REVIEW: Structural–Functional Domains of the Eukaryotic Genome
S. V. Razin1,2* and A. A. Gavrilov1
1Institute of Gene Biology, Russian Academy of Sciences, 119334 Moscow, Russia; E-mail: sergey.v.razin@usa.net2Lomonosov Moscow State University, Biological Faculty, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received October 19, 2017; Revision received November 27, 2017
It is well known that DNA folding in the eukaryotic cell nucleus is tightly coupled with the operation of epigenetic mechanisms defining the repertoires of the genes expressed in different types of cells. To understand these mechanisms, it is important to know how DNA is packaged in chromatin. About 30 years ago a hypothesis was formulated, according to which epigenetic mechanisms operate not at the level of individual genes, but rather groups of genes localized in structurally and functionally isolated genomic segments that were called structural and functional domains. The question of what exactly these domains constitute has been re-examined multiple times as our knowledge of principles of chromatin folding has changed. In this review, we discuss structural and functional genomic domains in light of the current model of interphase chromosome organization based on the results of analysis of spatial proximity between remote genomic elements.
KEY WORDS: chromatin, cell nucleus, topologically associating domain, Hi-C, noncoding RNA, gene activationDOI: 10.1134/S0006297918040028
Abbreviations: eRNA, enhancer RNA; Hi-C, high-throughput chromosome conformation capture; TAD, topologically associating domain.
The question of existence of structural and functional domains in the
eukaryotic genome that are targets for the action of regulatory
mechanisms has been discussed in the literature for many years. The
discussion began after the discovery of differential sensitivity of
long genomic regions to DNases [1] and was further
developed after the discovery of the locus control region of the
β-globin gene domain [2, 3]. The formulated domain model of eukaryotic genome
organization postulated that the experssion of one or several gene(s)
can be controled by changing the chromatin folding pattern of a long
genomic region – the domain where these genes are localized [4, 5]. The length of such domains
was supposedly limited by matrix attachment regions or by specific
genomic elements – insulators [6]. It
should be noted that the domain model of genome organization was based
on the results of studying the vertebrates β-globin gene domains
and some other genomic domains, which are now usually referred to as
domains with distinct boundaries [6, 7]. The increase in the number of characterized
genomic domains in the epoch of whole genome sequencing has shown that
organization of the domain of β-globin genes in vertebrates is
unique rather than typical of the genome in general. Most of the
tissue-specific genes are surrounded by continuously expressed
(“housekeeping”) genes and, hence, such genes are the
components of transcriptionally active (DNase-sensitive) chromatin in
cells of different lineages. As an example, one can mention the domain
of α-globin genes in vertebrates whose major regulatory element
is located in one of the introns of the housekeeping gene situated
upstream to the domain [8]. It is clear that the
domain model of eukaryotic genome organization based on the hypothesis
that activation of tissue-specific gene transcription requires cardinal
chromatin reconfiguration in the long genomic region [9] cannot be used for describing the regulatory
mechanisms of the domain of α-globin genes and other similar
domains. Considering the fact that such domains are predominant in the
genome, one can state that the domain model of eukaryotic genome
organization has lost its revance in the original version. At the same
time, research results suggest that animal genomes contain structural
and functional domains of a different type, which restrict the area of
enhancer activity. Being originally identified by functional tests,
these domains were named regulatory domains, landscapes, or
archipelages [10-12]. They
were demonstrated to coincide with the topologically associating
chromatin domains identified in the study of three-dimensional genome
organization in the cell nucleus [11, 13-15]. This review is devoted
to the modern concepts of chromatin packaging in the cell nucleus and
dicussion of the role of three-dimensional genome organization in the
regulation of gene activity.
HIGHER LEVELS OF CHROMATIN PACKAGING IN THE CELL NUCLEUS
For many years it was generally accepted that there was a certain hierarchy of the levels of DNA packaging in chromatin [16]. The first two levels of packaging – DNA wrapping around nucleosomal globules, with formation of the so-called 10-nm chromatin fibril, and subsequent folding of this fibril into a 30-nm fibril – seemed to be most comprehensible. Regarding the next level of chromatin packaging, the opinions of different researchers were at variance with each other. One of the popular models postulates that the 30-nm fibril is arranged into extended (50-250 kb) loops fixed at the nuclear matrix [16, 17]. Then they postulated the existence of loop clusters containing ~2500 kb DNA and forming chromomeres that can be observed in the meiotic prophase [18]. Another model postulated that the 30-nm fibril is coiled into several hierarchic solenoids [19, 20].
In recent years, the existence of a 30-nm chromatin fibril in living cells was questioned [21-25]. The novel methodological approaches have not shown any regular 30-nm fibrils in cell nuclei. Chromatin masses seemed to consist of densely associated nucleosomal filaments (10-nm fibrils). The densities of nucleosome packaging in the euchromatin and heterochromatin regions were different; however, no regular supernucleosomal structures have been found [25-27]. These results are in good agreement with the observations of other authors, who have demonstarted that the density of DNA packaging, even in the transcriptionally acitve chromatin fraction, substantially exceeds the density that can be reached through formation of a 30-nm fibril [28]. At present, most authors agree that there are no regulary supernucleosomal structures in the nuclei of living cells. At the same time, DNA is packaged through association of nucleosomal filaments with the formation of various types of aggregates. Under certain conditions, this process can be easily simulated in vitro [29].
Studies employing the so-called C-methods based on the analysis of physical closeness of different parts of the genome in the three-dimensional space of the cell nucleus have made a substantial contribution to understanding the principles of chromatin packaging. These techniques are based on the proximity ligation procedure [30]. The most informative method for analyzing the general principles of chromatin packaging is the Hi-C technique (high-throughput chromosome conformation capture), which allows analysis of the physical proximity of different DNA fragments on the whole-genome scale [31]. This experimental approach allowed to demonstrate the spatial segregation of active (A) and inactive (B) compartments in mammalian chromatin, which correspond to euchromatin and heterochromatin in the first approximation [31]. The analysis of higher-resolution Hi-C maps showed the presence of topologically associating domains (TADs) (Fig. 1). The main property of TADs is that spatial contacts between genomic elements are established much more frequently within a TAD than between the TADs [32-34]. The TADs and chromatin compartments have been found in mammals [32, 33], insects [34, 35], and birds [36]. Some contact domains can also be revealed in the genomes of plants and lower eukaryotes [37-40]. However, they are substantially different from the TADs of mammals and Drosophila both in size and in the levels of insulation and genome coverage. In original works, it was emphasized that the profiles of chromosomes partitioning into TADs are rather conservative both between the cells of different lineages and within the syntenic regions in closely related species [32, 41, 42]. However, this conservatism is limited [34, 43]. The profiles of chromosome partitioning into TADs are substantially different, inter alia, due to the differences in transcription profiles typical of specialized cells [43]. It should be noted that TADs per se are arranged hierarchically, i.e., may include several levels of smaller contact domains separated by weaker boundaries [43-45] (Fig. 1). The higher-resolution Hi-C maps have shown that mammalian TADs include contact subdomains, most of them being chromatin loops with CTCF-binding sites at their bases, where cohesin enrichment is detected [46]. The question as to the domains of which level should be referred to as TADs is still under discussion and answered differently [41, 43]. Most authors believe that the TADs of mammalian cells are one to several millions of base pairs in size [32, 33, 41], while the average size of TADs in Drosophila is hundreds of thousands of base pairs [34, 47]. At the same time, it should be noted that TADs can be distinguished from the contact domains of other levels by functional (the correspondence to replication-timing domains [48] and regulatory domains [11]) rather than structural criteria [49]. In a recently published work by Rowley et al. [35], the existence of TADs in the Drosophila genome is disputed. Those authors believe that the major structural units of Drosophila chromosomes are the compartments that may vary in length from several kilobases to several hundreds of kilobases [35].
