REVIEW: Genetic and Epigenetic Mechanisms of β-Globin Gene Switching
O. V. Iarovaia1,2*, A. P. Kovina1,2,3, N. V. Petrova1,2, S. V. Razin1,2,3, E. S. Ioudinkova1,2, Y. S. Vassetzky2,4, and S. V. Ulianov1,2,3
1Institute of Gene Biology, Russian Academy of Sciences, 119334 Moscow, Russia; E-mail: iarovaia@inbox.ru2French–Russian Laboratory for Research in Oncology LIA1066, Russia–France
3Lomonosov Moscow State University, Biological Faculty, 119991 Moscow, Russia
4Institut Gustave Roussy, 39 rue Camille-Desmoulins, 94805 Villejuif, France
* To whom correspondence should be addressed.
Received November 20, 2017; Revision received December 8, 2017
Vertebrates have multiple forms of hemoglobin that differ in the composition of their polypeptide chains. During ontogenesis, the composition of these subunits changes. Genes encoding different α- and β-polypeptide chains are located in two multigene clusters on different chromosomes. Each cluster contains several genes that are expressed at different stages of ontogenesis. The phenomenon of stage-specific transcription of globin genes is referred to as globin gene switching. Mechanisms of expression switching, stage-specific activation, and repression of transcription of α- and β-globin genes are of interest from both theoretical and practical points of view. Alteration of balanced expression of globin genes, which usually occurs due to damage to adult β-globin genes, leads to development of severe diseases – hemoglobinopathies. In most cases, reactivation of the fetal hemoglobin gene in patients with β-thalassemia and sickle cell disease can reduce negative consequences of irreversible alterations of expression of the β-globin genes. This review focuses on the current state of research on genetic and epigenetic mechanisms underlying stage-specific switching of β-globin genes.
KEY WORDS: α- and β-globin genes domains, hemoglobin switching, transcriptional regulation, spatial organization of chromatinDOI: 10.1134/S0006297918040090
Abbreviations: CTCF, a transcription factor protein; GWAS, genome-wide association studies; Hb, hemoglobin; HS, hypersensitive site; LCR, locus control region; MBP, methyl-binding proteins.
Hemoglobin (Hb), which constitutes ~98% of the total protein in
erythrocyte cytoplasm, is responsible for oxygen transport in the
bloodstream of vertebrates. Vertebrate hemoglobin is a tetramer
consisting of two α- and two β-polypeptide chains that are
associated with heme. In vertebrates, there are typically several
hemoglobin forms that differ in composition of polypeptide chains and
are adjusted for oxygen transport under different conditions and in
different stages of ontogenesis. Requirement for different forms of
hemoglobin, which are specific for embryonic, fetal, and adult
development stages, is determined by specificity of blood circulation
in the organism during different ontogenic stages. The presence of
several forms of hemoglobin that differ in polypeptide chain
composition is provided by the expression of related but different
genes. This is true for both α- and β-globin gene families.
In the human genome, synthesis of α- and β-globin chains is
directed by two separate gene clusters: an α-globin gene domain
that contains an embryonic gene ζ and two adult genes α; and
a β-globin gene domain comprising an embryonic gene ε, two
fetal genes Gγ, Aγ, and two adult genes δ and β
[1]. During the first trimester of pregnancy,
embryonic α- and β-type genes (ζ-globin and
ε-globin, respectively) are expressed in erythrocytes and
erythroblasts in the first primitive wave of hematopoiesis. Embryonic
hemoglobin totally disappears by the end of the first trimester.
However, during the 6th week of development, even before the
disappearance of embryonic hemoglobin, expression of fetal
β-globin genes and adult α-globin genes is triggered. During
this time, fetal hemoglobin (HbF – α2γ2) appears in
fetal blood. During the neonatal period, fetal hemoglobin is
substituted by adult hemoglobin (HbA1 (α2β2) and HbA2
(α2δ2)). Synthesis of adult hemoglobin is provided by
expression the adult globin genes. The phenomenon of stage-specific
activation and repression of transcription is referred to as globin
gene switching.
In humans, three successive erythropoiesis waves occur during embryonic, fetal, and adult stages of ontogenesis [2-4]. Primitive erythroid cells originate from yolk sac hemangioblasts and differentiate into erythrocytes in the bloodstream. Later, during ontogenesis in the yolk sac, definitive erythropoiesis cell precursors begin to differentiate. They enter the bloodstream, colonize the embryonic liver, and provide fetal erythropoiesis. Bone marrow hematopoietic stem cells originating from the aorta-gonad-mesonephros (AGM) give rise to adult erythrocytes. Thus, embryonic, fetal, and adult erythrocytes originate from different precursor cells and differentiate independently of each other. The type of erythropoiesis (embryonic, fetal, or definitive) is determined at several regulatory levels: from modulation of proliferative status of precursor cells to differential utilization of mechanisms of transduction of erythroid differentiation signal depending on various transcriptional activators and repressors and their cofactors as well as cell microenvironment factors [5]. Decision on stage-specific activation or repression of globin gene expression is most probably made simultaneously with determining the type of erythropoiesis. Historically, change in a profile of globin gene expression is referred to as globin gene switching. We believe that it is more appropriate to say that there is successive change of cell lines, which originally possess different transcriptional programs, rather than a stage-specific switching transcriptional program of a single cell line during erythroid differentiation. However, until now the possibility of true switching of the expression from fetal to adult type in a population of common precursor cells for human fetal and adult erythroblasts is being discussed [1, 6, 7].
Along with the onset of the molecular biology era, genetic and epigenetic mechanisms of switching the stage-specific expression of human β-globin genes have drawn attention of researchers. The β-globin gene domain is one of the best-studied models in molecular biology [8, 9]. By now, dozens of transcription factors have been characterized that direct stage-specific expression of β-globin genes [6], and epigenetic mechanisms have been revealed that act at the level of chromatin organization [10, 11]. The role of the spatial organization of the domain in switching the expression of β-globin genes has been studied [12-14]. Along with the fundamental aspect, understanding of the mechanism of stage-specific β-globin gene switching has important practical value. The β-globin gene domain is most frequently damaged in patients with anemia. Deficiency in the adult β-globin leads to development of sickle cell disease and β-thalassemia in mutation carriers. Reactivation of the fetal β-globin gene can partially compensate for the deficiency in normal β-globin and relieve symptoms of these diseases [15, 16]. For this reason, studying mechanisms of stage-specific β-globin gene switching is of interest from both theoretical and practical points of view.
STRUCTURAL AND FUNCTIONAL STUDIES OF ORGANIZATION OF THE
β-GLOBIN GENE DOMAIN
The β-globin gene domains in vertebrates are typical representatives of a group of genomic domains that have different sensitivity to DNase treatment depending on the type of cell differentiation [9]. Human, murine, and chicken β-globin gene domains have been most thoroughly studied (figure) [17, 18]. The human β-globin gene domain can be considered as a typical representative of these domains. This domain is surrounded by CTCF-dependent insulators [19], it includes globin genes arranged according to their activation order during development [20], and it has a locus control region (LCR) that controls transcription state of the domain and transcription activity of the globin genes [21]. The domain is integrated into an extended locus of olfactory receptors. It is situated in inactive (DNase-resistant) chromatin in nonerythroid cells, whereas it is located in active (DNase-sensitive) chromatin in erythroid cells [18]. Deletion of the LCR leads to lack of active domain configuration in erythroid cells [22]. The LCR includes several enhancers that are required for correct order of activation of the globin genes during development [23]. Additional regulatory elements participating in the control of the β-globin gene domain are located within a cluster of olfactory receptors far beyond the area limited by the insulators. However, these regulatory elements are usually not considered as part of the β-globin gene domain [18]. The active state of the domain in erythroid cells correlates with high levels of histone acetylation. At the same time, the level of histone acetylation within the LCR remains equally high during all developmental stages, whereas acetylation in embryonic–fetal and adult subdomains correlates with transcriptional activity of the corresponding genes [24]. The high level of intergenic transcription in erythroid cells is an interesting feature of the domain [25, 26]. According to some data, intergenic transcription is required for activation of subdomains specific for the development stage [27]. In contrast, other data indicate that intergenic transcription may be required for inactivation of stage-specific subdomains via RNA interference [28].
