REVIEW: Mechanisms of Brain Glucocorticoid Resistance in Stress-Induced Psychopathologies
V. M. Merkulov, T. I. Merkulova, and N. P. Bondar*
Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, 630090 Novosibirsk, Russia; E-mail: nbondar@bionet.nsc.ru* To whom correspondence should be addressed.
Received October 18, 2016; Revision received November 30, 2016
Exposure to stress activates the hypothalamic–pituitary–adrenal axis and leads to increased levels of glucocorticoid (GC) hormones. Prolonged elevation of GC levels causes neuronal dysfunction, decreases the density of synapses, and impairs neuronal plasticity. Decreased sensitivity to glucocorticoids (glucocorticoid resistance) that develops as a result of chronic stress is one of the characteristic features of stress-induced psychopathologies. In this article, we reviewed the published data on proposed molecular mechanisms that contribute to the development of glucocorticoid resistance in brain, including changes in the expression of the glucocorticoid receptor (GR) gene, biosynthesis of GR isoforms, and GR posttranslational modifications. We also present data on alterations in the expression of the FKBP5 gene encoding the main component of cell ultra-short negative feedback loop of GC signaling regulation. Recent discoveries on stress- and GR-induced changes in epigenetic modification patterns as well as normalizing action of antidepressants are discussed. GR and FKBP5 gene polymorphisms associated with stress-induced psychopathologies are described, and their role in glucocorticoid resistance is discussed.
KEY WORDS: glucocorticoid resistance, stress-induced psychopathologies, glucocorticoid receptor, FK506 binding protein 5 (FKBP5), epigenetic modificationsDOI: 10.1134/S0006297917030142
Abbreviations: ACTH, adrenocorticotropic hormone (corticotropin); DNMT, DNA methyltransferase; FKBP4, FK506 binding protein 4 (immunophilin); FKBP5, FK506 binding protein 5 (immunophilin); GC, glucocorticoid hormones (glucocorticoids); GR, glucocorticoid receptors; GRE, glucocorticoid-responsive element; HPA, hypothalamic–pituitary–adrenal axis; IL1, interleukin 1; TNF, tumor necrosis factor; UTR, untranslated region.
Chronic stress contributes significantly to the development of various
psychopathologies, including major depressive disorder (MDD) and
bipolar disorder [1, 2].
Normally, short-term stress activates the
hypothalamic–pituitary–adrenal (HPA) axis, which results in
the release of glucocorticoid hormones (GCs) (cortisol in humans and
corticosterone in rodents) by the adrenal glands [3]. After entering a cell, these hormones bind to the
glucocorticoid receptor (GR, NR3C1); the GR is then translocated to the
nucleus, where it positively or negatively regulates gene expression by
binding to specific DNA regions and/or interacting with other
transcription factors [4-6].
The GC-induced changes in the expression of most of these genes provide
the organism’s adequate response to stress, as well as
termination of the stress response [3, 7]. In particular, GR activation in neurons of the
hypothalamic paraventricular nucleus and in adenohypophyseal
corticotrophs inhibits expression of the CRH
(corticotropin-releasing hormone) and POMC (proopiomelanocortin)
genes, respectively [8, 9], and
initiates a negative feedback mechanism that decreases the HPA axis
activity and prevents negative consequences of long-term increase in GC
levels [3, 7].
However, this HPA axis-regulating negative feedback mechanism is impaired in chronic stress and in several stress-induced psychiatric disorders. Clinical studies have demonstrated that these pathological conditions are usually accompanied by HPA axis hyperactivation with simultaneous attenuation of the inhibitory effect of GCs on the production of adrenocorticotropic hormone (ACTH) and cortisol (i.e. by GC resistance), as estimated by the dexamethasone test [10-15] or the combined Dex-CRH test that assesses suppression by dexamethasone/stimulation by CRH [16, 17]. The GC resistance, which is observed in 80% of MDD patients characterized by long-term depressed mood and considerable changes in the neurovegetative and cognitive functions, is considered the most reproducible physiological trait of this disorder [11, 14]. Impairments in the HPA axis functioning, including GC resistance, are also typical for patients with unipolar and bipolar affective disorders [18, 19].
It is known that prolonged increase in the GC levels in chronic stress significantly affects brain neuroplasticity. Deteriorations of neuron–neuron connections are a typical feature of stress-induced psychiatric disorders [20]. In particular, stress strongly affects dendrites and postsynaptic dendritic spines in many brain regions. In the hippocampus, chronic stress causes atrophy of apical dendrites of pyramidal neurons in the CA1 and CA3 regions and decreases the density of dendritic spines on the postsynaptic neurons [21-24]. In addition, chronic stress disturbs neurogenesis in the dentate gyrus of the hippocampus [25, 26]. In general, induction of resistance to stress hormones might be considered as a manifestation of brain plasticity, since it develops in response to chronic action and drastically changes the mechanism of cell response to external stimuli. Therefore, normalization of the HPA axis activity is an important prerequisite for successful treatment of stress-induced disorders and restoration of neuronal plasticity [11, 17, 20]. The mechanism of therapeutic action of many antidepressants involves the restoration of the GR-mediated negative feedback mechanism to decrease the HPA axis activity [14, 27, 28].
In this article, we review the molecular mechanisms of GC resistance formation in the brain induced by the long-term exposure to stress factors. These mechanisms include changes in GR expression, GR posttranslational modifications, changes in the expression of the GR co-chaperone protein FKBP5, and GR-mediated alterations in epigenetic modification patterns. We also described some examples of GC response normalization by antidepressants.
Suppression of GR expression and posttranslational GR modifications as possible mechanisms of glucocorticoid resistance development in stress-INduced psychIATRIC disorders
Glucocorticoid receptor (GR) is a ligand-dependent transcription factor that regulates expression of hundreds of genes. In the absence of the hormone, GR is retained in the cytoplasm by forming a complex with several molecular chaperones. After binding the hormone, GR is released from this complex and translocated to the nucleus, where it interacts with specific DNA regions (glucocorticoid-responsive elements, GREs) of the target genes and either activates or represses these genes [4-6]. Since GR plays a key role in GC regulation, changes in the levels of its expression were originally considered as the most probable cause for the development of GC resistance in psychiatric disorders [11, 14].
In the early stages of GC resistance research (from 1985 to 1997), many studies compared the GR contents in blood cells and fibroblasts of MDD patients and their healthy counterparts. The GR levels were estimated from the binding of labeled hormone to proteins in total cell lysates or cytosols. No differences between the GR contents were observed in the total cell lysates of healthy and depressed individuals, and only slight decrease in the GR levels in the cytosol was found in patients with depression, which was most probably due to the receptor translocation to the nuclei [14].
Later studies focused on the expression of GR-encoding gene by studying postmortem samples of various brain structures from patients with stress-induced disorders and healthy subjects. The results of these studies were contradictory and depended on the brain region studied. Thus, in situ hybridization revealed no differences in the levels of GR mRNA in the hippocampi of six MDD patients that committed suicide and the control subjects [29]. Real-time PCR demonstrated that the total contents of GR mRNA in the amygdala, hippocampus, gyrus frontalis inferior, cingulate gyrus, and nucleus accumbens were similar in six MDD patients and in six healthy control subjects [30]. However, in situ hybridization showed that the contents of GR mRNA in frontal cortex layers III-VI in patients with depression and in entorhinal cortex layers III and VI and subiculum in patients with bipolar disorder were decreased compared to the control (15 samples in each group) [31]. Similar decrease in GR mRNA levels was shown by in situ hybridization in basolateral and lateral nuclei of the amygdala in patients with bipolar disorder [32].
Another source of information on GR expression levels is the data of comparative studies of transcriptomes in postmortem samples of various brain regions from patients with psychiatric disorders and healthy individuals that are deposited in various databases. We analyzed data that were obtained by microarray assay and RNA-sequencing (RNA-seq, also called whole transcriptome shotgun sequencing). Comparison of frontal cortex samples from 25 MDD patients and 25 healthy controls (GEO NCBI database: GSE54570 and GSE54575 [33]) and from 18 bipolar disorder patients and 18 healthy counterparts (NCBI BioProject Accessions: PRJNA235930 and PRJNA231202 [34]) revealed no significant difference between the total GR mRNA levels in healthy and affected individuals.
Based on accumulated evidence, GC resistance is highly unlikely to be related to the total decrease in the GR mRNA levels in brain structures in most patients. However, this does not exclude the possibility that GR expression might be decreased in some affected individuals or that GR expression is drastically downregulated in some brain regions. This problem needs further investigation.
It is important to note that most studies on GR levels in cells estimate expression of the “classic” GRα isoform (777 a.a.). In the absence of the hormone, this isoform forms a complex with heat-shock proteins in the cytoplasm and acts as a ligand-dependent transcription factor. However, the structure of the GR gene implies the possibility of alternative mRNA splicing (Fig. 1) and the use of alternative translation start codons (Fig. 2), which would generate multiple functionally different isoforms [35, 36]. For example, the GRβ isoform (742 a.a.) is formed by using the alternative acceptor splicing site in exon 9 (Fig. 1). GRβ does not bind the hormone and acts as a dominant inhibitor of GRα [37]. In addition to GRα inhibition, GRβ exhibits regulatory functions and acts as an inducer/repressor of numerous genes not regulated by GRα [38, 39]. Normally, the content of GRβ in most tissues and cell lines is either ten times lower than that of GRα, or this isoform is not detected at all [40]. However, GRβ becomes dominant in cells treated with proinflammatory cytokines TNFα and IL1 and mediates the development of GC resistance in these cells [41]. High levels of GRβ are observed in some patients with hormonal therapy-insensitive forms of asthma, rheumatoid arthritis, systemic lupus erythematosus, acute lymphoblastic leukemia, etc. [42]. In theory, GC resistance might also be related to an increase in the production of other GR isoforms, such as GR-P and GR-A (Fig. 1), which are also incapable of hormone binding [43].
