Hyperexpression of Integrin α5β1 Promotes Resistance of MCF-7 Human Breast Carcinoma Cells to Doxorubicin via ERK Protein Kinase Down-regulation
G. E. Morozevich1, N. I. Kozlova1, O. Y. Susova2, A. Y. Lupatov1, and A. E. Berman1*
1Orekhovich Institute of Biomedical Chemistry, 119121 Moscow, Russia; E-mail: 1938berman@gmail.com2Blokhin Russian Oncological Center, 115478 Moscow, Russia; E-mail: susovaolga@gmail.com
* To whom correspondence should be addressed.
Received March 14, 2017; Revision received May 11, 2017
In MCF-7 human breast carcinoma cells, α5β1 integrin hyperexpression, which was accomplished by transduction of a full-length α5 integrin cDNA, increased by about 50-70% the number of cells, survived during 48-72 h cell treatment with doxorubicin. Up-regulation of α5β1 reduced the level of the apoptogenic p53 protein and p21 cell cycle inhibitor, but enhanced the activity of Akt and mTOR protein kinases. In addition to these findings, we observed a significant decrease in the activity of both isoforms of phosphokinase Erk1/2, which is known to play a key role in cell viability pathways, including pathways alleviating stress damages caused by distinct antitumor drugs. Diminished Erk activity accompanying the rise of drug resistance can be explained by an “atypical” function of this kinase, which, in the cells studied, promotes an enhanced rather than reduced sensitivity to doxorubicin. To verify this suggestion, the effect of a specific Erk inhibitor, PD98059, on the resistance to doxorubicin of control and α5 cDNA-transduced MCF-7 cells was investigated. The data showed that suppression of Erk activity increased the resistance of control cells (transduced with an “empty” vector) to a level higher than that demonstrated by the α5 cDNA-transduced cells. The highest level of resistance was observed in α5β1-trancduced cells treated with PD98059. Akt and mTOR kinase inhibitors had little if any effect on doxorubicin resistance of α5 cDNA-transduced MCF-7 cells. The data show for the first time that integrin α5β1 can stimulate drug resistance of tumor cells through a mechanism based on the inhibition of protein kinase Erk. From a more general view, the results of this investigation suggest that signal protein kinases can perform in tumor cells “non-canonical” functions, opposite to those, which are the basis for using kinase inhibitors in targeted cancer therapy. It follows that if a protein kinase is supposed to be used as a target for such therapy, it is important to explore its features in the particular tumor prior to the onset of treatment.
KEY WORDS: integrins, tumor growth, drug resistance, Erk protein kinase, signalingDOI: 10.1134/S0006297917090048
Drug resistance of cancer cells is one of the main problems in therapy of oncological diseases. It is known that interaction of cells with extracellular matrix (ECM) plays a substantial role in acquiring drug resistance in tumors [1, 2]. ECM mediates cell responses via cell surface receptors, integrins. They comprise a large family of glycoproteins integrated within the plasma membrane. Each integrin is a heterodimer consisting of α- and β-subunits connected by non-covalent bonds. At present, 18 α- and 8 β-subunits have been identified, which can assemble into 24 unique receptors, although theoretically they may form up to 100 individual receptors. The integrin family is divided into several subfamilies based on a β-subunit common for members of a particular subfamily. Among them, the β1-subfamily is the most abundant and widespread in vivo. The majority of integrins display ligand cross-specificity to ECM proteins, with a few exceptions including α5β1, which binds only fibronectin. Moreover, the α5-subunit dimerizes only with the β1-subunit [3, 4].
Several studies have shown that integrin-mediated signals affect the resistance of tumor cells to stress caused by antitumor cytostatics [5-7]. However, this information refers to a few receptors, and published results are ambiguous. In particular, this conclusion follows from studies of fibronectin-specific integrin α5β1, which is expressed by a number of tumors [8, 9]. Ambiguous are also data on signaling pathways, initiated by individual receptors, as well as the role of signaling molecules in drug resistance [10-12]. Here, we demonstrate for the first time that α5β1 integrin can stimulate drug resistance in cancer cells via a mechanism based on inhibition of protein kinase Erk.
MATERIALS AND METHODS
Cells and reagents. MCF-7 human breast carcinoma cell line was obtained from ATCC (USA). Cells were cultured in DMEM supplemented with 10% fetal calf serum, 2 mM L-glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml) and incubated at 37°C in the atmosphere with 5% CО2.
