REVIEW: Changes in Retinal Glial Cells with Age and during Development of Age-Related Macular Degeneration
D. V. Telegina1, O. S. Kozhevnikova1, and N. G. Kolosova1,a*
1Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, 630090 Novosibirsk, Russia* To whom correspondence should be addressed.
Received March 27, 2018; Revision received May 8, 2018
Age is the major risk factor in the age-related macular degeneration (AMD) which is a complex multifactor neurodegenerative disease of the retina and the main cause of irreversible vision loss in people over 60 years old. The major role in AMD pathogenesis belongs to structure-functional changes in the retinal pigment epithelium cells, while the onset and progression of AMD are commonly believed to be caused by the immune system dysfunctions. The role of retinal glial cells (Muller cells, astrocytes, and microglia) in AMD pathogenesis is studied much less. These cells maintain neurons and retinal vessels through the synthesis of neurotrophic and angiogenic factors, as well as perform supporting, separating, trophic, secretory, and immune functions. It is known that retinal glia experiences morphological and functional changes with age. Age-related impairments in the functional activity of glial cells are closely related to the changes in the expression of trophic factors that affect the status of all cell types in the retina. In this review, we summarized available literature data on the role of retinal macro- and microglia and on the contribution of these cells to AMD pathogenesis.
KEY WORDS: aging, retina, age-related macular degeneration, astrocytes, Muller cells, microgliaDOI: 10.1134/S000629791809002X
Abbreviations: AMD, age-related macular degeneration; CX3CR1, C-X3-C-motif of chemokine receptor 1; GFAP, glial fibrillar acidic protein; IL, interleukin; RPE, retinal pigment epithelium; TNF, tumor necrosis factor.
Age-related macular degeneration (AMD) is a multifactor
neurodegenerative disease of the retina that is the major cause of
irreversible vision loss in people over 60 years old. Although age is
the main risk factor of AMD, its incidence is also affected by genetic
and ecological factors. According to clinical manifestations, AMD can
occur in the dry or wet form. Dry AMD (~90% of all cases) is
characterized by the formation of drusen on the retina beneath the
macula, pigment redistribution, and defects of the retinal pigment
epithelium (RPE) and choroid capillaries. Wet (exudative) AMD develops
in 10% patients and is characterized by neovascularization and growing
of newly formed vessels under the RPE through the Bruch’s
membrane defects. Pathological permeability of newly formed vessels
leads to macular edema, exudate secretion, bleeding into the vitreous
body and retina and eventually results in vision loss [1, 2].
The incidence of AMD increases with the increasing population age. There is no efficient AMD treatment, partially because of insignificant knowledge of its pathogenesis. Structure-functional changes of the retina in normal aging (loss of melanin granules by the RPE cells, impairments in folding and atrophy of microvilli, increase in the number and density of lipofuscin granules, drusen formation, compaction of the Bruch’s membrane, sclerosis of choroid capillary walls, and narrowing of capillary lumen) are similar to changes observed at the early stages of AMD. These changes underlie the AMD pathogenesis but do not always lead to the disease development [3, 4]. The development of AMD and other neurodegenerative diseases is essentially contributed by age-related impairments in the immune system functions and associated changes in homeostasis. AMD-affected retina is characterized by an increased immune reactivity (i.e., tissue ability to develop immune response to any antigenic stimulus) due to the formation of drusen – amorphous deposits between the RPE cells and external collagen zone of the Bruch’s membrane mainly consisting of lipofuscin [5]. Pathological changes in the RPE and choroid result in the death of retinal neurons that, in its turn, triggers neuroinflammation. The integrity and homeostasis of neurons in the retina, as well in the entire central nervous system, are maintained in part by the glial cells [6]. Glial cells comprise only a small portion of the retina, but their influence on neurons, vascular cells, and other types of cells is very important. Using experimental models of eye diseases, it has been shown that experimentally induced or pathology-related dysfunctions of the glia lead to homeostasis disturbance and subsequent neuronal damage [7]. Glial cells are activated when the retina is damaged and releases various biologically active molecules aimed at the tissue repair. Chronic activation of the glia is accompanied by an increased release of proinflammatory factors. Retinal glial cells are usually subdivided into macroglia (Muller cells and astrocytes) and microglia with specific morphological, physiological, and antigenic characteristics.
ASTROCYTES
Astrocytes play an important role in the development and functioning of the retina: they perform the neurotrophic function, provide a mechanical support for damaged neurons, regulate the microenvironment, ensure optimal functioning of retinal neurons and vessels, and participate in the formation and maintenance of the blood-retinal barrier [7]. Astrocytes express on their plasma membrane a large number of potassium channels thereby contributing to the removal of excessive extracellular K+ during active synaptic transmission and preventing depolarization and overexcitation of neurons. Moreover, astrocytes modulate synaptic plasticity of neurons through regulating extracellular concentrations of neurotransmitters [8].
The bodies of astrocytes are located within the layer of nervous fibers, where they perform supplementary functions required for the development of the retinal vascular network (figure, panels (a) and (b), II) [9]. During the embryonic development of the retina, astrocytes migrate from the optic nerve to the growing blood vessels. During the retina maturation in the postnatal period, there is a correlation between vascularization and spatial distribution of astrocytes, which are absent from the vessel-free zones of the retina [10]. Astrocytes and Muller cells are involved in the regulation of local blood flow, i.e., its response to changes in the activity of neurons [11].