Fig. 1. GR isoforms generated by alternative splicing. Exons and introns are designated with Arabic and Roman numbers, respectively; open boxes, noncoding regions; α and β, exon 9 fragments coding for the C-termini of GRα and GRβ isoforms, respectively; NTD, N-terminal domain; DBD, DNA-binding domain; LBD, ligand-binding domain; numbers on the right indicate protein molecule length in a.a. Intron VII located between exons 7 and 8 is not spliced out (as shown with a crossed circle). The reading frame in the beginning of this intron contains a stop codon, resulting in the synthesis of GR-P isoform with the truncated by 101 a.a. ligand-binding domain incapable of GC binding.
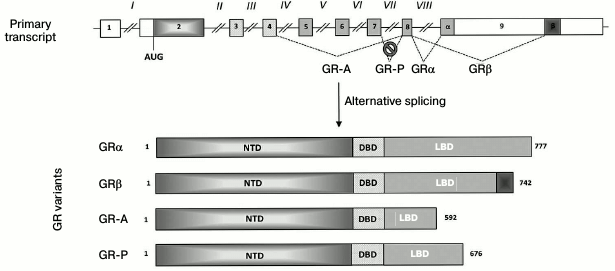
Fig. 2. Translational isoforms of GRα. Arrows with letters A-D designate positions of alternative AUG codons in the mRNA coding for the corresponding isoforms (the number of arrows corresponds to the number of AUG codons). Numbers inside the boxes, exons; NTD, N-terminal domain; DBD, DNA-binding domain; LBD, ligand-binding domain; numbers on the right indicate numbers of the last amino acid residue.
So far, studies on the occurrence of GR mRNA isoforms in brain structures of patients with psychiatric disorders are very scarce; however, the results of these studies are extremely interesting. Thus, it was found that the relative content of GRα mRNA in the postmortem samples of the amygdala and cingulate gyrus in six MDD patients was lower than in six healthy subjects, while the total contents of GR mRNA were the same in healthy and depressed individuals [30]. Similarly, studies of the mRNA levels for GRα and GRβ and the contents of the corresponding encoded proteins by real-time PCR and Western blotting, respectively, showed that the relative contents of GRα mRNA and GRα protein were lower in the prefrontal cortex and amygdala, but not hippocampus, of 24 suicide victims compared to the controls [44]. It was also found that polymorphism in the noncoding part of the GR gene exon 9 (rs6198, A → G) is associated with MDD and prevalence of depression symptoms in bipolar disorder patients [45]. It is known that this nucleotide substitution disturbs the destabilization site AUUUA in the 3′-UTR of the GRβ mRNA by converting it into GUUUA, which increases the lifetime of the corresponding mRNA and results in the accumulation of the GRβ isoform [46]. Since it is GRα that provides “correct” GC response, it is reasonable to assume that a decrease in its relative content might cause GC resistance.
The development of GC resistance in psychopathologies might also be related to the increased production of some GR translational isoforms (Fig. 2). In particular, exon 2, which is the first translated exon of the GR gene (Fig. 1), contains the ER22/23EK polymorphism that includes two linked oligonucleotide substitutions in codons 22 and 23. The first substitution (GAG → GAA) is synonymous, because both triplets code for glutamate (E). The second substitution (AGG → AAG) results in the replacement of conserved arginine (R) with lysine (K). Individuals with the affected gene are predisposed to depression [47]. They exhibit GC resistance in the dexamethasone test [48] and display more rapid response to antidepressants [47]. The described single-nucleotide substitutions alter the mRNA structure and, as a result, increase production of the GR translational form A that is less active than the B isoform, which appears to be the cause of GC resistance in ER22/23EK carriers [49]. It should be noted that the ratio between GR translational isoforms is affected by external factors, including various stressors [35, 50], and therefore might contribute to the sensitivity of target cells to GCs.
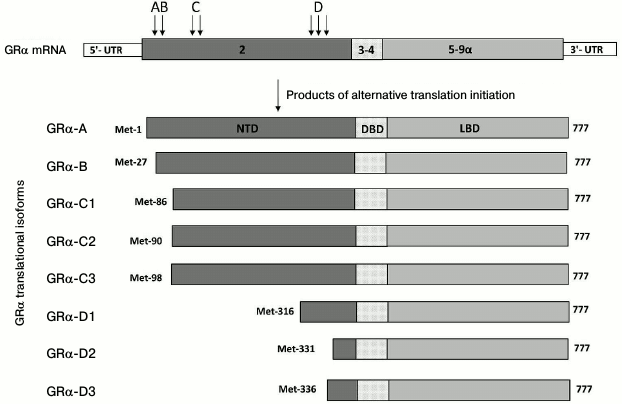
Although the mammalian genome contains a single gene for GR, this gene has at least nine alternative promoters with adjacent untranslated first exons, which results in the synthesis of a series of transcripts with different 5′-UTRs (Fig. 3) [51, 52]. The ratio between these transcripts can be altered in patients with psychiatric disorders. Thus, in several brain structures of individuals with MDD, the relative contents of transcripts containing 1B, 1C, and 1F exons were decreased, while the relative contents of transcripts with 1D and 1J exons were increased [30]. Interestingly, human GR exon 1F is an ortholog of exon 1(7) of mouse and rat GR genes. In these experimental animals, prenatal stresses or diminished maternal care in the early postnatal period result in increased methylation of the promoter upstream of the exon 1(7), decreased GR expression, and diminished efficiency of the GR-mediated negative feedback mechanism [53, 54]. However, Alt et al. [30] found no difference in the extent of methylation of the promoter upstream of the 1F exon in healthy and affected subjects. Nevertheless, since the 5′-UTR length and structure play an important role in the posttranscriptional regulation of the GR gene expression by affecting mRNA stability, translation effectiveness and forming protein isoforms [52], it is reasonable to assume that changes in the ratio between transcripts transcribed from different promoters of the same gene contribute to the development of GC resistance in psychiatric disorders.
Fig. 3. Alternative non-translated exons 1 of the human GR gene. Upper panel, schematic representation of exons (as open arrows); lower panel, alternative splicing as shown for the spicing of exons 1A3 and 1B to exon 2; AUG, translation start.
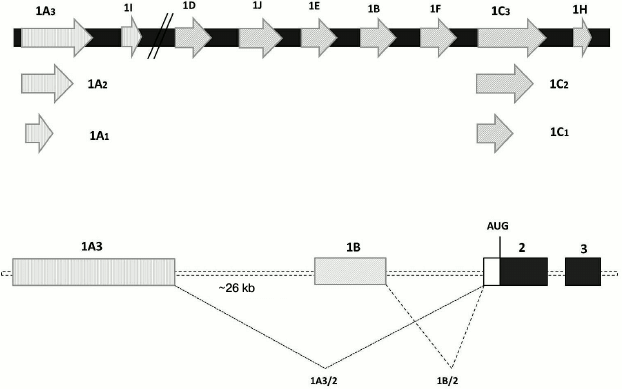
Another mechanism presumably involved in the formation of GC resistance is posttranslational modifications of GR. The best-studied of these modifications is phosphorylation. Human GR is phosphorylated predominantly on five serine residues (S203, S211, S113, S226, and S141). All these residues are in the N-terminal domain (Fig. 4) [55, 56] whose main function is regulation of the target gene transcription by interacting with a component of the basal transcriptional machinery [57] and/or protein cofactors [58]. It is known that the state of phosphorylation determines GR activation, subcellular localization, and recycling [56, 59]. GR is phosphorylated by cyclin-dependent protein kinases (S203, S211, S226), mitogen-activated protein kinases (S211, S226), casein kinase II (S113), and serum- and GC-activated kinase 1 (S203, S211) [56, 60-62].
Fig. 4. Serine residues undergoing phosphorylation in the GR N-terminal domain and the corresponding protein kinases. For designations, see Fig. 2.
Development of GC resistance in psychiatric disorders is now commonly believed to be related to increased GR phosphorylation at the serine S226 residue [63]. Phosphorylation by S226 inhibits the transactivator function of the receptor and promotes its exit from the nucleus [59, 61], which suggest that this modification might be a possible cause for GC resistance. Indirect evidences have been obtained that corroborate this hypothesis. In particular, in rats with an experimental model of depression-like behavior, chronic stress induced by social isolation caused an increase in the phosphorylation of S246 (the analog of human GR S226) in the hippocampus, whereas treatment with antidepressant fluoxetine restored normal phosphorylation levels and normalized the animals’ behavior [64]. Studies of the antidepressant effects of white mulberry (Morus alba L.) root extract and bee propolis showed that treatment of rats and mice with these substances decreased the levels of phosphorylation of GR residues S246 in rats and S234 in mice (analogs of human GR S226) in the hippocampi of treated animals. Simultaneously, the treatment increased phosphorylation of S232 in rats and S220 in mice [65] (residues corresponding to S211 in human GR). It is known that phosphorylation of human GR at S211 elevates it transactivation activity [62]. Therefore, both tested compounds caused GR activation, which presumably explains their antidepressant effect. Higher levels of S226 phosphorylation were also observed in leukocytes of MDD patients [66]. Taken together, these data suggest that S226 is involved in the mechanism of development of GC resistance, and it role in this process should be investigated further.
