REVIEW: Anti-amyloid Therapy of Alzheimer’s Disease: Current State and Prospects
S. A. Kozin1, E. P. Barykin1, V. A. Mitkevich1,a*, and A. A. Makarov1,b
1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, 119991 Moscow, Russia* To whom correspondence should be addressed.
Received May 17, 2018; Revision received June 4, 2018
Drug development for the treatment of Alzheimer’s disease (AD) has been for a long time focused on agents that were expected to support endogenous β-amyloid (Aβ) in a monomeric state and destroy soluble Aβ oligomers and insoluble Aβ aggregates. However, this strategy has failed over the last 20 years and was eventually abandoned. In this review, we propose a new approach to the anti-amyloid AD therapy based on the latest achievements in understanding molecular causes of cerebral amyloidosis in AD animal models.
KEY WORDS: Alzheimer’s disease, amyloid-β, isoaspartate, zinc, protein–protein complexes, cerebral β-amyloidosisDOI: 10.1134/S0006297918090079
Abbreviations: AD, Alzheimer’s disease; DMAs, disease-modifying agents; Aβ, β-amyloid; isoAβ, β-amyloid with isomerized Asp7 residue.
ALZHEIMER’S DISEASE: BACKGROUND
Alzheimer’s disease (AD) was for the first time described in 1906 [1]. Currently, it is the most common neurodegenerative pathology affecting more than 44 million people worldwide [2]. It was predicted that by 2050, the number of individuals with AD will increase up to 100 million. Only in Russia, ~1,000,000 patients have been diagnosed with AD [3]. From a psychiatric viewpoint, clinical AD signs are manifested as slowly progressing but steady decline in mental abilities, as well as social and cultural skills, that occurs over 3-10 years starting with inadequate behavior in everyday life (sudden unprovoked aggression, unreasonable emotions, disorientation in space and time, loss of short-term memory, etc.). AD is accompanied with brain tissue degradation that finally results in patient death due to respiratory failure.
Familial AD comprises less than 1% all AD cases and is associated with various mutations [4, 5] resulting in constitutively upregulated expression of β-amyloid (Aβ), a 39-to-43-amino acid peptide with heterogenous C-terminus [6]. The causes of sporadic AD (95% cases) remain unknown; however, it was found that they are closely related to aberrant aggregation of endogenous Aβ. Three major neuromorphological features that can confirm the post mortem diagnosis for all types of AD are (i) extracellular aggregates (amyloid plaques) found in certain brain regions and composed mainly of various Aβ isoforms and metal ions (zinc, copper, and iron), (ii) intracellular neurofibrillary tangles with hyperphosphorylated tau protein as the main component, and (iii) neuronal degeneration [7].
Currently, only symptomatic AD therapy is available that is aimed at overcoming neurotransmitter deficiencies and slowing (on average, for 1-3 years) transition from the state when patients are still capable of tending for their own needs to complete helplessness. There are five drugs approved by the US Food and Drug Administration (FDA) that are currently used in the AD therapy worldwide. They include four cholinesterase inhibitors (Tacrine, Donepezil, Rivastigmine, Galantamine) and one NMDA receptor antagonist (Memantine) [8]. Tacrine was approved by the FDA in 1993, Donepezil in 1996, Rivastigmine in 1998, Galantamine in 2001, and Memantine in 2003. Since 2003, no new anti-AD therapeutic agents have been introduced to the global market; however, the strategies for the AD treatment have been extensively developed based on the knowledge of fundamental mechanisms underlying AD emergence.
PROMISING AGENTS FOR AD THERAPY
At present, 105 potential agents for AD treatment are being investigated in clinical trials, among which 25 undergo phase I trial, 52 – phase II trial, and 28 – phase III trial [9]. Almost all of these trials are sponsored by leading pharmaceutical companies. The majority (70%) of these agents modulate molecular mechanisms underlying AD pathogenesis, i.e., act as disease-modifying agents (DMAs). The remaining 30% are symptom relieving drugs: 14% enhance cognitive functions, 13% act on emotional and behavioral functions, and 3% display a general revitalizing effect unrelated to any biological mechanism [9]. Twelve out of 25 phase I drugs affect aberrant Aβ aggregation; three of them act on tau protein aggregation; nine drugs ameliorate AD symptoms; and for one agent, no mechanism of action was found. Thirty-six out of 52 phase II agents are DMAs, among which 14 are aimed at preventing Aβ aggregation, four act against tau protein aggregation, and one exerts a combined action against both Aβ and tau. Eighteen out of 28 phase III drugs belong to DMAs; three agents improve cognitive functions, whereas seven drugs are aimed at correcting behavioral functions. Among phase III DMAs, 15 agents (including six antibody preparations) target Aβ aggregation, and one acts against tau protein aggregation. Therefore, the majority of currently proposed drug candidates are DMAs mostly represented by antibodies and low-molecular-weight compounds able to prevent aberrant Aβ aggregation via specific binding to endogenous Aβ.