Structure of the β-globin gene domain in mouse (a), chicken (b), and human (c) chromatin. Rectangles indicate the position of genes: white – embryonal, gray – fetal, black – adult. Location of cis-acting regulatory elements is indicated by circles. Model of spatial organization of the human β-globin gene domain at the stage of expression of embryonal (d), fetal (e), and adult (f) β-globin genes
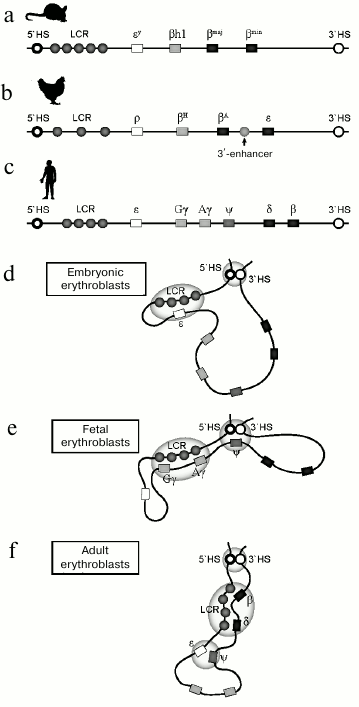
In mice, the β-globin gene domain includes two embryonic genes εy and βh1, and two adult genes βmaj and βmin. In contrast to humans, in mice during ontogenic expression switching from embryonic to adult only occurs once. Another important feature of the domain is low expression level of adult genes in embryonic erythrocytes [29]. This suggests the theoretical possibility of expression switching from embryonic to adult type in a single cell line. At the 5′-end of the β-globin locus there is also an LCR containing enhancers required for expression of all the β-globin genes [17]. For a long time, human β-globin genes integrated into the mouse genome remained the only model available for fundamental studies of the human β-globin gene domain. However, mechanisms of regulation of expression switching for human genes in transgenic mice is not a perfect model for regulation of human β-globin genes in their native genetic context. Human transgenes after integration acquire transcriptional regulation features that are specific for mice: transcription switching occurs only once. The human embryonic gene ε and the fetal genes are expressed concurrently with murine embryonic genes Ey and βh1, and human adult genes δ and β are expressed along with the murine adult genes [30]. An intermediate fetal erythropoiesis wave, when only fetal genes are expressed, is a late evolutionary acquisition and is only typical for primates.
The chicken β-globin gene domain is another popular model for studying regulation of transcriptional switching. This domain includes two adult genes, βH and βA, that are flanked with two embryonic genes, ρ and ε. In addition to LCR enhancers, the domain contains an additional enhancer located between the two genes, βA and ε [31].
Like the human genome, the β-globin gene domain in both mouse and chicken genomes is surrounded by olfactory receptor genes and is insulated from the genome by specialized regulatory elements. Thus, structural and functional organizations of the β-globin gene domain are conserved, and many features of transcriptional regulation and globin gene switching are shared by all endotherms.
MECHANISMS OF STAGE-SPECIFIC ACTIVATION OF β-GLOBIN
GENES
Stage-specific expression switching can occur through at least two mechanisms: by direct competition between a stage-specific gene promoter for shared regulatory elements, or by autonomous activation and/or autonomous silencing via specific activators and repressors as well as epigenetic mechanisms [1, 32]. Most probably, these mechanisms do not exclude, but rather supplement each other.
Enhancers within LCR direct transcription of both the adult β-globin gene and embryonic gene ε. A long time ago it was found that enhancer mutations that increase transcriptional activity of the adult gene led to proportional increase in transcriptional activity of gene ε. Based on this observation, it was suggested that the activity of both promoters is regulated by the same region of the enhancer. A hypothesis was proposed that promoters of adult and fetal genes compete for a common regulatory element that might be crucial for regulation of expression of these genes at corresponding stages of development [33, 34]. It was shown that at any moment an LCR interacts with only one promoter, and alternative interactions may determine differential expression of stage-specific genes [34]. Selection of the promoter that would interact with the LCR depends on the ontogenic stage.
The suggestion about possible promoter competition was confirmed using a modern approach of recruiting transcription factors to specific regulatory elements. Dimerization of a transcription factor LDB1 upon its binding to the enhancer and the promoter as a part of a composite protein complex provides rapprochement of these regulatory elements within the β-globin gene domain. Manual targeting of LDB1 to a fetal gene promoter in adult human erythroblasts reactivates the fetal gene and causes reciprocal repression of the adult genes. Increase in expression levels correlates with formation of a specific activator cluster of regulatory elements. These data indirectly confirm promoter competition as a leading mechanism of stage-specific activation [35].
As stated above, in addition to competition between regulatory elements, mechanisms of autonomous stage-specific activation and repression of corresponding genes may be involved in expression switching. Information for stage-specific expression activation is provided by both promoters of globin genes [36] and remote regulatory elements. Deletion analysis carried out on the transgenic murine model showed that active expression of the embryonic gene requires a core element including HS3 LCR, which binds to several transcription factors [37]. Activation of the adult and fetal genes depends on a core element of HS4 LCR [38]. Deletion of HS1 significantly decreased expression level of both embryonic and fetal genes at corresponding stages of development [39].
One of the mechanisms of autonomous differential activation could be low-level intergenic transcription of embryo-fetal or adult subdomains [26, 40, 41]. Deletion of a promoter controlling intergenic transcription of the adult subdomain results in repression of the adult genes even though promoters of these genes and LCR are not damaged. This suggests that the active state of the adult subdomain is provided by sequential histone acetylation within this subdomain, which is mediated by low-level transcription by RNA-polymerase II [42].
An interesting model was proposed that describes the balance of mechanisms of stage-specific activation and repression of β-globin genes in mice [43]. The histone methyltransferase G9a-GLP, which binds to promoters of both the adult and embryonic genes, plays a pivotal role in this mechanism. In adult murine erythroid cells both adult and embryonic genes bind simultaneously G9a-GLP and MLL2, which, according to the classical concept, provide divergent chromatin context. Changing balance in favor of either activation or repression activity occurs at the level of complex formation between G9a-GLP and MLL2 with specific demethylases. Concerted action of demethylase H3K4me3 Jarid1a and H3K9/K27 methyltransferase G9a-GLP at the promoter of the embryonic gene E(y) provides its repression, which goes along with appearance of repressive histone modifications H3K9me2 and H3K27me2. Simultaneous binding to the promoter of the adult gene βmaj demethylase UTX, which removes repressive modifications H3K27me2, and methyltransferase MLL2, which trimethylates histone H3 within the K4 region, activates the adult gene. G9a-dependent binding of components of the Mediator complex to chromatin within the locus plays an important role in the stage-specific activation of the adult gene (but not the embryonic gene) in mice. G9a stabilizes binding of the Mediator activator complex on the βmaj promoter, G9a methyltransferase activity being not required for manifestation of the activator function of the Mediator. Thus, repression of the embryonic gene E(y) (in mice) depends on the balance between coactivator and corepressor activities of G9a.
MECHANISMS OF STAGE-SPECIFIC REPRESSION OF β-GLOBIN
GENES
A great body of data has accumulated that speaks in favor of the existence of mechanisms of direct repression of the fetal and embryonic genes. So, an integrated construct containing the ε-globin gene under the LCR control is only expressed in the yolk sac. Thus, all genomic elements required not only for the stage-specific activation, but also for stage-specific repression are located in the embryonic gene or in the LCR. This observation suggests that the autonomous silencing of the embryonic gene plays a pivotal role in the mechanism of stage-specific expression switching.
Analysis of GWAS data revealed three loci whose mutations provided anomalously high levels of HbF (i.e. derepression of fetal β-globin genes) in adults [44, 45]. The first locus is situated in the β-globin gene domain as such, the second and the third were located in a BCL11A locus of chromosome 2 and intergenic region between genes HBS1L and MYB in chromosome 6, respectively.
A transcription factor with zinc finger-like DNA-recognition domain BCL11A (B-cell lymphoma/leukemia 11A, also referred to as EVI9, CTIP1) was cloned as a myeloid oncogene (in mice) and a B-cell oncogene (in humans) [46-48] required for normal lymphopoiesis. Genome-wide studies suggested that, in addition to lymphopoiesis, this factor directly participates in repression of the embryonic and fetal globin genes in adult erythroblasts. Knockdown of BCL11A in human erythroblasts leads to increase in expression of fetal hemoglobin [49]. In mice, BCL11A is only expressed in certain erythroid cells. These data suggest that BCL11A is one of the key transcription factors that provide switching [50]. BCL11A binds to regulatory elements within the LCR, with promoter of ε-globin gene, and also with an intergenic region between the fetal and adult genes, but it does not bind directly to either the adult or embryonic genes. In erythroid cells, BCL11A is integrated into a composite protein complex that comprises transcription factors GATA1, FOG1, RUNX1, KLF1, and SOX6 [51]. The repression mechanism also assumes recruitment of epigenetic factors that provide stage-specific repression of the fetal and embryonic globin genes. BCL11A binds to numerous modulators of chromatin structure (MBD3-NuRD, LSD1/CoREST, NCoR/SMRT, SIN3, and SWI/SNF), histone deacetylases HDAC1 and HDAC2, lysine-specific demethylase LSD1, and DNA methyltransferase DNMT1 [51].