ROLE of FKBP5 in development OF glucocorticoid resistance
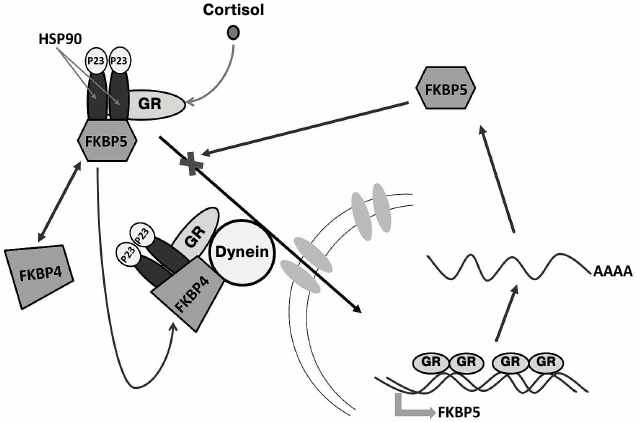
Immunophilin FKBP5 (FKBP51) is a component of the multiprotein complex that retains the cytoplasmic form of GR in the cytoplasm [67]. The complex includes one GR molecule, a dimer of a HSP90 heat-shock protein, and several other molecular chaperones and co-chaperones (HSP70, DnaJ/HSP40, p23, Hop, FKBP5, etc.) that keep GR in the hormone-binding conformation and protect it from proteolytic digestion [68, 69]. After GR binds the hormone, FKBP5 in the complex is rapidly replaced by another immunophilin – FKBP4 (FKBP52); the complex is then translocated into the nucleus [70] due to direct interaction between FKBP4 and the motor protein dynein that can move along the microtubules toward the nucleus [71].
Expression of the FKBP5 gene in humans and experimental animals is induced by GCs. In blood cells and bronchial epithelium, GCs increase the amounts of both FKBP5 mRNA and the corresponding protein product [72-75]. Experiments in animal models showed that FKBP5 expression is induced by GCs in all brain regions [76, 77]. The human FKBP5 gene contains numerous sites for GR binding that are located in introns 2, 5, and 7 [73, 78, 79]. Binding of the hormone-activated GR to these sites initiates an intracellular ultra-short feedback loop, when GC-induced activation of the FKBP5 gene transcription and increase in the FKBP5 protein content in the cytoplasm inhibit GR translocation to the nucleus, thereby diminishing the effect of GCs on the expression of GR target genes. Such genes include CRH and POMC, and impaired suppression of their expression by GCs might be the cause for development of GC resistance in chronic stress and stress-induced psychopathologies (Fig. 5) [18, 80, 81].
Fig. 5. FKBP5-mediated ultra-short feedback loop in the mechanism of GC regulation. In the absence of hormones, GR is retained in the cytoplasm in a multiprotein complex containing one GR molecule, HSP90 heat-shock protein dimer, HSP90-binding protein P23, and immunophilin FKBP5. After binding the hormone, FKBP5 is replaced by another immunophilin – FKBP4. This results in translocation of the complex to the nucleus due to the interaction between FKBP4 and the motor protein dynein that moves toward the nucleus along microtubules of the cytoskeleton. In the nucleus, GR induces biosynthesis of FKBP5, which blocks GR translocation to the nucleus due to competition with FKBP4.
Direct evidence for the existence of FKBP5-mediated GC resistance has been obtained in studies of South American squirrel monkeys (saimiri). Animals of this species are characterized by increased blood levels of cortisol that are 50-100 times higher than in the blood of other primates (including humans). However, squirrel monkeys display no symptoms of hypercortisolemia [82]. It was found that GC resistance of their target organs is provided by both upregulation of FKBP5 gene expression and specific features of the encoded protein. The FKBP5 content in the cytosol is an order of magnitude higher, which impedes FKBP5 exchange for FKBP4 in the complex and prevents GR translocation to the nucleus [83, 84]. Moreover, the presence of saimiri FKBP5 in the complex considerably decreases GR affinity to GCs [85].
The involvement of FKBP5 in GC resistance was demonstrated in animals with chronic stress-induced depression-like behavior. Thus, mild chronic stress upregulated FKBP5 gene expression and increased FKBP5 protein content in rat prefrontal cortex and ventral and dorsal regions of the hippocampus [86]. Simultaneously, the stress diminished GR translocation to the nuclei and suppressed GC induction of several target genes. When the animals were treated with duloxetine (a selective serotonin reuptake inhibitor), all the parameters reversed back to normal values, and the HPA axis functioning was normalized [86]. A similar effect was observed when putative antidepressant RO-05 (an inhibitor of serotonin, dopamine, and noradrenaline reuptake) was used [87].
In addition to its inhibitory effect on the GC-mediated signaling, FKBP5 blocks the Akt (protein kinase B) signaling pathway. FKBP5 acts as a scaffold protein that provides interaction between Akt and phosphatase PHLPP. PHLPP dephosphorylates Akt at the serine 473 residue, which inactivates Akt [88]. Since changes in Akt signaling are involved in the development of stress-induced psychiatric disorders and normalization of Akt signaling is one of the molecular effects of psychotropic medications [89-91], FKBP5 expression and its state in various psychopathologies require further study.
All these data have aroused considerable interest in the human FKBP5 gene as a factor involved in the development of various psychopathologies and accompanying GC resistance, as well as a gene that determines sensitivity/resistance to the action of psychotropic medications [14, 18, 27, 28]. Several single-nucleotide polymorphisms (SNPs) associated with psychiatric disorders have been identified in the noncoding FKBP5 gene regions [17, 92-96]. At least three of these (rs4713916, rs1360780, and rs3800737) have been associated with GC resistance [97]. The best-studied polymorphism is rs1360780 (G/A) located in FKBP5 intron 2 at a distance of 488 bp from the identified GRE [73]. Patients with MDD have a higher frequency of occurrence of the rs1360780 genotype AA(TT) [92, 95]. Patients with this genotype display poorer response to standard antidepressant therapy than individuals homozygous in the G allele [98] and exhibit some traits of GC resistance [99]. Moreover, in a psychosocial stress test, the levels of GC secretion in healthy individuals (homozygous in the A allele) did not restore completely after the stress [97, 100]. The data on the effect of G → A substitution on FKBP5 expression are contradictory. On one hand, experiments with genetic constructs bearing a fragment of the FKBP5 intron 2 containing rs1360780 and GRE showed that the A(T) allele considerably increases both basal and GC-induced expression of the reported gene in transfected HeLa cells [81]. On the other hand, studies on the effect of dexamethasone on FKBP5 mRNA level in blood cells of healthy individuals and MDD patients revealed that GCs downregulate FKBP5 expression in carriers of the A(T) allele [99, 100]. Since the same individuals exhibited lower sensitivity to GCs [99, 100], it might appear that these results contradict the commonly accepted notion that increased FKBP5 expression promotes GC resistance in stress-induced psychopathologies [18]. However, there are no data on the effect of GCs on FKBP5 expression in brain cells of subjects with different rs1360780 alleles. Considering current understanding of the mechanisms of tissue-specific GC regulation [101, 102], we suggest that in brain structures the effects of GCs on FKBP5 expression in the carriers of the risk allele would be opposite to the GC effects in blood cells. This problem can be elucidated by studying the effects of GCs on the FKBP5 mRNA levels in cultured neural cells obtained from carriers of alternative rs1360780 alleles.
Summarizing all the above said, further studies of human FKBP5 gene expression in brain structures will contribute much to revealing the mechanisms of GC resistance in stress-induced psychopathologies and to more comprehensive understanding of molecular causes of the development of such pathologies, as well as allow the development of new therapeutic approaches for their treatment. An example of such studies is a recent discovery of FKBP5 inhibitors SAFit1 and SAFit2 that activate axon growth in neural cell cultures and normalize HPA axis functioning in mice subjected to acute stress [103].
Chronic stress-induced changes in the chromatin epigenetic landscape as a possible cause for glucocorticoid resistance
When binding to DNA, GRs and other transcription factors recruit various cofactor proteins (including histone acetylase and deacetylase complexes) and chromatin-remodelling complexes, which results in multilocus reorganization of chromatin structure [6, 104-107]. Besides, GRs (and several dozens of other transcription factors) interact with DNA methyltransferases (DNMTs), thereby providing locus-specific DNA methylation [108]. Hence, perhaps GC resistance is a result of GR-mediated changes in chromatin epigenetic landscape that take place during chronic stress and in stress-induced psychiatric disorders. Epigenetic modifications of DNA and histones affect regulatory regions of GR target genes and can drastically alter the ability of these genes to respond to GCs, e.g. by causing loss in their sensitivity to GCs. For example, long-term GC treatment was found to attenuate the sensitivity of several GR target genes in human UL3 osteosarcoma cell line [109]. After the cells had been cultured for a prolonged period in dexamethasone-containing medium, expression of endogenous SGK1, CEBP, and PLZF genes and genome-integrated UL3 luciferase reporter gene under control of the MMTV promoter could not be induced by dexamethasone anymore. Chromatin immunoprecipitation showed that long-term treatment of cells with the hormone considerably decreased the ability of GREs of these genes to bind GRs, although exact alterations in the chromatin structure that caused this effect remained unknown [109].
A large body of evidence indicates that an increase in GC levels caused by direct CG administration or induced by chronic/acute stress can change the extent of DNA methylation, as well as the levels and patterns of histone modification in cells from different brain regions [110, 111]. The most striking results were obtained in studies of DNA methylation in animals subjected to prenatal and early postnatal stress [112]. In particular, in mouse hypothalamus, prenatal stress stably increased the level of CpG dinucleotide methylation at the site of NGF1-A transcription factor binding in promoter (7) of the GR-encoding gene Nr3c1 and decreased it in the promoter region of the Crh gene. These modifications suppressed the transcriptional activity of the Nr3c1 gene and activated expression of the Crh gene with following decrease in the efficiency of the HPA axis-regulating negative feedback loop [54]. Increased CpG methylation at the NGF1-A-binding site of the Nr3c1 gene promoter 1(7) was also observed in the hypothalamus of rat pups whose mothers displayed diminished maternal care [53]. Microarray analysis of a 6.5·106 bp DNA fragment including the Nr3c1 locus demonstrated that this type of early postnatal stress affects methylation of the whole-genome, and not of the GR-encoding gene alone [113]. There are also numerous data indicating that activity of DNA methylases is altered in chronic stress and stess-related psychopathologies, as well as that inhibitors of DNA methylases produce antidepressant effect [110, 111]. In particular, mice with chronic social defeat stress had increased expression of the de novo methyltransferase Dnmt3a [114] in nucleus accumbens, while DNMT inhibitor RG108 exhibited an antidepresant effect in the stressed animals [115]. Other DNMT inhibitors, such as 5-aza-2-deoxycytidine and 5-azacytidine, also produced antidepressant effect in rats with depression-like behavior [116, 117]. Expression of Dnmt3b, another enzyme with de novo methylase activity, was upregulated in various brain regions in postmortem samples of suicide victims [118]. Analysis of mRNA levels for four DNMT isoforms in the blood of MDD patients revealed considerable activation of Dnmt3b gene expression during the depressive phase [119]. So far, there are no data on how acute/chronic stress alters the patterns of whole-genome methylation in brain [110]. Bose et al. investigated effects of the synthetic GC, dexamethasone, on the methylome of rat embryonic neural stem cells (NSCs) [120]. Earlier, the same authors had demonstrated that dexamethasone treatment decreased NSC proliferative activity, inhibited differentiation of these cells, and significantly altered their transcriptome [121]. In addition, dexamethasone considerably decreased the total number of methylated loci and altered the pattern of methylation. Analysis of differentially methylated loci revealed that these loci contain many genes involved in the regulation of proliferation, differentiation, cell migration, aging, DNA methylation, mitochondria functioning, and oxidative stress response [120].