Polyclonal antibodies to the α5-integrin subunit and monoclonal antibodies to the integrin α5β1 were, respectively, from Chemicon and BD Pharmingen (USA). Polyclonal antibodies to Akt, Erk and their phosphorylated forms (pAkt Ser473 and pErk Thr202/Tyr204), phosphorylated mTOR (pmTOR), proteins p53 and Bcl-2 were from Cell Signaling Tech (USA). Erk inhibitor, PD98059, and Akt inhibitor, LY294002, were from Calbiochem (USA) and other chemicals were from Sigma (USA).
Full-length cDNA cell transduction. Lentivirus plasmid vector Ex-A0280-Lv21 containing full-length α5-integrin cDNA and neomycin-resistance gene, control vector Ex-EGFP-Lv21, as well as a packaging plasmid mix were purchased from GeneCopoeia (USA). Cells were transduced with cDNA, according to the manufacturer’s protocol, followed by selection in geneticin-containing culture medium (800 µg/ml) for 7 days.
Drug resistance. Cells (2·104) were cultured in 100 µl of complete DMEM in 96-well plates for 24 h followed by incubation with various concentrations of doxorubicin for 48-72 h. The percent of surviving cells was determined by MTT assay. Here, drug resistance was defined as cell survival under standard cytostatic treatment (concentration, incubation time). The half-maximal inhibitory concentration (IC50), taken as a measure of cell survival, is the cytostatic concentration causing 50% death of treated cells.
Flow cytometry. Cells ((3-5)·105) were fixed with 70% ethanol, washed with PBS, resuspended in 1 ml citrate buffer containing 50 μg/ml propidium iodide and 50 µl RNase A solution (10 μg/ml) and incubated for 3 h at 4°C. Cell surface expression of integrins was assessed by treating the cells with primary mAb specific to human integrin dimers (BD Pharmingen) followed by staining with FITC-conjugated secondary antibody and fixation with 2% formaldehyde. The cells were analyzed using Becton Dickinson (USA) flow cytometer.
SDS-PAGE and Western blotting. Cells were extracted with the buffer (50 mM Tris-HCl, pH 7.5, 1% Triton X-100, 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS) containing protease/phosphatase inhibitor cocktails (Santa Cruz Biotech, USA), each 1 µl/106 cells, and centrifuged for 10 min at 13,000g. Further procedures were performed as described elsewhere [13].
Statistical analysis. Differences between groups were assessed using Student’s t-test. Significance level was set at p < 0.05.
RESULTS
Hyperexpression of α5β1 potentiates resistance of MCF-7 cells to doxorubicin. We have shown previously that, in drug-resistant human breast carcinoma line MCF-7Dox, down-regulation of fibronectin-specific integrin α5β1 resulted in 2-fold reduced resistance to doxorubicin, thereby suggesting that this receptor is involved in mechanisms underlying drug resistance [14]. A characteristic feature of the MCF-7Dox line is high level of expression of the α5β1 receptor with almost complete suppression of other collagen and fibronectin-binding integrins, α2β1, α3β1 and ανβ3, which are the most specific integrins for epithelial cells [15]. Therefore, it seemed interesting to assess the implication of this receptor in drug resistance in a closely related model, expressing, like most tumor cells, a more abundant set of integrins.
To this end, we investigated doxorubicin resistance in MCF-7 cell line, in relation to which MCF-7Dox line is a derivative. Unlike MCF-7Dox, the parent cells are characterized by much lower resistance to doxorubicin, high expression of integrin α2β1, moderate expression of integrins α3β1 and ανβ3, while they are almost inactive in expression of α5β1 [15]. Based on these data, we analyzed doxorubicin resistance of MCF-7 cells in which expression of α5β1 was restored by transduction with a plasmid vector carrying the full-length α5 subunit cDNA. Restored α5β1 expression (hyperexpression) was quantified by Western blotting assay of cell lysate proteins and by FACS analysis of transduced cells.