BCL11A not only recruits repressing modulators of chromatin structure, but it also participates in organization of stage-specific interactions between promoters of the adult genes and the LCR [52]. Formation of the adult-type activator cluster occurs so that the fetal gene promoters are displaced from the cluster. Thus, BCL11A provides both direct repression of the embryonic and fetal genes and advantage of the adult genes in a competition for a common enhancer in adult erythroblasts.
Transcription factor SOX6 is yet another participant of stage-specific switching of globin gene expression. SOX6 determines differentiation pathways for many cell types [4, 53]. SOX6 deficiency not only results in neonatal pathologies, but also causes substantial increase in expression levels of embryonic genes βh1 and Ey in mice. Repression by SOX6 is perhaps due to its interaction with the key factor of stage-specific repression, BCL11A, within the chromatin of the promoter of the fetal genes [52]. In regulatory pathway networks, SOX6 can affect different mechanisms. In particular, SOX6 enhances definitive erythropoiesis by stimulating survival, proliferation, and terminal differentiation of erythroid cells [54].
Proteins of the GATA family (GATA1-6) are transcription factors having a zinc finger-like DNA-recognition domain. GATA-binding sites are located within regulatory elements of many erythroid-specific genes, in particular in the α- and β-globin gene domains. These transcription factors can act either as repressors or as transcription activators for the genes they control. The action of GATA1 may be dual. On one hand, GATA1 promotes expression of the adult β-globin genes in mice via recruitment of RNA-polymerase II to a promoter. On the other hand, GATA1 binds to a region located upstream of the fetal genes and recruits the repressor NuRD chromatin remodeling complex to chromatin. It is believed that this region is required for efficient repression of the fetal genes. GATA-1-binding sites are present within the human ε-globin promoter. Using transgenic mice that carry the human β-globin gene domain, it was shown that mutations in them that affect GATA-1 binding lead to lack of ε-globin repression in adult animals. Perhaps, GATA-1 represses the embryonic gene when in a complex with transcription factors YY1 and SP1. Promoter mutations in embryonic gene ε that are in the binding sites for these transcription factors impair stage-specific repression of the embryonic gene [55].
The role of erythroid-specific Krüppel-like factors KLF1 and -2 in expression switching is ambiguous. KLF1 was found to be a transcriptional activator of adult β-globin genes. However, KLF1 and -2 also positively regulate transcription of fetal and embryonic genes [56]. This transcription factor binds to a CACCC motif, which is frequent in regulatory elements of globin genes. KLF1 binds to HS1-HS3 in the LCR, to promoter of the adult βmaj gene in murine definitive erythroid cells, and also to promoters of embryonic genes Ey and βh1 in embryonic erythroid cells [57]. The pattern of binding of this factor by HS1-HS4-elements within the LCR in the human β-globin gene domain also varies in a stage-specific manner, which suggests that it participates in expression switching of globin genes. KLF1 interacts with CBP/p300 and BRG1, which is a component of the chromatin remodeling complex SWI/SNF [58]. In addition to that, KLF1 promotes transcription of BCL11A by binding to the BCL11A gene promoter. Thus, at the same time, it can activate transcription of adult genes and mediate repression of embryonic and fetal genes at higher regulation levels.
The promoters of embryonic and fetal genes, but not adult globin genes, contain direct repeats binding protein factors, which are similar to COUP-TF nuclear orphan receptors. Mutations in these regions provide active expression of the embryonic gene in adult erythroblasts, which indicates a possible role of the corresponding protein factors in repression of the embryonic gene [59]. COUP-TFII is expressed in embryonic and fetal erythroid cells, yolk sac, and in fetal liver; it is not expressed in adult erythroid cells. COUP-TFII interacts with BCL11A. However, it is still unclear whether this interaction is required for repression of the embryonic and fetal genes [60].
DR1 repeats in the embryonic gene promoter bind transcription factor DRED, which competes with the activating transcription factor KLF1 for promoter binding and provides repression of the embryonic and fetal genes. DRED contains TR2 and TR4 nuclear orphan receptors, which form a heterodimer upon binding to promoters of the embryonic and fetal genes [61]. The adult genes do not contain DR1; DR1 mutations are associated with high levels of fetal hemoglobin in adult erythroid cells. Conserved DR-elements within the hematopoietic enhancer GATA1 (G1HE) bind TR2/TR4, providing repression of GATA1 during terminal stages of differentiation [62]. This complex interaction network also provides stage-specific repression of the embryonic and fetal genes in adult erythroblasts [63].
A pivotal role of transcription factor NF-E4 in globin gene expression switching in chickens was demonstrated long ago [64]. The human homolog of the chicken NF-E4, p22NF-E4, is required for stage-specific activation of fetal genes. This protein along with a transcription factor CP2 forms the SSP (stage selector protein) protein complex. SSP is specific for the fetal stage of the ontogenesis. It provides stage-specific activation of fetal genes by binding to their promoters. Upon ectopic expression, this factor stimulates transcription of fetal genes and slows transcription switching in transgenic mice that carry the human β-globin gene domain [65].
The list of transcription factors that directly or indirectly participate in the stage-specific repression of β-globin genes grows continuously, and complete understanding of their role in transcription switching of β-globin genes still requires time.
EPIGENETIC MECHANISMS OF β-GLOBIN GENE SWITCHING
Transcription factors can alter the transcription state of a gene by recruiting modulators of the epigenetic landscape of chromatin. Epigenetic modifications in turn mobilize regulatory protein complexes, which finally determine accessibility of chromatin to RNA-polymerase II. Mechanisms of epigenetic transcriptional regulation include: posttranslational modification of histones; DNA methylation that impedes binding of transcription factors and recruits corepressors, for instance, histone deacetylases; RNA interference leading to transcriptional inactivation [66]; spatial rearrangement of loci (see below) and gene relocation between transcriptionally-active and repressed compartments of the cell nucleus [67].
DNA methylation is a well-studied epigenetic marker associated with the repression state of chromatin. De novo CpG island methylation is catalyzed by two DNA methyltransferases, DNMT3A and DNMT3B, which symmetrically methylate cytosine residues in both strands. The methylation state is maintained after replication by DNMT1, which methylates CpG islands of the nascent DNA strand. Effectors of DNA methylation are proteins recognizing symmetrically methylated CpG dinucleotides – MBD1, MBD2, MECP2, and MBD4 [68]. DNA methylation is one of the key mechanisms of ontogenic regulation and cell differentiation [69]. Promoters of all the β-globin genes are methylated in nonerythroid cells. It was found long ago that promoter methylation in erythroid cells is stage-specific: in adult erythroid cells of endotherms, promoters of the inactive fetal and embryonic β-globin genes are methylated [70]. Silencing of embryonic and fetal genes in adult cells is mediated by MBD2, which recruits histone deacetylases HDAC1/2 included into NuRD remodeling complex to methylated DNA [71, 72]. The DNA methylation inhibitor 5-azacytidine reactivates the fetal genes. 5-Azacytidine is utilized for therapy in patients with thalassemia and sickle cell disease. Indeed, it causes accumulation of fetal hemoglobin in erythrocytes. The same effect is observed during clinical application of the DNMT1 inhibitor decitabine [73].
Another important epigenetic mechanism that determines transcriptional activity of a gene is modification of histones within chromatin. The transcriptionally active chromatin configuration is maintained first of all by a high level of histone acetylation [74], which is catalyzed by P300/CBP and the other histone acetylases. On the contrary, histone deacetylases are important for gene repression. Activation of the β-globin genes is associated with the formation of the corresponding chromatin context specific for different stages of ontogenesis. Acetylation of regulatory elements occurs in a clear stage-specific manner [24]. In fetal liver erythroblasts, activating histone modifications (pan acetylation of histone H3 and its dimethylation at K4) are predominantly located in a region of the embryonic gene ε and the fetal genes Gγ and Aγ. In bone marrow erythroblasts, pan acetylation of H3 and its dimethylation at K4 is observed in a segment containing the adult β-globin genes δ and β, whereas the fetal genes Gγ and Aγ virtually lack these activating modifications. In chromatin, LCR-activating modifications are present at all stage of ontogenesis [75-78]. During early stages of erythroid differentiation, HS4 in LCR is perhaps a platform for assembly of histone acetyltransferase complexes and a nucleation point for extension of the acetylated domain [79]. Stage-specific repression of fetal genes is associated with active deacetylation of histones in a corresponding part of the locus.
Classical histone deacetylase inhibitors (butyrate and trichostatin) similarly to DNA methyltransferase inhibitors can induce significant derepression of the fetal genes. Histone deacetylase inhibitors are successfully applied for therapy of β-thalassemia and sickle cell disease [80, 81]. However, the mechanism of histone deacetylase inhibitor action in vivo is not limited to a direct effect on regulatory elements of the β-globin gene domain. Histone deacetylases modulate expression of transcription factors and signal proteins that guide primitive and definitive erythropoiesis [82, 83]. For this reason, inducing transcription of the fetal genes by inhibitors may be mediated by a complex network of interacting mechanisms.
