status epilepticus enhances depotentiation after
fully established LTP in an NMDAR-dependent but GluN2B-independent
manner, Neural Plast., 6592038.
13.O’Leary, H., Bernard, P. B., Castano, A.
M., and Benke, T. A. (2016) Enhanced long term potentiation and
decreased AMPA receptor desensitization in the acute period following a
single kainate induced early life seizure, Neurobiol. Dis.,
87, 134-144.
14.Monyer, H., Burnashev, N., Laurie, D. J.,
Sakmann, B., and Seeburg, P. H. (1994) Developmental and regional
expression in the rat brain and functional properties of four NMDA
receptors, Neuron, 12, 529-540.
15.Cull-Candy, S., Brickley, S., and Farrant, M.
(2001) NMDA receptor subunits: diversity, development and disease,
Curr. Opin. Neurobiol., 11, 327-335.
16.Naylor, D. E., Liu, H., Niquet, J., and
Wasterlain, C. G. (2013) Rapid surface accumulation of NMDA receptors
increases glutamatergic excitation during status epilepticus,
Neurobiol. Dis., 54, 225-238.
17.Di Maio, R., Mastroberardino, P. G., Hu, X.,
Montero, L., and Greenamyre, J. T. (2011) Pilocarpine alters NMDA
receptor expression and function in hippocampal neurons: NADPH oxidase
and ERK1/2 mechanisms, Neurobiol. Dis., 42, 482-495.
18.Bartlett, T. E., Bannister, N. J., Collett, V.
J., Dargan, S. L., Massey, P. V., Bortolotto, Z. A., Fitzjohn, S. M.,
Bashir, Z. I., Collingridge, G. L., and Lodge, D. (2007) Differential
roles of NR2A- and NR2B-containing NMDA receptors in LTP and LTD in the
CA1 region of two-week-old rat hippocampus, Neuropharmacology,
52, 60-70.
19.Wang, D., Cui, Z., Zeng, Q., Kuang, H., Wang, L.
P., Tsien, J. Z., and Cao, X. (2009) Genetic enhancement of memory and
long-term potentiation but not CA1 long-term depression in NR2B
transgenic rats, PLoS One, 4, e7486.
20.Swijsen, A., Nelissen, K., Janssen, D., Rigo, J.
M., and Hoogland, G. (2012) Validation of reference genes for
quantitative real-time PCR studies in the dentate gyrus after
experimental febrile seizures, BMC Res. Notes, 5,
685.
21.Livak, K. J., and Schmittgen, T. D. (2001)
Analysis of relative gene expression data using real-time quantitative
PCR and the 2−ΔΔCt method, Methods,
25, 402-408.
22.Zaitsev, A. V., and Anwyl, R. (2012) Inhibition
of the slow afterhyperpolarization restores the classical spike
timing-dependent plasticity rule obeyed in layer 2/3 pyramidal cells of
the prefrontal cortex, J. Neurophysiol., 107,
205-215.
23.Buonomano, D. V. (1999) Distinct functional types
of associative long-term potentiation in neocortical and hippocampal
pyramidal neurons, J. Neurosci., 19, 6748-6754.
24.Ben-Ari, Y., and Gho, M. (1988) Long-lasting
modification of the synaptic properties of rat CA3 hippocampal neurones
induced by kainic acid, J. Physiol., 404, 365-384.
25.Amakhin, D. V., Ergina, J. L., Chizhov, A. V.,
and Zaitsev, A. V. (2016) Synaptic conductances during interictal
discharges in pyramidal neurons of rat entorhinal cortex, Front.
Cell. Neurosci., 10, 233.
26.Zaitsev, A. V., Kim, K. K., Vasilev, D. S.,
Lukomskaya, N. Y., Lavrentyeva, V. V., Tumanova, N. L., Zhuravin, I.
A., and Magazanik, L. G. (2015) N-methyl-D-aspartate receptor channel
blockers prevent pentylenetetrazole-induced convulsions and
morphological changes in rat brain neurons, J. Neurosci. Res.,
93, 454-465.
27.Aniol, V. A., Stepanichev, M. Y., Lazareva, N.
A., and Gulyaeva, N. V. (2011) An early decrease in cell proliferation
after pentylenetetrazole-induced seizures, Epilepsy Behav.,
22, 433-441.
28.Ahmed, M. M., Arif, M., Chikuma, T., and Kato, T.
(2005) Pentylenetetrazol-induced seizures affect the levels of prolyl
oligopeptidase, thimet oligopeptidase and glial proteins in rat brain
regions, and attenuation by MK-801 pretreatment, Neurochem.
Int., 47, 248-259.
29.Zhvania, M. G., Ksovreli, M., Japaridze, N. J.,
and Lordkipanidze, T. G. (2015) Ultrastructural changes to rat
hippocampus in pentylenetetrazol- and kainic acid-induced status
epilepticus: a study using electron microscopy, Micron,
74, 22-29.
30.Ahmadirad, N., Shojaei, A., Javan, M.,
Pourgholami, M. H., and Mirnajafi-Zadeh, J. (2014) Effect of
minocycline on pentylenetetrazol-induced chemical kindled seizures in
mice, Neurol. Sci., 35, 571-576.
31.Davoudi, M., Shojaei, A., Palizvan, M. R., Javan,
M., and Mirnajafi-Zadeh, J. (2013) Comparison between standard protocol
and a novel window protocol for induction of pentylenetetrazol kindled
seizures in the rat, Epilepsy Res., 106, 54-63.
32.Wasterlain, C. G., Naylor, D. E., Liu, H.,
Niquet, J., and Baldwin, R. (2013) Trafficking of NMDA receptors during
status epilepticus: therapeutic implications, Epilepsia,
54, 78-80.
33.Paoletti, P., Bellone, C., and Zhou, Q. (2013)
NMDA receptor subunit diversity: impact on receptor properties,
synaptic plasticity and disease, Nat. Rev. Neurosci., 14,
383-400.
34.Fox, C. J., Russell, K. I., Wang, Y. T., and
Christie, B. R. (2006) Contribution of NR2A and NR2B NMDA subunits to
bidirectional synaptic plasticity in the hippocampus in vivo,
Hippocampus, 16, 907-915.
35.Xu, Z., Chen, R. Q., Gu, Q. H., Yan, J. Z., Wang,
S. H., Liu, S. Y., and Lu, W. (2009) Metaplastic regulation of
long-term potentiation/long-term depression threshold by
activity-dependent changes of NR2A/NR2B ratio, J. Neurosci.,
29, 8764-8773.
36.Frasca, A., Aalbers, M., Frigerio, F.,
Fiordaliso, F., Salio, M., Gobbi, M., Cagnotto, A., Gardoni, F.,
Battaglia, G. S., Hoogland, G., Di Luca, M., and Vezzani, A. (2011)
Misplaced NMDA receptors in epileptogenesis contribute to
excitotoxicity, Neurobiol. Dis., 43, 507-515.
37.Parsons, M. P., and Raymond, L. A. (2014)
Extrasynaptic NMDA receptor involvement in central nervous system
disorders, Neuron, 82, 279-293.