Monoclonal anti-Aβ antibodies currently tested in clinical trials [10, 11] target one of the three epitope classes. Aducanumab [12], Bapineuzumab [13], and GSK933776 [14] recognize Aβ N-terminus; Solanezumab [15] and Crenezumab [16, 17] target the central region of Aβ; Ponezumab [18] binds to the Aβ C-terminus. Gantenerumab [19] recognizes an epitope composed of amino acids located at the N-terminus and the central fragment of Aβ. These antibodies have different binding specificity toward Aβ aggregates. In particular, Aducanumab and Gantenerumab bind mainly to aggregated Aβ, whereas Solanezumab selectively binds to soluble Aβ monomers. In contrast, Bapineuzumab and Crenezumab bind with a high affinity to oligomeric Aβ. X-ray crystallography analysis of antibody complexes with Aβ revealed additional epitopes that could be also considered promising pharmaceutical targets [20]. Depending on the Aβ oligomerization pathway, the structure of Aβ aggregates may vary [21, 22]. However, the primary building unit in any type of aggregates is a U-shaped (hairpin) Aβ molecule. Hydrophobic residues including Phe19, Phe20 and Ile34, connect two strands of the polypeptide chain, while a salt bridge between Asp23 and Lys28 stabilizes the structure [23-26]. So far, Crenezumab is the only antibody targeting the middle portion of the Aβ peptide that can bind to several Aβ aggregate forms and cause their dissociation.
Various short peptides and peptidomimetics are also used for targeted Aβ binding and prevention of its aggregation [27]. However, unlike the monoclonal antibody-binding sites, the Aβ regions responsible for the binding of these compounds have not been precisely identified [28-30]. Propanesulfonic acid derivative Alzhemed (Tramiprosate) [31] is the most studied low-molecular-weight anti-aggregation peptidomimetic used for AD treatment. It has been designed to bind with a high affinity to the HHQK motif (a.a. 13-16) responsible for the Aβ interaction with microglial cells and presumably, for Aβ aggregation [32]. Despite the fact that Alzhemed demonstrated high efficacy in experiments [33], it turned out to be inapplicable as an anti-AD agent [34].
ANTI-AMYLOID THERAPY TARGETING THE 1-16 FRAGMENT OF
Aβ
According to the commonly accepted amyloid hypothesis, AD pathogenesis is triggered by the formation of soluble neurotoxic oligomers from physiologically monomeric Aβ molecules followed by the generation of insoluble polymeric Aβ aggregates eventually accumulated as amyloid plaques [35]. Prevention of cerebral amyloidogenesis via inhibiting pathological Aβ oligomerization (anti-amyloid therapy) is considered the most promising strategy for AD treatment [36]. Dimerization of monomeric Aβ is a prerequisite for aggregate formation. It was found that Aβ dimers are neurotoxic, and their serum levels in AD patients correlate with clinical manifestation of the disease [37, 38]. Therefore, targeted blockade of Aβ dimerization might be the most efficient way for preventing amyloidogenesis. Obviously, Aβ in its natural conformation is not a pathogenic molecule, thereby suggesting that AD pathological cascade is initiated by some additional factors.
The driving forces behind Aβ pathological aggregation remain unknown; however, it was found that a crucial role in this process belongs to zinc ions [39]. Human Aβ binds zinc ion via its metal-binding domain 1-16 (Aβ1-16) [40, 41]. This domain contains the 11-14 motif required for zinc-induced Aβ dimerization [42] resulting in the formation of stable Aβ aggregates with parallel β-sheet arrangement of the monomers [43]. Note that in amyloid plagues, Aβ amino acids 17-42 (Aβ17-42) constitute a hydrophobic core consisting of β-sheets and β-turns, whereas residues 1-16 are located outside this core and do not participate in the amyloid plaque stabilization [44]. However, the hydrophobic fragment Aβ17-42 per se does not form amyloid aggregates in vivo [45, 46]. The metal-binding domain in rat and mouse Aβ contains three amino acid substitutions (Arg5Gly, Tyr10Phe, and His13Arg) that distinguish this protein from other mammalian β-amyloids and are presumably associated with the resistance of these rodents to AD-like neurodegenerative pathologies [47]. Altogether, these facts indicate the key role of the metal-binding domain 1-16 in cerebral amyloidogenesis in AD [41, 48-55].
