Interaction between RAD51 and MCM Complex Is Essential for RAD51 Foci Forming in Colon Cancer HCT116 Cells
Jun Huang, Hong-Liang Luo, Hua Pan, Cheng Qiu, Teng-Fei Hao, and Zheng-Ming Zhu*
Second Affiliated Hospital of Nanchang University, Department of Gastrointestinal Surgery, Nanchang 330006, China; E-mail: zhuzhengming7088@126.com* To whom correspondence should be addressed.
Received May 8, 2017; Revision received October 25, 2017
Colon cancer remains one of the most common digestive system malignancies in the World. This study investigated the possible interaction between RAD51 and minichromosome maintenance proteins (MCMs) in HCT116 cells, which can serve as a model system for forming colon cancer foci. The interaction between RAD51 and MCMs was detected by mass spectrometry. Silenced MCM vectors were transfected into HTC116 cells. The expressions of RAD51 and MCMs were detected using Western blotting. Foci forming and chromatin fraction of RAD51 in HCT116 cells were also analyzed. The results showed that RAD51 directly interacted with MCM2, MCM3, MCM5, and MCM6 in colon cancer HTC116 cells. Suppression of MCM2 or MCM6 by shRNA decreased the chromatin localization of RAD51 in HTC116 cells. Moreover, silenced MCM2 or MCM6 decreased the foci forming of RAD51 in HTC116 cells. Our study suggests that the interaction between MCMs and RAD51 is essential for the chromatin localization and foci forming of RAD51 in HCT116 cell DNA damage recovery, and it may be a theoretical basis for analysis of RAD51 in tumor samples of colon cancer patients.
KEY WORDS: colon cancer, HTC116 cells, RAD51, MCM complex, homologous recombinationDOI: 10.1134/S0006297918010091
Abbreviations: DSBs, DNA double-stranded breaks; HR, homologous recombination; MCM, minichromosome maintenance (proteins); shRNA, short hairpin RNA.
Colon cancer remains one of the most common digestive system
malignancies in the World [1]. The pathogenic
mechanism of colon cancer is very complex, which results in high
morbidity and high mortality of colon cancer [2].
Previous studies have demonstrated that the failure of DNA damage
recovery contributes to the occurrence of colon cancer [3, 4], in which DNA
double-stranded breaks (DSBs) are most common, threatening genome
stability seriously [5]. Moreover, it has also been
reported that homologous recombination (HR), ionizing radiation, DNA
ruptures, and anti-cancer drugs cause DSBs during the development of
cancer [6-8].
HR is a pivotal repair pathway for the maintenance of genome integrity, which is essential for the repair of DNA breaks including DSBs [9]. Previous studies have shown that the major HR proteins including RAD51, RAD52, and RAD54 depend on DSBs through complex mechanisms [10, 11]. For example, RAD52 was shown to interact with histone H2AX in breast cancer [12, 13], and RAD54 was correlated with ionizing radiation-induced foci forming [14]. Recent studies revealed that foci formation of RAD51 in response to ionizing radiation is an important step in the repair of DNA DSBs [15, 16]. Besides, a recent study suggested a spatiotemporal regulation model of RAD51 that was essential for homologous recombination and associated with chromatin in the S phase [17].
It has been shown that minichromosome maintenance (MCM) complex is the core component of eukaryotic DNA replication machinery, and it plays a critical role in activating DNA replication and facilitating possessive DNA synthesis [18]. The MCM complex is composed of eight highly conserved proteins, MCM2, MCM3, MCM4, MCM5, MCM6, MCM7, MCM8, and MCM9, which form the MCM protein family [19]. Shukla et al. indicated that the MCM complex had strong propensity to be physically recruited to the sites where hRAD51 and hRAD52-mediated homologously aligned ends need to be replicationally repaired [20]. Park et al. demonstrated that MCM8-MCM9 promoted RAD51 recruitment at DNA damage sites to facilitate the HR process [21]. However, the possible molecular mechanisms of the interaction of RAD51 with MCM2-7 remain unclear.