a) Changes in the state of retinal glial cell during AMD development. Central panel, healthy retina (II); left panel, classical variant of AMD development (I) including glia activation and migration into the inner retinal layers; right panel, AMD development on the background of immune system failure (III) with pronounced gliosis of Muller cells and astrocytes in the absence of adequate migration of macrophage cells. b) Changes in the retinal glia during AMD development in OXYS rats on the background of immune system failure. Immunohistochemical staining of the microglia (Iba1+ cells) (I); astrocytes and Muller cells (GFAP+ and Vimentin+ cells, respectively) (II) in the retina of Wistar rats without pathological changes; activated microglia (CD68+ cells) (III); migration of CD68+ macrophage cells (IV); gliosis of astrocytes (V) and Muller cells (VI) in the retina of OXYS rats during development of retinopathy (according to Telegina et al. [39], unpublished data). Scale, 50 μm; RPE, retinal pigment epithelium; GL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; PhL, photosensory layer
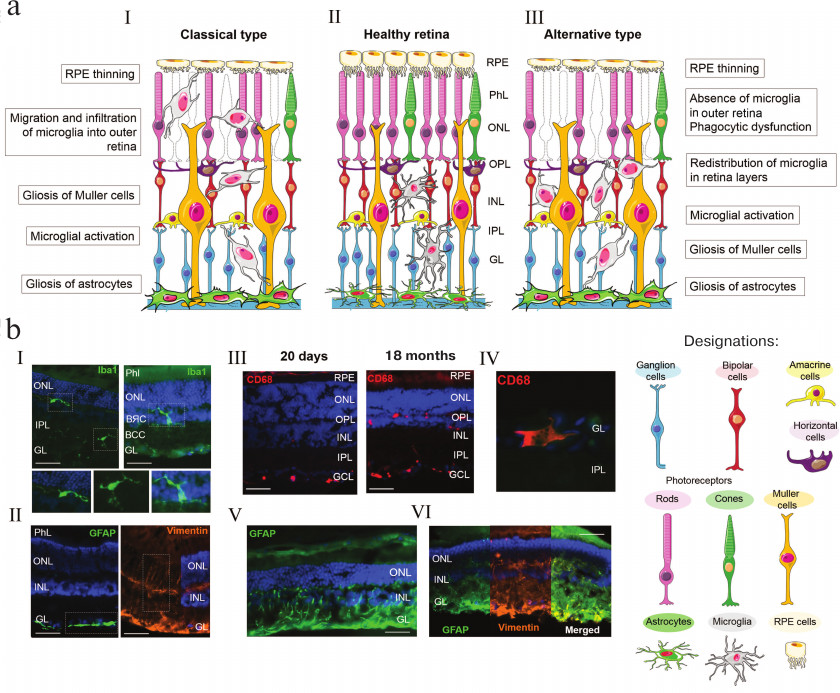
Astrogliosis (reactive astrocytosis) is an evolutionary conserved defense mechanism that involves activation of thousands of genes in a stress stimulus-dependent manner. Activation of astrocytes and astrogliosis accompany the majority of neurodegenerative diseases of the brain and retina. The typical signs of astrogliosis are sharp increase of the astrocyte content (hyperplasia/proliferation), increase in the number and length of astroglial cell processes, cell body hypertrophy, migration and upregulated expression of cytoskeleton components, such as glial fibrillar acidic protein (GFAP), vimentin, and nestin [12]. Trauma or disease induce secretion by astrocytes of proteins capable of disturbing the integrity of the retinal-brain barrier. These proteins upregulate expression of various genes encoding cytokines, chemokines, and elements of the complement cascade, thereby promoting degeneration of the retina [10, 13]. Activated retinal astrocytes express increased levels of matrix metalloproteinase 9 (MMP9) and urokinase type plasminogen activator (uPA). This has a negative effect on the extracellular matrix associated with the internal limiting membrane and promotes the loss of the retinal ganglion cells through apoptosis [14]. In wet AMD, diabetic retinopathy, and retinopathy of prematurity, astrocytes and Muller cells in the mature retina are involved in pathological neovascularization by expressing proangiogenic factors in response to pathogenic stimuli [7]. Analysis of postmortem retinal samples from AMD patients by electron microscopy showed a large number of reactive and hypertrophic astrocytes that phagocytized dead cells in the ganglion cell layer. Together with Muller cells, these astrocytes formed “glial membranes” situated between the vitreous body and the internal limiting membrane [15].
MULLER CELLS
Muller cells are radial glial cells of the retina and have a bipolar morphology characteristic of the radial glia. Muller cells comprise 90% of all retinal glial cells. Muller cells originate from pluripotent precursors [6]. Although this process is studied insufficiently, it is known to involve activation of the Notch, Rax, and Janus-activated kinase (Jak) signaling pathways [16-19]. Muller cells are the only type of glial cells whose bodies are located within the inner nuclear layer. Their processes penetrate all layers of the retina thereby promoting contacts between the neighboring neurons and participating in the formation of the external and internal limiting membranes (figure, panels (a) and (b), II) [7]. It was shown that apoptosis of neurons and degeneration of the retina during the early postnatal development in mice occur only at the sites where the death of Muller cells was induced experimentally [20]. Unlike the retinas of bird and amphibians capable of photoreceptor regeneration, the retina of adult mammals has a limited ability for regeneration and is nearly incapable of de novo neurogenesis [21]. A growing body of evidence obtained during the last few years showed that Muller cells are a hidden population of retinal stem cells [22]. A few first attempts have been made to pharmacologically stimulate regeneration and reprogramming of Muller cells into precursor cells capable of differentiating to the neuronal cells of the retina [23].
Muller cells maintain the viability of photoreceptors and neurons. They direct light onto photoreceptors, provide structural stabilization of the retina, and modulate immune and inflammatory responses [24]. They perform a trophic function by supplying neurons and photoreceptors with nutrients, such as glucose, pyruvate, lactate, and amino acids [6]. Directly or indirectly, Muller cells maintain homeostasis of neurons by promoting production of trophic factors, antioxidants, cytokines, and growth factors, such as brain-derived neurotrophic factor (BDNF), basic fibroblast growth factor (bFGF), ciliary neurotrophic factor (CNTF), insulin-like growth factor 1 (IGF1), glial neurotrophic factor (GDNF), leukemia-inhibiting factor (LIF), neurotrophin 3 (NT3), and pigment epithelium-derived factor (PEDF) [25, 26]. Muller cells also express transporters and enzymes that capture and utilize neurotransmitters [27]; they can protect neurons against oxidative stress via increasing secretion of antioxidants (glutathione, ferroxidase, and heme oxygenase) [28]. Muller cells maintain the homeostasis of retina microenvironment by regulating the extracellular medium ionic composition. Highly active neurons of the retina secret K+ and water into the extracellular space, thereby promoting the influx of Na+ and Ca2+ into the cells. Muller cells restore the physiological values of pH and ion concentrations by expressing on the plasma membrane potassium ion channels (in particular, Kir4.1) that deport K+ ions from the regions with high K+ concentration into the regions with low or stable K+ concentration, such as subretinal space, vitreous body, and blood vessels [24]. Aquaporins (e.g., aquaporin 4) regulating water content in the retina function in a similar manner [29].