For the first time, the ability to induce cerebral amyloidogenesis of Aβ aggregates isolated post mortem from AD patients was demonstrated in monkeys injected intracerebrally with homogenates of the autopsied AD patients’ brain [56, 57] (Fig. 1). Later, a series of studies in animal AD models [58-61] found that the molecular agent causing accumulation of pathological amyloid plaques in brain tissues is one of the conformational (or chemically modified) Aβ variants [62-64]. However, the precise molecular nature of this variant remained unknown [65]. It was suggested that it can be Aβ phosphorylated at serine 8 residue, pyroglutamylated at position 3 [66, 67], or non-covalently bound to other biomolecules [35]. Chemically modified Aβ isoforms with altered ability to interact with zinc ions [68] could be of special interest as potential molecular targets and/or biomarkers in AD therapy and diagnostics [48-51, 69-71].
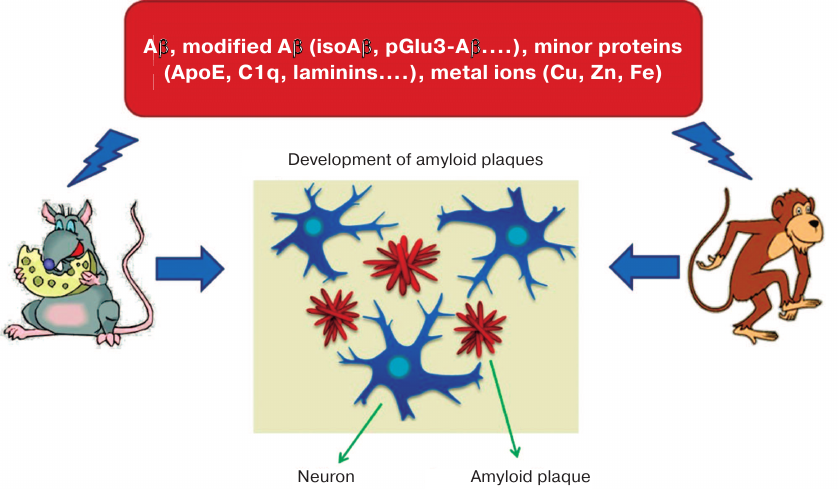
Fig. 1. Animals (monkeys and transgenic mice) intracerebrally injected with brain homogenate from AD patients develop Alzheimer-like neuropathology [56-63]. Amyloid plaques contained Aβ and its modified species, other proteins (ApoE, C1q, laminins, etc.), and metals (Cu, Zn, Fe). Intracerebral administration of Aβ, Aβ dimers, Aβ oligomers, Aβ + Zn/Cu, or Aβ + ApoE fails to induce amyloidosis in the injected animals [64], whereas isoAβ administered intravenously accelerates development of cerebral β-amyloidosis in mice [91].
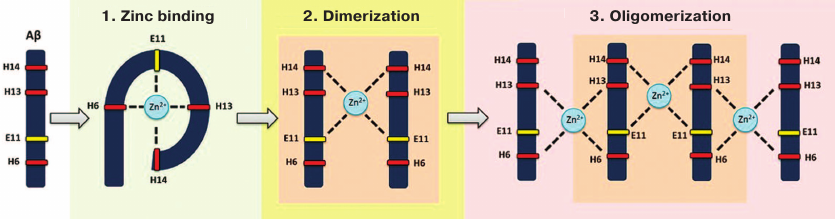
After the Aβ fragment 1-16 was identified as a zinc-binding domain [41, 72-74], its conformation in the absence or presence of zinc ions has been established [75]. It was found that interaction of zinc ions with Aβ proceeds in two stages [76]. First, zinc ions are bound by the side chains of Glu11, His13, and His14 of the prestructured motif 10-15. Next, the side chain of His6 enters the zinc ion coordination sphere, which results in the emergence of ordered compact structure including the entire fragment 1-16 complexed with a single zinc ion. It was determined that the Aβ motif 11-14 (Glu-Val-His-His) not only acts as a primary zinc-ion recognition site [75, 76], but also controls zinc-induced Aβ oligomerization [42, 77, 78], while His13 plays a crucial role in the zinc-induced Aβ aggregation [79]. The spatial structure of the 11-14 motif is very rigid and remains virtually unchanged in both intact and zinc-bound Aβ [75, 80]. The secondary structure of this motif is represented by the left-handed polyproline II helix [81] (left-handed helices are known to be involved in protein–protein interactions [82]). Collectively, these properties (zinc binding, conserved structure, and predisposition to interact with biomolecules) make the motif 11-14 an important structural and functional determinant of Aβ.