Histone methylation can determine both the active state of genes and their inactivation [2, 84]. Repression of β-globin genes in nonerythroid tissues and perhaps in stem cells is mediated by the histone methyltransferase G9a, which provides accumulation of the modified form of histone H3K9me2. This modification is not only distributed in transcriptional units, but also in the LCR, thus forming several chromatin domains [85]. The nucleation center of the repressive chromatin domain is the HS2 enhancer, which is a part of the LCR. HS2-bound repressor complex MafK-Bach1 recruits histone methyltransferase G9a to the enhancer. In erythroblast precursors and nonerythroid cells, methyltransferase forms a large G9a-dependent domain H3K9me2, which comprises the entire β-globin gene locus [86]. During erythroid differentiation, Bach1 leaves the complex and it is degraded via the ubiquitin-proteasome pathway [87]. According to certain data, repressive modification H3K9me2 also plays a role in providing the stage-specific expression pattern. Inhibition analysis suggests participation of histone methyltransferase G9a/KMT1C and repressive modification H3K9me2 in the repression of fetal genes in adult erythroblasts [88]. Activation of fetal gene expression upon the action of an inhibitor correlates with the absence of H3K9me2 within the entire domain, appearance of LDB1 on promoters of the fetal genes, and formation of a contact between the LCR and promoters of the fetal genes [7].
According to other data, dimethylation of H3 (H3K9me2) does not determine selection of the stage-specific transcriptional state, as it is present in promoters of adult and embryonic genes regardless of their activity. The presence of H3K27me2 modification, which recruits a Polycomb repressor complex in the fetal gene promoter of adult erythroblasts, is crucial for stage-specific repression. In chromatin of adult erythroid cells, promoters of active adult genes carry both repressive modification H3K9me2 and activating modification H3K4me3, whereas H3K27me2 modification is absent [89].
Maintenance of the DNA methylation the pattern and spread of repressive histone modifications are interconnected and interdependent processes. Histone acetylation within chromatin impedes DNA methylation. In some cases, DNA methylation and MBP (methyl-binding proteins) recruit corepressors possessing a SET domain and catalyzing H3K9 methylation. On the other hand, repressive histone modifications recruit DNA methyltransferases. Thus, mechanisms with positive feedback regulation enhance regulatory signal [90].
Arginine methyltransferases (PRMTs) play an important role in the regulation of switching. H4 histone methylation at arginine 3 (H4R3Me2s) catalyzed by arginine methyltransferase PRMT5 targets DNMT3A DNA methyltransferase to the fetal globin gene promoters [91]. This modification is associated with repression. The complex of DNA methyltransferase and arginine methyltransferase on promoters of embryonic genes is additionally bound by NuRD and the methyltransferase Suv4-20h1, which methylates histones H3 and H4. On the other hand, PRMT1 activity through interaction with partner proteins induces chromatin acetylation (Lys9/Lys14C) within promoters of the adult genes. This provides advantage to acetylated promoters of the adult genes in competition for a common enhancer [92]. Thus, PRMT participates in expression switching by repressing fetal and activating adult genes at the same time.
Chromatin remodeling factors play a pivotal role in regulation of stage-specific expression of the globin genes. One of these factors is NuRD (nucleosome remodeling and deacetylase), which is one of the best-studied ones. The complex comprises six proteins, including histone deacetylases 1/2, ATP-dependent helicase, which modulates accessibility of chromatin to transcription factors, and MBD2/MBD3. NuRD can act both as repressor and activator of gene expression [93]. Binding to a promoter of the fetal gene γA and recruiting histone deacetylases to methylated DNA, GATA1/FOG-1/NuRD provides its repression in adult erythroid cells [94]. Recruitment of NuRD to the repression complex is also performed by transcription factors BCL11A, LRF, and TR2/TR4 [95]. The same complex is associated with active transcription of adult genes. The activator role of NuRD in a complex with IKAROS and P-TEFb may be determined by participation of the complex in transcription elongation [96].
The SWI/SNF complex is an ATP-dependent chromatin remodeling factor and it activates transcription though destabilization of DNA–histone interaction within nucleosomes. SWI/SNF is a part of the so-called PYR complex that participates in expression switching in mice. The complex is only present in adult erythroid cells. In addition to SWI/SNF, it comprises NuRD, IKAROS, and the transcription factor PYR [97, 98]. Transcription factor PYR binds to a pyrimidine-rich 250-bp-long sequence located between the stage-specific subdomains [99]. It is believed that PYR associates with chromatin at the moment of transcription switching. Thus, NuRD provides repression of fetal genes, whereas SWI/SNF opens the adult subdomain for binding of activating transcription factors [97, 98].
SPATIAL ORGANIZATION OF CHROMATIN AS AN EPIGENETIC FACTOR OF
STAGE-SPECIFIC EXPRESSION SWITCHING IN THE β-GLOBIN GENE
DOMAIN
In recent years, evidence has appeared indicating that mechanisms directing differential gene expression in specialized cells may act by setting the spatial organization of the genome, which provides the possibility for direct contacts between the enhancer and promoters of activated genes. Works on the mouse β-globin gene domain played an important role in establishing this concept. Utilizing fixation of chromosome conformation [100], it was shown that in erythroid cells producing globins, the LCR of the globin gene domain is located in the vicinity (perhaps forming direct contact) of active genes [101, 102]. Hence, in fetal erythroid cells, DNA fragments are looped that separate the LCR and promoters of the fetal genes, whereas in adult erythroid cells, DNA fragments are looped that separate LCR and promoters of the adult genes. Further studies have shown that such arrangement is rather universal and can be seen in other vertebrates [13, 103]. Mechanisms maintaining alternative spatial arrangements of the domain are virtually mechanisms of globin gene expression switching. Indeed, it was shown that manual formation of a chromatin loop that approaches the LCR to the fetal globin genes in adult erythroblasts is sufficient for activation of transcription of these genes [14, 35]. Hence, the number of cells producing fetal globins grows significantly, whereas duration of transcription pulses is not changed. This suggest that in this situation reactivation of genes that were switched off by epigenetic mechanisms occurs rather than increase in transcription activity of already active genes. This phenomenon is not specific for the fetal genes. Manual formation of a chromatin loop that brings the LCR to the adult globin genes in cells expressing the fetal genes results in activation of transcription of the adult genes [104]. Mechanisms establishing and maintaining function-dependent spatial organization of the genome are being extensively studied. Recently, a new inducer of fetal gene expression was discovered – SIRT1 deacetylase. SIRT1 not only represses transcription of suppressors of fetal genes BCL11A, KLF1, HDAC1, and HDAC2, but it also enhances binding of RNA-polymerase II to a promoter, increases H4K16Ac copy number in chromatin, and directly participates in formation of contacts between regulatory elements by binding to fetal gene promoters and LCR. However, the key role in bringing LCR to globin gene promoters is played by specific transcription factors. Virtually simultaneously it found that transcription factors KLF1 and GATA1 and cofactor GATA1 FOG1 participate in formation of the loop [105, 106]. Somewhat later, it was shown that important regulators of hematopoiesis, TAL1 [107] and BCL11A [52], participate in formation of a contact between LCR and promoters of fetal and embryonic genes. TAL1 binds to regulatory elements of the β-globin gene domain in a complex containing GATA-1, LMO2, and Ldb1. All these transcription factors bind to both LCR and globin gene promoters. However, the exact mechanism providing the interaction remained obscure. Rather long ago an assumption was made that interaction between the regulatory elements may be assured by homodimerization of DNA-binding transcription factors [108]. Functional analysis has demonstrated that the key role in providing interaction between the regulatory elements and loop formation is played by LDB1 protein. Highly conserved protein LDB1 (LIM domain-binding factor 1), which is essential for erythroid differentiation, does not possess any DNA-binding or enzymatic activity. Nevertheless, it is capable of binding to LMO2, GATA1, and TAL1 and of dimerization in vitro. The LDB1/GATA1/TAL1/LMO2 complex binds to the LCR and to the globin gene promoters. LMO2 mediates association of this complex with transcription factors GATA1 and TAL1, which in turn interact with E-box/GATA motifs that are frequent in regulatory elements of erythroid-specific genes, in particular in the β-globin gene domain. LDB1 dimerization assures colocation of the LCR and the β-globin gene promoters. Selection of interacting promoter and subsequent activation of the β-globin genes is determined by differential stage-specific LDB1 binding to promoters of the fetal or adult genes [109, 110]. It was shown for several erythroid-specific genes that LDB1 and an architectural protein CTCF together participate in formation of a chromatin loop that puts enhancer and promoter in close proximity. However, neither Mediator nor cohesin are required for formation of the loop within the β-globin gene domain [111].