In this study, we analyzed the interaction between RAD51 and the MCM complex in colon cancer HCT116 cells to reveal the possible mechanism of RAD51 and MCM complex formation in the development of colon cancer. Our study might provide a theoretical basis for illustrating one of the molecular mechanisms involved in colon cancer development.
MATERIALS AND METHODS
Cell culture. Human colon cancer HCT116 cells and normal human embryonic kidney 293T cells were obtained from the Chinese Academy of Sciences Type Culture Collection (Shanghai, China). The cells were maintained in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and streptomycin/penicillin in 100 U/ml concentration. Cultures were incubated at 37°C under 5% CO2.
Chromatin fraction. Cell chromatin fraction was prepared as described previously [22]. HCT116 cells were lysed in 100 µl of buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 mM sucrose, and 10% glycerol) containing 0.2% Triton X-100, 1 mM dithiothreitol (DTT), and protease inhibitors on ice for 8 min. The cell suspension was centrifuged at 1000 rpm for 5 min at 4°C. The supernatant contained the cytoplasmic fraction. The pellet was resuspended in 100 µl of buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, and protease inhibitors), placed on ice for 30 min, and centrifuged at 1500 rpm for 5 min in a microcentrifuge at 4°C. The supernatant was combined with cytoplasmic fraction as the non-chromatin fraction. The pellet was resuspended in 150 µl of buffer C (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 1% NP-40, 1 mM DTT, and protease inhibitors) on ice for 10 min, briefly sonicated, and centrifuged (13,000 rpm for 10 min in a microcentrifuge at 4°C). The supernatant was a chromatin-enriched fraction. Proteins in these fractions were quantitated and analyzed using SDS-PAGE. Anti-RAD51 (ab63801; Abcam Biotechnology, USA), anti-Flag (Sigma-Aldrich, USA), and anti-HA (3F10; Roche Diagnostics, Switzerland) antibodies were used.
Immunoprecipitation and immunoblot analysis. Cells were suspended in 1% Nonidet P-40 lysis buffer (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% Nonidet P-40, 10 mM NaF, 1 mM Na3VO3, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride, 10 µg/ml leupeptin, and 10 µg/ml pepstatin) and then incubated on ice for 30 min. After centrifugation at 1500 rpm for 5 min, the supernatant was immunoprecipitated with 5 µg anti-FLAG, anti-HA or anti-Myc (Cell Signaling Technology, USA) cross-linked to protein A- (Amersham Biosciences, USA) or -G-Sepharose (Zymed Laboratories, USA). The immunoprecipitates were washed three times with 0.1% Nonidet P-40 lysis buffer subjected to immunoblot analysis. Cell lysates or immunoprecipitated proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore, USA). The membranes were then incubated with anti-FLAG, anti-HA, and anti-Myc antibodies, respectively. Immunoreactive protein bands were visualized using chemiluminescence (Perkin Elmer, USA). Anti-GST (Nacalai Tesque, Inc., Japan) and anti-α-tubulin (Sigma-Aldrich) antibodies were used.
In vitro binding assays. GST fusion proteins were produced in Escherichia coli strain BL21 by standard methods using glutathione agarose (Sigma-Aldrich) affinity chromatography. RAD51 was cloned into the pGEX4T1 vector (Amersham Biosciences) for protein expression and purification. GST pull-down assays were performed as described previously [23]. For coimmunoprecipitation, 293T cells (1·106) were seeded into a 10-cm dish before transfection. Transfection of 2.5 g of expression vector was performed with Bio-T (Bioland Scientific, Germany) according to the manufacturer’s protocol. Whole cell extracts were made in 1 ml of radioimmune precipitation assay (RIPA) buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS). Coimmunoprecipitation and immunoblotting were performed as follows: the lysate was pre-cleared by incubation with agarose protein A/G beads for 1 h and then immunoprecipitated overnight with 1-2 g of the indicated antibodies and agarose protein A/G beads. Immunoblotting was performed with the indicated antibodies. For endogenous co-immunoprecipitation, confluent 293T cells maintained in DMEM supplemented with 10% FBS were used. Whole cell extracts (WCE) were prepared from 1 ml of RIPA buffer and immunoprecipitated with 2 µg of antibody and agarose protein A/G beads overnight at 4°C. Immunoblotting was performed with the target antibody.