Recently, the molecular mechanism for the zinc-dependent oligomerization of human Aβ metal-binding domains was described [83, 84] (Fig. 2). First, a monomeric zinc–metal-binding domain complex is assembled, in which zinc ion is coordinated by His6, Glu11, His13, and His14. Then, the two domains form a dimer with a single zinc ion coordinated by Glu11 and His14 of the interacting domains. This dimerization is followed by the conformational transition of motifs 6-14 in each subunit resulting in the formation of the second zinc-binding site involving His6 and His13, so the dimer becomes a seed for further zinc-dependent oligomerization proceeding in a chain reaction manner. Each domain in the developing oligomer preserves its initial conformation [83]. Such domain structure allows each Aβ molecule to bind two other Aβ molecules through zinc ions. Isomerization of Asp7 plays an important role in this potentially pathogenic process [51], because it significantly increases the ability of Aβ metal-binding domain 1-16 to undergo zinc-dependent oligomerization due to intramolecular conformational changes that provide a steric opportunity for the motif 11-14 to interact with other Aβ molecules [68, 85]. Zinc-induced oligomerization of intact Aβ may be initiated by the formation of zinc-dependent heterodimers between intact and isomerized Aβ molecules via motifs 11-14 of the interacting subunits [85]. Therefore, targeted blockade of this motif should significantly slow down or even prevent zinc-induced Aβ oligomerization and, subsequently, cerebral amyloidogenesis.
Fig. 2. Zinc-induced oligomerization of the Aβ metal-binding domain. Zinc ion binding by Aβ is followed by the zinc-induced formation of Aβ dimer that serves as a seed for zinc-induced Aβ oligomerization.
Development of amyloid plaques in the brain (cerebral amyloidogenesis) is one of the major symptoms of AD pathogenesis. In experimental animals, cerebral amyloidogenesis can be significantly accelerated by injecting brain homogenates from AD patients [86]. More than 50% Aβ molecules in amyloid plaques contain isomerized Asp7 residue (isoAβ molecules) [87, 88]. IsoAβ can spontaneously form from synthetic Aβ peptides [75, 89]. Based on the results of [65, 90], it may be expected that isoAβ acts as a seed of pathological oligomerization and aggregation of physiologically normal endogenous Aβ molecules. Indeed, it was shown that intravenous administration of synthetic isoAβ1-42 results in significantly accelerated cerebral amyloidogenesis in B6C3-Tg(APPswe,PSEN1-dE9)85Dbo/j mice (AD model) [91] known to develop a significant amount of cerebral amyloid plaques at the age of 4 to 6 months. In these mice, accumulation of amyloid plaques correlates with the development of cognitive dysfunctions [92, 93]. It was found that isoAβ1-42 is toxic for neuronal cells [94, 95]. Therefore, isoAβ1-42 is the most plausible AD pathogenic agent, and its appearance in the blood serum (presumably, due to spontaneous protein aging) results in the formation of neurotoxic oligomers of endogenous Aβ [91]. The isoAβ1-42 domain 1-16 in was found to be necessary and sufficient for inducing cerebral amyloidogenesis after its administration to experimental animals [52, 91]. The data on the crucial role of motif 1-16 in Aβ oligomerization [43, 47, 83, 85] and the fact that it represents an independent structural domain in Aβ [41, 44, 75, 80] make the motif 1-16 the major pharmaceutical target in the anti-amyloid AD therapy [55].