Formation of specific epigenetic context during differentiation of adult erythroid cells precedes LDB1 binding and formation of contacts between the genomic elements through dimerization of this transcription factor. So, inhibition of G9a methyltransferase, which methylates histone H3 (H3K9me2), activates fetal genes and represses adult genes in adult erythroid cells. Changes in the expression pattern are accompanied by the loss of H3K9me2 in the chromatin locus, appearance of LDB1 on fetal gene promoters, and de novo formation of contact between fetal gene promoters and the LCR. These observations suggest that G9a methyltransferase binding and shaping the epigenetic landscape precedes the loop formation in the sequence of regulatory events [7].
Activation and repression of the fetal globin genes is accompanied by bringing together regulatory elements apart from promoters and enhancers. So, in fetal erythroblasts, a genomic element separating stage-specific subdomains (HBBP1) contacts with domain-flanking DNase I hypersensitivity sites HS5 and 3′HS1 (figure). Transcription switching is accompanied by loss of HBBP1 contacts with the domain-flanking genomic elements and emergence of HBBP1 contact with embryonic globin gene. It is believed that this is how the fetal genes are isolated from the enhancer. HBBP1 deletion leads to significant increase in expression activity of the fetal genes and by its impact it is virtually identical to reactivation of the fetal genes upon removal of suppressor BCL11A [112].
Fundamental aspects of the mechanisms of stage-specific expression switching are of clear interest in terms of studying common features of transcription regulation. Stage-specific expression of the β-globin genes is ensured by a complex network of interacting factors and divergent mechanisms. These include promoter competition for common enhancer, binding of specific activators and repressors of transcription, DNA methylation, and establishment of a complex stage-specific histone context within chromatin that alters spatial organization of the domain during ontogenesis. It cannot be ruled out that selection of transcriptional state of stage-specific genes occurs simultaneously with selection of either a primitive or definitive erythropoiesis pathway much before beginning of active expression of globin genes. However, the complexity and multicomponent nature of the regulatory systems of expression switching does not ensure its sustainability. Utilization of a low molecular weight inhibitor affecting a single component of the hierarchical regulatory network may disrupt the entire multilevel system that provides stage-specific repression and activation [113]. On the other hand, direct triggering of transcription of a repressed gene may be easily achieved via bypassing any mechanisms by manual approaching of an enhancer to a promoter.
Studying the mechanisms of repression of fetal genes has one important practical aspect. In the USA, treatment of 3.5 million patients with severe forms of anemia requires over 14 million units of red blood cells donated each year [114] (~$1.5 billions). Reactivation of fetal β-globin genes allows partial compensation for deficiency of the adult β-globin and amelioration of the condition of patients with β-thalassemia and sickle cell disease. For this reason, studies on the mechanisms of β-globin gene switching and developing inducers for derepression of the fetal genes are necessary for finding new approaches in drug therapy of anemia.
Acknowledgments
This work was supported by the Russian Science Foundation (grant No. 14-24-00022).
REFERENCES
1.Stamatoyannopoulos, G. (2005) Control of globin
gene expression during development and erythroid differentiation,
Exp. Hematol., 33, 259-271.
2.Wozniak, R. J., and Bresnick, E. H. (2008)
Epigenetic control of complex loci during erythropoiesis, Curr. Top.
Dev. Biol., 82, 55-83.
3.Jagannathan-Bogdan, M., and Zon, L. I. (2013)
Hematopoiesis, Development, 140, 2463-2467.
4.Palis, J. (2014) Primitive and definitive
erythropoiesis in mammals, Frontiers Physiol., 5,
3.
5.Vandekerckhove, J., Courtois, G., Coulon, S.,
Ribeil, J., and Hermine, O. (2009) Regulation of erythropoiesis, in
Disorders of Erythropoiesis, Erythrocytes and Iron Metabolism
(Beaumont, C., and Brugnara, C., eds.) European School of Haematology,
pp. 44-86.
6.Sankaran, V. G., Xu, J., and Orkin, S. H. (2010)
Advances in the understanding of haemoglobin switching, Br. J.
Haematol., 149, 181-194.
7.Krivega, I., Byrnes, C., de Vasconcellos, J. F.,
Lee, Y. T., Kaushal, M., Dean, A., and Miller, J. L. (2015) Inhibition
of G9a methyltransferase stimulates fetal hemoglobin production by
facilitating LCR/gamma-globin looping, Blood, 126,
665-672.
8.Cao, A., and Moi, P. (2002) Regulation of the
globin genes, Pediatr. Res., 51, 415-421.
9.Razin, S. V., Ulianov, S. V., Ioudinkova, E. S.,
Gushchanskaya, E. S., Gavrilov, A. A., and Iarovaia, O. V. (2012)
Domains of alpha- and beta-globin genes in the context of the
structural-functional organization of the eukaryotic genome,
Biochemistry (Moscow), 77, 1409-1423.
10.Ginder, G. D. (2015) Epigenetic regulation of
fetal globin gene expression in adult erythroid cells, Transl.
Res., 165, 115-125.
11.Lee, W. S., McColl, B., Maksimovic, J., and
Vadolas, J. (2017) Epigenetic interplay at the beta-globin locus,
Biochim. Biophys. Acta, 1860, 393-404.
12.Palstra, R. J., de Laat, W., and Grosveld, F.
(2008) Beta-globin regulation and long-range interactions, Adv.
Genetics, 61, 107-142.
13.Noordermeer, D., and de Laat, W. (2008) Joining
the loops: beta-globin gene regulation, IUBMB Life,
60, 824-833.
14.Breda, L., Motta, I., Lourenco, S., Gemmo, C.,
Deng, W., Rupon, J. W., Abdulmalik, O. Y., Manwani, D., Blobel, G. A.,
and Rivella, S. (2016) Forced chromatin looping raises fetal hemoglobin
in adult sickle cells to higher levels than pharmacologic inducers,
Blood, 128, 1139-1143.
15.Sankaran, V. G., and Orkin, S. H. (2013) The
switch from fetal to adult hemoglobin, Cold Spring Harb. Perspect.
Med., 3, a011643.
16.Faruqi, M. (2013) Blood disorders: epigenetically
enhancing haemoglobin production, Nat. Rev. Drug Discov.,
12, 262-263.
17.Recillas-Targa, F., and Razin, S. V. (2001)
Chromatin domains and regulation of gene expression: familiar and
enigmatic clusters of chicken globin genes, Crit. Rev. Eukaryot.
Gene Expr., 11, 227-242.
18.Razin, S. V., Farrell, C. M., and Recillas-Targa,
F. (2003) Genomic domains and regulatory elements operating at the
domain level, Int. Rev. Cytol., 226, 63-125.
19.Burgess-Beusse, B., Farrell, C., Gaszner, M.,
Litt, M., Mutskov, V., Recillas-Targa, F., Simpson, M., West, A., and
Felsenfeld, G. (2002) The insulation of genes from external enhancers
and silencing chromatin, Proc. Natl. Acad. Sci. USA,
99, 16433-16437.
20.Hanscombe, O., Whyatt, D., Fraser, P.,
Yannoutsos, N., Greaves, D., Dillon, N., and Grosveld, F. (1991)
Importance of globin gene order for correct developmental expression,
Genes Dev., 5, 1387-1394.
21.Talbot, D., Collis, P., Antoniou, M., Vidal, M.,
Grosveld, F., and Greaves, D. R. (1989) A dominant control region from
the human beta-globin locus conferring integration site-independent
gene expression, Nature, 338, 352-355.
22.Forrester, W. C., Epner, E., Driscoll, M. C.,
Enver, T., Brice, M., Papayannopoulou, T., and Groudine, M. (1990) A
deletion of the human beta-globin locus activation region causes a
major alteration in chromatin structure and replication across the
entire beta-globin locus, Genes Dev., 4,
1637-1649.
23.Fraser, P., Pruzina, S., Antoniou, M., and
Grosveld, F. (1993) Each hypersensitive site of the human beta-globin
locus control region confers a different developmental pattern of
expression on the globin genes, Genes Dev., 7,
106-113.
24.Yin, W., Barkess, G., Fang, X., Xiang, P., Cao,
H., Stamatoyannopoulos, G., and Li, Q. (2007) Histone acetylation at
the human beta-globin locus changes with developmental age,
Blood, 110, 4101-4107.
25.Plant, K. E., Routledge, S. J., and Proudfoot, N.
J. (2001) Intergenic transcription in the human beta-globin gene
cluster, Mol. Cell. Biol., 21, 6507-6514.