Fig. 1. Chromosome organization into topologically associating domains. The contact map of a hypothetic genomic region is shown, and the annotation of TADs for this region demonstrates the presence of several hierarchic levels of domains. Each pixel (rhombus) on the contact map indicates the total number of contacts of the respective chromosome regions expressed in colors, from blue (few contacts) to dark red (many contacts). For example, the number of contacts between chromosome regions A and B is shown by the pixel marked with the black dotted lines. TADs are usually interpreted as chromatin globules, as is shown schematically in the lower part of the figure.
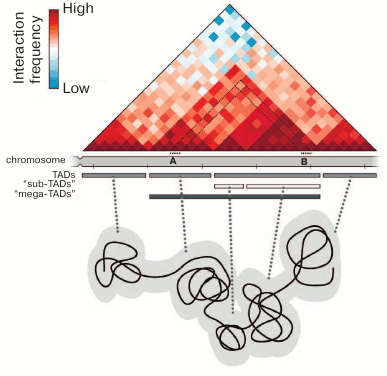
The physical nature of TADs is not quite clear. They are usually interpreted as chromatin globules that can be visualized by different microscopic techniques [50-52] (Fig. 1). This interpretation is confirmed by the results of hybridization in situ with the samples distributed along an individual TAD [53-55]. It should be mentioned that microscopic techniques make it possible to analyze single cells, whereas biochemical methods have been used until recently only for cell population analysis. It is clear that the analysis of populations can reveal only some general regularities. It creates certain difficulties in the direct comparison of results obtained by microscopic and biochemical approaches. The problem can be partially solved by averaging the microscopic observations of a great number of cells. In particular, this approach applied to the analysis of the super-resolution microscopy images allowed to confirm the existence of active and inactive chromatin compartments revealed by analysis of the contact maps of genomic interactions [55]. On the other hand, the experimental protocols developed in recent years allow construction of Hi-C maps of spatial genome organization in single cells [56-58]. The results obtained using these protocols can be directly compared with the data of super-resolution microscopy and altogether demonstrate good agreement between observations in both approaches [59]. Among the conceptually important results, it should be noted that the genomic positions of TADs in single cells do not always coincide with the positions of “statistically average TADs” predicted by population data analysis [58]. Another important observation made during the Hi-C analysis of single cells is that the same TAD may adopt different configurations, beginning from a highly extended stretch to a completely condensed globule [60]. Both observations are indicative of the dynamics of chromatin fibril and considerable variability of spatial organization of separate genomic segments in single cells.
TOPOLOGICALLY ASSOCIATING DOMAINS ARE FUNCTIONAL UNITS OF THE
GENOME
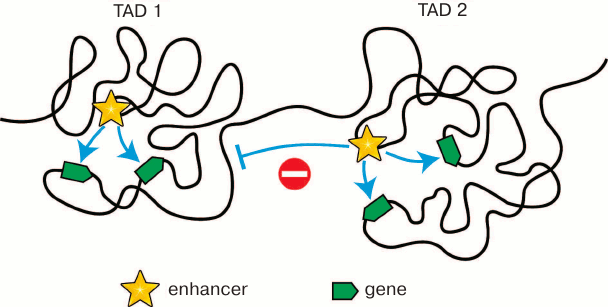
According to the almost universally accepted model, an enhancer can activate a gene only if it contacts the promoter of the gene. Most enhancers are located at a considerable distance from the genes they activate. Accordingly, for promoter activation, an enhancer must contact this promoter via looping of an intervening DNA segment [61] (see below Fig. 3). The existence of such spatial contacts between remote enhancers and promoters can be really found by both biochemical (Hi-C and other C techniques) and microscopic (fluorescent in situ hybridization) approaches [62-66]. The significance of enhancer–promoter communication for transcription activation has been directly demonstrated in experiments on forced chromatin looping [67]. Spatial contacts between remote genomic elements are established mostly inside the TADs. Thereby, TAD bounadires must restrict the scope of enhancer activity as has been demonstrated in some studies [11, 68, 69] (Fig. 2). The fusion of TADs due to deletion of the intervening spacer region leads to changes in scope of enhancer activity, which in some cases results in emergence of various pathologies due to the impaired regulation of gene expression within the fused TADs [13-15]. All these results show that TADs are the structural and functional genome units playing the key role in the work of transcription regulation systems. That is probably why the profile of chromosome partitioning into TADs is conservative within syntenic regions in different biological species [32, 41, 42]. In addition to restriction of the scope of enhancer activity, the chromosome partitioning into TADs reduces the time necessasry for establishing enhancer–promoter communication. Eukaryotic cells have no mechanisms providing the directional movement of the enhancer toward the promoter. The local movements of different regions of the chromatin fibril are stochastic due to the energy of thermal motion of molecules [70]. The establishment of enhancer–promoter communication will depend on how soon the enhacer and the controlled promoter will meet in the nuclear space. It has been experimentally determined that a certain randomly chosen genomic locus can examine 0.5-0.8 μm in 1 h [71]. Restriction of the search area by an individual TAD (Fig. 2) undoubtedly shortens the time needed for establishing the enhancer–promoter communication. In this context, it is essential that the TAD structure is not rigid. Inside the TAD, there is a continuous alternation of chromatin configurations [60, 72].
Fig. 2. Restriction of the scope of activity of enhancers by topologically associating domains. Localization within the same TAD increases the probability of spatial contacts between the enhancer and the controlled promoters. On the contrary, localization in different TADs prevents communication between “wrong” partners.
With reference to functional genome units, it would be wrong to consider only the transcriptional regulatory domains. Replication domains play a key role in the work of the eukaryotic genome. Such domains include replicons and replication time zones. The profile of replicons is rather dynamic and varies from cycle to cycle due to the presence of a considerable number of alternative replication origin regions in eukaryotic genomes [73]. On the other hand, DNA replication time zones are sufficiently stable in each particular type of cells [74]. Several researchers have demonstrated a strong correlation between these zones and TADs [48, 75].
MECHANISMS OF TAD FORMATION
The ability to form various compact structures is a basic property of nucleosomal fibrils. This ability is determined by the possibility of establishing electrostatic interactions between the positively charged N-terminal domains of histones (especially histone H4) and the negatively charged domain on the surface of nucleosomal globules [29, 76]. Experiments in vitro have demonstrated that at low chromatin concentrations the interactions occur mainly between nucleosomes within the nucleosomal chain, with the formation of a 30-nm fibril [77, 78]. At high chromatin concentrations, the interactions more commonly occur between the nucleosomes of different chains (or different regions of the same chain), resulting in the formation of various condensed structures. The ability to establish electrostatic contacts between nucleosomes is controlled by the levels of histone acetylation. At high levels of acetylation, the positive charge of histone N-terminal domains decreases and, hence, the opportunity of establishing internucleosomal contacts is lost [79, 80]. The high level of histone acetylation is typical of active chromatin. In Drosophila, active genes are localized mainly between the TADs, while inactive genes are localized inside the TADs [43]. We have demonstrated by computer modeling that a chromatin fibril composed of alternating active and more extended inactive regions is folded into compact globules (TADs) containing mostly inactive chromatin separated by less compact regions (inter-TADs) containing mostly active chromatin [43]. It is clear that the TADs formed thereby have functions in the storage of repressed genes. Indeed, by comparing TAD profiles in different cell lines, we have demonstrated that the activation of transcription of tissue-specific genes correlates with the decompaction of TADs and in some cases leads to the emergence of new inter-TADs [43]. TADs must have more complex organization to play the role of functional domains of the genome, which is typical primarily of TADs in mammalian chromosomes. The architectural proteins that can arrange the genome into loops play a key role in establishment and maintenance of this organization. In mammals, CTCF and cohesin play the key role in the contacts between remote genomic elements [81-84]. As noted in some works, CTCF and cohesin are localized preferentially at TAD boundaries [32]. Moreover, deletions of CTCF-binding sites at TAD boundaries result in the weakening of TAD insulation and, in some cases, in the fusion of neighboring TADs [13, 85-87]. The inhibition of experssion and induced degradation of CTCF have the same effect [88, 89].