α4 NICOTINIC ACETYLCHOLINE RECEPTOR FRAGMENT AS A POTENTIAL
ANTI-AMYLOID DRUG
Considering that zinc-induced oligomerization of intact Aβ may be initiated by the formation of zinc-dependent heterodimers between intact and isomerized Aβ molecules via the motifs 11-14 (Glu11-Val12-His13-His14) of the interacting subunits [85], targeted blockade of this motif should significantly slow down or even prevent zinc-induced Aβ oligomerization and, therefore, cerebral amyloidogenesis. Potential agents capable of specific binding of motif 11-14 include antibodies, low-molecular-weight compounds, peptides, peptidomimetics, and other substances. Passive immunotherapy is widely used in today medicine; for a long time, it has been considered a promising approach to the anti-amyloid therapy in AD. Currently, several antibodies (Bapineuzumab, Gantenerumab, Aducanumab) are available that hinder interactions of the motif 11-14, thereby preventing Aβ aggregation and causing disintegration of already existing amyloid plaques [96]. However, clinical trials demonstrated that these antibodies are toxic and can even aggravate neuronal dysfunction [97].
It is known that Aβ interacts with acetylcholine receptors. The critical role in this interaction belongs to the motif 11-14; although the Aβ-binding site in the receptor has not been identified yet [98]. Bioinformatic analysis showed that the extracellular domain of the nicotinic acetylcholine receptor (α4 subunit) contains a potential interaction partner of Aβ – the fragment His-Ala-Glu-Glu (HAEE) complementary to the Aβ motif 11-14 and conserved in humans, mice, and chicken [99, 100].
It was found that synthetic peptide acetyl-His-Ala-Glu-Glu-NH2 (Ac-HAEE-NH2) specifically binds to the motif 11-14 in the Aβ metal-binding domain 1-16, thereby blocking zinc-induced domain dimerization and hindering aggregation of the full-size Aβ in vitro [99, 100]. Intravenously injected Ac-HAEE-NH2 slowed down cerebral amyloidogenesis in B6C3-Tg(APPswe,PSEN1-dE9)85Dbo/j mice (AD model): average number of amyloid plaques per brain section decreased from 14.2 ± 3.1 (control group) to 5.8 ± 2.1 (treated group) [100]. Based on this parameter, the efficacy of Ac-HAEE-NH2 was significantly higher than that of the anti-amyloid drug Alzhemed (Tramiprosate), one of the best-known candidates for AD treatment [101]. It was found that in TgCRND8 mice, Alzhemed decreased the amount of amyloid plaques by 30% as compared to the control group [31].
INHIBITION OF METAL-DEPENDENT INTERACTIONS OF Aβ ISOFORMS
WITH PARTNER PROTEINS AS A NEW APPROACH TO THE ANTI-AGGREGATION THERAPY
OF AD
We believe that the failure of clinical trials with the anti-amyloid agents developed over the last 20 years is related to the shortcomings in our understanding of molecular mechanism involved in the initiation of pathological aggregation of endogenous Aβ. According to the commonly accepted hypothesis, the primary process triggering pathological AD cascade is the appearance of soluble toxic Aβ oligomers [54] (Fig. 3). It is believed that any molecule of endogenous Aβ can spontaneously adopt the pathological conformation, so that its interaction with other Aβ molecules would result in the formation of dimers and oligomers serving as seeds of pathological amyloid aggregation [65, 86, 90, 102]. Hence, prototypic anti-amyloid agents were expected to maintain Aβ in its monomeric state and/or to destroy existing amyloid plaques [103]. However, the inefficacy of standard anti-aggregation therapy indicates that neurotoxic oligomers do not appear spontaneously, but rather emerge under the influence of certain factors. The data indicating the existence of such amyloidogenesis mechanisms have been obtained during the last few years: (i) cerebral amyloidogenesis is caused by structurally and/or chemically modified Aβ isoforms [52, 64, 91]; (ii) the level of isomerized Asp7-bearing Aβ in patients with AD is elevated [104]; (iii) pathogenic prion-like seeds of Aβ aggregation can spread via peripheral circulation [91, 105]; (iv) interaction with zinc ions is necessary for cerebral amyloidogenesis [39, 106]; (v) metal-mediated interactions of Aβ with neuronal receptors may play a substantial role in neurodegeneration [98, 107-113].