26.Miles, J., Mitchell, J. A., Chakalova, L.,
Goyenechea, B., Osborne, C. S., O’Neill, L., Tanimoto, K., Engel,
J. D., and Fraser, P. (2007) Intergenic transcription, cell-cycle and
the developmentally regulated epigenetic profile of the human
beta-globin locus, PloS One, 2, e630.
27.Gribnau, J., Diderich, K., Pruzina, S.,
Calzolari, R., and Fraser, P. (2000) Intergenic transcription and
developmental remodeling of chromatin subdomains in the human
beta-globin locus, Mol. Cell, 5, 377-386.
28.Haussecker, D., and Proudfoot, N. J. (2005)
Dicer-dependent turnover of intergenic transcripts from the human
beta-globin gene cluster, Mol. Cell. Biol., 25,
9724-9733.
29.Trimborn, T., Gribnau, J., Grosveld, F., and
Fraser, P. (1999) Mechanisms of developmental control of transcription
in the murine alpha- and beta-globin loci, Genes Dev.,
13, 112-124.
30.Gaensler, K. M., Kitamura, M., and Kan, Y. W.
(1993) Germ-line transmission and developmental regulation of a 150-kb
yeast artificial chromosome containing the human beta-globin locus in
transgenic mice, Proc. Natl. Acad. Sci. USA, 90,
11381-11385.
31.Nickol, J. M., and Felsenfeld, G. (1988)
Bidirectional control of the chicken beta- and epsilon-globin genes by
a shared enhancer, Proc. Natl. Acad. Sci. USA, 85,
2548-2552.
32.Bank, A. (2006) Regulation of human fetal
hemoglobin: new players, new complexities, Blood,
107, 435-443.
33.Enver, T., Raich, N., Ebens, A. J.,
Papayannopoulou, T., Costantini, F., and Stamatoyannopoulos, G. (1990)
Developmental regulation of human fetal-to-adult globin gene switching
in transgenic mice, Nature, 344, 309-313.
34.Wijgerde, M., Grosveld, F., and Fraser, P. (1995)
Transcription complex stability and chromatin dynamics in vivo,
Nature, 377, 209-213.
35.Deng, W., Rupon, J. W., Krivega, I., Breda, L.,
Motta, I., Jahn, K. S., Reik, A., Gregory, P. D., Rivella, S., Dean,
A., and Blobel, G. A. (2014) Reactivation of developmentally silenced
globin genes by forced chromatin looping, Cell,
158, 849-860.
36.Li, Q., Blau, C. A., Clegg, C. H., Rohde, A., and
Stamatoyannopoulos, G. (1998) Multiple epsilon-promoter elements
participate in the developmental control of epsilon-globin genes in
transgenic mice, J. Biol. Chem., 273,
17361-17367.
37.Navas, P. A., Peterson, K. R., Li, Q., Skarpidi,
E., Rohde, A., Shaw, S. E., Clegg, C. H., Asano, H., and
Stamatoyannopoulos, G. (1998) Developmental specificity of the
interaction between the locus control region and embryonic or fetal
globin genes in transgenic mice with an HS3 core deletion, Mol.
Cell. Biol., 18, 4188-4196.
38.Navas, P. A., Peterson, K. R., Li, Q., McArthur,
M., and Stamatoyannopoulos, G. (2001) The 5′HS4 core element of
the human beta-globin locus control region is required for high-level
globin gene expression in definitive but not in primitive
erythropoiesis, J. Mol. Biol., 312, 17-26.
39.Fedosyuk, H., and Peterson, K. R. (2007) Deletion
of the human beta-globin LCR 5′HS4 or 5′HS1 differentially
affects beta-like globin gene expression in beta-YAC transgenic mice,
Blood Cells Mol. Dis., 39, 44-55.
40.Ashe, H. L., Monks, J., Wijgerde, M., Fraser, P.,
and Proudfoot, N. J. (1997) Intergenic transcription and transinduction
of the human beta-globin locus, Genes Dev., 11,
2494-2509.
41.Wong, H., Winn, P. J., and Mozziconacci, J.
(2009) A molecular model of chromatin organisation and transcription:
how a multi-RNA polymerase II machine transcribes and remodels the
beta-globin locus during development, BioEssays,
31, 1357-1366.
42.Travers, A. (1999) Chromatin modification by DNA
tracking, Proc. Natl. Acad. Sci. USA, 96,
13634-13637.
43.Chaturvedi, C. P., Somasundaram, B., Singh, K.,
Carpenedo, R. L., Stanford, W. L., Dilworth, F. J., and Brand, M.
(2012) Maintenance of gene silencing by the coordinate action of the
H3K9 methyltransferase G9a/KMT1C and the H3K4 demethylase
Jarid1a/KDM5A, Proc. Natl. Acad. Sci. USA, 109,
18845-18850.
44.Menzel, S., Garner, C., Gut, I., Matsuda, F.,
Yamaguchi, M., Heath, S., Foglio, M., Zelenika, D., Boland, A., Rooks,
H., Best, S., Spector, T. D., Farrall, M., Lathrop, M., and Thein, S.
L. (2007) A QTL influencing F cell production maps to a gene encoding a
zinc-finger protein on chromosome 2p15, Nature Genet.,
39, 1197-1199.
45.Nuinoon, M., Makarasara, W., Mushiroda, T.,
Setianingsih, I., Wahidiyat, P. A., Sripichai, O., Kumasaka, N.,
Takahashi, A., Svasti, S., Munkongdee, T., Mahasirimongkol, S.,
Peerapittayamongkol, C., Viprakasit, V., Kamatani, N., Winichagoon, P.,
Kubo, M., Nakamura, Y., and Fucharoen, S. (2010) A genome-wide
association identified the common genetic variants influence disease
severity in beta0-thalassemia/hemoglobin E, Hum. Genet.,
127, 303-314.
46.Fell, H. P., Smith, R. G., and Tucker, P. W.
(1986) Molecular analysis of the t(2;14) translocation of childhood
chronic lymphocytic leukemia, Science, 232,
491-494.
47.Nakamura, T., Yamazaki, Y., Saiki, Y., Moriyama,
M., Largaespada, D. A., Jenkins, N. A., and Copeland, N. G. (2000) Evi9
encodes a novel zinc finger protein that physically interacts with
BCL6, a known human B-cell proto-oncogene product, Mol. Cell.
Biol., 20, 3178-3186.
48.Suzuki, T., Shen, H., Akagi, K., Morse, H. C.,
Malley, J. D., Naiman, D. Q., Jenkins, N. A., and Copeland, N. G.
(2002) New genes involved in cancer identified by retroviral tagging,
Nat. Genet., 32, 166-174.
49.Sankaran, V. G., Menne, T. F., Xu, J., Akie, T.
E., Lettre, G., Van Handel, B., Mikkola, H. K., Hirschhorn, J. N.,
Cantor, A. B., and Orkin, S. H. (2008) Human fetal hemoglobin
expression is regulated by the developmental stage-specific repressor
BCL11A, Science, 322, 1839-1842.
50.Sankaran, V. G., Xu, J., Ragoczy, T., Ippolito,
G. C., Walkley, C. R., Maika, S. D., Fujiwara, Y., Ito, M., Groudine,
M., Bender, M. A., Tucker, P. W., and Orkin, S. H. (2009) Developmental
and species-divergent globin switching are driven by BCL11A,
Nature, 460, 1093-1097.
51.Xu, J., Bauer, D. E., Kerenyi, M. A., Vo, T. D.,
Hou, S., Hsu, Y. J., Yao, H., Trowbridge, J. J., Mandel, G., and Orkin,
S. H. (2013) Corepressor-dependent silencing of fetal hemoglobin
expression by BCL11A, Proc. Natl. Acad. Sci. USA,
110, 6518-6523.
52.Xu, J., Sankaran, V. G., Ni, M., Menne, T. F.,
Puram, R. V., Kim, W., and Orkin, S. H. (2010) Transcriptional
silencing of gamma-globin by BCL11A involves long-range interactions
and cooperation with SOX6, Genes Dev., 24,
783-798.
53.Hagiwara, N. (2011) Sox6, jack of all trades: a
versatile regulatory protein in vertebrate development, Dev.
Dynamics, 240, 1311-1321.
54.Dumitriu, B., Patrick, M. R., Petschek, J. P.,
Cherukuri, S., Klingmuller, U., Fox, P. L., and Lefebvre, V. (2006)
Sox6 cell-autonomously stimulates erythroid cell survival,
proliferation, and terminal maturation and is thereby an important
enhancer of definitive erythropoiesis during mouse development,
Blood, 108, 1198-1207.
55.Raich, N., Clegg, C. H., Grofti, J., Romeo, P.
H., and Stamatoyannopoulos, G. (1995) GATA1 and YY1 are developmental
repressors of the human epsilon-globin gene, EMBO J.,
14, 801-809.