Activated Muller cells can either exhibit a neuroprotective effect or contribute to degenerative changes, thus inhibiting repair of the retinal neurons [12, 30].
GLIOSIS
Almost all pathogenic stimuli are able to activate retinal macroglia (astrocytes and Muller cells) and induce gliosis, i.e., reactive changes aimed at protection of the retina against further damages and maintenance of homeostasis by increasing expression of cytoprotective factors, limiting retina remodeling, and restoring the balance between neuromediators, ions, and water [24]. Gliosis can be induced in the retina by degenerative processes, mechanical injury, inflammation, and/or aging [31]. The degree of gliosis-specific physiological, biochemical, and morphological changes depends on the damage severity. Unlike reactive gliosis, proliferative gliosis can accelerate neurodegeneration in chronic diseases of the retina, such as AMD, glaucoma, diabetic retinopathy, and pigmented retinitis, via direct and indirectly damage of neurons and vessels [4].
The specific feature of reactive gliosis is upregulated expression of intermediate filaments, such as vimentin, nestin, and GFAP [7]. The regulation of GFAP expression is so sensitive that it could be used as an index of retinal stress and damage, as well as activation of Muller cells [32]. Among the retinal cells, Muller cells are the main subjects of gliosis that develops through the following stages: cell hypertrophy, loss of functionality, neuroprotection, inflammation, proliferation, and remodeling [33]. The majority of retinal diseases are associated with gliosis of Muller cells and astrocytes [3]. Gliosis of Muller cells becomes massive or proliferative with the damage of the blood-retinal barrier [28]. During proliferative gliosis, Muller cells can contribute to the death of neurons by promoting the synthesis and secretion of tumor necrosis factor (TNF), monocytic chemotactic type 1 protein (MCP-1 or Ccl-2), interleukins, interferon, intercellular adhesion molecule 1 (CD54), and nitric oxide (NO). Excessive synthesis of NO leads to the activation of free-radical processes and protein nitrosylation, which are toxic for neurons [30].
Changes in the astrocyte morphology and biochemistry in the retina during reactive gliosis are similar to those in Muller cells and include increase in the expression of intermediate filament GFAP and vimentin (figure, panels (a) and (b), V and VI) [32, 34]. Similar to Muller cells undergoing gliosis, reactive astroglia can expresses neurotrophic factors supporting cell viability or molecules inhibiting axon regeneration and repair via promoting neurotoxicity or secondary damage of adjacent neurons and glial cells [35]. AMD is characterized by a large amount of hypertrophic reactive astrocytes phagocytizing dead ganglion cells resulted from necrosis or apoptosis [36]. On the other hand, reactive gliosis is associated with the regulation of enzymatic and non-enzymatic antioxidant defense systems that can increase the ability of astrocytes to protect neurons against free radicals [37].
Studies in animal models of retinal diseases allowed to reveal changes in the glia at the early disease stages and to estimate their contribution to the development of neurodegeneration. Thus, the retina detachment in GFAP- and vimentin-deficient mice is characterized by lower levels of gliosis and monocyte migration leading to decreased cell death and prevention of photoreceptor degeneration [35]. Chemical inhibition of gliosis can protect ganglion cells from excitotoxicity [38]. In rds (retinal degradation slow) mice, an increase in the GFAP expression precedes neuronal apoptosis, whereas in rd (retinal degradation) mice, apoptosis of neurons was observed earlier than upregulation of expression of intermediate filaments [39]. Our studies showed that active AMD progression in senescence-accelerated OXYS rats was characterized by increased GFAP levels in the retina and gliosis of Muller cells (figure, panels (a) and (b), V and VI) [40] but also by impairments in autophagy, activated programmed neurosis, and only a few cases of neuronal apoptosis [41]. At the same time, the number of TUNEL+ cells at the preclinical symptom-free stage of the disease was higher and the level of the GFAP protein was lower in OXYS rats than in the control Wistar rats [40, 42].
Hence, retinal macroglia performs numerous functions to provide and maintain normal homeostasis of neurons and neural tissue as a whole. Any impairments in the morphology and functions of Muller cells and astrocytes increase the susceptibility of neurons to stress stimuli causing and/or aggravating the loss of retinal neurons and, resulting, as a consequence, in neurodegeneration development. Gliosis in the retina, on the one hand, is aimed at maintaining the viability of neurons; on the other hand, it can accelerate degenerative changes of neurons and AMD progression.
MICROGLIA
Microglia is the third type of retinal glial cells that includes residential macrophages of the central nervous system expressing many typical macrophage markers, such as CD11b, CD14, CD68, and EMR [43], as well as the calcium-binding protein Iba1, a specific marker of the microglia [44]. The major function of the retinal microglia is participation in the immunity-mediated protective mechanisms. Microglial cells act as phagocytes and, together with perivascular cells, form a network of immune effector cells in the central nervous system [4]. Microglia activation is a common pathophysiological mechanism in the development of various degenerative diseases of the retina and often happens in parallel or before active death of neuronal cells [45]. Like other macrophages, brain and retinal microglial cells can have different phenotypes. Under physiological conditions, they are inactive and have a small body with branched processes. In the retina, microglial cells are located in the plexiform layers. They have a branched structure with small body and long processes capable of penetrating into the nuclear layers. Ramified microglia actively participate in the cell–cell interactions with neurons and macroglia, as well in phagocytosis and homeostasis maintenance in the retina [46]. When responding to injury, microglial cells acquire an ameba-like shape, migrate into the damaged region, and accumulate there. Activated microglial cells have a high ability for phagocytosis and express a number of pro- and anti-inflammatory molecules [47]. It has been shown that activated microglia can be neurotoxic and lead to the degeneration of photoreceptors thereby contributing (in combination with choroid macrophages) to chronic inflammation typical of some age-related diseases, including AMD (figure, panels (a) and (b), III and IV) [48].