Fig. 3. Classical and isoAβ-dependent models of amyloidogenesis. According to the classical model, amyloid plaque development results from the oligomerization of soluble Aβ with subsequent formation of insoluble Aβ fibrils on the neuronal cell surface [54]. The amyloidogenesis model proposed by us assumes the presence of a pathogenic agent – modified isoAβ peptide, whose percentage content increases with age [104]. IsoAβ induces amyloidogenesis in model mouse strains [52, 91] due to the formation of amyloid matrix, which is an isoAβ–Zn2+–neuronal receptor complex that serves as a seed for developing amyloid aggregates [100].
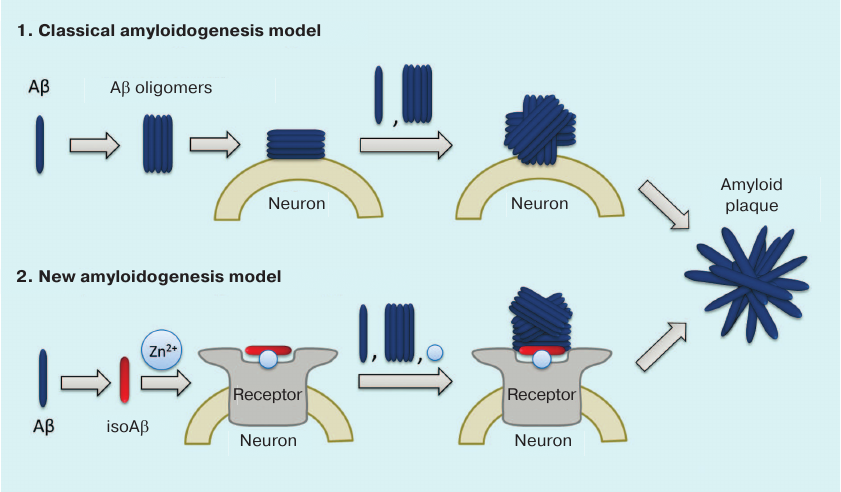
Based on these data, the following molecular mechanism for the initiation of pathological Aβ aggregation in AD was proposed (Fig. 3). The process starts with the emergence of chemically modified peptide species – isoAβ – in the blood serum (e.g., due to spontaneous aging or neurotrauma). Then, isoAβ enters brain tissues and interacts with neuronal cell receptors in a zinc-dependent manner. As a result, isoAβ forms high-affinity zinc-containing complexes with neuronal receptors (amyloid matrices) that serve as seeds of pathological Aβ aggregation. When interacting with the amyloid matrix, endogenous Aβ loses its native conformation and forms aggregates via the zinc-dependent oligomerization mechanism resulting in the formation of insoluble amyloid plaques that exist in a dynamic equilibrium with soluble Aβ oligomers. These oligomers serve as the amyloid matrix on the neuronal cell surface, as well as independent seeds of aggregation in the extracellular space.
Based on the proposed mechanism, we suggest a new strategy for AD anti-amyloid therapy (Fig. 4). Typically, anti-aggregation therapy with monoclonal antibodies is aimed at the destruction of amyloid plaques and Aβ oligomers. The new strategy considers amyloid matrix as the major therapeutic target and implies disruption of zinc-dependent complexes between Aβ and receptors. The efficacy of such therapy was demonstrated in the experiments using peptide HAEE, a competitive inhibitor of interaction between Aβ and α4 nicotinic acetylcholine receptor [100].
Fig. 4. Anti-amyloid strategy for AD therapy based on amyloid matrix destruction. The isoAβ–Zn2+–α4β2 nicotinic acetylcholine receptor (α4β2 nAChR) complex acts as the amyloid matrix and can be destroyed by the exogenous peptide HAEE.
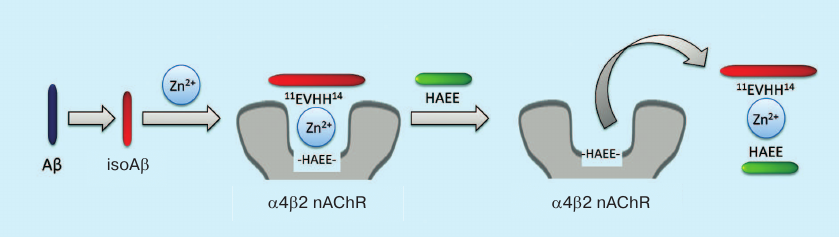
Recent advances in understanding the molecular causes of the emergence of AD pathological conditions have led to the hypothesis on the crucial role of zinc-mediated interactions between Aβ isoforms and neuronal receptors in the AD development. These interactions result in the formation of complexes on the neuronal membrane that serve as an amyloid matrix. Binding of endogenous Aβ to the amyloid matrix results in its transformation into neurotoxic oligomers and amyloid plaques. Amyloid matrix is a conceptually new type of pharmaceutical target for the development of anti-amyloid agents for AD therapy.