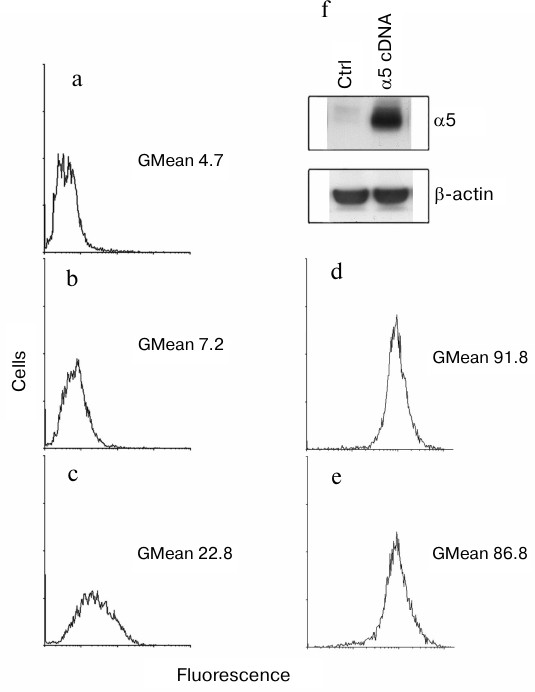
We found that surface expression of α5β1-dimer on MCF-7 cells was increased 3-fold after transduction with α5 cDNA assessed by measuring the geometric mean of fluorescence intensity (GMean) (Fig. 1, a-c). A sharply increased amount of α5-integrin detected in cell lysate of α5- versus control vector-transduced cells is consistent with this finding (Fig. 1f). To assess an effect of α5 cDNA transduction on expression of other β1-integrin subfamily members, a flow cytometry assay was applied to analyze surface expression of receptor α2β1 as the most abundant integrin for this cell line [15]. We observed that transduction with vector containing α5-subunit cDNA had virtually no effect on surface expression of α2β1 integrin (Fig. 1, d and e) as evidenced by the lack of significant differences in GMean in control versus α5 cDNA-transduced cells. Thus, we concluded that de novo formation of α5β1 dimer did not deplete the intracellular β1-subunit pool and did not affect the expression of endogenous β1-integrins.
Fig. 1. Efficient hyperexpression of α5β1 integrin in MCF-7 cells transduced with full-length cDNA α5-subunit. Cells were infected with lentivirus carrying α5 cDNA vector or none specific sequence (empty vector) followed by selection in geneticin as described in “Materials and Methods”. a-e) FACS analysis of surface expression of various integrins: a) cells were transduced with an empty vector followed by staining with anti-mouse Ig FITC-conjugated antibodies (see “Materials and Methods”); b) cells transduced with an empty vector were treated with human α5β1 antibodies (Pharmingen, 555615; 1 : 1000 dilution), followed by staining with anti-mouse Ig FITC-conjugated antibodies; c) cells transduced with α5 cDNA vector were treated and stained as in (b); d) cells transduced with an empty vector were treated with human α2β1 antibodies (Pharmingen, 555668; 1 : 1000 dilution), followed by staining with anti-mouse Ig FITC-conjugated antibodies; e) cells transduced with α5 cDNA vector were treated and stained as in (d); f) Western blotting assay of cell lysate proteins. In brief, 30 µg lysate proteins were run on PAGE, electroblotted and probed with primary and secondary antibodies (see “Materials and Methods”). Ctrl, cells transduced with control vector; α5 cDNA, cells transduced with vector encoding full-length cDNA α5-subunit.
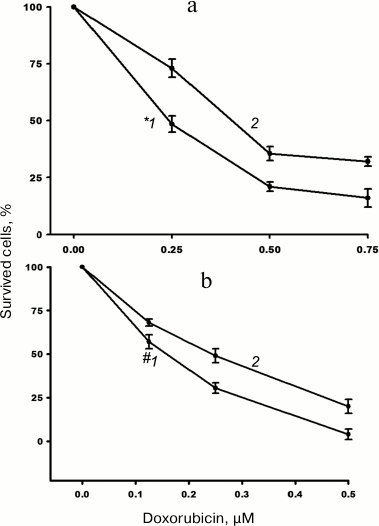
The impact of α5β1 hyperexpression on resistance of MCF-7 cells to doxorubicin was checked by assessing changes in IC50. We found that in the cells transduced with α5 cDNA, IC50 increased 1.5-1.7 times as compared to control vector-transduced cells (Fig. 2).
Fig. 2. Hyperexpression of α5β1 promotes resistance of MCF-7 cells to doxorubicin. Cells infected with lentivirus carrying control (1) or α5 cDNA-containing (2) vectors were cultured in DMEM in the presence of various concentrations of doxorubicin. IC50 was calculated as described in “Materials and Methods”. a) Curves: 1) IC50 = 0.24 µM; 2) IC50 = 0.41 µM; incubation for 48 h. b) Curves: 1) IC50 = 0.16 µM; 2) IC50 = 0.24 µM; incubation for 72 h. Data from three independent experiments are displayed as M ± SEM; *1 – p < 0.02 compared to curve 2; #1 – p < 0.05 compared to curve 2.