shRNA transfection. Short hairpin RNA directed against MCM2 and MCM6 were ligated into U6/GFP/Neo plasmids (GenePharm, China) following the manufacturer’s protocol. Cells transfection was performed using Lipofectamine 3000 reagent (Invitrogen, USA) according to the manufacturer’s instructions. The shRNA sequence against MCM2 was: 5′-TCATCGGAATCCTTCACCA-3. The shRNA sequence against MCM6 was: 5-TGAGATGAGTCAAGATAAA-3. Cells transfected with empty vector were used as control.
Immunofluorescence microscopy. Cells were fixed with 3.7% formaldehyde solution for 10 min on ice and permeabilized sequentially with 50, 75, and 95% ethanol on ice for 5 min each. The slides were blocked with PBS-containing blocking solution for 30 min at room temperature and then incubated with primary antibody for 1 h at room temperature and washed for 5 min with PBS three times. After that, the cells were incubated with Alexa Fluor 488- or -594-conjugated secondary antibody (Molecular Probes, USA) for 1 h at room temperature, washed three times with PBS, and preserved in Vectashield (Vector Inc., USA). DNA was stained with 1 µg/ml bis-benzimide (Hoechst33258; Sigma-Aldrich) for 20 min at room temperature. The samples were visualized using an Olympus Power BX51 fluorescence microscope (Olympus, Co., Japan).
Identification of RAD51-interacting proteins by mass spectrometry. HCT116 cells stably expressing FLAG-RAD51 were fractionated into cytoplasmic and nuclear compartments. RAD51-interacting proteins were isolated by FLAG MCM2 (Sigma-Aldrich) beads and eluted with 5 mg/ml 3× FLAG peptides in PBS. The eluate was first denatured in 8 M urea and then reduced and alkylated with 10 mM Tris-(2-carboxyethyl)-phosphine hydrochloride (Roche Diagnostics) and 55 mM iodoacetamide (Sigma-Aldrich), respectively. The samples were digested overnight with trypsin (Promega, USA) and pressure-loaded onto a 250-µm silica capillary column (Polymicro Technologies, USA). After desalting, each biphasic column was connected to a 100 µm silica capillary analytical C18 column. Each MudPIT column was placed in line with an 1100 quaternary HPLC pump (Agilent Technologies, USA), and the eluted peptides were electrosprayed directly into an LTQ mass spectrometer (Thermo Fisher Scientific, USA). MS/MS spectra were extracted using RawXtract (version 1.9.9) and searched with the SEQUEST algorithm against the human International Protein Index (IPI) database. SEQUEST search results were assembled and filtered using the DTASelect (version 2.0) algorithm, requiring peptides to be at least half-tryptic and a minimum of two peptides per protein identification. The protein identification false positive rate was kept below 5%.
Statistical analysis. All experiments were carried out in triplicate and the results are expressed as mean ± standard error of mean (SEM) in this study. Statistical analysis was performed using Graph Prism 5.0 software (GraphPad Prism, USA). Statistical differences between groups were analyzed by Student’s t-test and ANOVA for parametric data; p < 0.05 or p < 0.01 was set as the level of statistical significance.