56.Alhashem, Y. N., Vinjamur, D. S., Basu, M.,
Klingmuller, U., Gaensler, K. M., and Lloyd, J. A. (2011) Transcription
factors KLF1 and KLF2 positively regulate embryonic and fetal
beta-globin genes through direct promoter binding, J. Biol.
Chem., 286, 24819-24827.
57.Im, H., Grass, J. A., Johnson, K. D., Kim, S. I.,
Boyer, M. E., Imbalzano, A. N., Bieker, J. J., and Bresnick, E. H.
(2005) Chromatin domain activation via GATA-1 utilization of a small
subset of dispersed GATA motifs within a broad chromosomal region,
Proc. Natl. Acad. Sci. USA, 102, 17065-17070.
58.Zhang, W., Kadam, S., Emerson, B. M., and Bieker,
J. J. (2001) Site-specific acetylation by p300 or CREB binding protein
regulates erythroid Kruppel-like factor transcriptional activity via
its interaction with the SWI–SNF complex, Mol. Cell.
Biol., 21, 2413-2422.
59.Filipe, A., Li, Q., Deveaux, S., Godin, I.,
Romeo, P. H., Stamatoyannopoulos, G., and Mignotte, V. (1999)
Regulation of embryonic/fetal globin genes by nuclear hormone
receptors: a novel perspective on hemoglobin switching, EMBO J.,
18, 687-697.
60.Avram, D., Fields, A., Pretty On Top, K.,
Nevrivy, D. J., Ishmael, J. E., and Leid, M. (2000) Isolation of a
novel family of C(2)H(2) zinc finger proteins implicated in
transcriptional repression mediated by chicken ovalbumin upstream
promoter transcription factor (COUP-TF) orphan nuclear receptors, J.
Biol. Chem., 275, 10315-10322.
61.Tanabe, O., Katsuoka, F., Campbell, A. D., Song,
W., Yamamoto, M., Tanimoto, K., and Engel, J. D. (2002) An
embryonic/fetal beta-type globin gene repressor contains a nuclear
receptor TR2/TR4 heterodimer, EMBO J., 21,
3434-3442.
62.Tanabe, O., Shen, Y., Liu, Q., Campbell, A. D.,
Kuroha, T., Yamamoto, M., and Engel, J. D. (2007) The TR2 and TR4
orphan nuclear receptors repress Gata1 transcription, Genes
Dev., 21, 2832-2844.
63.Tanabe, O., McPhee, D., Kobayashi, S., Shen, Y.,
Brandt, W., Jiang, X., Campbell, A. D., Chen, Y. T., Chang, C.,
Yamamoto, M., Tanimoto, K., and Engel, J. D. (2007) Embryonic and fetal
beta-globin gene repression by the orphan nuclear receptors, TR2 and
TR4, EMBO J., 26, 2295-2306.
64.Choi, O. R., and Engel, J. D. (1988)
Developmental regulation of beta-globin gene switching, Cell,
55, 17-26.
65.Zhou, W., Zhao, Q., Sutton, R., Cumming, H.,
Wang, X., Cerruti, L., Hall, M., Wu, R., Cunningham, J. M., and Jane,
S. M. (2004) The role of p22 NF-E4 in human globin gene switching,
J. Biol. Chem., 279, 26227-26232.
66.Verdel, A., Vavasseur, A., Le Gorrec, M., and
Touat-Todeschini, L. (2009) Common themes in siRNA-mediated epigenetic
silencing pathways, Int. J. Dev. Biol., 53,
245-257.
67.Allis, C. D., and Jenuwein, T. (2016) The
molecular hallmarks of epigenetic control, Nat. Rev. Genet.,
17, 487-500.
68.Hendrich, B., and Bird, A. (1998) Identification
and characterization of a family of mammalian methyl-CpG binding
proteins, Mol. Cell. Biol., 18, 6538-6547.
69.Khavari, D. A., Sen, G. L., and Rinn, J. L.
(2010) DNA methylation and epigenetic control of cellular
differentiation, Cell. Cycle, 9, 3880-3883.
70.Mabaera, R., Richardson, C. A., Johnson, K., Hsu,
M., Fiering, S., and Lowrey, C. H. (2007) Developmental- and
differentiation-specific patterns of human gamma- and beta-globin
promoter DNA methylation, Blood, 110,
1343-1352.
71.Ng, H. H., Zhang, Y., Hendrich, B., Johnson, C.
A., Turner, B. M., Erdjument-Bromage, H., Tempst, P., Reinberg, D., and
Bird, A. (1999) MBD2 is a transcriptional repressor belonging to the
MeCP1 histone deacetylase complex, Nat. Genet.,
23, 58-61.
72.Feng, Q., and Zhang, Y. (2001) The MeCP1 complex
represses transcription through preferential binding, remodeling, and
deacetylating methylated nucleosomes, Genes Dev.,
15, 827-832.
73.Olivieri, N. F., Saunthararajah, Y.,
Thayalasuthan, V., Kwiatkowski, J., Ware, R. E., Kuypers, F. A., Kim,
H. Y., Trachtenberg, F. L., and Vichinsky, E. P. (2011) A pilot study
of subcutaneous decitabine in beta-thalassemia intermedia,
Blood, 118, 2708-2711.
74.Verdin, E., and Ott, M. (2015) 50 years of
protein acetylation: from gene regulation to epigenetics, metabolism
and beyond, Nat. Rev. Mol. Cell Biol., 16,
258-264.
75.Hebbes, T. R., Clayton, A. L., Thorne, A. W., and
Crane-Robinson, C. (1994) Core histone hyperacetylation co-maps with
generalized DNase I sensitivity in the chicken beta-globin chromosomal
domain, EMBO J., 13, 1823-1830.
76.Forsberg, E. C., Downs, K. M., Christensen, H.
M., Im, H., Nuzzi, P. A., and Bresnick, E. H. (2000) Developmentally
dynamic histone acetylation pattern of a tissue-specific chromatin
domain, Proc. Natl. Acad. Sci. USA, 97,
14494-14499.
77.Fu, X. H., Liu, D. P., and Liang, C. C. (2002)
Chromatin structure and transcriptional regulation of the beta-globin
locus, Exp. Cell Res., 278, 1-11.
78.Bulger, M., Schubeler, D., Bender, M. A.,
Hamilton, J., Farrell, C. M., Hardison, R. C., and Groudine, M. (2003)
A complex chromatin landscape revealed by patterns of nuclease
sensitivity and histone modification within the mouse beta-globin
locus, Mol. Cell. Biol., 23, 5234-5244.
79.Litt, M. D., Simpson, M., Recillas-Targa, F.,
Prioleau, M. N., and Felsenfeld, G. (2001) Transitions in histone
acetylation reveal boundaries of three separately regulated neighboring
loci, EMBO J., 20, 2224-2235.
80.Ley, T. J., DeSimone, J., Anagnou, N. P., Keller,
G. H., Humphries, R. K., Turner, P. H., Young, N. S., Keller, P., and
Nienhuis, A. W. (1982) 5-Azacytidine selectively increases gamma-globin
synthesis in a patient with beta+ thalassemia, N. Engl.
J. Med., 307, 1469-1475.
81.Migliaccio, A. R., Rotili, D., Nebbioso, A.,
Atweh, G., and Mai, A. (2008) Histone deacetylase inhibitors and
hemoglobin F induction in beta-thalassemia, Int. J. Biochem. Cell
Biol., 40, 2341-2347.
82.Boosalis, M. S., Bandyopadhyay, R., Bresnick, E.
H., Pace, B. S., Van DeMark, K., Zhang, B., Faller, D. V., and Perrine,
S. P. (2001) Short-chain fatty acid derivatives stimulate cell
proliferation and induce STAT-5 activation, Blood,
97, 3259-3267.
83.Sangerman, J., Lee, M. S., Yao, X., Oteng, E.,
Hsiao, C. H., Li, W., Zein, S., Ofori-Acquah, S. F., and Pace, B. S.
(2006) Mechanism for fetal hemoglobin induction by histone deacetylase
inhibitors involves gamma-globin activation by CREB1 and ATF-2,
Blood, 108, 3590-3599.
84.Jenuwein, T., and Allis, C. D. (2001) Translating
the histone code, Science, 293, 1074-1080.
85.Litt, M. D., Simpson, M., Gaszner, M., Allis, C.
D., and Felsenfeld, G. (2001) Correlation between histone lysine
methylation and developmental changes at the chicken beta-globin locus,
Science, 293, 2453-2455.
86.Hosey, A. M., Chaturvedi, C. P., and Brand, M.
(2010) Crosstalk between histone modifications maintains the
developmental pattern of gene expression on a tissue-specific locus,
Epigenetics, 5, 273-281.