Signaling pathways mediating effects caused by α5β1 hyperexpression. To clarify the mechanisms mediating the effect of integrin α5β1 on drug resistance we analyzed the expression of proteins, which are known to be involved in signal transduction and regulation of the diverse functions of cells. As can be seen from Fig. 3, hyperexpression of α5β1 downregulates expression of proapoptotic protein p53 and p21 cell cycle inhibitor while stimulates expression of antiapoptotic Bcl-2 protein.
Fig. 3. Effect of α5β1 hyperexpression on expression of signaling proteins in MCF-7 cells. Cells were transduced with control (Ctrl) or α5 cDNA-containing vectors and lysed, and then 30 µg lysate proteins were run on 7.5% SDS-PAGE followed by Western blotting. Membranes were probed with antibodies to indicated proteins at dilution 1 : 1000.
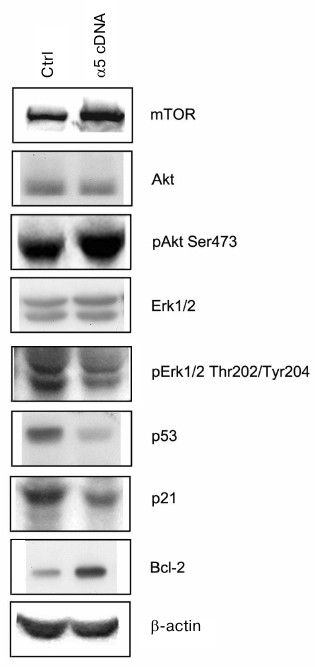
The proteins mentioned above control events occurring inside the cell nucleus, i.e. they are involved at the final steps of signal transduction triggered by, apart from integrins, various cell surface receptors and intracellular metabolites. Upstream events induced within the cell membrane are more specific to integrins. Among them are the most fully characterized pathways mediated by protein kinases IP3-K/Akt and MAPK family protein kinases including protein kinase Erk [16-18]. To elucidate whether such pathways are involved in α5β1 integrin-induced signaling in MCF-7 cells hyperexpressing α5β1, we examined changes in expression and activity of protein kinases Akt and Erk1/2 (Erk isomers with molecular weight 42 and 44 kDa, respectively). To this end, expression of protein kinases was assessed by Western blotting assay in cell lysate using antibodies to the total enzyme protein, whereas their activity was determined by probing with antibodies specifically recognizing their active (phosphorylated) forms. We found that hyperexpression of α5β1 had no effect on the total protein expression of Akt and Erk1/2 in transduced cells, but sharply increased activity of protein kinase Akt and its downstream substrate, protein kinase mTOR (Fig. 3). These signaling proteins are involved in various metabolic and cell survival mechanisms. At the same time, the activity of both isoforms of protein kinase Erk1/2 which, like Akt and mTOR, control cell proliferation and play a key role in cell survival and stress alleviation, was markedly down-regulated.
Reduced activity of Erk occurring in response to α5β1 hyperexpression-induced increase of drug resistance may be interpreted as “atypical” function of this protein kinase in MCF-7 cells. We hypothesized that in these cells, Erk mediates signals aimed at attenuation rather than promotion of drug resistance. If this is the case, the inhibition of Erk in control cells would result in an effect similar to that observed in cells transduced with α5 cDNA, i.e. enhanced drug resistance. Therefore, this result would corroborate our previous data demonstrating stimulation of substrate-dependent apoptosis (anoikis) in response to downregulation of α2β1 integrin in MCF-7 cells [19].
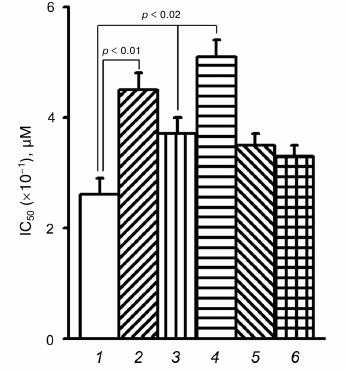
To verify these possibilities, the effect of PD98059, a specific Erk inhibitor, on resistance to doxorubicin was determined in control and α5 cDNA-transduced cells (Fig. 4). In control cells, the mean IC50 increased by 70% (4.5 versus 2.6) after their treatment with PD98059. In α5 cDNA-transduced cells, IC50 increased by 40% (up to 3.7). The highest IC50 (5.1) was observed in α5 cDNA-transduced cells treated with PD98059, i.e. it increased by approximately 100% compared to control. Hence, the overall effect induced by α5β1 cDNA hyperexpression together with Erk inhibition is equal to the sum of the effects of each of them and, therefore, is additive.