Microglia cells sense the environment through surface receptors, including receptors of cytokines, chemokines, complement components, antibodies, etc. (for details, see review [49]). Normally, the activity of microglial cells is also regulated by a number of inhibitory mechanisms. Thus, CX3CL1 chemokine, lectin CD22, and other membrane proteins, including CD200, CD47, and neuronal cell adhesion molecules, attenuate microglia activation [50]. An important role in this process belong to RPE cells, because transforming growing factor beta (TGFβ) secreted by these cells induces release of interleukin 10 (IL-10) from the microglia. IL-10 downregulates expression of antigen-presenting proteins CD80 and CD86 of the main histocompatibility complex II (MHCII) [49]. Analysis of the expression profile of microglial genes revealed that TGFβ could contribute to microglia transition to the anti-inflammatory phenotype [51].
To ensure optimal homeostasis in healthy retina, it is necessary to maintain constant level of neurotrophic factors responsible for the interaction between the microglia and Muller cells. Microglial cells are able to directly activate secretion by Muller cells of some neurotrophic factors, such as glia-derived neurotrophic factor (GDNF), leukemia-inhibiting factor (LIF), ciliary neurotrophic factor (CNTF), nerve growth factor (NGF), neurotrophin 3 (NT-3), and basic fibroblast growth factor (bFGF). These factors provide the preservation of photoreceptors under stress and promote their recovery in the case of damage [49]. Activated Muller cells also secrete diazepam-binding inhibitor (DBI), a ligand of the 18-kDa translocator protein (TSPO). TSPO is expressed in the activated microglia and modulates microglial inflammation and phagocytosis via suppressing excessive activation of the microglia [52, 53]. Moreover, microglial cells activated as a result of retinal damage synthesize and secrete immature forms of neurotrophins (proBDNF and proNGF) that bind to the complex of p75NTR-receptor and sortilin and trigger cell apoptosis [54].
Microglia activation is also controlled by another important mechanism – direct physical interactions between the microglia and other retinal cells [10]. The CX3CL1 protein (fractalkine) is constitutively secreted by healthy retinal neurons and endothelial cells. It binds to the fractalkine receptor, the C-X3-C-motif of the chemokine receptor 1 (CX3CR1), on the microglia cells and thus prevents the neurotoxicity [55]. The CX3CR1 signaling pathway plays an important role in the regulation, activation, and migration of microglial cells [56]. Under physiological conditions, fractalkine inhibits excessive activation of the microglia; however, during inflammation, fractalkine promotes activation of the microglia and astrocytes thereby promoting both neuroprotective and neurotoxic effects of the microglia [57]. The CX3CR1 deficit inhibits the response of the microglia leading to neurotoxicity and degeneration [4].
Recent data show that age-associated alterations in the microglia include changes in the number and location of microglial cells in the retina, morphology of individual cells and their motility (decrease in the migration rate), and expression levels of some genes. The density of microglial cells in the outer and inner plexiform layers slightly increases (by ~19-21%) with aging (figure, panel (b), IV). Note that microglial cells are able to migrate from the inner to the outer plexiform layer and to accumulate in the subretinal space [58]. Such redistribution results in the microglia contact with photoreceptors and RPE cells that induces in them secretion of immune factors specific for pathological AMD phenotypes (figure, panel (a)) [59, 60].
In addition to changes in cell morphology, the activity and reactivity of microglial changes also change with age. Many researchers have shown that during normal aging, microglial cells permanently exist in a state of moderate activation with upregulated expression of the main histocompatibility complex II (MHCII) component CD11b and inflammatory cytokines (IL-1β, TNF, IL-6). This indicates that age-related specific changes in the microglia can promote para-inflammation [61] which increases the probability of neurodegenerative diseases characterized by chronic neuroinflammation.
Activation of microglial cells is a common feature of many retinal diseases, because microglial cells promptly respond to various triggers associated with apoptosis and neuronal degeneration. Although an exact stimulus responsible for the activation of microglial cells in neurodegenerative diseases remains unknown, it has been shown that microglia activation and secretion of chemokines and TNF are preceded by astrogliosis, changes in the physiology of neurons, apoptosis of photoreceptors, and degeneration of the retina [62]. At the first stages of neurodegeneration, the microglia trigger the repair mechanisms, for instance, formation of glial scar [3]. However, excessive or prolonged activation of the microglia can promote chronic inflammation with severe pathological side effects that can lead to the irreversible loss of neurons [45]. Activated microglial cells express a complex of neurotoxic and neurotrophic mediators. It is possible that changes in neurons and/or glial cells enhance the response of microglia that eventually influences the survival of neurons. On the other hand, dysfunction of the microglia and the loss of its protective functions (secretion of trophic factors, antioxidants, and cytokines, as well as phagocytosis) also can lead to neuronal death [63].
Microglia activation in AMD is associated with changes in RPE cells and formation of drusen and amyloid plaques. If at the early AMD stages this activation is aimed at the removal of cell debris, including extracellular deposits of β-amyloid and drusen, pro-inflammatory cytokines induced by the microglia at the later stages contribute to the development of neurodegeneration [3]. Patients with geographic atrophy of the retina have in the inner and outer nuclear layers ameba-like microglial cells that actively phagocytize dead photoreceptor cells [64].
It has been shown in many mouse AMD models that an increase in the number of microglial cells in the subretinal space activates degenerative processes in the retina [65]. For instance, drusen-like deposits were observed in the RPE in CEP mice (mice immunized with carboxyethyl pyrrole (CEP)-conjugated albumin; model of geographic atrophy) [67]. In these animals, proinflammatory macrophages and the microglia are located between the RPE and external segments of photoreceptors. It was shown that the C–C-receptor of chemokine 2 (CCR2) is necessary for migration of immunocompetent cells and associated with the retinal damage, since no retinal damage development was observed in CEP-immunized CCR2–/– mice that also contained no reactive phagocytes [68]. The number of macrophages and microglial cells is increased in the subretinal space of transgenic mice expressing regulator of the complement alternative pathway – complement factor H (CFH) with a single Y402H polymorphism. Mice of this strain display abnormalities similar to those observed in humans with AMD [69]. Accumulation of microglia in the subretinal space is observed in Cx3cr1−/− mice already at the age of 12 months [70]. In Ccl2-deficient mice, activated macrophages are accumulated in the subretinal space [71]. Double knockout Ccl2−/−Cx3cr1−/− mice demonstrate thickening of the Bruch’s membrane, increase in the A2E levels, microglia infiltration, and atrophy of photoreceptor cells [72]. These data show that chemokine deficiency leads to the development of AMD-like symptoms and suggest that dysfunction of phagocytes can play a key role in AMD pathogenesis [73].