RESULTS
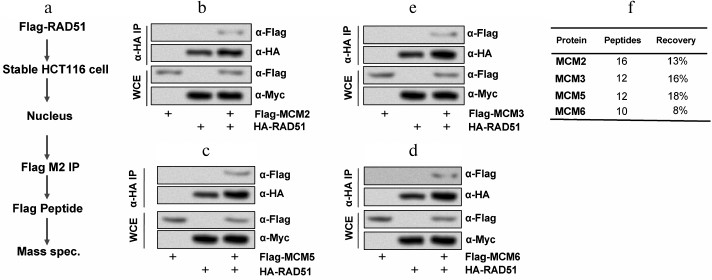
RAD51 interacted with MCMs. To understand the functional roles of RAD51 in colon cancer HCT116 cells, the specific RAD51-interacting proteins were identified. Firstly, we produced a stable HCT116 cell line expressing differently tagged RAD51 and MCM complex. Then the nuclear extracts from a stable clone were immunoprecipitated with anti-Flag M2 beads eluted with 3× Flag peptide, and mass spectrometric analysis was carried out (Fig. 1a). Co-immunoprecipitation showed interaction between RAD51 and MCM complex in HCT116 cells (Fig. 1, b-e). The peptide recovery of RAD51 from the nuclear IPs was 13, 16, 0, 18, 8, and 0% for MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7 (Fig. 1f). MCM2, MCM3, MCM5, and MCM6 play significant roles in DNA replication and replication checkpoint in HCT116 cells. These results suggest that MCM2, MCM3, MCM5, and MCM6 might be the interactive proteins of RAD51 in HCT116 cells.
Fig. 1. RAD51 interacts with MCMs in HCT116 cells. a) The procedure for detecting the interaction mechanism between RAD51 and MCM; b) RAD51 interacted with MCM2; c) RAD51 interacted with MCM3; d) RAD51 interacted with MCM5; e) RAD51 interacted with MCM6; f) statistical analysis of the four kinds of MCMs. WCE, whole cell extracts.
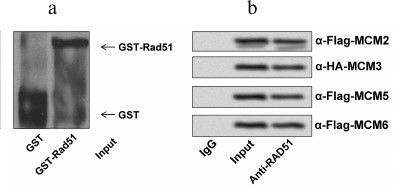
RAD51 interacted directly with MCMs in HCT116 cells. To determine if RAD51 interacts with MCM complex directly or not, we performed a GST pull-down assay using GST-RAD51 bound on glutathione-agarose beads to pull down MCM proteins individually (Fig. 2a). As displayed in Fig. 2b, the interactions between RAD51 and MCM2, MCM3, MCM5, or MCM6 in HCT116 cells indicate a multi-interface association between RAD51 and the MCM complex.
Fig. 2. RAD51 directly interacts with MCMs. a) GST or GST-RAD51 proteins bound on glutathione-agarose beads were purified and used to pull down MCM proteins. b) MCM2, MCM3, MCM5, and MCM6 were detected by GST-RAD51 beads.
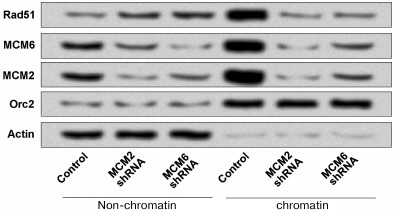
Downregulation of MCMs decreased the RAD51 chromatin fraction. Previous studies have demonstrated that a substantial portion of RAD51 is located on chromatin in the absence of DNA damage [15, 16]. In addition, the MCM complex is the core component of eukaryotic DNA replication machinery and has recently been indicated as an important player in replication checkpoint [22]. To determine whether MCMs are essential for RAD51 chromatin localization, we used a gene silencing-mediated method (MCMs shRNA) to downregulate the expressions of MCM2 and MCM6 in HCT116 cells and then performed chromatin fraction. As shown in Fig. 3, the chromatin fraction was significantly depleted in MCM2 shRNA and MCM6 shRNA compared to the non-chromatin. Orc2 and Actin are marker proteins for chromatin and non-chromatin fractions, respectively.
Fig. 3. Downregulation of MCM2 and MCM6 decreased the RAD51 chromatin formation in HCT116 cells. The expressions of MCM2 and MCM6 were downregulated by using shRNA and the chromatin fraction of RAD51 was detected. Orc2 and Actin are respectively marker proteins for chromatin and non-chromatin fractions. shRNA, short hairpin RNA.