Fig. 4. Down-regulation of protein kinase Erk augments resistance of MCF-7 cells to doxorubicin. Cells transduced with control (1, 2) or α5 cDNA vector (3-6) were incubated for 24 h with 25 µM Erk inhibitor PD98059 (2, 4), 25 µM Akt inhibitor LY294002 (5), or 200 nM mTOR inhibitor rapamycin (6), followed by treatment with various concentrations of doxorubicin for 48 h with subsequent IC50 determination. The data from three independent experiments are displayed as M ± SEM.
The data in Fig. 3 demonstrate that re-expression of α5β1 is accompanied by a marked up-regulation of protein kinases Akt and mTOR of which the latter is an Akt down-stream effector [20]. Involvement of Akt in mechanisms neutralizing stress caused by anticancer agents has been demonstrated in various cell lines [21, 22]. So, the observed increase in the activity of these protein kinases could evidence about their involvement in enhancing the resistance to doxorubicin of cells transduced with α5 cDNA.
However, as seen in Fig. 4, treatment of the transduced cells with specific Akt and mTOR inhibitors (LY294002 and rapamycin, respectively) did not result in marked decrease in drug resistance, suggesting, therefore, that these protein kinases did not contribute much in increasing drug resistance caused by α5β1 hyperexpression.
DISCUSSION
As mentioned above, information on the role of integrin-binding integrins in the drug resistance of tumor cells is ambiguous. The growth of acute myeloleukemia cells on the fibronectin substrate blocked the cytostatic effects of daunorubicin and cytosine arabinoside, with the blocking effect being manifested when the cell–substrate interaction was mediated by fibronectin-specific integrin α4β1, but not with α5β1 integrin, which has the same ligand specificity [23]. In line with these results is elevated sensitivity to temozolomide in glioblastoma cells in response to down-regulation of α5β1 [8]. However, adhesion to fibronectin and activation of α5β1 receptor did not affect sensitivity to doxorubicin in lymphoblastic leukemia T cells, while their interaction with collagen attenuated cytostatic effect [9]. Clinical observations of patients with ovary cancer demonstrated high correlation between expression of α5β1 integrin and resistance to antitumor drugs [24].
The ambiguity of these findings seems to result from differences in the signaling pathways that mediate the effect of integrins in different types of tumors. For instance, high drug resistance found in multiple myeloma cell line was shown to be caused by activation of Src/Syk/STAT3 and Akt signaling pathway via α5β1 integrin [10]. In the lines of prostate carcinoma and non-small cell lung cancer, the neutralizing effect of α5β1, directed against the cytostatic effect of SAHA (a hydroxamic acid derivative), is due to the activation of receptor tyrosine protein kinase MET [11], while in a leukemia line, similar effect of α5β1 is mediated via activation protein kinase GSK3β [12].
Here, we present for the first time the data suggesting that the anti-cytostatic function of α5β1 integrin can be mediated by suppression rather than stimulation of Erk-dependent signaling pathway, and inhibition of this protein kinase can enhance resistance to cytostatic agents. These results are not in line with a number of publications. For example, in glioma cells, the resistance to temozolomide, induced by integrin β1, depends on the activation of Erk protein kinase [25]. Investigation of two colon cancer cell lines demonstrated direct dependence between β6 integrin-induced resistance to 5-fluorouracil and activity of Erk [26]. These results corroborate the increased resistance to doxorubicin found in the acute T-cell lymphoblastic leukemia lines with MAP/Erk signaling cascade activated by integrin α2β1 [9]. However, it is noteworthy that in this report, as well as in another study [27], activation of Erk occurred only upon increase in signaling activity of collagen-binding integrins and was not observed when fibronectin-binding receptors, including α5β1, were activated. Also of important is that modifications of the Akt signaling pathway did not affect drug resistance of the cells.
On the other hand, the data presented above on the “atypical” function of Erk in the mechanism of drug resistance of MCF-7 cells are consistent with our previous investigation made on the same cells demonstrating paradoxical role of Erk in the activity of integrin α2β1 directing against anoikis, apoptosis induced by disruption of cell–matrix contacts [19]. These data agree with
markedly diminished cytostatic effect of anticancer drug taxol upon blocking Ras/Raf/Erk signaling pathway observed in MCF-7 cell line [28]. Immortalized embryonic fibroblasts transformed with v-H-ras or c-src oncogenes as well as colon adenocarcinoma cells expressing active oncogene K-ras display reduced resistance to cisplatin, etoposide, and siramesine, which is sharply upregulated upon inhibiting Erk activity [29].