Light-induced injury of the retina imitates photooxidative damages in AMD. Light promotes the development of many diseases of the retina, as it has been confirmed by studies in animal models, in which retinal degeneration was accelerated by an increase in the intensity of illumination, since the light-induced damage caused apoptosis more quickly than it occurred in AMD [62]. Despite the differences in the light intensity and duration of exposure, the injury of the retina is accompanied by the loss of photoreceptors and increase in the reactivity and motility of microglial cells. Thus, it was shown that immediately after the exposure to light, the ameba-like microglial cells of the microglia rapidly translocate to the outer nuclear layer and the subretinal space. The density of phagocytizing microglia increased within two day after the exposure and then decreased to the initial level [74]. Interesting, even that ameba-like microglial cells acquired the branched shape 10 days later, they still exhibited the hidden immunophenotypical profile of activation [74].
Therefore, microglial cells rapidly respond to various types of damage and degeneration of the retinal neurons by migrating into the nuclear layers and subretinal space that separates photoreceptors and RPE and lacks innate and adaptive immune cells under physiological conditions [48]. Such changes in the distribution of immune cells in the external retina can lead to pathological changes in the immune environment of photoreceptors and RPE cells. As a result, degenerative changes in RPE cells cause a vicious circle promoting chronic inflammation in the retina and choroid. Age-related changes in the immune system contribute to this destructive process by changing the functions of immune cells [75, 76]. It should be noted that accumulation of microglia and macrophages in the subretinal space has been detected in histological retinal specimens from both AMD patients and elderly people without AMD [59].
Activation of retinal microglial cells, i.e., their transition into the ameba-like state and increased accumulation in the subretinal space, is considered an important component of AMD pathogenesis. However, in the studies on OXYS rats, we found that the symptoms of AMD development are associated with a decrease in the phagocytic function (elimination of dead cells) of the microglia. We compared age-associated changes in the distribution of activated macrophages, microglia, and macroglia in different retinal layers in OXYS and Wistar (control) rats and found no migration of activated macrophages and microglial cells into the photoreceptor-containing layer during manifestation of AMD-like clinical signs of retinopathy or during disease progression in OXYS rats (figure, panel (a)) [40]. Moreover, at the preclinical stage of the disease (at the age of 20 days), the number of activated macrophages in the retina of OXYS rats was lower than in the retina of control Wistar rats. Migration of activated macrophages and microglia into the inner layers of the neuroretina increased in the rats of both strains, but only in the OXYS rats, the levels of these cells were elevated in the ganglion and inner nuclear layers and lowered in the plexiform layers. Based on the data obtained, we assumed that the retinopathy in OXYS rats develops in parallel to phagocytosis dysfunction and decreased elimination of dead cells [40]. We believe that these processes are determined by the imbalance in the immune responses specific for OXYS rats, including inflammation, on the background of accelerated thymus involution and decrease in the activity of the T cell components of the immune system [77]. Indeed, an increasing number of studies indicate that genetically determined dysfunctions of the immune system and the associated changes in the inflammatory homeostasis can contribute to the AMD development and determine specific features of disease progression and organism’s response to therapy [77-80].
Although the paradigm on the secondary role of glia is outdated, the contribution of changes in the glial cells to AMD pathogenesis is still insufficiently studied. However, the functions of these cells determine the interactions of retinal neurons and adequate immune responses to external factors, stress, and aging. Impairments in the phagocytic functions of the microglia lead to accumulation of damaged cells, whereas gliosis of Muller cells and astrocytes decreases the bioavailability of neurotrophic factors. The understanding of age-related changes in the state and reactivity of retinal glial cells is necessary for revealing causes and mechanisms of development of age-related neurodegenerative diseases, including AMD. Moreover, recent publications suggest that glia can be a target for prophylaxis of degenerative changes in the retina and for recovery of retina functions in AMD.
Funding
The work was supported by the Russian Foundation for Basic Research (project no. 18-315-00216) and the Government of the Russian Federation (project no. 2017-220-06-735576001).
REFERENCES
1.Shao, J., Choudhary, M. M., and Schachat, A. P.
(2016) Neovascular age-related macular degeneration, in Retinal
Pharmacotherapeutics, Karger Publishers, Vol. 55, pp. 125-136.
2.Telegina, D. V., Kozhevnikova, O. S., and Kolosova,
N. G. (2017) Molecular mechanisms of cell death in retina during
development of age-related macular degeneration, Adv. Gerontol.,
7, 17-24.
3.Ardeljan, D., and Chan, C. C. (2013) Aging is not a
disease: distinguishing age-related macular degeneration from aging,
Prog. Retin. Eye Res., 37, 68-89.
4.Cuenca, N., Fernandez-Sanchez, L., Campello, L.,
Maneu, V., De la Villa, P., Lax, P., and Pinilla, I. (2014) Cellular
responses following retinal injuries and therapeutic approaches for
neurodegenerative diseases, Prog. Retin. Eye Res., 43,
17-75.
5.Bora, N. S., Matta, B., Lyzogubov, V. V., and Bora,
P. S. (2015) Relationship between the complement system, risk factors
and prediction models in age-related macular degeneration, Mol.
Immunol., 63, 176-183.
6.Goldman, D. (2014) Muller glial cell reprogramming
and retina regeneration, Nat. Rev. Neurosci., 15,
431-442.
7.Coorey, N. J., Shen, W., Chung, S. H., Zhu, L., and
Gillies, M. C. (2012) The role of glia in retinal vascular disease,
Clin. Exp. Optometry, 95, 266-281.
8.Rossi, D. (2015) Astrocyte physiopathology: at the
crossroads of intercellular networking, inflammation and cell death,
Prog. Neurobiol., 130, 86-120.
9.Kur, J., Newman, E. A., and Chan-Ling, T. (2012)
Cellular and physiological mechanisms underlying blood flow regulation
in the retina and choroid in health and disease, Prog. Retin. Eye
Res., 31, 377-406.