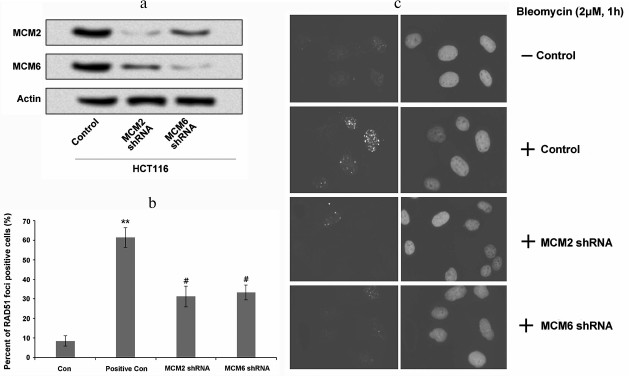
Suppression of MCMs decreased RAD51 foci forming. We further wanted to determine whether the association of RAD51 with MCMs was essential for RAD51 foci forming in response to DNA damage. The expression of MCMs was downregulated in HCT116 cells using shRNA and the foci forming of RAD51 was analyzed. As indicated in Fig. 4a, the expression levels of MCM2 and MCM6 were decreased after MCM2 shRNA and MCM6 shRNA transfection. Studies have revealed that bleomycin is an inducer of nuclear foci forming in tumor cells [24, 25]. Therefore, in this study we used bleomycin to evaluate nuclear foci forming in HCT116 cells. Our staining data show that compared to the controls, the percentage of RAD51 foci forming was significantly higher in the positive control cells (cells without MCM suppression; Fig. 4, b and c, p < 0.01), whereas the percentage of RAD51 foci forming was decreased after MCM2 shRNA or MCM6 shRNA transfection. These data revealed that suppression of MCMs decreased the percentage of RAD51 foci forming to respond to DNA damage.
Fig. 4. Suppression of MCMs decreased foci forming of RAD51 in HCT116 cells. a) MCM2 and MCM6 expressions in HCT116 cells were decreased by MCM2 shRNA or MCM6 shRNA transfection; b) percentage of RAD51 foci forming was significantly increased by the treatment of bleomycin compared to the control, whereas it was significantly decreased by the suppression of MCM2 or MCM6; c) foci forming in each group was detected using immunofluorescence microscopy consistent with that in panel (b). ** p < 0.01 compared to control, and # p < 0.05 compared to positive control (cells treated with bleomycin).
DISCUSSION
Previous studies have demonstrated the crucial roles of DNA damage recovery failure in the carcinogenesis of tumor development [26, 27]. RAD family proteins have been reported to be involved in the process of DNA damage recovery [28]. In the present study, we provide new evidence for the interaction between RAD51 and MCM complex in colon cancer HCT116 cell DNA damage recovery. Our results indicated that RAD51 directly interacted with MCM2, MCM3, MCM5, and MCM6 in HCT116 cells. Suppression of gene expression of MCM2 and MCM6 clearly decreased the RAD51 chromatin fraction and foci forming. These findings suggest that the interactions between RAD51 and MCM complex are essential for RAD51 chromatin fraction, foci forming, and DNA damage recovery in HCT116 cells.