Although when discussing the role of CTCF and other architectural proteins in spatial genome organization the emphasis is often placed on the ability of these proteins to make loops [90, 91], chromatin loop formation per se cannot lead to the appearance of a topologically associating domain, within which the spatial contacts between remote genomic elements are preferentially established. Chromatin loop formation guarantees only preferential spatial interactions between the chromatin segments localized at the loop base. For explaining the mechainsm of TAD formation, it has been postulated that there is a continuous processive looping of different chromatin segments within a region limited by convergent CTCF-binding sites. At the same time, looping may begin at random sites, and the period of loop existence is limited. This model termed “the mechanism of loop extrusion” [92] and confirmed by the results of computer modeling provides a rational explanation for quite a number of experimental observations [87, 92, 93]. The model postulates the existence of a certain molecular motor supporting DNA looping. Some indirect evidence suggests that cohesins perform the function of a motor [92-94]. However, the DNA-looping ability of cohesins has not been demonstrated directly. The nature of the extrusion motor, as well as the nature of loop-bordering insulator, is not fundamentally important for the loop extrusion model. For example, the anchored RNA polymerase molecules can function as extrusion motors [95]. As for the restrictive element that prevents loop spreading, its function can be performed, in addition to CTCF-binding sites, by promoters or some noncanonical DNA structures.
TAD organization as a series of dynamic (appearing and disappearing) chromatin loops must favor the reduction of time necessary for that contact between different regions of chromatin fibrils within a TAD, because scanning in this case will be performed in not three- but in one-dimensional space (along the DNA strand).
INVOLVEMENT OF NONCODING RNA IN SPATIAL GENOME
ORGANIZATION
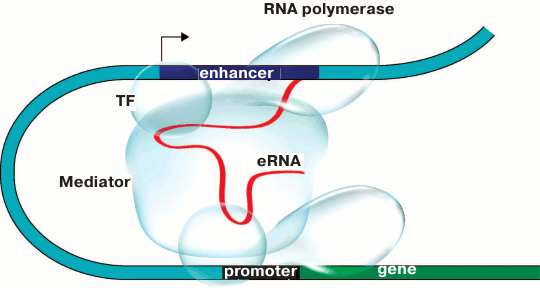
The results of some works show that enhancers are brought to the controlled promoters with either direct or indirect involvement of enhancer RNA (eRNA) transcribed on either side of the enhancer [95-97]. The particular role of eRNA in enhancer–promoter communication is yet to be investigated. According to one of the scenarios (Fig. 3), eRNA can maintain contacts between the enhancer and the Mediator coactivator complex. Indeed, for some noncoding RNAs transcribed from enhancers (noncoding activator RNA, ncRNA-a) it has been demonstrated that their ability to stimulate transcription depends on the interaction between such RNAs and components of the Mediator coactivator complex. At the same time, a chromatin loop was formed between the enhancer, from which ncRNA-a is transcribed, and the activated gene. Deletions of both ncRNA-a and Mediator components impaired chromatin loop formation and suppressed the ability of the enhancer to activate the controlled gene [98, 99]. It is interesting that the architectural protein CTCF also binds a broad range of noncoding RNAs on the whole-genome scale [100, 101]. CTCF contains an RNA-binding domain at the C-end, and CTCF multimerization probably important for DNA looping [102] depends on the presence of RNA [101]. Moreover, recently it has been shown that YY1 – the universally expressed transcription factor that is bound to CTCF [103] and enriched at the base of the DNA loops [46] – is also bound to RNA, thereby increasing the affinity of this factor to its binding sites in the genome [104].
Fig. 3. Model of promoter–enhancer communication. Enhancer RNA (eRNA) plays the key role in establishing contacts between proteins bound to the enhancer and the promoter and facilitates the assembly of activator chromatin protein that triggers gene transcription. TF is the transcription factor.
The spatial genome organization on a larger scale can also be regulated with the involvement of noncoding RNA (as discussed in review [105]). It has been shown, for example, that the long noncoding RNA Firre is able to direct the colocalization of several genomic loci located on different chromosomes [106]. The XIST-RNA functioning under conditions of dosage compensation covers the entire X-chromosome, providing its heterochromatization and inactivation [107]. The noncoding RNA MALAT1 acts as a molecular “scaffold” for the assembly of speckles – the nuclear bodies containing components of the splicing apparatus, to which the active genes are attracted [105]. Currently, it is difficult to assess how common is the phenomenon of RNA involvement in the contacts of remote genomic elements and large-scale genome organization. This is a very young research field, and the available data concern the study of a rather limited set of noncoding RNAs and genomic loci.
Concluding remarks. The idea that the eukaryotic genome is arranged into loops of 50-150 kb is not new. A lot of evidence supporting the existence of such loops appeared as early as in the 1970s [108-111]. At that time it was believed that chromatin loops are fixed on some skeletal structure referred to as nuclear matrix, nuclear skeleton, or scaffold of metaphase chromosome [112]. Now it is clear that there is no such structure. It appears during the saline extraction of nuclei as a result of aggregation of different proteins, first of all, the proteins of RNP particles, in the interchromatin compartment [113]. At the same time, the procedure for obtaining nuclear matrix allows the fixation of particular elements of intranuclear organization, whereby the study of the nuclear matrix contributed to disclosing some principles of spatial organization of chromatin fibril in interphase chromosomes [113]. According to the nuclear matrix model, the DNA present within this structure must consist mainly of DNA fragments localized at the bases of chromatin loops [111]. It would be interesting to compare the properties of this DNA fraction with the currenly known data on the bases of topological DNA loops and inter-TADs. In a number of works, it has been demonstarted that the active genes and elongating complexes of RNA polymerase II are concentrated within the nuclear matrix-attached DNA [114-118]. This correlates well with the preferential localization of active genes in the inter-TADs [32, 43]. The nuclear matrix was shown to contain CTCF and the CTCF-dependent insulator from the domain of chicken β-globin genes [119, 120]. As it has been more than once mentioned above, the presence of CTCF-binding sites is typical of inter-TADs. The nuclear matrix-attached DNA was preferentially cleaved by DNA topoisomerase II [121, 122]. Currently, it was demonstrated that DNA topoisomerase II is colocalized with CTCF and cohesin at the TAD boundaries [123]. Finally, it has been reported that the inter-disks of polytene chromosomes of Drosophila, which coincide with inter-TADs [43, 124], are enriched in DNA sequences preferentially binding to the nuclear matrix [125]. All the above leads to a conclusion that the DNA fraction isolated within the nuclear matrix consists mainly of the marginal areas of TADs and, probably, contact domains of other levels. In light of this conclusion, it seems important to reconsider the entire array of experimental data on the spatial organization of eukaryotic DNA that were obtained in the last quarter of the 20th century and attempt to integrate these data into the moderm models of structural and functional organization of the eukaryotic genome.