Mechanisms responsible for opposed functions of Erk-induced signals in different cell types have been poorly examined. It is assumed that the intracellular localization of Erk is of great importance, and during translocation from the cytoplasm into the nucleus, this protein kinase induces signals directed to cell death [30].
Results of this study and data published elsewhere [19, 28, 29] demonstrating “noncanonical” functions of Erk, are of interest from view of targeted therapy designed to act on a particular signaling protein kinases in distinct neoplasms. The action of almost all currently used targeted antitumor drugs is aimed at suppressing the activity of distinct links of signal chains, which provide the tumor cell with the ability to survive in stressful situations. Many of these pathways “cross” at Erk, and attempts to use this protein kinase as a target in treatment of cancer are presented in a number of publications. For instance, a clear-cut therapeutic effect was demonstrated in patients with renal cell carcinoma treated with Erk inhibitor sorafenib, and based on clinical studies it was recommended as the first-line therapy [31, 32]. Another Erk inhibitor, cobimetinib, substantially enhances therapeutic effect of vemurafenib (BRAF inhibitor) in treatment of melanoma [33]. Obviously, ignoring the ability of Erk to exert noncanonical “inverted” properties in some neoplasms may not only abrogate effects of targeted therapy, but even contribute to development of the disease.
Given the fact that the cell line discussed in this study is a model of estrogen dependent breast cancer, it would be interesting to consider the possible significance of changes in Erk protein kinase in development of hormonal resistance of this neoplasia. The success of hormonal therapy in patients with breast cancer is determined by the level of expression of the estrogen receptor ER-α by tumor cells. Probability of successful therapy of patients with ER-α-positive tumor is 7-8 times higher than with ER-α-negative tumor [34]. To this end, of special interest are the studies demonstrating that expression of ER-α inversely correlates with Erk activity [35, 36]. These results indicate that success of breast cancer therapy with anti-estrogen drugs (e.g. tamoxifen), as well as by cytostatics, can be substantially determined by the properties (conventional or “non-canonical”) of protein kinase Erk.
Acknowledgments
This study was performed within the framework of the 2013-2020 Program of Basic Scientific Research of the Russian National Academies of Sciences, subject “Development of cell models of molecular processes in organs and tissues” (Orekhovich Institute of Biomedical Chemistry) and with financial support by the Russian Foundation for Basic Research (projects No. 15-04-05511, 17-04-00716).
REFERENCES
1.Elliott, T., and Sethi, T. (2002) Integrins and
extracellular matrix: a novel mechanism of multidrug resistance,
Expert. Rev. Anticancer Ther., 2, 449-459.
2.Hazlehurst, L. A., Landowski, T. H., and Dalton, W.
S. (2003) Role of the tumor microenvironment in mediating de
novo resistance to drugs and physiological mediators of cell death,
Oncogene, 22, 7396-7402.
3.Flier, A. V., and Sonnenberg, A. (2001) Function
and interactions of integrins, Cell Tis. Rev., 305,
285-298.
4.Hynes, R. O. (2002) Integrins: bidirectional,
allosteric signaling machines, Cell, 110, 673-687.
5.Hodkinson, P. S., Mackinnon, A. C., and Sethi, T.
(2007) Extracellular matrix regulation of drug resistance in small-cell
lung cancer, Int. J. Radiat. Biol., 83, 733-741.
6.Long, Q. Z., Zhou, M., Liu, X. G., Du, Y. F., Fan,
J. H., Li, X., and He, D. L. (2013) Interaction of CCN1 with
ανβ3 integrin induces P-glycoprotein and confers
vinblastine resistance in renal cell carcinoma cells, Anticancer
Drugs, 24, 810-817.
7.Liu, C. C., Leclair, P., Yap, S. Q., and Lim, C. J.
(2013) The membrane-proximal KXGFFKR motif of α-integrin mediates
chemoresistance, Mol. Cell. Biol., 33, 4334-4345.
8.Janouskova, H., Maglott, A., Leger, D. Y., Bossert,
C., Noulet, F., Guerin, E., Guenot, D., Pinel, S., Chastagner, P.,
Plenat, F., Entz-Werle, N., Lehmann-Che, J., Martin, S., Teisinger, J.,
and Dontenwill, M. (2012) Integrin α5β1 plays a critical
role in resistance to temozolomide by interfering with the p53 pathway
in high-grade glioma, Cancer Res., 14, 3463-3470.