The data on the effects of stress on histone modifications in brain are so abundant that their discussion requires a separate review article. These data include information on changes in the activity of histone-modifying enzymes and therapeutic effects of their inhibitors [122], as well as the results of studies on the alterations in the levels and patterns of histone modification marks [110, 111]. Here are some examples. Estimation of the total content of H3K4me3, H3K9me3, and H3K27me3 marks (related to transcription activation, heterochromatinization, and transcription repression, respectively) [123] in the rat hippocampus by Western blotting found that the level of trimethylation of the Lys9 residue (H3K9me3) increased, while the levels of the H3K27me3 and H3K4me3 marks decreased or remained unchanged, respectively. On the contrary, during chronic stress, the level of H3K9me3 slightly decreased, while the level of H3K4me3 increased [124]. Considerable increase in H3 histone methylation of Lys9 (heterochromatin mark) in rat hippocampus during acute stress was found by chromatin immunoprecipitation (ChIP) coupled with next generation sequencing (ChIP-Seq). Analysis of genomic localization of the H3K9me3 response revealed that this mark was enriched at transposable element loci and caused retrotransposon silencing [125]. Social defeat stress downregulated histone H4 acetylation of Lys8 in the rat ventral hippocampus, as demonstrated by Western blotting. At the same time, in a subgroup of rats less resilient to this type of stress, acetylation of histone H3 by Lys18 was increased in the prefrontal cortex and ventral hippocampus, and acetylation of histone H4 by Lys12 was increased in the ventral hippocampus [126]. Chronic social defeat stress also decreased the level of H3K9me2 mark (repressive histone modification) in nucleus accumbens, which correlated with downregulation of expression of histone methyltransferase G9a and G9a-like protein responsible for such modification [127]. It was demonstrated using the same stress model that the antidepressant effect of fluoxetine is related to its ability to increase histone H3 Lys9 dimethylation in the promoter regions of several genes [128]. On the other hand, histone deacetylase inhibitors that increase the level of transcription-activating histone modifications act as antidepressants [122, 129, 130].
Despite a large body of experimental data, no full picture describing the effects of stress on DNA and histone modifications has emerged [110, 111]. Largely, this could be because different brain structures have been studied for different histone modifications using different experimental approaches. Another probably more substantial reason is the deficit of whole-genome data that could be obtained by modern technologies of next generation sequencing (ChIP-seq, Me-DIP). Widespread application of these methods will allow an integrated view on the epigenetic landscape modifications caused by stress-induced pathologies. Moreover, comparison of epigenetic data to the results of transcriptome analysis (RNA-seq) will identify systems of genes and their key components essential for the development of psychopathologies. Complex study of changes in the epigenetic landscape in various brain regions caused by long-term stress-induced elevated concentrations of GCs is a promising approach to elucidating the mechanisms of GC resistance, since, as demonstrated for certain genes, reorganization of chromatin structure at the sites of GRE localization is an obligatory component of GR regulatory activity [6, 104-107]. One of the consequences of such reorganization might be the loss of sensitivity to GCs for some sets of GR target genes.
GCs regulate many vital processes in vertebrates such as coordinated growth, differentiation, reproduction, adaptation, and behavior. These hormones are involved in the regulation of carbohydrate, protein, and lipid metabolisms, maintenance of water and electrolyte balance; they control proliferation, differentiation, and apoptosis of many types of cells and exhibit antiinflammatory and immunosuppressive properties [131, 132]. One of the major GC functions is their participation in the organism’s adaptation to various types of physical and psychoemotional stress and in the organism’s response to stress by initiating the negative feedback mechanism aimed to downregulated the HPA axis activity [133, 134]. However, chronic stress and many stress-related psychiatric disorders result in permanent HPA axis activation due to the development of GC resistance, the most pronounced manifestation of which is a diminished inhibitory effect of GCs on the production of CRH, ACTH, and, a result, cortisol [10, 12-15, 135].
It is important to emphasize that GC resistance and associated HPA axis hyperactivity are closely related to neuroinflammatory processes. GCs play an antiinflammatory role, in particular during neuroinflammation induced by physiological or psychological stress. Numerous studies have demonstrated that GCs increase the concentrations of antiinflammatory cytokines and decrease the concentrations of proinflammatory ones [136-138]. Therefore, the dysfunctions of the GC regulation induced by chronic stress promote neuroinflammation [137] that is often observed in stress-induced psychopathologies [139-141].
Because GC resistance is one of the most typical features of psychiatric disorders such as MDD [12, 13] and bipolar disorder [15], the molecular mechanisms of its formation in brain regions have been a subject of numerous studies. The main topics of these studies are: (i) expression of the GR gene, biosynthesis of GR isoforms, and posttranslational GR modifications; (ii) expression of the FKBP5 gene, a component of the ultra-short negative feedback loop of GC signaling in cells; (iii) GR and FKBP5 genetic polymorphisms associated with stress-related psychiatric disorders; (iv) changes in the pattern of epigenetic modifications caused by GC activity and stress factors and/or typical stress-related psychopathologies.
Taken together, the results of these studies suggest that development of GC resistance in response to chronic stress does not involve a single universal cause, and that molecular mechanism of GC resistance are very diverse. Virtually every factor studied has been proven to be an important element in the formation of GC resistance. The least-studied among these factors are alterations in the epigenetic landscape of brain cells caused by prolonged increase in GC levels resulting from chronic stress and observed in stress-induced psychiatric disorders. Until recently, such studies have focused mostly on changes in the total content of particular epigenetic modifications or on the search for such modifications in the loci of particular genes [110, 111]. In the last few years, development of whole-genome techniques for identification of epigenetic modifications (ChIP-seq, MeDIP-seq) has raised these studies to a qualitatively new level that is not limited by the choice of presumed molecular determinants and might allow identification of key gene systems responsible for the development of stress-related pathologies. The informative value of such studies will be even higher in a combination with modern methods of transcriptome analysis (RNA-seq). Because GCs are essential regulators of various aspects of central nervous system functioning (growth, differentiation, neuron survival, synaptic plasticity) and play an important role in behavioral and cognitive disorders [142-145], we expect that the use of whole-genome methods in studies of the mechanisms underlying GC system dysfunctions in stress-related psychopathologies will significantly contribute to understanding of the molecular mechanisms of these processes.
Acknowledgements
This work was supported by the Russian Science Foundation (project No. 16-15-10131).
REFERENCES
1.Pittenger, C., and Duman, R. S. (2008) Stress,
depression, and neuroplasticity: a convergence of mechanisms,
Neuropsychopharmacology, 33, 88-109.
2.Miklowitz, D. J. (2011) Functional impairment,
stress, and psychosocial intervention in bipolar disorder, Curr.
Psychiatry Rep., 13, 504-512.
3.McEwen, B. S. (2007) Physiology and neurobiology of
stress and adaptation: central role of the brain, Physiol. Rev.,
87, 873-904.
4.Kumar, R., and Thompson, E. B. (2005) Gene
regulation by the glucocorticoid receptor: structure-function
relationship, J. Steroid Biochem. Mol. Biol., 94,
383-394.
5.Merkulov, V. M., and Merkulova, T. I. (2009)
Structural variants of glucocorticoid receptor binding sites and
different versions of positive glucocorticoid responsive elements:
analysis of GR-TRRD database, J. Steroid Biochem. Mol. Biol.,
115, 1-8.
6.Meijsing, S. H. (2015) Mechanisms of
glucocorticoid-regulated gene transcription, Adv. Exp. Med.
Biol., 872, 59-81.
7.Cattaneo, A., and Riva, M. A. (2016) Stress-induced
mechanisms in mental illness: a role for glucocorticoid signalling,
J. Steroid Biochem. Mol. Biol., 160, 169-174.
8.Drouin, J., Sun, Y. L., Chamberland, M., Gauthier,
Y., De Lean, A., Nemer, M., and Schmidt, T. J. (1993) Novel
glucocorticoid receptor complex with DNA element of the
hormone-repressed POMC gene, EMBO J., 12,
145-156.
9.Malkoski, S. P., and Dorin, R. I. (1999) Composite
glucocorticoid regulation at a functionally defined negative
glucocorticoid response element of the human corticotropin-releasing
hormone gene, Mol. Endocrinol., 13, 1629-1644.
10.Cohen, S., Janicki-Deverts, D., Doyle, W. J.,
Miller, G. E., Frank, E., Rabin, B. S., and Turner, R. B. (2012)
Chronic stress, glucocorticoid receptor resistance, inflammation, and
disease risk, Proc. Natl. Acad. Sci. USA, 109,
5995-5999.
11.Holsboer, F. (2000) The corticosteroid receptor
hypothesis of depression, Neuropsychopharmacology, 23,
477-501.