With reference to the modern models, it is clear that they will also be improved and modified with the accumulation of new experimental data. Among the most topical current trends in the study of spatial genome organization, we should mention the transition from cell population to single cell studies [59]. The emergence of a wide range of so-called C techniques [126] has substantially extended our insight into the spatial organization of eukaryotic genomes. However, observations have been made so far mostly in cell population studies. Hence, the findings concern only the most probable chromatin configurations [59]. The question to what extent, e.g., TADs or A/B chromatin compartments are the result of averaging the panel of different configurations adopted in single cells needs further investigation.
It seems equally important to study chromatin fibril dynamics, inter alia, by different methods of in vivo visualization of individual genomic loci [127]. Dynamic organization is an important characteristic of biological systems. Continuous alternation of different chromatin configurations offers an opportunity for cell differentiation and adaptation to changing external conditions via temporary fixation of configurations required for the expression of various groups of genes [128]. The particular mechanisms underlying all these processes are yet to be elucidated in further studies.
Acknowledgments
This work was supported by the Russian Science Foundation (project No. 14-24-00022).
REFERENCES
1.Stalder, J., Larsen, A., Engel, J. D., Dolan, M.,
Groudine, M., and Weintraub, H. (1980) Tissue-specific DNA cleavages in
the globin chromatin domain introduced by DNAase I, Cell,
20, 451-460.
2.Grosveld, F., van Assandelt, G. B., Greaves, D. R.,
and Kollias, B. (1987) Position-independent, high-level expression of
the human β-globin gene in transgenic mice, Cell,
51, 975-985.
3.Forrester, W. C., Epner, E., Driscoll, M. C.,
Enver, T., Brice, M., Papayannopoulou, T., and Groudine, M. (1990) A
deletion of the human β-globin locus activation region causes a
major alteration in chromatin structure and replication across the
entire β-globin locus, Genes Dev., 4, 1637-1649.
4.Bodnar, J. W. (1988) A domain model for eukaryotic
DNA organization: a molecular basis for cell differentiation and
chromosome evolution, J. Theor. Biol., 132, 479-507.
5.Goldman, M. A. (1988) The chromatin domain as a
unit of gene regulation, BioEssays, 9, 50-55.
6.Razin, S. V., Farrell, C. M., and Recillas-Targa,
F. (2003) Genomic domains and regulatory elements operating at the
domain level, Int. Rev. Cytol., 226, 63-125.
7.Dillon, N., and Sabbatini, P. (2000) Functional
gene expression domains: defining the functional units of eukaryotic
gene regulation, BioEssays, 22, 657-665.
8.Flint, J., Tufarelli, C., Peden, J., Clark, K.,
Daniels, R. J., Hardison, R., Miller, W., Philipsen, S., Tan-Un, K. C.,
McMorrow, T., Frampton, J., Alter, B. P., Frischauf, A. M., and Higgs,
D. R. (2001) Comparative genome analysis delimits a chromosomal domain
and identifies key regulatory elements in the alpha globin cluster,
Hum. Mol. Genet., 10, 371-382.
9.Razin, S. V., Iarovaia, O. V., Sjakste, N.,
Sjakste, T., Bagdoniene, L., Rynditch, A. V., Eivazova, E. R.,
Lipinski, M., and Vassetzky, Y. S. (2007) Chromatin domains and
regulation of transcription, J. Mol. Biol., 369,
597-607.
10.Symmons, O., and Spitz, F. (2013) From remote
enhancers to gene regulation: charting the genome’s regulatory
landscapes, Philos. Trans. R. Soc. Lond. B. Biol. Sci.,
368, 20120358.
11.Symmons, O., Uslu, V. V., Tsujimura, T., Ruf, S.,
Nassari, S., Schwarzer, W., Ettwiller, L., and Spitz, F. (2014)
Functional and topological characteristics of mammalian regulatory
domains, Genome Res., 24, 390-400.
12.Montavon, T., Soshnikova, N., Mascrez, B., Joye,
E., Thevenet, L., Splinter, E., de Laat, W., Spitz, F., and Duboule, D.
(2011) A regulatory archipelago controls Hox genes transcription
in digits, Cell, 147, 1132-1145.
13.Lupianez, D. G., Kraft, K., Heinrich, V.,
Krawitz, P., Brancati, F., Klopocki, E., Horn, D., Kayserili, H.,
Opitz, J. M., Laxova, R., Santos-Simarro, F., Gilbert-Dussardier, B.,
Wittler, L., Borschiwer, M., Haas, S. A., Osterwalder, M., Franke, M.,
Timmermann, B., Hecht, J., Spielmann, M., Visel, A., and Mundlos, S.
(2015) Disruptions of topological chromatin domains cause pathogenic
rewiring of gene–enhancer interactions, Cell, 161,
1012-1025.
14.Franke, M., Ibrahim, D. M., Andrey, G.,
Schwarzer, W., Heinrich, V., Schopflin, R., Kraft, K., Kempfer, R.,
Jerkovic, I., Chan, W. L., Spielmann, M., Timmermann, B., Wittler, L.,
Kurth, I., Cambiaso, P., Zuffardi, O., Houge, G., Lambie, L., Brancati,
F., Pombo, A., Vingron, M., Spitz, F., and Mundlos, S. (2016) Formation
of new chromatin domains determines pathogenicity of genomic
duplications, Nature, 538, 265-269.
15.Valton, A. L., and Dekker, J. (2016) TAD
disruption as oncogenic driver, Curr. Opin. Genet. Dev.,
36, 34-40.
16.Getzenberg, R. H., Pienta, K. J., Ward, W. S.,
and Coffey, D. S. (1991) Nuclear structure and the three-dimensional
organization of DNA, J. Cell. Biochem., 47, 289-299.
17.Jackson, D. A., Dickinson, P., and Cook, P. R.
(1990) The size of chromatin loops in HeLa cells, EMBO J.,
9, 567-571.
18.Wanner, G., and Formanek, H. (2000) A new
chromosome model, J. Struct. Biol., 132, 147-161.
19.Sedat, J., and Manuelidis, L. (1978) A direct
approach to the structure of eukaryotic chromosomes, Cold Spring
Harb. Symp. Quant. Biol., 42, 331-350.
20.Kireeva, N., Lakonishok, M., Kireev, I., Hirano,
T., and Belmont, A. S. (2004) Visualization of early chromosome
condensation: a hierarchical folding, axial glue model of chromosome
structure, J. Cell. Biol., 166, 775-785.
21.Fussner, E., Strauss, M., Djuric, U., Li, R.,
Ahmed, K., Hart, M., Ellis, J., and Bazett-Jones, D. P. (2012) Open and
closed domains in the mouse genome are configured as 10-nm chromatin
fibres, EMBO Rep., 13, 992-996.
22.Gan, L., Ladinsky, M. S., and Jensen, G. J.
(2013) Chromatin in a marine picoeukaryote is a disordered assemblage
of nucleosomes, Chromosoma, 122, 377-386.
23.Eltsov, M., Maclellan, K. M., Maeshima, K.,
Frangakis, A. S., and Dubochet, J. (2008) Analysis of cryo-electron
microscopy images does not support the existence of 30-nm chromatin
fibers in mitotic chromosomes in situ, Proc. Natl. Acad. Sci.
USA, 105, 19732-19737.
24.Maeshima, K., Imai, R., Hikima, T., and Joti, Y.
(2014) Chromatin structure revealed by X-ray scattering analysis and
computational modeling, Methods, 70, 154-161.
25.Maeshima, K., Imai, R., Tamura, S., and Nozaki,
T. (2014) Chromatin as dynamic 10-nm fibers, Chromosoma,
123, 225-237.