RAD51 is a highly conserved protein that catalyzes DNA repair via HR and is a major DNA repair pathway that directly modulates cellular sensitivity to DNA-damaging treatments [29]. For example, Zhao et al. indicated that inhibition of RAD51 sensitized the cytotoxicity of olaparib for breast cancer cells through wild-type phosphatase and tensin homolog deleted on chromosome ten (PTEN) [30]. The MCM complex was found to participate in activating DNA replication and facilitating DNA synthesis [18, 31]. Restricting MCM loading to the G1 phase ensures that initiation and elongation occur just once per cell cycle [32]. Liu et al. pointed out that silencing of MCM2 by shRNA led to cell cycle arrest and apoptosis in colon cancer HCT116 cells [33]. Giaginis et al. presented evidence that MCM2 and MCM5 were associated with clinicopathological parameters and colon cancer proliferative capacity [34]. In our study, we found that RAD51 interact with MCM2, MCM3, MCM5, and MCM6 in HCT116 cells. MCM4 and MCM7 did not directly interact with RAD51 in HCT116, which implies that MCM4 and MCM7 do not participate in the formation of RAD51 foci in HCT116 cells or that MCM4 and MCM7 interacts with other RAD family proteins such as RAD52 and RAD54 in HCT116 cells. More studies are needed to further explore the relationship between MCM4, MCM7, and RAD family proteins.
The chromatin localization and foci-forming process of RAD51 are essential for RAD51-mediated repair of DNA DSBs [35]. Reh et al. indicated that RAD51 protected the genome from large deletions and chromosomal abnormalities [36]. In this study, we then explored whether MCMs are involved in the chromatin localization and foci forming of RAD51 using gene silencing method. We found that the chromatin fraction and foci forming were both decreased after suppression of MCM2 and MCM6, which suggests that MCMs participate in the chromatin localization and foci forming of RAD51 in HCT116 cells. Because MCMs are essential for eukaryotic DNA replication [37], we suggest that MCMs interact with RAD51 and play an important role in cell DNA damage recovery. In addition, our results are consistent with a previous study that showed that MCM complex has propensity to recruit RAD51 [20].
In conclusion, the data presented in this study reveals that RAD51 directly interacts with MCM2, MCM3, MCM5, and MCM6 in colon cancer HCT116 cells, and this kind of interaction is essential for the chromatin localization and foci forming of RAD51 in HCT116 cells. Our study provides a theoretical basis for analysis of RAD51 in tumor samples of colon cancer patients and might be applied for colon cancer localizing. Further studies are still needed to explore the detailed mechanism.
REFERENCES
1.Kim, E. Y., Kwon, K. A., Choi, I. J., Ryu, J. K.,
and Hahm, K. B. (2014) International Digestive Endoscopy Network 2014:
turnpike to the future, Clin. Endosc., 47, 371-382.
2.Bakker, I. S., Snijders, H. S., Grossmann, I.,
Karsten, T. M., Havenga, K., and Wiggers, T. (2016) High mortality
rates after non-elective colon cancer resection: results of a national
audit, Col. Dis., 18, 612-621.
3.Yuriko, T., Sachiko, I., Reina, O., Nana, C.,
Naomi, N., and Ken-Ichi, I. (2015) Effects of growth arrest and DNA
damage-inducible protein 34 (GADD34) on inflammation-induced colon
cancer in mice, Br. J. Cancer, 113, 669-679.
4.Hochster, H. S., and Sargent, D. J. (2016) One good
DNA-damage deserves another: oxaliplatin in MSI-high colon cancer,
J. Natl. Cancer Inst., 108, doi: 10.1093/jnci/djw011.
5.Seiwert, N., Neitzel, C., Stroh, S., Frisan, T.,
Audebert, M., Toulany, M., Kaina, B., and Fahrer, J. (2017) AKT2
suppresses pro-survival autophagy triggered by DNA double-strand breaks
in colorectal cancer cells, Cell Death Dis., 8,
e3019.
6.Mahaney, B. L., Katheryn, M., and Lees-Miller, S.
P. (2009) Repair of ionizing radiation-induced DNA double-strand breaks
by non-homologous end-joining, Biochem. J., 417,
639650.
7.Thompson, L. H. (2012) Recognition, signaling, and
repair of DNA double-strand breaks produced by ionizing radiation in
mammalian cells: the molecular choreography, Mutat. Res.,
751, 158-246.
8.Barnard, S., Bouffler, S., and Kai, R. (2013) The
shape of the radiation dose response for DNA double-strand break
induction and repair, Gen. Integr., 4, 1.