87.Brand, M., Ranish, J. A., Kummer, N. T.,
Hamilton, J., Igarashi, K., Francastel, C., Chi, T. H., Crabtree, G.
R., Aebersold, R., and Groudine, M. (2004) Dynamic changes in
transcription factor complexes during erythroid differentiation
revealed by quantitative proteomics, Nat. Struct. Mol. Biol.,
11, 73-80.
88.Renneville, A., Van Galen, P., Canver, M. C.,
McConkey, M., Krill-Burger, J. M., Dorfman, D. M., Holson, E. B.,
Bernstein, B. E., Orkin, S. H., Bauer, D. E., and Ebert, B. L. (2015)
EHMT1 and EHMT2 inhibition induces fetal hemoglobin expression,
Blood, 126, 1930-1939.
89.Chaturvedi, C. P., Hosey, A. M., Palii, C.,
Perez-Iratxeta, C., Nakatani, Y., Ranish, J. A., Dilworth, F. J., and
Brand, M. (2009) Dual role for the methyltransferase G9a in the
maintenance of beta-globin gene transcription in adult erythroid cells,
Proc. Natl. Acad. Sci. USA, 106, 18303-18308.
90.Vaissiere, T., Sawan, C., and Herceg, Z. (2008)
Epigenetic interplay between histone modifications and DNA methylation
in gene silencing, Mutat. Res., 659, 40-48.
91.Zhao, Q., Rank, G., Tan, Y. T., Li, H., Moritz,
R. L., Simpson, R. J., Cerruti, L., Curtis, D. J., Patel, D. J., Allis,
C. D., Cunningham, J. M., and Jane, S. M. (2009) PRMT5-mediated
methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA
methylation in gene silencing, Nat. Struct. Mol. Biol.,
16, 304-311.
92.Li, X., Hu, X., Patel, B., Zhou, Z., Liang, S.,
Ybarra, R., Qiu, Y., Felsenfeld, G., Bungert, J., and Huang, S. (2010)
H4R3 methylation facilitates beta-globin transcription by regulating
histone acetyltransferase binding and H3 acetylation, Blood,
115, 2028-2037.
93.Miccio, A., Wang, Y., Hong, W., Gregory, G. D.,
Wang, H., Yu, X., Choi, J. K., Shelat, S., Tong, W., Poncz, M., and
Blobel, G. A. (2010) NuRD mediates activating and repressive functions
of GATA-1 and FOG-1 during blood development, EMBO J.,
29, 442-456.
94.Hong, W., Nakazawa, M., Chen, Y. Y., Kori, R.,
Vakoc, C. R., Rakowski, C., and Blobel, G. A. (2005) FOG-1 recruits the
NuRD repressor complex to mediate transcriptional repression by GATA-1,
EMBO J., 24, 2367-2378.
95.Cui, S., Kolodziej, K. E., Obara, N.,
Amaral-Psarris, A., Demmers, J., Shi, L., Engel, J. D., Grosveld, F.,
Strouboulis, J., and Tanabe, O. (2011) Nuclear receptors TR2 and TR4
recruit multiple epigenetic transcriptional corepressors that associate
specifically with the embryonic beta-type globin promoters in
differentiated adult erythroid cells, Mol. Cell. Biol.,
31, 3298-3311.
96.Bottardi, S., Mavoungou, L., and Milot, E. (2015)
IKAROS: a multifunctional regulator of the polymerase II transcription
cycle, Trends Genet., 31, 500-508.
97.O’Neill, D., Yang, J., Erdjument-Bromage,
H., Bornschlegel, K., Tempst, P., and Bank, A. (1999) Tissue-specific
and developmental stage-specific DNA binding by a mammalian SWI/SNF
complex associated with human fetal-to-adult globin gene switching,
Proc. Natl. Acad. Sci. USA, 96, 349-354.
98.O’Neill, D. W., Schoetz, S. S., Lopez, R.
A., Castle, M., Rabinowitz, L., Shor, E., Krawchuk, D., Goll, M. G.,
Renz, M., Seelig, H. P., Han, S., Seong, R. H., Park, S. D., Agalioti,
T., Munshi, N., Thanos, D., Erdjument-Bromage, H., Tempst, P., and
Bank, A. (2000) An IKAROS-containing chromatin-remodeling complex in
adult-type erythroid cells, Mol. Cell. Biol., 20,
7572-7582.
99.O’Neill, D., Bornschlegel, K., Flamm, M.,
Castle, M., and Bank, A. (1991) A DNA-binding factor in adult
hematopoietic cells interacts with a pyrimidine-rich domain upstream
from the human delta-globin gene, Proc. Natl. Acad. Sci. USA,
88, 8953-8957.
100.Dekker, J., Rippe, K., Dekker, M., and
Kleckner, N. (2002) Capturing chromosome conformation, Science,
295, 1306-1311.
101.Tolhuis, B., Palstra, R. J., Splinter, E.,
Grosveld, F., and de Laat, W. (2002) Looping and interaction between
hypersensitive sites in the active beta-globin locus, Mol. Cell,
10, 1453-1465.
102.Palstra, R. J., Tolhuis, B., Splinter, E.,
Nijmeijer, R., Grosveld, F., and de Laat, W. (2003) The beta-globin
nuclear compartment in development and erythroid differentiation,
Nat. Genet., 35, 190-194.
103.Ulianov, S. V., Galitsyna, A. A., Flyamer, I.
M., Golov, A. K., Khrameeva, E. E., Imakaev, M. V., Abdennur, N. A.,
Gelfand, M. S., Gavrilov, A. A., and Razin, S. V. (2017) Activation of
the alpha-globin gene expression correlates with dramatic upregulation
of nearby non-globin genes and changes in local and large-scale
chromatin spatial structure, Epigenetics Chromatin,
10, 35.
104.Morgan, S. L., Mariano, N. C., Bermudez, A.,
Arruda, N. L., Wu, F., Luo, Y., Shankar, G., Jia, L., Chen, H., Hu, J.
F., Hoffman, A. R., Huang, C. C., Pitteri, S. J., and Wang, K. C.
(2017) Manipulation of nuclear architecture through CRISPR-mediated
chromosomal looping, Nat. Commun., 8, 15993.
105.Drissen, R., Palstra, R. J., Gillemans, N.,
Splinter, E., Grosveld, F., Philipsen, S., and de Laat, W. (2004) The
active spatial organization of the beta-globin locus requires the
transcription factor EKLF, Genes Dev., 18,
2485-2490.
106.Vakoc, C. R., Letting, D. L., Gheldof, N.,
Sawado, T., Bender, M. A., Groudine, M., Weiss, M. J., Dekker, J., and
Blobel, G. A. (2005) Proximity among distant regulatory elements at the
beta-globin locus requires GATA-1 and FOG-1, Mol. Cell,
17, 453-462.
107.Yun, W. J., Kim, Y. W., Kang, Y., Lee, J.,
Dean, A., and Kim, A. (2014) The hematopoietic regulator TAL1 is
required for chromatin looping between the beta-globin LCR and human
gamma-globin genes to activate transcription, Nucleic Acids
Res., 42, 4283-4293.
108.Van der Vliet, P. C., and Verrijzer, C. P.
(1993) Bending of DNA by transcription factors, BioEssays,
15, 25-32.
109.Song, S. H., Hou, C., and Dean, A. (2007) A
positive role for NLI/Ldb1 in long-range beta-globin locus control
region function, Mol. Cell, 28, 810-822.
110.Kiefer, C. M., Lee, J., Hou, C., Dale, R. K.,
Lee, Y. T., Meier, E. R., Miller, J. L., and Dean, A. (2011) Distinct
Ldb1/NLI complexes orchestrate gamma-globin repression and reactivation
through ETO2 in human adult erythroid cells, Blood,
118, 6200-6208.
111.Krivega, I., and Dean, A. (2017) LDB1-mediated
enhancer looping can be established independent of mediator and
cohesin, Nucleic Acids Res., 45, 8255-8268.
112.Huang, P., Keller, C. A., Giardine, B., Grevet,
J. D., Davies, J. O. J., Hughes, J. R., Kurita, R., Nakamura, Y.,
Hardison, R. C., and Blobel, G. A. (2017) Comparative analysis of
three-dimensional chromosomal architecture identifies a novel fetal
hemoglobin regulatory element, Genes Dev., 31,
1704-1713.
113.Fard, A. D., Hosseini, S. A., Shahjahani, M.,
Salari, F., and Jaseb, K. (2013) Evaluation of novel fetal hemoglobin
inducer drugs in treatment of beta-hemoglobinopathy disorders, Int.
J. Hematol. Oncol. Stem Cell Res., 7, 47-54.
114.Ansari, S., and Szallasi, A. (2012) Blood
management by transfusion triggers: when less is more, Blood
Transf., 10, 28-33.