26.Maeshima, K., Rogge, R., Tamura, S., Joti, Y.,
Hikima, T., Szerlong, H., Krause, C., Herman, J., Seidel, E., DeLuca,
J., Ishikawa, T., and Hansen, J. C. (2016) Nucleosomal arrays
self-assemble into supramolecular globular structures lacking 30-nm
fibers, EMBO J., 35, 1115-1132.
27.Ou, H. D., Phan, S., Deerinck, T. J., Thor, A.,
Ellisman, M. H., and O’Shea, C. C. (2017) ChromEMT: visualizing
3D chromatin structure and compaction in interphase and mitotic cells,
Science, 357, pii: eaag0025.
28.Hu, Y., Kireev, I., Plutz, M., Ashourian, N., and
Belmont, A. S. (2009) Large-scale chromatin structure of inducible
genes: transcription on a condensed, linear template, J. Cell.
Biol., 185, 87-100.
29.Pepenella, S., Murphy, K. J., and Hayes, J. J.
(2014) Intra- and inter-nucleosome interactions of the core histone
tail domains in higher-order chromatin structure, Chromosoma,
123, 3-13.
30.Dekker, J., Rippe, K., Dekker, M., and Kleckner,
N. (2002) Capturing chromosome conformation, Science,
295, 1306-1311.
31.Lieberman-Aiden, E., van Berkum, N. L., Williams,
L., Imakaev, M., Ragoczy, T., Telling, A., Amit, I., Lajoie, B. R.,
Sabo, P. J., Dorschner, M. O., Sandstrom, R., Bernstein, B., Bender, M.
A., Groudine, M., Gnirke, A., Stamatoyannopoulos, J., Mirny, L. A.,
Lander, E. S., and Dekker, J. (2009) Comprehensive mapping of
long-range interactions reveals folding principles of the human genome,
Science, 326, 289-293.
32.Dixon, J. R., Selvaraj, S., Yue, F., Kim, A., Li,
Y., Shen, Y., Hu, M., Liu, J. S., and Ren, B. (2012) Topological
domains in mammalian genomes identified by analysis of chromatin
interactions, Nature, 485, 376-380.
33.Nora, E. P., Lajoie, B. R., Schulz, E. G.,
Giorgetti, L., Okamoto, I., Servant, N., Piolot, T., van Berkum, N. L.,
Meisig, J., Sedat, J., Gribnau, J., Barillot, E., Bluthgen, N., Dekker,
J., and Heard, E. (2012) Spatial partitioning of the regulatory
landscape of the X-inactivation centre, Nature, 485,
381-385.
34.Sexton, T., Yaffe, E., Kenigsberg, E.,
Bantignies, F., Leblanc, B., Hoichman, M., Parrinello, H., Tanay, A.,
and Cavalli, G. (2012) Three-dimensional folding and functional
organization principles of the Drosophila genome, Cell,
148, 458-472.
35.Rowley, M. J., Nichols, M. H., Lyu, X.,
Ando-Kuri, M., Rivera, I. S. M., Hermetz, K., Wang, P., Ruan, Y., and
Corces, V. G. (2017) Evolutionarily conserved principles predict 3D
chromatin organization, Mol. Cell., 67, 837-852.
36.Ulianov, S. V., Galitsyna, A. A., Flyamer, I. M.,
Golov, A. K., Khrameeva, E. E., Imakaev, M. V., Abdennur, N. A.,
Gelfand, M. S., Gavrilov, A. A., and Razin, S. V. (2017) Activation of
the alpha-globin gene expression correlates with dramatic upregulation
of nearby non-globin genes and changes in local and large-scale
chromatin spatial structure, Epigenet. Chromat., 10,
35.
37.Wang, C., Liu, C., Roqueiro, D., Grimm, D.,
Schwab, R., Becker, C., Lanz, C., and Weigel, D. (2015) Genome-wide
analysis of local chromatin packing in Arabidopsis thaliana,
Genome Res., 25, 246-256.
38.Hsieh, T. H., Weiner, A., Lajoie, B., Dekker, J.,
Friedman, N., and Rando, O. J. (2015) Mapping nucleosome resolution
chromosome folding in yeast by micro-C, Cell, 162,
108-119.
39.Eser, U., Chandler-Brown, D., Ay, F., Straight,
A. F., Duan, Z., Noble, W. S., and Skotheim, J. M. (2017) Form and
function of topologically associating genomic domains in budding yeast,
Proc. Natl. Acad. Sci. USA, 114, 3061-3070.
40.Nikolaou, C. (2017) Invisible cities: segregated
domains in the yeast genome with distinct structural and functional
attributes, Curr. Genet., doi: 10.1007/s00294-017-0731-6.
41.Dixon, J. R., Gorkin, D. U., and Ren, B. (2016)
Chromatin domains: the unit of chromosome organization, Mol.
Cell., 62, 668-680.
42.Vietri Rudan, M., Barrington, C., Henderson, S.,
Ernst, C., Odom, D. T., Tanay, A., and Hadjur, S. (2015) Comparative
Hi-C reveals that CTCF underlies evolution of chromosomal domain
architecture, Cell. Rep., 10, 1297-1309.
43.Ulianov, S. V., Khrameeva, E. E., Gavrilov, A.
A., Flyamer, I. M., Kos, P., Mikhaleva, E. A., Penin, A. A., Logacheva,
M. D., Imakaev, M. V., Chertovich, A., Gelfand, M. S., Shevelyov, Y.
Y., and Razin, S. V. (2016) Active chromatin and transcription play a
key role in chromosome partitioning into topologically associating
domains, Genome Res., 26, 70-84.
44.Fraser, J., Ferrai, C., Chiariello, A. M.,
Schueler, M., Rito, T., Laudanno, G., Barbieri, M., Moore, B. L.,
Kraemer, D. C., Aitken, S., Xie, S. Q., Morris, K. J., Itoh, M.,
Kawaji, H., Jaeger, I., Hayashizaki, Y., Carninci, P., Forrest, A. R.,
Consortium, F., Semple, C. A., Dostie, J., Pombo, A., and Nicodemi, M.
(2015) Hierarchical folding and reorganization of chromosomes are
linked to transcriptional changes in cellular differentiation, Mol.
Syst. Biol., 11, 852.
45.Weinreb, C., and Raphael, B. J. (2016)
Identification of hierarchical chromatin domains,
Bioinformatics, 32, 1601-1609.
46.Rao, S. S., Huntley, M. H., Durand, N. C.,
Stamenova, E. K., Bochkov, I. D., Robinson, J. T., Sanborn, A. L.,
Machol, I., Omer, A. D., Lander, E. S., and Aiden, E. L. (2014) A 3D
map of the human genome at kilobase resolution reveals principles of
chromatin looping, Cell, 159, 1665-1680.
47.Hou, C., Li, L., Qin, Z. S., and Corces, V. G.
(2012) Gene density, transcription, and insulators contribute to the
partition of the Drosophila genome into physical domains,
Mol. Cell., 48, 471-484.
48.Pope, B. D., Ryba, T., Dileep, V., Yue, F., Wu,
W., Denas, O., Vera, D. L., Wang, Y., Hansen, R. S., Canfield, T. K.,
Thurman, R. E., Cheng, Y., Gulsoy, G., Dennis, J. H., Snyder, M. P.,
Stamatoyannopoulos, J. A., Taylor, J., Hardison, R. C., Kahveci, T.,
Ren, B., and Gilbert, D. M. (2014) Topologically associating domains
are stable units of replication-timing regulation, Nature,
515, 402-405.
49.Zhan, Y., Mariani, L., Barozzi, I., Schulz, E.
G., Bluthgen, N., Stadler, M., Tiana, G., and Giorgetti, L. (2017)
Reciprocal insulation analysis of Hi-C data shows that TADs represent a
functionally but not structurally privileged scale in the hierarchical
folding of chromosomes, Genome Res., 27, 479-490.