9.Rong, Y. S., and Golic, K. G. (2004) The homologous
chromosome is an effective template for the repair of mitotic DNA
double-strand breaks in Drosophila, Genetics, 165,
1831-1842.
10.Yi-Hsuan, L., Chia-Ching, C., Chui-Wei, W., and
Shu-Chun, T. (2009) Recruitment of Rad51 and Rad52 to short telomeres
triggers a Mec1-mediated hypersensitivity to double-stranded DNA breaks
in senescent budding yeast, PLoS One, 4, e8224.
11.Jasin, M. (2001) Double-Strand Break Repair
and Homologous Recombination in Mammalian Cells, Humana Press.
12.Sharma, A., Singh, K., and Almasan, A. (2012)
Histone H2AX phosphorylation: a marker for DNA damage, Methods Mol.
Biol., 920, 613-626.
13.Kyungsoo, H., Warren, F., Soon, C. D., Srividya,
B., Leandro, C., Devaraj, S. G. T., Bhavin, S., Sunil, S., Chang, J.
C., and Melnick, A. M. (2014) Histone deacetylase inhibitor treatment
induces “BRCAness” and synergistic lethality with PARP
inhibitor and cisplatin against human triple negative breast cancer
cells, Oncotarget, 5, 5637-5650.
14.Veelen, L. R. V., Essers, J., Van de Rakt, M. W.,
Odijk, H., Pastink, A., Zdzienicka, M. Z., Paulusma, C. C., and Kanaar,
R. (2005) Ionizing radiation-induced foci formation of mammalian Rad51
and Rad54 depends on the Rad51 paralogs, but not on Rad52, Mutat.
Res., 574, 34-49.
15.Mladenov, E., Anachkova, B., and Tsaneva, I.
(2006) Sub-nuclear localization of Rad51 in response to DNA damage,
Genes Cells, 11, 513-524.
16.Madalena, T., Derek, D., and West, S. C. (2003)
BRCA2-dependent and independent formation of RAD51 nuclear foci,
Oncogene, 22, 1115-1123.
17.Bindra, R. S., Schaffer, P. J., Meng, A., Woo,
J., Maseide, K., Roth, M. E., Lizardi, P., Hedley, D. W., Bristow, R.
G., and Glazer, P. M. (2004) Down-regulation of Rad51 and decreased
homologous recombination in hypoxic cancer cells, Mol. Cell.
Biol., 24, 8504-8518.
18.Lei, M. (2005) The MCM complex: its role in DNA
replication and implications for cancer therapy, Curr. Cancer Drug
Targets, 5, 365-380.
19.Hyrien, O. (2016) How MCM loading and spreading
specify eukaryotic DNA replication initiation sites, F1000Res.,
5.
20.Shukla, A., Navadgi, V. M., Mallikarjuna, K., and
Rao, B. J. (2005) Interaction of hRad51 and hRad52 with MCM complex: a
cross-talk between recombination and replication proteins, Biochem.
Biophys. Res. Commun., 329, 1240-1245.
21.Park, J., Long, D. T., Lee, K. Y., Abbas, T.,
Shibata, E., Negishi, M., Luo, Y., Schimenti, J. C., Gambus, A.,
Walter, J. C., and Dutta, A. (2013) The MCM8–MCM9 complex
promotes RAD51 recruitment at DNA damage sites to facilitate homologous
recombination, Mol. Cell. Biol., 33, 1632-1644.
22.Xiangzi, H., Aaron, A., Kang, F., Toshiya, T.,
and Youwei, Z. (2014) The interaction between checkpoint kinase 1
(Chk1) and the minichromosome maintenance (MCM) complex is required for
DNA damage-induced Chk1 phosphorylation, J. Biol. Chem.,
289, 24716-24723.
23.Purcell, D. J., Chauhan, S., Jimenez-Stinson, D.,
Elliott, K. R., Tsewang, T. D., Lee, Y. H., Marples, B., and Lee, D. Y.