12.Jarcho, M. R., Slavich, G. M., Tylova-Stein, H.,
Wolkowitz, O. M., and Burke, H. M. (2013) Dysregulated diurnal cortisol
pattern is associated with glucocorticoid resistance in women with
major depressive disorder, Biol. Psychol., 93,
150-158.
13.Pariante, C. M. (2009) Risk factors for
development of depression and psychosis. Glucocorticoid receptors and
pituitary implications for treatment with antidepressant and
glucocorticoids, Ann. NY Acad. Sci., 1179, 144-152.
14.Pariante, C. M., and Miller, A. H. (2001)
Glucocorticoid receptors in major depression: relevance to
pathophysiology and treatment, Biol. Psychiatry, 49,
391-404.
15.Naughton, M., Dinan, T. G., and Scott, L. V.
(2014) Corticotropin-releasing hormone and the
hypothalamic-pituitary-adrenal axis in psychiatric disease, Handbook
Clin. Neurol., 124, 69-91.
16.Watson, S., Gallagher, P., Del-Estal, D., Hearn,
A., Ferrier, I. N., and Young, A. H. (2002)
Hypothalamic-pituitary-adrenal axis function in patients with chronic
depression, Psychol. Med., 32, 1021-1028.
17.Zobel, A. W., Nickel, T., Sonntag, A., Uhr, M.,
Holsboer, F., and Ising, M. (2001) Cortisol response in the combined
dexamethasone/CRH test as predictor of relapse in patients with
remitted depression. A prospective study, J. Psychiatr. Res.,
35, 83-94.
18.Binder, E. B. (2009) The role of FKBP5, a
co-chaperone of the glucocorticoid receptor in the pathogenesis and
therapy of affective and anxiety disorders,
Psychoneuroendocrinology, 34, Suppl. 1, S186-195.
19.Pariante, C. M., and Lightman, S. L. (2008) The
HPA axis in major depression: classical theories and new developments,
Trends Neurosci., 31, 464-468.
20.Hall, B. S., Moda, R. N., and Liston, C. (2015)
Glucocorticoid mechanisms of functional connectivity changes in
stress-related neuropsychiatric disorders, Neurobiol. Stress,
1, 174-183.
21.Jacobson, L., and Sapolsky, R. (1991) The role of
the hippocampus in feedback regulation of the
hypothalamic-pituitary-adrenocortical axis, Endocr. Rev.,
12, 118-134.
22.Sousa, N., Lukoyanov, N. V., Madeira, M. D.,
Almeida, O. F., and Paula-Barbosa, M. M. (2000) Reorganization of the
morphology of hippocampal neurites and synapses after stress-induced
damage correlates with behavioral improvement, Neuroscience,
97, 253-266.
23.Vyas, A., Mitra, R., Shankaranarayana Rao, B. S.,
and Chattarji, S. (2002) Chronic stress induces contrasting patterns of
dendritic remodeling in hippocampal and amygdaloid neurons, J.
Neurosci., 22, 6810-6818.
24.Vyas, S., Rodrigues, A. J., Silva, J. M.,
Tronche, F., Almeida, O. F., Sousa, N., and Sotiropoulos, I. (2016)
Chronic stress and glucocorticoids: from neuronal plasticity to
neurodegeneration, Neural Plast., 2016, 6391686.
25.Gould, E., McEwen, B. S., Tanapat, P., Galea, L.
A., and Fuchs, E. (1997) Neurogenesis in the dentate gyrus of the adult
tree shrew is regulated by psychosocial stress and NMDA receptor
activation, J. Neurosci., 17, 2492-2498.
26.Shors, T. J. (2006) Significant life events and
the shape of memories to come: a hypothesis, Neurobiol. Learn.
Mem., 85, 103-115.
27.Anacker, C., Zunszain, P. A., Carvalho, L. A.,
and Pariante, C. M. (2011) The glucocorticoid receptor: pivot of
depression and of antidepressant treatment?
Psychoneuroendocrinology, 36, 415-425.
28.Pariante, C. M. (2006) The glucocorticoid
receptor: part of the solution or part of the problem? J.
Psychopharmacol., 20, 79-84.
29.Lopez, J. F., Chalmers, D. T., Little, K. Y., and
Watson, S. J. (1998) A. E. Bennett Research Award. Regulation of
serotonin 1A, glucocorticoid, and mineralocorticoid receptor in rat and
human hippocampus: implications for the neurobiology of depression,
Biol. Psychiatry, 43, 547-573.
30.Alt, S. R., Turner, J. D., Klok, M. D., Meijer,
O. C., Lakke, E. A., Derijk, R. H., and Muller, C. P. (2010)
Differential expression of glucocorticoid receptor transcripts in major
depressive disorder is not epigenetically programmed,
Psychoneuroendocrinology, 35, 544-556.
31.Webster, M. J., Knable, M. B., O’Grady, J.,
Orthmann, J., and Weickert, C. S. (2002) Regional specificity of brain
glucocorticoid receptor mRNA alterations in subjects with schizophrenia
and mood disorders, Mol. Psychiatry, 7, 924, 985-994.
32.Perlman, W. R., Webster, M. J., Kleinman, J. E.,
and Weickert, C. S. (2004) Reduced glucocorticoid and estrogen receptor
alpha messenger ribonucleic acid levels in the amygdala of patients
with major mental illness, Biol. Psychiatry, 56,
844-852.
33.Chang, L. C., Jamain, S., Lin, C. W., Rujescu,
D., Tseng, G. C., and Sibille, E. (2014) A conserved BDNF, glutamate-
and GABA-enriched gene module related to human depression identified by
coexpression meta-analysis and DNA variant genome-wide association
studies, PLoS One, 9, e90980.
34.Akula, N., Barb, J., Jiang, X., Wendland, J. R.,
Choi, K. H., Sen, S. K., Hou, L., Chen, D. T., Laje, G., Johnson, K.,
Lipska, B. K., Kleinman, J. E., Corrada-Bravo, H., Detera-Wadleigh, S.,
Munson, P. J., and McMahon, F. J. (2014) RNA-sequencing of the brain
transcriptome implicates dysregulation of neuroplasticity, circadian
rhythms and GTPase binding in bipolar disorder, Mol. Psychiatry,
19, 1179-1185.
35.Oakley, R. H., and Cidlowski, J. A. (2011)
Cellular processing of the glucocorticoid receptor gene and protein:
new mechanisms for generating tissue-specific actions of
glucocorticoids, J. Biol. Chem., 286, 3177-3184.
36.Merkulov, V. M., and Merkulova, T. I. (2011)
Isoforms of glucocorticoid receptor formed by alternative splicing and
usage of alternative mRNA translation starts, Vavilov Zh. Genet.
Selekt., 4, 621-632.
37.Oakley, R. H., Sar, M., and Cidlowski, J. A.
(1996) The human glucocorticoid receptor beta isoform. Expression,
biochemical properties, and putative function, J. Biol. Chem.,
271, 9550-9559.
38.Kino, T., Manoli, I., Kelkar, S., Wang, Y., Su,
Y. A., and Chrousos, G. P. (2009) Glucocorticoid receptor (GR) beta has
intrinsic, GRalpha-independent transcriptional activity, Biochem.
Biophys. Res. Commun., 381, 671-675.
39.Lewis-Tuffin, L. J., Jewell, C. M., Bienstock, R.
J., Collins, J. B., and Cidlowski, J. A. (2007) Human glucocorticoid
receptor beta binds RU-486 and is transcriptionally active, Mol.
Cell Biol., 27, 2266-2282.
40.Pujols, L., Mullol, J., Roca-Ferrer, J., Torrego,
A., Xaubet, A., Cidlowski, J. A., and Picado, C. (2002) Expression of
glucocorticoid receptor alpha- and beta-isoforms in human cells and
tissues, Am. J. Physiol. Cell Physiol., 283,
1324-1331.
41.Webster, J. C., Oakley, R. H., Jewell, C. M., and
Cidlowski, J. A. (2001) Proinflammatory cytokines regulate human
glucocorticoid receptor gene expression and lead to the accumulation of
the dominant negative beta isoform: a mechanism for the generation of
glucocorticoid resistance, Proc. Natl. Acad. Sci. USA,
98, 6865-6870.
42.Lewis-Tuffin, L. J., and Cidlowski, J. A. (2006)
The physiology of human glucocorticoid receptor beta (hGRbeta) and
glucocorticoid resistance, Ann. NY Acad. Sci., 1069,
1-9.
43.Moalli, P. A., Pillay, S., Krett, N. L., and
Rosen, S. T. (1993) Alternatively spliced glucocorticoid receptor
messenger RNAs in glucocorticoid-resistant human multiple myeloma
cells, Cancer Res., 53, 3877-3879.
44.Pandey, G. N., Rizavi, H. S., Ren, X., Dwivedi,
Y., and Palkovits, M. (2013) Region-specific alterations in
glucocorticoid receptor expression in the postmortem brain of teenage
suicide victims, Psychoneuroendocrinology, 38,
2628-2639.
45.Szczepankiewicz, A., Leszczynska-Rodziewicz, A.,
Pawlak, J., Rajewska-Rager, A., Dmitrzak-Weglarz, M., Wilkosc, M.,
Skibinska, M., and Hauser, J. (2011) Glucocorticoid receptor
polymorphism is associated with major depression and predominance of
depression in the course of bipolar disorder, J. Affect.
Disord., 134, 138-144.
46.Derijk, R. H., Schaaf, M. J., Turner, G., Datson,
N. A., Vreugdenhil, E., Cidlowski, J., De Kloet, E. R., Emery, P.,
Sternberg, E. M., and Detera-Wadleigh, S. D. (2001) A human
glucocorticoid receptor gene variant that increases the stability of
the glucocorticoid receptor beta-isoform mRNA is associated with
rheumatoid arthritis, J. Rheumatol., 28, 2383-2388.