50.Markaki, Y., Gunkel, M., Schermelleh, L.,
Beichmanis, S., Neumann, J., Heidemann, M., Leonhardt, H., Eick, D.,
Cremer, C., and Cremer, T. (2010) Functional nuclear organization of
transcription and DNA replication: a topographical marriage between
chromatin domains and the interchromatin compartment, Cold Spring
Harb. Symp. Quant. Biol., 75, 475-492.
51.Smeets, D., Markaki, Y., Schmid, V. J., Kraus,
F., Tattermusch, A., Cerase, A., Sterr, M., Fiedler, S., Demmerle, J.,
Popken, J., Leonhardt, H., Brockdorff, N., Cremer, T., Schermelleh, L.,
and Cremer, M. (2014) Three-dimensional super-resolution microscopy of
the inactive X chromosome territory reveals a collapse of its active
nuclear compartment harboring distinct Xist RNA foci, Epigenet.
Chromat., 7, 8.
52.Kolbl, A. C., Weigl, D., Mulaw, M., Thormeyer,
T., Bohlander, S. K., Cremer, T., and Dietzel, S. (2012) The radial
nuclear positioning of genes correlates with features of megabase-sized
chromatin domains, Chromosome Res., 20, 735-752.
53.Nora, E. P., Dekker, J., and Heard, E. (2013)
Segmental folding of chromosomes: a basis for structural and regulatory
chromosomal neighborhoods? BioEssays, 35, 818-828.
54.Fabre, P. J., Benke, A., Joye, E., Nguyen Huynh,
T. H., Manley, S., and Duboule, D. (2015) Nanoscale spatial
organization of the HoxD gene cluster in distinct
transcriptional states, Proc. Natl. Acad. Sci. USA, 112,
13964-13969.
55.Wang, S., Su, J. H., Beliveau, B. J., Bintu, B.,
Moffitt, J. R., Wu, C. T., and Zhuang, X. (2016) Spatial organization
of chromatin domains and compartments in single chromosomes,
Science, 353, 598-602.
56.Nagano, T., Lubling, Y., Stevens, T. J.,
Schoenfelder, S., Yaffe, E., Dean, W., Laue, E. D., Tanay, A., and
Fraser, P. (2013) Single-cell Hi-C reveals cell-to-cell variability in
chromosome structure, Nature, 502, 59-64.
57.Nagano, T., Lubling, Y., Varnai, C., Dudley, C.,
Leung, W., Baran, Y., Mendelson Cohen, N., Wingett, S., Fraser, P., and
Tanay, A. (2017) Cell-cycle dynamics of chromosomal organization at
single-cell resolution, Nature, 547, 61-67.
58.Flyamer, I. M., Gassler, J., Imakaev, M.,
Brandao, H. B., Ulianov, S. V., Abdennur, N., Razin, S. V., Mirny, L.
A., and Tachibana-Konwalski, K. (2017) Single-nucleus Hi-C reveals
unique chromatin reorganization at oocyte-to-zygote transition,
Nature, 544, 110-114.
59.Ulianov, S. V., Tachibana-Konwalski, K., and
Razin, S. V. (2017) Single-cell Hi-C bridges microscopy and genome-wide
sequencing approaches to study 3D chromatin organization,
BioEssays, 39, doi: 10.1002/bies.201700104.
60.Stevens, T. J., Lando, D., Basu, S., Atkinson, L.
P., Cao, Y., Lee, S. F., Leeb, M., Wohlfahrt, K. J., Boucher, W.,
O’Shaughnessy-Kirwan, A., Cramard, J., Faure, A. J., Ralser, M.,
Blanco, E., Morey, L., Sanso, M., Palayret, M. G. S., Lehner, B., Di
Croce, L., Wutz, A., Hendrich, B., Klenerman, D., and Laue, E. D.
(2017) 3D structures of individual mammalian genomes studied by
single-cell Hi-C, Nature, 544, 59-64.
61.Vernimmen, D., and Bickmore, W. A. (2015) The
hierarchy of transcriptional activation: from enhancer to promoter,
Trends Genet., 31, 696-708.
62.Chepelev, I., Wei, G., Wangsa, D., Tang, Q., and
Zhao, K. (2012) Characterization of genome-wide enhancer–promoter
interactions reveals co-expression of interacting genes and modes of
higher order chromatin organization, Cell. Res., 22,
490-503.
63.Mifsud, B., Tavares-Cadete, F., Young, A. N.,
Sugar, R., Schoenfelder, S., Ferreira, L., Wingett, S. W., Andrews, S.,
Grey, W., Ewels, P. A., Herman, B., Happe, S., Higgs, A., LeProust, E.,
Follows, G. A., Fraser, P., Luscombe, N. M., and Osborne, C. S. (2015)
Mapping long-range promoter contacts in human cells with
high-resolution capture Hi-C, Nat. Genet., 47,
598-606.
64.Williamson, I., Berlivet, S., Eskeland, R.,
Boyle, S., Illingworth, R. S., Paquette, D., Dostie, J., and Bickmore,
W. A. (2014) Spatial genome organization: contrasting views from
chromosome conformation capture and fluorescence in situ
hybridization, Genes Dev., 28, 2778-2791.
65.Williamson, I., Lettice, L. A., Hill, R. E., and
Bickmore, W. A. (2016) Shh and ZRS enhancer colocalisation is specific
to the zone of polarising activity, Development, 143,
2994-3001.
66.Heidari, N., Phanstiel, D. H., He, C., Grubert,
F., Jahanbani, F., Kasowski, M., Zhang, M. Q., and Snyder, M. P. (2014)
Genome-wide map of regulatory interactions in the human genome,
Genome Res., 24, 1905-1917.
67.Morgan, S. L., Mariano, N. C., Bermudez, A.,
Arruda, N. L., Wu, F., Luo, Y., Shankar, G., Jia, L., Chen, H., Hu, J.
F., Hoffman, A. R., Huang, C. C., Pitteri, S. J., and Wang, K. C.
(2017) Manipulation of nuclear architecture through CRISPR-mediated
chromosomal looping, Nat. Commun., 8, 15993.
68.Ibn-Salem, J., Muro, E. M., and Andrade-Navarro,
M. A. (2016) Co-regulation of paralog genes in the three-dimensional
chromatin architecture, Nucleic Acids Res., 45,
81-91.
69.Symmons, O., Pan, L., Remeseiro, S., Aktas, T.,
Klein, F., Huber, W., and Spitz, F. (2016) The Shh topological domain
facilitates the action of remote enhancers by reducing the effects of
genomic distances, Dev. Cell., 39, 529-543.
70.Lucas, J. S., Zhang, Y., Dudko, O. K., and Murre,
C. (2014) 3D trajectories adopted by coding and regulatory DNA
elements: first-passage times for genomic interactions, Cell,
158, 339-352.
71.Dekker, J., and Mirny, L. (2016) The 3D genome as
moderator of chromosomal communication, Cell, 164,
1110-1121.
72.Tiana, G., Amitai, A., Pollex, T., Piolot, T.,
Holcman, D., Heard, E., and Giorgetti, L. (2016) Structural
fluctuations of the chromatin fiber within topologically associating
domains, Biophys. J., 110, 1234-1245.
73.Fragkos, M., Ganier, O., Coulombe, P., and
Mechali, M. (2015) DNA replication origin activation in space and time,
Nat. Rev. Mol. Cell. Biol., 16, 360-374.
74.Rhind, N., and Gilbert, D. M. (2013) DNA
replication timing, Cold Spring Harb. Perspect Biol., 5,
a010132.