10.Vecino, E., Rodriguez, F. D., Ruzafa, N.,
Pereiro, X., and Sharma, S. C. (2016) Glia–neuron interactions in
the mammalian retina, Prog. Retin. Eye Res., 51,
1-40.
11.Newman, E. A. (2015) Glial cell regulation of
neuronal activity and blood flow in the retina by release of
gliotransmitters, Phil. Trans. R. Soc. B, 370,
20140195.
12.De Hoz, R., Rojas, B., Ramirez, A. I., Salazar,
J. J., Gallego, B. I., Trivino, A., and Ramirez, J. M. (2016) Retinal
macroglial responses in health and disease, BioMed. Res. Int.,
2016, 2954721.
13.Kim, J. H., Kim, J. H., Park, J., Lee, S. W.,
Kim, W. J., Yu, Y. S., and Kim, K. W. (2006) Blood–neural
barrier: intercellular communication at glio-vascular interface, J.
Biochem. Mol. Biol., 39, 339-345.
14.Zhang, X., Cheng, M., and Chintala, S. K. (2004)
Kainic acid-mediated upregulation of matrix metalloproteinase-9
promotes retinal degeneration, Invest. Ophthalm. Vis. Sci.,
45, 2374-2383.
15.Ramirez, J. M., Ramirez, A. I., Salazar, J. J.,
de Hoz, R., and Trivino, A. (2001) Changes of astrocytes in retinal
ageing and age-related macular degeneration, Exp. Eye Res.,
73, 601-615.
16.Jadhav, A. P., Cho, S. H., and Cepko, C. L.
(2006) Notch activity permits retinal cells to progress through
multiple progenitor states and acquire a stem cell property, Proc.
Natl. Acad. Sci. USA, 103, 18998-19003.
17.Jadhav, A. P., Roesch, K., and Cepko, C. L.
(2009) Development and neurogenic potential of Muller glial cells in
the vertebrate retina, Prog. Retin. Eye Res., 28,
249-262.
18.Goureau, O., Do Rhee, K., and Yang, X. J. (2004)
Ciliary neurotrophic factor promotes Muller glia differentiation from
the postnatal retinal progenitor pool, Dev. Neurosci.,
26, 359-370.
19.Bhattacharya, S., Das, A. V., Mallya, K. B., and
Ahmad, I. (2008) Ciliary neurotrophic factor-mediated signaling
regulates neuronal versus glial differentiation of retinal stem
cell/progenitors by concentration-dependent recruitment of
mitogens-activated protein kinase and Janus kinase-signal transducer
and activator of transcription pathways in conjunction with Notch
signaling, Stem Cells, 26, 2611-2624.
20.Dubois-Dauphin, M., Poitry-Yamate, C., De Bilbao,
F., Julliard, A. K., Jourdan, F., and Donati, G. (1999) Early postnatal
Muller cell death leads to retinal but not optic nerve degeneration in
NSE-Hu-Bcl-2 transgenic mice, Neuroscience, 95, 9-21.
21.Xia, X., and Ahmad, I. (2016) Unlocking the
neurogenic potential of mammalian Muller glia, Int. J. Stem
Cells, 9, 169-175.
22.Hamon, A., Roger, J. E., Yang, X. J., and Perron,
M. (2016) Muller glial cell-dependent regeneration of the neural
retina: an overview across vertebrate model systems, Develop.
Dynam., 245, 727-738.
23.Webster, M. K., Cooley-Themm, C., Barnett, J. D.,
Bach, H. B., Vainner, J. M., Webster, S. E., and Linn, C. L. (2017)
Evidence of BrdU-positive retinal neurons after application of an
Alpha7 nicotinic acetylcholine receptor agonist, Neuroscience,
346, 437-446.
24.Bringmann, A., and Wiedemann, P. (2012) Muller
glial cells in retinal disease, Ophthalmologica, 227,
1-19.
25.Gallina, D., Todd, L., and Fischer, A. J. (2014)
A comparative analysis of Muller glia-mediated regeneration in the
vertebrate retina, Exp. Eye Res., 123, 121-130.
26.Kolomeyer, A. M., and Zarbin, M. A. (2014)
Trophic factors in the pathogenesis and therapy for retinal
degenerative diseases, Survey Ophthalmol., 59,
134-165.
27.Hurley, J. B., Chertov, A. O., Lindsay, K.,
Giamarco, M., Cleghorn, W., Du, J., and Brockerhoff, S. (2014) Energy
metabolism in the vertebrate retina, in Vertebrate
Photoreceptors, Springer, Japan, pp. 91-137.
28.Reichenbach, A., and Bringmann, A. (2013) New
functions of Muller cells, Glia, 61, 651-678.
29.Schey, K. L., Wang, Z., Wenke, J. L., and Qi, Y.
(2014) Aquaporins in the eye: expression, function, and roles in ocular
disease, Biochim. Biophys. Acta, 1840, 1513-1523.
30.Hippert, C., Graca, A. B., Barber, A. C., West,
E. L., Smith, A. J., Ali, R. R., and Pearson, R. A. (2015) Muller glia
activation in response to inherited retinal degeneration is highly
varied and disease-specific, PLoS One, 10, e0120415.
31.Luna, G., Lewis, G. P., Banna, C. D., Skalli, O.,
and Fisher, S. K. (2010) Expression profiles of nestin and synemin in
reactive astrocytes and Muller cells following retinal injury: a
comparison with glial fibrillar acidic protein and vimentin, Mol.
Vis., 16, 2511-2523.
32.Belecky-Adams, T. L., Chernoff, E. C., Wilson, J.
M., and Dharmarajan, S. (2013) Reactive Muller glia as potential
retinal progenitors, in Neural Stem Cells – New
Perspectives, InTech.
33.Hol, E. M., and Pekny, M. (2015) Glial fibrillary
acidic protein (GFAP) and the astrocyte intermediate filament system in
diseases of the central nervous system, Curr. Opin. Cell Biol.,
32, 121-130.
34.Nakazawa, T., Takeda, M., Lewis, G. P., Cho, K.
S., Jiao, J., Wilhelmsson, U., Fisher, S. K., Pekny, M., Chen, D. F.,
and Miller, J. W. (2007) Attenuated glial reactions and photoreceptor
degeneration after retinal detachment in mice deficient in glial
fibrillary acidic protein and vimentin, Invest. Ophthalmol. Vis.