(2015) Novel CARM1-interacting protein, DZIP3, is a transcriptional
coactivator of estrogen receptor-alpha, Mol. Endocrinol.,
29, 1708-1719.
24.Lu, J., Kashaev, N., and Huber, N. (2001) Werner
helicase relocates into nuclear foci in response to DNA damaging agents
and co-localizes with RPA and Rad51, Genes Cells, 6,
421-430.
25.Tianju, L., Hong, J., Matthew, U., Biao, H.,
Naozumi, H., Bethany, M., Andrew, M. K., Lukacs, N. W., and Phan, S. H.
(2004) Regulation of found in inflammatory zone 1 expression in
bleomycin-induced lung fibrosis: role of IL-4/IL-13 and mediation via
STAT-6, J. Immunol., 173, 3425-3431.
26.Wang, H., Zhang, X., Teng, L., and Legerski, R.
J. (2015) DNA damage checkpoint recovery and cancer development,
Exp. Cell Res., 334, 350-358.
27.Glover, K. P., Markell, L. K., Donner, E. M., and
Xan, H. (2014) Protein kinase C-activating tumor promoters modulate the
DNA damage response in UVC-irradiated TK6 cells, Toxicol. Lett.,
229, 210-219.
28.Delmas, S., Shunburne, L., Ngo, H. P., and
Allers, T. (2009) Mre11–Rad50 promotes rapid repair of DNA damage
in the polyploid archaeon Haloferax volcanii by restraining
homologous recombination, PLoS Genet., 5, e1000552.
29.Gachechiladze, M., Skarda, J., Soltermann, A.,
and Joerger, M. (2017) RAD51 as a potential surrogate marker for DNA
repair capacity in solid malignancies, Int. J. Cancer,
141, 1286-1294.
30.Zhao, Q., Guan, J., Zhang, Z., Lv, J., Wang, Y.,
Liu, L., Zhou, Q., and Mao, W. (2017) Inhibition of Rad51 sensitizes
breast cancer cells with wild-type PTEN to olaparib, Biomed.
Pharmacother., 94, 165-168.
31.Simon, N. E., and Schwacha, A. (2014) The Mcm2-7
replicative helicase: a promising chemotherapeutic target, 549719.
32.Labib, K., Tercero, J. A., and Diffley, J. F.
(2000) Uninterrupted MCM2-7 function required for DNA replication fork
progression, Science, 288, 1643-1647.
33.Liu, Y., He, G., Wang, Y., Guan, X., Pang, X.,
and Zhang, B. (2013) MCM-2 is a therapeutic target of trichostatin A in
colon cancer cells, Toxicol. Lett., 221, 23-30.
34.Giaginis, C., Georgiadou, M., Dimakopoulou, K.,
Tsourouflis, G., Gatzidou, E., Kouraklis, G., and Theocharis, S. (2009)
Clinical significance of MCM-2 and MCM-5 expression in colon cancer:
association with clinicopathological parameters and tumor proliferative
capacity, Dig. Dis. Sci., 54, 282-291.
35.Courilleau, C., Chailleux, C., Jauneau, A.,
Grimal, F., Briois, S., Boutet-Robinet, E., Boudsocq, F., Trouche, D.,
and Canitrot, Y. (2012) The chromatin remodeler p400 ATPase facilitates
Rad51-mediated repair of DNA double-strand breaks, J. Cell
Biol., 199, 1067-1081.
36.Reh, W. A., Nairn, R. S., Lowery, M. P., and
Vasquez, K. M. (2017) The homologous recombination protein RAD51D
protects the genome from large deletions, Nucleic Acids Res.,
45, 1835-1847.
37.Han, X., Aslanian, A., Fu, K., Tsuji, T., and
Zhang, Y. (2014) The interaction between checkpoint kinase 1 (Chk1) and
the minichromosome maintenance (MCM) complex is required for DNA
damage-induced Chk1 phosphorylation, J. Biol. Chem., 289,
24716-24723.