75.Moindrot, B., Audit, B., Klous, P., Baker, A.,
Thermes, C., de Laat, W., Bouvet, P., Mongelard, F., and Arneodo, A.
(2012) 3D chromatin conformation correlates with replication timing and
is conserved in resting cells, Nucleic Acids Res., 40,
9470-9481.
76.Kalashnikova, A. A., Porter-Goff, M. E.,
Muthurajan, U. M., Luger, K., and Hansen, J. C. (2013) The role of the
nucleosome acidic patch in modulating higher order chromatin structure,
J. R. Soc. Interface, 10, 20121022.
77.Luger, K., Mader, A. W., Richmond, R. K.,
Sargent, D. F., and Richmond, T. J. (1997) Crystal structure of the
nucleosome core particle at 2.8 Å resolution, Nature,
389, 251-260.
78.Sinha, D., and Shogren-Knaak, M. A. (2010) Role
of direct interactions between the histone H4 tail and the H2A core in
long range nucleosome contacts, J. Biol. Chem., 285,
16572-16581.
79.Shogren-Knaak, M., Ishii, H., Sun, J. M., Pazin,
M. J., Davie, J. R., and Peterson, C. L. (2006) Histone H4-K16
acetylation controls chromatin structure and protein interactions,
Science, 311, 844-847.
80.Allahverdi, A., Yang, R., Korolev, N., Fan, Y.,
Davey, C. A., Liu, C. F., and Nordenskiold, L. (2011) The effects of
histone H4 tail acetylations on cation-induced chromatin folding and
self-association, Nucleic Acids Res., 39, 1680-1691.
81.Dowen, J. M., Fan, Z. P., Hnisz, D., Ren, G.,
Abraham, B. J., Zhang, L. N., Weintraub, A. S., Schuijers, J., Lee, T.
I., Zhao, K., and Young, R. A. (2014) Control of cell identity genes
occurs in insulated neighborhoods in mammalian chromosomes,
Cell, 159, 374-387.
82.Hanssen, L. L. P., Kassouf, M. T., Oudelaar, A.
M., Biggs, D., Preece, C., Downes, D. J., Gosden, M., Sharpe, J. A.,
Sloane-Stanley, J. A., Hughes, J. R., Davies, B., and Higgs, D. R.
(2017) Tissue-specific CTCF-cohesin-mediated chromatin architecture
delimits enhancer interactions and function in vivo, Nat.
Cell. Biol., 19, 952-961.
83.Hansen, A. S., Pustova, I., Cattoglio, C., Tjian,
R., and Darzacq, X. (2017) CTCF and cohesin regulate chromatin loop
stability with distinct dynamics, Elife, 6, pii:
e25776.
84.Merkenschlager, M., and Nora, E. P. (2016) CTCF
and cohesin in genome folding and transcriptional gene regulation,
Annu. Rev. Genom. Hum. Genet., 17, 17-43.
85.Narendra, V., Bulajic, M., Dekker, J., Mazzoni,
E. O., and Reinberg, D. (2016) CTCF-mediated topological boundaries
during development foster appropriate gene regulation, Genes
Dev., 30, 2657-2662.
86.Narendra, V., Rocha, P. P., An, D., Raviram, R.,
Skok, J. A., Mazzoni, E. O., and Reinberg, D. (2015) CTCF establishes
discrete functional chromatin domains at the Hox clusters during
differentiation, Science, 347, 1017-1021.
87.Sanborn, A. L., Rao, S. S., Huang, S. C., Durand,
N. C., Huntley, M. H., Jewett, A. I., Bochkov, I. D., Chinnappan, D.,
Cutkosky, A., Li, J., Geeting, K. P., Gnirke, A., Melnikov, A.,
McKenna, D., Stamenova, E. K., Lander, E. S., and Aiden, E. L. (2015)
Chromatin extrusion explains key features of loop and domain formation
in wild-type and engineered genomes, Proc. Natl. Acad. Sci. USA,
112, 6456-6465.
88.Zuin, J., Dixon, J. R., van der Reijden, M. I.,
Ye, Z., Kolovos, P., Brouwer, R. W., van de Corput, M. P., van de
Werken, H. J., Knoch, T. A., van, Ijcken. W. F., Grosveld, F. G., Ren,
B., and Wendt, K. S. (2014) Cohesin and CTCF differentially affect
chromatin architecture and gene expression in human cells, Proc.
Natl. Acad. Sci. USA, 111, 996-1001.
89.Nora, E. P., Goloborodko, A., Valton, A. L.,
Gibcus, J. H., Uebersohn, A., Abdennur, N., Dekker, J., Mirny, L. A.,
and Bruneau, B. G. (2017) Targeted degradation of CTCF decouples local
insulation of chromosome domains from genomic compartmentalization,
Cell, 169, 930-944.
90.Vietri Rudan, M., and Hadjur, S. (2015) Genetic
tailors: CTCF and cohesin shape the genome during evolution, Trends
Genet., 31, 651-660.
91.Holwerda, S., and de Laat, W. (2012) Chromatin
loops, gene positioning, and gene expression, Front Genet.,
3, 217.
92.Fudenberg, G., Imakaev, M., Lu, C., Goloborodko,
A., Abdennur, N., and Mirny, L. A. (2016) Formation of chromosomal
domains by loop extrusion, Cell Rep., 15, 2038-2049.
93.Haarhuis, J. H. I., van der Weide, R. H., Blomen,
V. A., Yanez-Cuna, J. O., Amendola, M., van Ruiten, M. S., Krijger, P.
H. L., Teunissen, H., Medema, R. H., van Steensel, B., Brummelkamp, T.
R., de Wit, E., and Rowland, B. D. (2017) The cohesin release factor
WAPL restricts chromatin loop extension, Cell, 169,
693-707.
94.Rao, S. S. P., Huang, S. C., Glenn St Hilaire,
B., Engreitz, J. M., Perez, E. M., Kieffer-Kwon, K. R., Sanborn, A. L.,
Johnstone, S. E., Bascom, G. D., Bochkov, I. D., Huang, X., Shamim, M.
S., Shin, J., Turner, D., Ye, Z., Omer, A. D., Robinson, J. T.,
Schlick, T., Bernstein, B. E., Casellas, R., Lander, E. S., and Aiden,
E. L. (2017) Cohesin loss eliminates all loop domains, Cell,
171, 305-320.
95.Ulianov, S. V., Gavrilov, A. A., and Razin, S. V.
(2015) Nuclear compartments, genome folding, and enhancer-promoter
communication, Int. Rev. Cell. Mol. Biol., 315,
183-244.
96.Wang, K. C., Yang, Y. W., Liu, B., Sanyal, A.,
Corces-Zimmerman, R., Chen, Y., Lajoie, B. R., Protacio, A., Flynn, R.
A., Gupta, R. A., Wysocka, J., Lei, M., Dekker, J., Helms, J. A., and
Chang, H. Y. (2011) A long noncoding RNA maintains active chromatin to
coordinate homeotic gene expression, Nature, 472,
120-124.
97.Xiang, J. F., Yin, Q. F., Chen, T., Zhang, Y.,
Zhang, X. O., Wu, Z., Zhang, S., Wang, H. B., Ge, J., Lu, X., Yang, L.,
and Chen, L. L. (2014) Human colorectal cancer-specific CCAT1-L lncRNA
regulates long-range chromatin interactions at the MYC locus,
Cell Res., 24, 513-531.
98.Trimarchi, T., Bilal, E., Ntziachristos, P.,
Fabbri, G., Dalla-Favera, R., Tsirigos, A., and Aifantis, I. (2014)
Genome-wide mapping and characterization of notch-regulated long
noncoding RNAs in acute leukemia, Cell, 158, 593-606.