Sci., 48, 2760-2768.
35.Edwards, M. M., McLeod, D. S., Bhutto, I. A.,
Villalonga, M. B., Seddon, J. M., and Lutty, G. A. (2016) Idiopathic
preretinal glia in aging and age-related macular degeneration, Exp.
Eye Res., 150, 44-61.
36.Verkhratsky, A., Rodriguez, J. J., and Parpura,
V. (2014) Neuroglia in ageing and disease, Cell Tissue Res.,
357, 493-503.
37.Ganesh, B. S., and Chintala, S. K. (2011)
Inhibition of reactive gliosis attenuates excitotoxicity-mediated death
of retinal ganglion cells, PloS One, 6, e18305.
38.Kalloniatis, M., Nivison-Smith, L., Chua, J.,
Acosta, M. L., and Fletcher, E. L. (2016) Using the rd1 mouse to
understand functional and anatomical retinal remodelling and treatment
implications in retinitis pigmentosa: a review, Exp. Eye Res.,
150, 106-121.
39.Telegina, D. V., Kozhevnikova, O. S., Bayborodin,
S. I., and Kolosova, N. G. (2017) Contributions of age-related
alterations of the retinal pigment epithelium and of glia to the
AMD-like pathology in OXYS rats, Sci. Rep., 7, 41533.
40.Kozhevnikova, O. S., Telegina, D. V., Devyatkin,
V. A., and Kolosova, N. G. (2018) Involvement of the autophagic pathway
in the progression of AMD-like retinopathy in senescence-accelerated
OXYS rats, Biogerontology, 19, 223-235.
41.Telegina, D. V., Korbolina, E. E., Ershov, N. I.,
Kolosova, N. G., and Kozhevnikova, O. S. (2015) Identification of
functional networks associated with cell death in the retina of OXYS
rats during the development of retinopathy, Cell Cycle,
14, 3544-3556.
42.Kettenmann, H., Hanisch, U. K., Noda, M., and
Verkhratsky, A. (2011) Physiology of microglia, Physiol. Rev.,
91, 461-553.
43.Ohsawa, K., Imai, Y., Sasaki, Y., and Kohsaka, S.
(2004) Microglia/macrophage-specific protein Iba1 binds to fibrin and
enhances its actin-binding activity, J. Neurochem., 88,
844-856.
44.Langmann, T. (2007) Microglia activation in
retinal degeneration, J. Leukoc. Biol., 81,
1345-1351.
45.Nimmerjahn, A., Kirchhoff, F., and Helmchen, F.
(2005) Resting microglial cells are highly dynamic surveillants of
brain parenchyma in vivo, Science, 308,
1314-1318.
46.Fu, R., Shen, Q., Xu, P., Luo, J. J., and Tang,
Y. (2014) Phagocytosis of microglia in the central nervous system
diseases, Mol. Neurobiol., 49, 1422-1434.
47.Xu, H., Chen, M., and Forrester, J. V. (2009)
Para-inflammation in the aging retina, Progr. Retin. Eye Res.,
28, 348-368.
48.Karlstetter, M., Scholz, R., Rutar, M., Wong, W.
T., Provis, J. M., and Langmann, T. (2015) Retinal microglia: just
bystander or target for therapy? Prog. Retin. Eye Res.,
45, 30-57.
49.Perry, V. H., and Teeling, J. (2013) Microglia
and macrophages of the central nervous system: the contribution of
microglia priming and systemic inflammation to chronic
neurodegeneration, Semin. Immunopathol., 35, 601-612.
50.Paglinawan, R., Malipiero, U., Schlapbach, R.,
Frei, K., Reith, W., and Fontana, A. (2003) TGFβ directs gene
expression of activated microglia to an anti-inflammatory phenotype
strongly focusing on chemokines genes and cell migratory genes,
Glia, 44, 219-231.
51.Karlstetter, M., Nothdurfter, C., Aslanidis, A.,
Moeller, K., Horn, F., Scholz, R., Neumann, H., Weber, B. H.,
Rupprecht, R., and Langmann, T. (2014) Translocator protein
(18 kDa) (TSPO) is expressed in reactive retinal microglia and
modulates microglial inflammation and phagocytosis, J.
Neuroinflamm., 11, 3.
52.Wang, M., Wang, X., Zhao, L., Ma, W., Rodriguez,
I. R., Fariss, R. N., and Wong, W. T. (2014) Macroglia-microglia
interactions via TSPO signaling regulates microglial activation in the
mouse retina, J. Neurosci., 34, 3793-3806.
53.Kimura, A., Namekata, K., Guo, X., Harada, C.,
and Harada, T. (2016) Neuroprotection, growth factors and BDNF-TrkB
signalling in retinal degeneration, Int. J. Mol. Sci.,
17, E1584.
54.Cardona, A. E., Pioro, E. P., Sasse, M. E.,
Kostenko, V., Cardona, S. M., Dijkstra, I. M., Huang, D., Kidd, G.,
Dombrowski, S., Dutta, R., Lee, J. C., Cook, D. N., Jung, S., Lira, S.
A., Littman, D. R., and Ransohoff, R. M. (2006) Control of microglial
neurotoxicity by the fractalkine receptor, Nat. Neurosci.,
9, 917-924.
55.Liang, K. J., Lee, J. E., Wang, Y. D., Ma, W.,
Fontainhas, A. M., Fariss, R. N., and Wong, W. T. (2009) Regulation of
dynamic behavior of retinal microglia by CX3CR1 signaling, Invest.
Ophthalmol. Vis. Sci., 50, 4444-4451.
56.Sheridan, G. K., and Murphy, K. J. (2013)
Neuron–glia crosstalk in health and disease: fractalkine and
CX3CR1 take centre stage, Open Biol., 3, 130181.
57.Damani, M. R., Zhao, L., Fontainhas, A. M.,
Amaral, J., Fariss, R. N., and Wong, W. T. (2011) Age-related
alterations in the dynamic behavior of microglia, Aging Cells,
10, 263-276.
58.Ma, W., Coon, S., Zhao, L., Fariss, R. N., and
Wong, W. T. (2013) A2E accumulation influences retinal microglial
activation and complement regulation, Neurobiol. Aging,
34, 943-960.