9.Naci, D., El Azreq, M. A., Chetoui, N., Lauden, L.,
Sigaux, F., Charron, D., Al-Daccak, R., and Aoudjit, F. (2012)
α2β1 integrin promotes chemoresistance against doxorubicin
in cancer cells through extracellular signal-regulated kinase (ERK),
J. Biol. Chem., 287, 17065-17076.
10.Lin, L., Yan, F., Zhao, D., Lv, M., Liang, X.,
Dai, H., Qin, X., Zhang, Y., Hao, J., Sun, X., Yin, Y., Huang, X.,
Zhang, J., Lu, J., and Ge, Q. (2016) Reelin promotes the adhesion and
drug resistance of multiple myeloma cells via integrin β1
signaling and STAT3, Oncotarget, 7, 9844-9858.
11.Ding, L., Zhang, Z., Liang, G., Yao, Z., Wu, H.,
Wang, B., Zhang, J., Tariq, M., Ying, M., and Yang, B. (2015) SAHA
triggered MET activation contributes to SAHA tolerance in solid cancer
cells, Cancer Lett., 356, 828-836.
12.De Toni-Costes, F., Despeaux, M., Bertrand, J.,
Bourogaa, E., Ysebaert, L., Payrastre, B., and Racaud-Sultan, C. (2010)
A new α5β1 integrin-dependent survival pathway through
GSK3β activation in leukemic cells, PLoS One, 5, 3,
e9807.
13.Morozevich, G. E., Kozlova, N. I., Cheglakov, I.
B., Ushakova, N. A., Preobrazhenskaya, M. E., and Berman, A. E. (2008)
Implication of α5β1 integrin in invasion of drug-resistant
MCF-7/ADR breast carcinoma cells: a role for MMP-2 collagenase,
Biochemistry (Moscow), 73, 791-796.
14.Morozevich, G. E., Kozlova, N. I., Ushakova, N.
A., Preobrazhenskaia, M. E., and Berman, A. E. (2011) Implication of
integrin α5β1 in human breast carcinoma apoptosis and drug
resistance, Biomed. Khim., 57, 77-84.
15.Morozevich, G. E., Kozlova, N. I., Ushakova N.
A., and Berman, A. E. (2009) Integrin α5β1 controls invasion
of human breast carcinoma cells by direct and indirect modulation of
MMP-2 collagenase, Cell Cycle, 8, 2219-2225.
16.Morozevich, G. E., Kozlova, N. I., Ushakova, N.
A., Preobrazhenskaya, M. E., and Berman, A. E. (2012) Integrin
α5β1 simultaneously controls EGFR-dependent proliferation
and Akt-dependent pro-survival signaling in epidermoid carcinoma cells,
Aging (Albany NY), 4, 368-374.
17.King, W. G., Mattaliano, M. D., Chan, T. O.,
Tsichlis, P. N., and Brugge, J. S. (1997) Phosphatidylinositol 3-kinase
is required for integrin-stimulated AKT and Raf-1/mitogen-activated
protein kinase pathway activation, Mol. Cell. Biol., 17,
4406-4418.
18.Paoli, P., Giannoni, E., and Chiarugi, P. (2013)
Anoikis molecular pathways and its role in cancer progression,
Biochim. Biophys. Acta, 12, 3481-3498.
19.Morozevich, G. E., Kozlova, N. I., Susova, O. Y.,
Karalkin, P. A., and Berman, A. E. (2015) Implication of
α2β1 integrin in anoikis of MCF-7 human breast carcinoma
cells, Biochemistry (Moscow), 80, 97-104.
20.Sarbassov, D. D., Guertin, D. A., Ali, S. M., and
Sabatini, D. M. (2005) Phosphorylation and regulation of Akt/PKB by the
RICTOR–mTOR complex, Science, 307, 1098-1101.
21.Pommier, Y., Sordet, O., Antony, S., Hayward, R.
L., and Kohn, K. W. (2004) Apoptosis defects and chemotherapy
resistance: molecular interaction maps and networks, Oncogene,
23, 2934-2949.
22.Layani-Bazar, A., Skornick, I., Berrebi, A.,
Pauker, M. H., Noy, E., Silberman, A., Albeck, M., Longo, D. L.,
Kalechman, Y., and Sredni, B. (2014) Redox modulation of adjacent
thiols in VLA-4 by AS101 converts myeloid leukemia cells from a
drug-resistant to drug-sensitive state, Cancer Res., 74,
3092-3103.