99.Lai, F., Orom, U. A., Cesaroni, M., Beringer, M.,
Taatjes, D. J., Blobel, G. A., and Shiekhattar, R. (2013) Activating
RNAs associate with mediator to enhance chromatin architecture and
transcription, Nature, 494, 497-501.
100.Kung, J. T., Kesner, B., An, J. Y., Ahn, J. Y.,
Cifuentes-Rojas, C., Colognori, D., Jeon, Y., Szanto, A., del Rosario,
B. C., Pinter, S. F., Erwin, J. A., and Lee, J. T. (2015)
Locus-specific targeting to the X chromosome revealed by the RNA
interactome of CTCF, Mol. Cell., 57, 361-375.
101.Saldana-Meyer, R., Gonzalez-Buendia, E.,
Guerrero, G., Narendra, V., Bonasio, R., Recillas-Targa, F., and
Reinberg, D. (2014) CTCF regulates the human p53 gene through
direct interaction with its natural antisense transcript, Wrap53,
Genes Dev., 28, 723-734.
102.Ong, C. T., and Corces, V. G. (2014) CTCF: an
architectural protein bridging genome topology and function, Nat.
Rev. Genet., 15, 234-246.
103.Donohoe, M. E., Zhang, L. F., Xu, N., Shi, Y.,
and Lee, J. T. (2007) Identification of a Ctcf cofactor, Yy1, for the X
chromosome binary switch, Mol. Cell., 25, 43-56.
104.Sigova, A. A., Abraham, B. J., Ji, X., Molinie,
B., Hannett, N. M., Guo, Y. E., Jangi, M., Giallourakis, C. C., Sharp,
P. A., and Young, R. A. (2015) Transcription factor trapping by RNA in
gene regulatory elements, Science, 350, 978-981.
105.Engreitz, J. M., Ollikainen, N., and Guttman,
M. (2016) Long non-coding RNAs: spatial amplifiers that control nuclear
structure and gene expression, Nat. Rev. Mol. Cell Biol.,
17, 756-770.
106.Hacisuleyman, E., Goff, L. A., Trapnell, C.,
Williams, A., Henao-Mejia, J., Sun, L., McClanahan, P., Hendrickson, D.
G., Sauvageau, M., Kelley, D. R., Morse, M., Engreitz, J., Lander, E.
S., Guttman, M., Lodish, H. F., Flavell, R., Raj, A., and Rinn, J. L.
(2014) Topological organization of multichromosomal regions by the long
intergenic noncoding RNA Firre, Nat. Struct. Mol. Biol.,
21, 198-206.
107.Engreitz, J. M., Pandya-Jones, A., McDonel, P.,
Shishkin, A., Sirokman, K., Surka, C., Kadri, S., Xing, J., Goren, A.,
Lander, E. S., Plath, K., and Guttman, M. (2013) The Xist lncRNA
exploits three-dimensional genome architecture to spread across the X
chromosome, Science, 341, 1237973.
108.Cook, P. R., Brazell, I. A., and Jost, E.
(1976) Characterization of nuclear structures containing superhelical
DNA, J. Cell. Sci., 22, 303-324.
109.Paulson, J. R., and Laemmli, U. K. (1977) The
structure of histone-depleted metaphase chromosomes, Cell,
12, 817-828.
110.Hancock, R., and Hughes, M. E. (1982)
Organization of DNA in the eukaryotic nucleus, Biol. Cell,
44, 201-212.
111.Razin, S. V., Mantieva, V. L., and Georgiev, G.
P. (1979) The similarity of DNA sequences remaining bound to scaffold
upon nuclease treatment of interphase nuclei and metaphase chromosomes,
Nucleic Acids Res., 7, 1713-1735.
112.Razin, S. V., Gromova, I. I., and Iarovaia, O.
V. (1995) Specificity and functional significance of DNA interaction
with the nuclear matrix: new approaches to clarify the old questions,
Int. Rev. Cytol., 162B, 405-448.
113.Razin, S. V., Iarovaia, O. V., and Vassetzky,
Y. S. (2014) A requiem to the nuclear matrix: from a controversial
concept to 3D organization of the nucleus, Chromosoma,
123, 217-224.
114.Cook, P. R. (1989) The nucleoskeleton and the
topology of transcription, Eur. J. Biochem., 185,
487-501.
115.Cook, P. R. (2002) Predicting three-dimensional
genome structure from transcriptional activity, Nat. Genet.,
32, 347-352.
116.Robinson, S. I., Small, D., Idzerda, R.,
McKnight, G. S., and Vogelstein, B. (1983) The association of active
genes with the nuclear matrix of the chicken oviduct, Nucleic Acids
Res., 15, 5113-5130.
117.Razin, S. V., and Yarovaya, O. V. (1985)
Initiated complexes of RNA polymerase II are concentrated in the
nuclear skeleton associated DNA, Exp. Cell Res., 158,
273-275.
118.Ciejek, E. M., Tsai, M.-J., and O’Malley,
B. W. (1983) Actively transcribed genes are associated with the nuclear
matrix, Nature, 306, 607-609.
119.Dunn, K. L., Zhao, H., and Davie, J. R. (2003)
The insulator binding protein CTCF associates with the nuclear matrix,
Exp. Cell Res., 288, 218-223.
120.Yusufzai, T. M., and Felsenfeld, G. (2004) The
5′-HS4 chicken beta-globin insulator is a CTCF-dependent nuclear
matrix-associated element, Proc. Natl. Acad. Sci. USA,
101, 8620-8624.
121.Razin, S. V., Hancock, R., Iarovaia, O.,
Westergaard, O., Gromova, I., and Georgiev, G. P. (1993)
Structural-functional organization of chromosomal DNA domains, Cold
Spring Harbor Symp. Quant. Biol., 58, 25-35.
122.Gromova, I. I., Thomsen, B., and Razin, S. V.
(1995) Different topoisomerase II antitumor drugs direct similar
specific long-range fragmentation of an amplified c-MYC gene
locus in living cells and in high-salt-extracted nuclei, Proc. Natl.
Acad. Sci. USA, 92, 102-106.
123.Uuskula-Reimand, L., Hou, H.,
Samavarchi-Tehrani, P., Rudan, M. V., Liang, M., Medina-Rivera, A.,
Mohammed, H., Schmidt, D., Schwalie, P., Young, E. J., Reimand, J.,
Hadjur, S., Gingras, A. C., and Wilson, M. D. (2016) Topoisomerase II
beta interacts with cohesin and CTCF at topological domain borders,
Genome Biol., 17, 182.
124.Eagen, K. P., Hartl, T. A., and Kornberg, R. D.
(2015) Stable chromosome condensation revealed by chromosome
conformation capture, Cell, 163, 934-946.
125.Schwartz, Y. B., Ioudinkova, E. S., Demakov, S.
A., Razin, S. V., and Zhimulev, I. F. (1999) Interbands of
Drosophila melanogaster polytene chromosomes contain matrix
association regions, J. Cell. Biochem., 72, 368-372.
126.De Wit, E., and de Laat, W. (2012) A decade of
3C technologies: insights into nuclear organization, Genes Dev.,
26, 11-24.
127.Bystricky, K. (2015) Chromosome dynamics and
folding in eukaryotes: insights from live cell microscopy, FEBS
Lett., 589, 3014-3022.
128.Razin, S. V., Gavrilov, A. A., Ioudinkova, E.
S., and Iarovaia, O. V. (2013) Communication of genome regulatory
elements in a folded chromosome, FEBS Lett., 587,
1840-1847.