59.Ma, W., and Wong, W. T. (2016) Aging changes in
retinal microglia and their relevance to age-related retinal disease,
Adv. Exp. Med. Biol., 854, 73-78.
60.Medzhitov, R. (2008) Origin and physiological
roles of inflammation, Nature, 454, 428-435.
61.Karlstetter, M., Ebert, S., and Langmann, T.
(2010) Microglia in the healthy and degenerating retina: insights from
novel mouse models, Immunobiology, 215, 685-691.
62.Polazzi, E., and Monti, B. (2010) Microglia and
neuroprotection: from in vitro studies to therapeutic
applications, Progr. Neurobiol., 92, 293-315.
63.Gupta, N., Brown, K. E., and Milam, A. H. (2003)
Activated microglia in human retinitis pigmentosa, late-onset retinal
degeneration, and age-related macular degeneration, Exp. Eye
Res., 76, 463-471.
64.Luhmann, U. F., Lange, C. A., Robbie, S., Munro,
P. M., Cowing, J. A., Armer, H. E., Luong, V., Carvalho, L. S.,
MacLaren, R. E., Fitzke, F. W., Bainbridge, J. W., and Ali, R. R.
(2012) Differential modulation of retinal degeneration by Ccl2 and
Cx3cr1 chemokine signaling, PLoS One, 7, e35551.
65.Hollyfield, J. G., Bonilha, V. L., Rayborn, M.
E., Yang, X., Shadrach, K. G., Lu, L., Ufret, R. L., Salomon, R. G.,
and Perez, V. L. (2008) Oxidative damage-induced inflammation initiates
age-related macular degeneration, Nat. Med., 14,
194-198.
66.Hollyfield, J. G., Perez, V. L., and Salomon, R.
G. (2010) A hapten generated from an oxidation fragment of
docosahexaenoic acid is sufficient to initiate age-related macular
degeneration, Mol. Neurobiol., 41, 290-298.
67.Cruz-Guilloty, F., Saeed, A. M., Echegaray, J.
J., Duffort, S., Ballmick, A., Tan, Y., Betancourt, M., Viteri, E.,
Ramkhellawan, G. C., Ewald, E., Feuer, W., Huang, D., Wen, R., Hong,
L., Wang, H., Laird, J. M., Sene, A., Apte, R. S., Salomon, R. G.,
Hollyfield, J. G., and Perez, V. L. (2013) Infiltration of
proinflammatory m1 macrophages into the external retina precedes damage
in a mouse model of age-related macular degeneration, Int. J.
Inflamm., 2013, 503725.
68.Ufret-Vincenty, R. L., Aredo, B., Liu, X.,
McMahon, A., Chen, P. W., Sun, H., Niederkorn, J. Y., and Kedzierski,
W. (2010) Transgenic mice expressing variants of complement factor H
develop AMD-like retinal findings, Invest. Ophthalmol. Vis.
Sci., 51, 5878-5887.
69.Combadiere, C., Feumi, C., Raoul, W., Keller, N.,
Rodero, M., Pezard, Lavalette, S., Houssier, M., Jonet, L., Picard, E.,
Debre, P., Sirinyan, M., Deterre, P., Ferroukhi, T., Cohen, S. Y.,
Chauvaud, D., Jeanny, J. C., Chemtob, S., Behar-Cohen, F., and
Sennlaub, F. (2007) CX3CR1-dependent subretinal microglia cell
accumulation is associated with cardinal features of age-related
macular degeneration, J. Clin. Invest., 117,
2920-2928.
70.Luhmann, U. F., Robbie, S., Munro, P. M., Barker,
S. E., Duran, Y., Luong, V., Fitzke, F. W., Bainbridge, J. W., Ali, R.
R., and MacLaren, R. E. (2009) The drusen-like phenotype in aging
Ccl2-knockout mice is caused by an accelerated accumulation of swollen
autofluorescent subretinal macrophages, Invest. Ophthalmol. Vis.
Sci., 50, 5934-5943.
71.Chan, C. C., Ross, R. J., Shen, D., Ding, X.,
Majumdar, Z., Bojanowski, C. M., Zhou, M., Salem, N., Jr., Bonner, R.,
and Tuo, J. (2008) Ccl2/Cx3cr1-deficient mice: an animal model for
age-related macular degeneration, Ophthalm. Res., 40,
124-128.
72.Pennesi, M. E., Neuringer, M., and Courtney, R.
J. (2012) Animal models of age-related macular degeneration, Mol.
Aspects Med., 33, 487-509.
73.Santos, A. M., Martin-Oliva, D., Ferrer-Martin,
R. M., Tassi, M., Calvente, R., Sierra, A., Carrasco, M. C.,
Marin-Teva, J. L., Navascues, J., and Cuadros, M. A. (2010) Microglial
response to light-induced photoprotector degeneration in the mouse
retina, J. Comp. Neurol., 518, 477-492.
74.Ma, W., Zhao, L., Fontainhas, A. M., Fariss, R.
N., and Wong, W. T. (2009) Microglia in the mouse retina alter the
structure and function of retinal pigmented epithelial cells: a
potential cellular interaction relevant to AMD, PloS One,
4, e7945.
75.Ma, W., Zhao, L., and Wong, W. T. (2012)
Microglia in the outer retina and their relevance to pathogenesis of
age-related macular degeneration, Adv. Exp. Med. Biol.,
732, 37-42.
76.Kozhevnikova, O. S., Korbolina, E. E., Ershov, N.
I., and Kolosova, N. G. (2013) Rat retinal transcriptome: effects of
aging and AMD-like retinopathy, Cell Cycle, 12,
1745-1761.
77.Perez, V. L., and Caspi, R. R. (2015) Immune
mechanisms in inflammatory and degenerative eye disease, Trends
Immunol., 36, 354-363.
78.Shaw, P. X., Stiles, T., Douglas, C., Ho, D.,
Fan, W., Du, H., and Xiao, X. (2016) Oxidative stress, innate immunity,
and age-related macular degeneration, AIMS Mol. Sci., 3,
196-221.
79.Xu, H., and Chen, M. (2016) Targeting the
complement system for the management of retinal inflammatory and
degenerative diseases, Eur. J. Pharmacol., 787,
94-104.