23.Matsunaga, T., Takemoto, N., Sato, T., Takimoto,
R., Tanaka, I., Fujimi, A., Akiyama, T., Kuroda, H., Kawano, Y.,
Kobune, M., Kato, J., Hirayama, Y., Sakamaki, S., Kohda, K., Miyake,
K., and Niitsu, Y. (2003) Interaction between leukemic cell VLA-4 and
stromal fibronectin is a decisive factor for minimal residual disease
of acute myelogenous leukemia, Nat. Med., 9,
1158-1165.
24.Hu, Z., Gao, S., Gao, J., Hou, R., Liu, C., Liu,
J., Li, B., Liu, D., Zhang, S., and Lin, B. (2012) Elevated levels of
Lewisy and integrin α5β1 correlate with
chemotherapeutic drug resistance in epithelial ovarian carcinoma,
Int. J. Mol. Sci., 13, 15588-15600.
25.Han, S., Li, Z., Master, L. M., Master, Z. W.,
and Wu, A. (2014) Exogenous IGFBP-2 promotes proliferation, invasion,
and chemoresistance to temozolomide in glioma cells via the integrin
β1-ERK pathway, Br. J. Cancer, 111, 1400-1409.
26.Liu, S., Wang, J., Niu, W., Liu, E., Wang, J.,
Peng, C., Lin, P., Wang, B., Khan, A. Q., Gao, H., Liang, B., Shahbaz,
M., and Niu, J. (2013) The β6-integrin-ERK/MAP kinase pathway
contributes to chemo resistance in colon cancer, Cancer Lett.,
328, 325-334.
27.El Azreq, M. A., Naci, D., and Aoudjit, F. (2012)
Collagen/β1 integrin signaling up-regulates the ABCC1/MRP-1
transporter in an ERK/MAPK-dependent manner, Mol. Biol. Cell,
17, 3473-3484.
28.Blagosklonny, M. V., Schulte, T., Nguyen, P.,
Trepel, J., and Neckers, L. M. (1996) Taxol-induced apoptosis and
phosphorylation of Bcl-2 protein involves c-Raf-1 and represents a
novel c-Raf-1 signal transduction pathway, Cancer Res.,
56, 1851-1854.
29.Fehrenbache, N., Bastholm, L.,
Kirkegaard-Sorensen, T., Rafn, B., Bottzauw, T., Nielsen, C., Weber,
E., Shirasawa, S., Kallunki, T., and Jaattela, M. (2008) Sensitization
to the lysosomal cell death pathway by oncogene-induced down-regulation
of lysosome-associated membrane proteins 1 and 2, Cancer Res.,
68, 6623-6633.
30.Cagnol, S., and Chambard, J. C. (2009) ERK and
cell death: mechanisms of ERK induced cell death – apoptosis,
autophagy and senescence, FEBS J., 277, 2-21.
31.Mai, H., Huang, J., Zhang, Y., Qu, N., Qu, H.,
Mei, G. H., Liu, J., Xu, X., and Chen, L. (2017) In vivo
relation between plasma concentration of sorafenib and its safety in
Chinese patients with metastatic renal cell carcinoma: a single center
clinical study, Oncotarget, doi: 10.18632/oncotarget.16465.
32.Matveev, V. B., and Chernyaev, V. A. (2015)
Sorafenib is the first targeted agent to treat metastatic kidney
cancer, Cancer Urol., 11, 73-78.
33.Roskoski, R., Jr. (2017) Allosteric MEK1/2
inhibitors including cobimetanib and trametinib in the treatment of
cutaneous melanomas, Pharmacol. Res., 117, 20-31.
34.Duffy, M. J. (2006) Estrogen receptors: role in
breast cancer, Crit. Rev. Lab. Sci., 43, 325-347.
35.Oh, A. S., Lorant, L. A., Holloway, J. N.,
Miller, D. L., Kern, F. G., and El-Ashry, D. (2001) Hyperactivation of
MAPK induces loss of ERα expression in breast cancer cells,
Mol. Endocrinol., 15, 1344-1359.
36.Jelovac, D., Sabnis, G., Long, B. J., Macedo, L.,
Goloubeva, O. G., and Brodie, A. M. (2005) Activation of
mitogen-activated protein kinase in xenografts and cells during
prolonged treatment with aromatase inhibitor letrozole, Cancer
Res., 65, 5380-5389.