47.Van Rossum, E. F., Binder, E. B., Majer, M.,
Koper, J. W., Ising, M., Modell, S., Salyakina, D., Lamberts, S. W.,
and Holsboer, F. (2006) Polymorphisms of the glucocorticoid receptor
gene and major depression, Biol. Psychiatry, 59,
681-688.
48.Van Rossum, E. F., and Lamberts, S. W. (2004)
Polymorphisms in the glucocorticoid receptor gene and their
associations with metabolic parameters and body composition, Recent
Prog. Horm. Res., 59, 333-357.
49.Russcher, H., Van Rossum, E. F., De Jong, F. H.,
Brinkmann, A. O., Lamberts, S. W., and Koper, J. W. (2005) Increased
expression of the glucocorticoid receptor-A translational isoform as a
result of the ER22/23EK polymorphism, Mol. Endocrinol.,
19, 1687-1696.
50.Kochetov, A. V., Merkulova, T. I., and Merkulov,
V. M. (2012) Possible link between the synthesis of GR alpha isoforms
and eIF2 alpha phosphorylation state, Med. Hypotheses,
79, 709-712.
51.Cao-Lei, L., Leija, S. C., Kumsta, R., Wust, S.,
Meyer, J., Turner, J. D., and Muller, C. P. (2011) Transcriptional
control of the human glucocorticoid receptor: identification and
analysis of alternative promoter regions, Hum. Genet.,
129, 533-543.
52.Turner, J. D., Vernocchi, S., Schmitz, S., and
Muller, C. P. (2014) Role of the 5′-untranslated regions in
post-transcriptional regulation of the human glucocorticoid receptor,
Biochim. Biophys. Acta, 1839, 1051-1061.
53.Weaver, I. C., Cervoni, N., Champagne, F. A.,
D’Alessio, A. C., Sharma, S., Seckl, J. R., Dymov, S., Szyf, M.,
and Meaney, M. J. (2004) Epigenetic programming by maternal behavior,
Nat. Neurosci., 7, 847-854.
54.Mueller, B. R., and Bale, T. L. (2008)
Sex-specific programming of offspring emotionality after stress early
in pregnancy, J. Neurosci., 28, 9055-9065.
55.DeRijk, R. H., Schaaf, M., and De Kloet, E. R.
(2002) Glucocorticoid receptor variants: clinical implications, J.
Steroid Biochem. Mol. Biol., 81, 103-122.
56.Ismaili, N., and Garabedian, M. J. (2004)
Modulation of glucocorticoid receptor function via phosphorylation,
Ann. NY Acad. Sci., 1024, 86-101.
57.Ford, J., McEwan, I. J., Wright, A. P., and
Gustafsson, J. A. (1997) Involvement of the transcription factor IID
protein complex in gene activation by the N-terminal transactivation
domain of the glucocorticoid receptor in vitro, Mol.
Endocrinol., 11, 1467-1475.
58.Robyr, D., Wolffe, A. P., and Wahli, W. (2000)
Nuclear hormone receptor coregulators in action: diversity for shared
tasks, Mol. Endocrinol., 14, 329-347.
59.Chen, W., Dang, T., Blind, R. D., Wang, Z.,
Cavasotto, C. N., Hittelman, A. B., Rogatsky, I., Logan, S. K., and
Garabedian, M. J. (2008) Glucocorticoid receptor phosphorylation
differentially affects target gene expression, Mol. Endocrinol.,
22, 1754-1766.
60.Anacker, C., Cattaneo, A., Musaelyan, K.,
Zunszain, P. A., Horowitz, M., Molteni, R., Luoni, A., Calabrese, F.,
Tansey, K., Gennarelli, M., Thuret, S., Price, J., Uher, R., Riva, M.
A., and Pariante, C. M. (2013) Role for the kinase SGK1 in stress,
depression, and glucocorticoid effects on hippocampal neurogenesis,
Proc. Natl. Acad. Sci. USA, 110, 8708-8713.
61.Itoh, M., Adachi, M., Yasui, H., Takekawa, M.,
Tanaka, H., and Imai, K. (2002) Nuclear export of glucocorticoid
receptor is enhanced by c-Jun N-terminal kinase-mediated
phosphorylation, Mol. Endocrinol., 16, 2382-2392.
62.Miller, A. L., Webb, M. S., Copik, A. J., Wang,
Y., Johnson, B. H., Kumar, R., and Thompson, E. B. (2005) p38
mitogen-activated protein kinase (MAPK) is a key mediator in
glucocorticoid-induced apoptosis of lymphoid cells: correlation between
p38 MAPK activation and site-specific phosphorylation of the human
glucocorticoid receptor at serine 211, Mol. Endocrinol.,
19, 1569-1583.
63.Jovicic, M. J., Lukic, I., Radojcic, M., Adzic,
M., and Maric, N. P. (2015) Modulation of c-Jun N-terminal
kinase signaling and specific glucocorticoid receptor phosphorylation
in the treatment of major depression, Med. Hypotheses,
85, 291-294.
64.Mitic, M., Simic, I., Djordjevic, J., Radojcic,
M. B., and Adzic, M. (2013) Gender-specific effects of fluoxetine on
hippocampal glucocorticoid receptor phosphorylation and behavior in
chronically stressed rats, Neuropharmacology, 70,
100-111.
65.Lee, M. S., Park, W. S., Kim, Y. H., Kwon, S. H.,
Jang, Y. J., Han, D., Morita, K., and Her, S. (2013)
Antidepressant-like effects of cortex mori radicis extract via
bidirectional phosphorylation of glucocorticoid receptors in the
hippocampus, Behav. Brain Res., 236, 56-61.
66.Simic, I., Maric, N. P., Mitic, M., Soldatovic,
I., Pavlovic, Z., Mihaljevic, M., Andric, S., Radojcic, M. B., and
Adzic, M. (2013) Phosphorylation of leukocyte glucocorticoid receptor
in patients with current episode of major depressive disorder, Prog.
Neuropsychopharmacol. Biol. Psychiatry, 40, 281-285.
67.Echeverria, P. C., and Picard, D. (2010)
Molecular chaperones, essential partners of steroid hormone receptors
for activity and mobility, Biochim. Biophys. Acta, 1803,
641-649.
68.Cheung, J., and Smith, D. F. (2000) Molecular
chaperone interactions with steroid receptors: an update, Mol.
Endocrinol., 14, 939-946.
69.Pratt, W. B., Galigniana, M. D., Morishima, Y.,
and Murphy, P. J. (2004) Role of molecular chaperones in steroid
receptor action, Essays Biochem., 40, 41-58.
70.Davies, T. H., Ning, Y. M., and Sanchez, E. R.
(2002) A new first step in activation of steroid receptors:
hormone-induced switching of FKBP51 and FKBP52 immunophilins, J.
Biol. Chem., 277, 4597-4600.
71.Harrell, J. M., Murphy, P. J., Morishima, Y.,
Chen, H., Mansfield, J. F., Galigniana, M. D., and Pratt, W. B. (2004)
Evidence for glucocorticoid receptor transport on microtubules by
dynein, J. Biol. Chem., 279, 54647-54654.
72.Billing, A. M., Fack, F., Renaut, J., Olinger, C.
M., Schote, A. B., Turner, J. D., and Muller, C. P. (2007) Proteomic
analysis of the cortisol-mediated stress response in THP-1 monocytes
using DIGE technology, J. Mass Spectrom., 42,
1433-1444.
73.U, M., Shen, L., Oshida, T., Miyauchi, J.,
Yamada, M., and Miyashita, T. (2004) Identification of novel direct
transcriptional targets of glucocorticoid receptor, Leukemia,
18, 1850-1856.
74.Vermeer, H., Hendriks-Stegeman, B. I., Van der
Burg, B., Van Buul-Offers, S. C., and Jansen, M. (2003)
Glucocorticoid-induced increase in lymphocytic FKBP51 messenger
ribonucleic acid expression: a potential marker for glucocorticoid
sensitivity, potency, and bioavailability, J. Clin. Endocrinol.
Metab., 88, 277-284.
75.Woodruff, P. G., Boushey, H. A., Dolganov, G. M.,
Barker, C. S., Yang, Y. H., Donnelly, S., Ellwanger, A., Sidhu, S. S.,
Dao-Pick, T. P., Pantoja, C., Erle, D. J., Yamamoto, K. R., and Fahy,
J. V. (2007) Genome-wide profiling identifies epithelial cell genes
associated with asthma and with treatment response to corticosteroids,
Proc. Natl. Acad. Sci. USA, 104, 15858-15863.
76.Lee, R. S., Tamashiro, K. L., Yang, X., Purcell,
R. H., Harvey, A., Willour, V. L., Huo, Y., Rongione, M., Wand, G. S.,
and Potash, J. B. (2010) Chronic corticosterone exposure increases
expression and decreases deoxyribonucleic acid methylation of Fkbp5 in
mice, Endocrinology, 151, 4332-4343.
77.Scharf, S. H., Liebl, C., Binder, E. B., Schmidt,
M. V., and Muller, M. B. (2011) Expression and regulation of the
Fkbp5 gene in the adult mouse brain, PLoS One, 6,
e16883.
78.Hubler, T. R., and Scammell, J. G. (2004)
Intronic hormone response elements mediate regulation of FKBP5 by
progestins and glucocorticoids, Cell Stress Chaperones,
9, 243-252.
79.Reddy, T. E., Pauli, F., Sprouse, R. O., Neff, N.
F., Newberry, K. M., Garabedian, M. J., and Myers, R. M. (2009) Genomic
determination of the glucocorticoid response reveals unexpected
mechanisms of gene regulation, Genome Res., 19,
2163-2171.
80.Denny, W. B., Prapapanich, V., Smith, D. F., and
Scammell, J. G. (2005) Structure-function analysis of squirrel monkey
FK506-binding protein 51, a potent inhibitor of glucocorticoid receptor
activity, Endocrinology, 146, 3194-3201.
81.Klengel, T., Mehta, D., Anacker, C., Rex-Haffner,
M., Pruessner, J. C., Pariante, C. M., Pace, T. W., Mercer, K. B.,
Mayberg, H. S., Bradley, B., Nemeroff, C. B., Holsboer, F., Heim, C.
M., Ressler, K. J., Rein, T., and Binder, E. B. (2013) Allele-specific
FKBP5 DNA demethylation mediates gene-childhood trauma interactions,
Nat. Neurosci., 16, 33-41.
82.Chrousos, G. P., Renquist, D., Brandon, D., Eil,
C., Pugeat, M., Vigersky, R., Cutler, G. B., Jr., Loriaux, D. L., and
Lipsett, M. B. (1982) Glucocorticoid hormone resistance during primate
evolution: receptor-mediated mechanisms, Proc. Natl. Acad. Sci.
USA, 79, 2036-2040.
83.Reynolds, P. D., Ruan, Y., Smith, D. F., and
Scammell, J. G. (1999) Glucocorticoid resistance in the squirrel monkey
is associated with overexpression of the immunophilin FKBP51, J.
Clin. Endocrinol. Metab., 84, 663-669.
84.Scammell, J. G., Denny, W. B., Valentine, D. L.,
and Smith, D. F. (2001) Overexpression of the FK506-binding
immunophilin FKBP51 is the common cause of glucocorticoid resistance in
three New World primates, Gen. Comp. Endocrinol., 124,
152-165.
85.Denny, W. B., Valentine, D. L., Reynolds, P. D.,
Smith, D. F., and Scammell, J. G. (2000) Squirrel monkey immunophilin
FKBP51 is a potent inhibitor of glucocorticoid receptor binding,
Endocrinology, 141, 4107-4113.
86.Guidotti, G., Calabrese, F., Anacker, C.,
Racagni, G., Pariante, C. M., and Riva, M. A. (2013) Glucocorticoid
receptor and FKBP5 expression is altered following exposure to chronic
stress: modulation by antidepressant treatment,
Neuropsychopharmacology, 38, 616-627.
87.Xing, Y., Hou, J., Meng, Q., Yang, M., Kurihara,
H., and Tian, J. (2015) Novel antidepressant candidate RO-05 modulated
glucocorticoid receptors activation and FKBP5 expression in chronic
mild stress model in rats, Neuroscience, 290,
255-265.
88.Pei, H., Li, L., Fridley, B. L., Jenkins, G. D.,
Kalari, K. R., Lingle, W., Petersen, G., Lou, Z., and Wang, L. (2009)
FKBP51 affects cancer cell response to chemotherapy by negatively
regulating Akt, Cancer Cell, 16, 259-266.
89.Beaulieu, J. M., Gainetdinov, R. R., and Caron,
M. G. (2009) Akt/GSK3 signaling in the action of psychotropic drugs,
Annu. Rev. Pharmacol. Toxicol., 49, 327-347.
90.Charney, D. S., and Manji, H. K. (2004) Life
stress, genes, and depression: multiple pathways lead to increased risk
and new opportunities for intervention, Science’s STKE,
2004, re5.
91.Duman, R. S., and Voleti, B. (2012) Signaling
pathways underlying the pathophysiology and treatment of depression:
novel mechanisms for rapid-acting agents, Trends Neurosci.,
35, 47-56.
92.Binder, E. B., Salyakina, D., Lichtner, P.,
Wochnik, G. M., Ising, M., Putz, B., Papiol, S., Seaman, S., Lucae, S.,
Kohli, M. A., Nickel, T., Kunzel, H. E., Fuchs, B., Majer, M., Pfennig,
A., Kern, N., Brunner, J., Modell, S., Baghai, T., Deiml, T., Zill, P.,
Bondy, B., Rupprecht, R., Messer, T., Kohnlein, O., Dabitz, H., Bruckl,
T., Muller, N., Pfister, H., Lieb, R., Mueller, J. C., Lohmussaar, E.,
Strom, T. M., Bettecken, T., Meitinger, T., Uhr, M., Rein, T.,
Holsboer, F., and Muller-Myhsok, B. (2004) Polymorphisms in FKBP5 are
associated with increased recurrence of depressive episodes and rapid
response to antidepressant treatment, Nat. Genet., 36,
1319-1325.
93.Brent, D., Melhem, N., Ferrell, R., Emslie, G.,
Wagner, K. D., Ryan, N., Vitiello, B., Birmaher, B., Mayes, T.,
Zelazny, J., Onorato, M., Devlin, B., Clarke, G., DeBar, L., and
Keller, M. (2010) Association of FKBP5 polymorphisms with suicidal
events in the Treatment of Resistant Depression in Adolescents (TORDIA)
study, Am. J. Psychiatry, 167, 190-197.
94.Lavebratt, C., Aberg, E., Sjoholm, L. K., and
Forsell, Y. (2010) Variations in FKBP5 and BDNF genes are
suggestively associated with depression in a Swedish population-based
cohort, J. Affect. Disord., 125, 249-255.
95.Lekman, M., Laje, G., Charney, D., Rush, A. J.,
Wilson, A. F., Sorant, A. J., Lipsky, R., Wisniewski, S. R., Manji, H.,
McMahon, F. J., and Paddock, S. (2008) The FKBP5 gene in
depression and treatment response – an association study in
the Sequenced Treatment Alternatives to Relieve Depression (STAR*D)
cohort, Biol. Psychiatry, 63, 1103-1110.
96.Papiol, S., Arias, B., Gasto, C., Gutierrez, B.,
Catalan, R., and Fananas, L. (2007) Genetic variability at HPA axis in
major depression and clinical response to antidepressant treatment,
J. Affect. Disord., 104, 83-90.
97.Ising, M., Depping, A. M., Siebertz, A., Lucae,
S., Unschuld, P. G., Kloiber, S., Horstmann, S., Uhr, M.,
Muller-Myhsok, B., and Holsboer, F. (2008) Polymorphisms in the
FKBP5 gene region modulate recovery from psychosocial stress in
healthy controls, Eur. J. Neurosci., 28, 389-398.
98.Stamm, T. J., Rampp, C., Wiethoff, K., Stingl,
J., Mossner, R., O’Malley, G., Ricken, R., Seemuller, F., Keck,
M., Fisher, R., Gaebel, W., Maier, W., Moller, H. J., Bauer, M., and
Adli, M. (2016) The FKBP5 polymorphism rs1360780 influences the effect
of an algorithm-based antidepressant treatment and is associated with
remission in patients with major depression, J.
Psychopharmacol., 30, 40-47.
99.Menke, A., Klengel, T., Rubel, J., Bruckl, T.,
Pfister, H., Lucae, S., Uhr, M., Holsboer, F., and Binder, E. B. (2013)
Genetic variation in FKBP5 associated with the extent of stress hormone
dysregulation in major depression, Genes Brain Behav.,
12, 289-296.
100.Hohne, N., Poidinger, M., Merz, F., Pfister,
H., Bruckl, T., Zimmermann, P., Uhr, M., Holsboer, F., and Ising, M.
(2015) FKBP5 genotype-dependent DNA methylation and mRNA regulation
after psychosocial stress in remitted depression and healthy controls,
Int. J. Neuropsychopharmacol., 18.
101.Abramovitz, L., Shapira, T., Ben-Dror, I.,
Dror, V., Granot, L., Rousso, T., Landoy, E., Blau, L., Thiel, G., and
Vardimon, L. (2008) Dual role of NRSF/REST in activation and repression
of the glucocorticoid response, J. Biol. Chem., 283,
110-119.
102.Merkulova, T. I., Ananko, E. A.,
Ignat’eva, E. V., and Kolchanov, N. A. (2013) Regulatory
transcription codes in eukaryotic genomes, Genetika, 49,
37-54.
103.Gaali, S., Kirschner, A., Cuboni, S., Hartmann,
J., Kozany, C., Balsevich, G., Namendorf, C., Fernandez-Vizarra, P.,
Sippel, C., Zannas, A. S., Draenert, R., Binder, E. B., Almeida, O. F.,
Ruhter, G., Uhr, M., Schmidt, M. V., Touma, C., Bracher, A., and
Hausch, F. (2015) Selective inhibitors of the FK506-binding protein 51
by induced fit, Nat. Chem. Biol., 11, 33-37.
104.Nicolaides, N. C., Galata, Z., Kino, T.,
Chrousos, G. P., and Charmandari, E. (2010) The human glucocorticoid
receptor: molecular basis of biologic function, Steroids,
75, 1-12.
105.King, H. A., Trotter, K. W., and Archer, T. K.
(2012) Chromatin remodeling during glucocorticoid receptor regulated
transactivation, Biochim. Biophys. Acta, 1819,
716-726.
106.Wu, J. N., Pinello, L., Yissachar, E.,
Wischhusen, J. W., Yuan, G. C., and Roberts, C. W. (2015) Functionally
distinct patterns of nucleosome remodeling at enhancers in
glucocorticoid-treated acute lymphoblastic leukemia, Epigenetics
Chromatin, 8, 53.
107.Swinstead, E. E., Paakinaho, V., Presman, D.
M., and Hager, G. L. (2016) Pioneer factors and ATP-dependent chromatin
remodeling factors interact dynamically: a new perspective –
multiple transcription factors can effect chromatin pioneer functions
through dynamic interactions with ATP-dependent chromatin remodeling
factors, Bioessays, 38, 1150-1157.
108.Hervouet, E., Vallette, F. M., and Cartron, P.
F. (2010) Dnmt1/transcription factor interactions: an alternative
mechanism of DNA methylation inheritance, Genes Cancer,
1, 434-443.
109.Burkhart, B. A., Ivey, M. L., and Archer, T. K.
(2009) Long-term low level glucocorticoid exposure induces persistent
repression in chromatin, Mol. Cell Endocrinol., 298,
66-75.
110.Nestler, E. J., Pena, C. J., Kundakovic, M.,
Mitchell, A., and Akbarian, S. (2016) Epigenetic basis of mental
illness, Neuroscientist, 22, 447-463.
111.Pena, C. J., Bagot, R. C., Labonte, B., and
Nestler, E. J. (2014) Epigenetic signaling in psychiatric disorders,
J. Mol. Biol., 426, 3389-3412.
112.Kundakovic, M., and Champagne, F. A. (2015)
Early-life experience, epigenetics, and the developing brain,
Neuropsychopharmacology, 40, 141-153.
113.McGowan, P. O., Suderman, M., Sasaki, A.,
Huang, T. C., Hallett, M., Meaney, M. J., and Szyf, M. (2011) Broad
epigenetic signature of maternal care in the brain of adult rats,
PLoS One, 6, e14739.
114.Lo, P. K., and Sukumar, S. (2008) Epigenomics
and breast cancer, Pharmacogenomics, 9, 1879-1902.
115.LaPlant, Q., Vialou, V., Covington, H. E., 3rd,
Dumitriu, D., Feng, J., Warren, B. L., Maze, I., Dietz, D. M., Watts,
E. L., Iniguez, S. D., Koo, J. W., Mouzon, E., Renthal, W., Hollis, F.,
Wang, H., Noonan, M. A., Ren, Y., Eisch, A. J., Bolanos, C. A., Kabbaj,
M., Xiao, G., Neve, R. L., Hurd, Y. L., Oosting, R. S., Fan, G.,
Morrison, J. H., and Nestler, E. J. (2010) Dnmt3a regulates emotional
behavior and spine plasticity in the nucleus accumbens, Nat.
Neurosci., 13, 1137-1143.
116.Sales, A. J., Biojone, C., Terceti, M. S.,
Guimaraes, F. S., Gomes, M. V., and Joca, S. R. (2011)
Antidepressant-like effect induced by systemic and intra-hippocampal
administration of DNA methylation inhibitors, Br. J. Pharmacol.,
164, 1711-1721.
117.Xing, B., Liu, P., Xu, W. J., Xu, F. Y., and
Dang, Y. H. (2014) Effect of microinjecting of 5-aza-2-deoxycytidine
into ventrolateral orbital cortex on depressive-like behavior in rats,
Neurosci. Lett., 574, 11-14.
118.Poulter, M. O., Du, L., Weaver, I. C.,
Palkovits, M., Faludi, G., Merali, Z., Szyf, M., and Anisman, H. (2008)
GABAA receptor promoter hypermethylation in suicide brain:
implications for the involvement of epigenetic processes, Biol.
Psychiatry, 64, 645-652.
119.Higuchi, F., Uchida, S., Yamagata, H., Otsuki,
K., Hobara, T., Abe, N., Shibata, T., and Watanabe, Y. (2011)
State-dependent changes in the expression of DNA methyltransferases in
mood disorder patients, J. Psychiatr. Res., 45,
1295-1300.
120.Bose, R., Spulber, S., Kilian, P., Heldring,
N., Lonnerberg, P., Johnsson, A., Conti, M., Hermanson, O., and
Ceccatelli, S. (2015) Tet3 mediates stable glucocorticoid-induced
alterations in DNA methylation and Dnmt3a/Dkk1 expression in neural
progenitors, Cell Death Dis., 6, e1793.
121.Bose, R., Moors, M., Tofighi, R., Cascante, A.,
Hermanson, O., and Ceccatelli, S. (2010) Glucocorticoids induce
long-lasting effects in neural stem cells resulting in
senescence-related alterations, Cell Death Dis., 1,
e92.
122.Covington, H. E., 3rd, Maze, I., Vialou, V.,
and Nestler, E. J. (2015) Antidepressant action of HDAC inhibition in
the prefrontal cortex, Neuroscience, 298, 329-335.
123.Jenuwein, T., and Allis, C. D. (2001)
Translating the histone code, Science, 293,
1074-1080.
124.Hunter, R. G., McCarthy, K. J., Milne, T. A.,
Pfaff, D. W., and McEwen, B. S. (2009) Regulation of hippocampal H3
histone methylation by acute and chronic stress, Proc. Natl. Acad.
Sci. USA, 106, 20912-20917.
125.Hunter, R. G., Murakami, G., Dewell, S.,
Seligsohn, M., Baker, M. E., Datson, N. A., McEwen, B. S., and Pfaff,
D. W. (2012) Acute stress and hippocampal histone H3 lysine 9
trimethylation, a retrotransposon silencing response, Proc. Natl.
Acad. Sci. USA, 109, 17657-17662.
126.Kenworthy, C. A., Sengupta, A., Luz, S. M., Ver
Hoeve, E. S., Meda, K., Bhatnagar, S., and Abel, T. (2014) Social
defeat induces changes in histone acetylation and expression of histone
modifying enzymes in the ventral hippocampus, prefrontal cortex, and
dorsal raphe nucleus, Neuroscience, 264, 88-98.
127.Covington, H. E., 3rd, Maze, I., Sun, H.,
Bomze, H. M., DeMaio, K. D., Wu, E. Y., Dietz, D. M., Lobo, M. K.,
Ghose, S., Mouzon, E., Neve, R. L., Tamminga, C. A., and Nestler, E. J.
(2011) A role for repressive histone methylation in cocaine-induced
vulnerability to stress, Neuron, 71, 656-670.
128.Robison, A. J., Vialou, V., Sun, H. S.,
Labonte, B., Golden, S. A., Dias, C., Turecki, G., Tamminga, C., Russo,
S., Mazei-Robison, M., and Nestler, E. J. (2014) Fluoxetine
epigenetically alters the CaMKIIalpha promoter in nucleus accumbens to
regulate DeltaFosB binding and antidepressant effects,
Neuropsychopharmacology, 39, 1178-1186.
129.Covington, H. E., 3rd, Maze, I., LaPlant, Q.
C., Vialou, V. F., Ohnishi, Y. N., Berton, O., Fass, D. M., Renthal,
W., Rush, A. J., 3rd, Wu, E. Y., Ghose, S., Krishnan, V., Russo, S. J.,
Tamminga, C., Haggarty, S. J., and Nestler, E. J. (2009) Antidepressant
actions of histone deacetylase inhibitors, J. Neurosci.,
29, 11451-11460.
130.Tsankova, N. M., Berton, O., Renthal, W.,
Kumar, A., Neve, R. L., and Nestler, E. J. (2006) Sustained hippocampal
chromatin regulation in a mouse model of depression and antidepressant
action, Nat. Neurosci., 9, 519-525.
131.Hierholzer, K., and Buhler, H. (1996)
Metabolism of cortical steroid hormones and their general mode of
action, in Comprehensive Human Physiology (Greger, R., and
Windhorst, U., eds.) Springer-Verlag, Heidelberg, pp. 79-93.
132.Sapolsky, R. M., Romero, L. M., and Munck, A.
U. (2000) How do glucocorticoids influence stress responses?
Integrating permissive, suppressive, stimulatory, and preparative
actions, Endocrin. Rev., 21, 55-89.
133.Charmandari, E., Tsigos, C., and Chrousos, G.
(2005) Endocrinology of the stress response, Annu. Rev.
Physiol., 67, 259-284.
134.Smith, S. M., and Vale, W. W. (2006) The role
of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses
to stress, Dialogues Clin. Neurosci., 8, 383-395.
135.Holsboer, F., and Barden, N. (1996)
Antidepressants and hypothalamic-pituitary-adrenocortical regulation,
Endocrin. Rev., 17, 187-205.
136.Arango-Lievano, M., and Jeanneteau, F. (2016)
Timing and crosstalk of glucocorticoid signaling with cytokines,
neurotransmitters and growth factors, Pharmacol. Res.,
113, 1-17.
137.Kim, Y. K., Na, K. S., Myint, A. M., and
Leonard, B. E. (2016) The role of pro-inflammatory cytokines in
neuroinflammation, neurogenesis and the neuroendocrine system in major
depression, Prog. Neuropsychopharmacol. Biol. Psychiatry,
64, 277-284.
138.Grigoryan, G. A., Dygalo, N. N., Gekht, A. B.,
Stepanichev, M. Yu., and Gulyaeva, N. V. (2014) Molecular and cellular
mechanisms of depression: role of glucocorticoids, cytokines,
nuerotransmitters, and trophic factors in genesis of depressive
disorders, Usp. Fiziol. Nauk, 45, 3-19.
139.Jeon, S. W., and Kim, Y. K. (2016)
Neuroinflammation and cytokine abnormality in major depression: cause
or consequence in that illness? World J. Psychiatry, 6,
283-293.
140.Reus, G. Z., Fries, G. R., Stertz, L., Badawy,
M., Passos, I. C., Barichello, T., Kapczinski, F., and Quevedo, J.
(2015) The role of inflammation and microglial activation in the
pathophysiology of psychiatric disorders, Neuroscience,
300, 141-154.
141.Hong, H., Kim, B. S., and Im, H. I. (2016)
Pathophysiological role of neuroinflammation in neurodegenerative
diseases and psychiatric disorders, Int. Neurourol. J.,
20, S2-7.
142.De Kloet, E. R., De Jong, I. E., and Oitzl, M.
S. (2008) Neuropharmacology of glucocorticoids: focus on emotion,
cognition and cocaine, Eur. J. Pharmacol., 585,
473-482.
143.Fietta, P., and Delsante, G. (2009) Central
nervous system effects of natural and synthetic glucocorticoids,
Psychiatry Clin. Neurosci., 63, 613-622.
144.Finsterwald, C., and Alberini, C. M. (2014)
Stress and glucocorticoid receptor-dependent mechanisms in long-term
memory: from adaptive responses to psychopathologies, Neurobiol.
Learn. Mem., 112, 17-29.
145.Kino, T. (2015) Stress, glucocorticoid
hormones, and hippocampal neural progenitor cells: implications to mood
disorders, Front. Physiol., 6, 230.