Funding
The study was supported by the Russian Science Foundation (project no. 14-24-00100).
REFERENCES
1.Alzheimer, A. (1906) Uber einen eigenartigen
schweren Erkrankungsprozebeta der Hirnrinde, Neurol.
Centralblatt, 23, 1129-1136.
2.Alzheimer’s Association (2014) 2014
Alzheimer’s disease facts and figures, Alzheimer’s
Dementia, 10, e47-e92.
3.Gavrilova, S. I. (2007) Pharmacotherapy of
Alzheimer’s Disease [in Russian], Pul’s, Moscow.
4.Rogaev, E. I., Sherrington, R., Rogaeva, E. A.,
Levesque, G., Ikeda, M., Liang, Y., Chi, H., Lin, C., Holman, K.,
Tsuda, T., Mar, L., Sorbi, S., Nacmias, B., Piacentini, S., Amaducci,
L., Chumakov, I., Cohen, D., Lannfelt, L., Fraser, P. E., Rommens, J.
M., and St. George-Hyslop, P. H. (1995) Familial Alzheimer’s
disease in kindreds with missense mutations in a gene on chromosome 1
related to the Alzheimer’s disease type 3 gene, Nature,
376, 775-778.
5.Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva,
E. A., Levesque, G., Ikeda, M., Chi, H., Lin, C., Li, G., Holman, K.,
Tsuda, T., Mar, L., Foncin, J. F., Bruni, A. C., Montesi, M. P., Sorbi,
S., Rainero, I., Pinessi, L., Nee, L., Chumakov, I., Pollen, D.,
Brookes, A., Sanseau, P., Polinsky, R. J., Wasco, W., Da Silva, H. A.
R., Haines, J. L., Pericak-Vance, M. A., Tanzi, R. E., Roses, A. D.,
Fraser, P. E., Rommens, J. M., and St. George-Hyslop, P. H. (1995)
Cloning of a gene bearing missense mutations in early-onset familial
Alzheimer’s disease, Nature, 375,
754-760.
6.Querfurth, H. W., and LaFerla, F. M. (2010)
Alzheimer’s disease, N. Engl. J. Med., 362,
329-344.
7.Cummings, J. L. (2004) Alzheimer’s disease,
N. Engl. J. Med., 351, 56-67.
8.Cummings, J., Morstorf, T., and Zhong, K. (2014)
Alzheimer’s disease drug-development pipeline: few candidates,
frequent failures, Alzheimer’s Res. Ther.,
6, 37.
9.Cummings, J., Lee, G., Mortsdorf, T., Ritter, A.,
and Zhong, K. (2017) Alzheimer’s disease drug development
pipeline: 2017, Alzheimer’s Dement. (N.Y.),
3, 367-384.
10.Rygiel, K. (2016) Novel strategies for
Alzheimer’s disease treatment: an overview of anti-amyloid beta
monoclonal antibodies, Indian J. Pharmacol., 48,
629-636.
11.Guell-Bosch, J., Montoliu-Gaya, L.,
Esquerda-Canals, G., and Villegas, S. (2016) Abeta immunotherapy for
Alzheimer’s disease: where are we? Neurodegen. Dis.
Manag., 6, 179-181.
12.Sevigny, J., Chiao, P., Bussiere, T., Weinreb, P.
H., Williams, L., Maier, M., Dunstan, R., Salloway, S., Chen, T., Ling,
Y., O’Gorman, J., Qian, F., Arastu, M., Li, M., Chollate, S.,
Brennan, M. S., Quintero-Monzon, O., Scannevin, R. H., Arnold, H. M.,
Engber, T., Rhodes, K., Ferrero, J., Hang, Y., Mikulskis, A., Grimm,
J., Hock, C., Nitsch, R. M., and Sandrock, A. (2016) The antibody
aducanumab reduces Abeta plaques in Alzheimer’s disease,
Nature, 537, 50-56.
13.Feinberg, H., Saldanha, J. W., Diep, L., Goel,
A., Widom, A., Veldman, G. M., Weis, W. I., Schenk, D., and Basi, G. S.
(2014) Crystal structure reveals conservation of amyloid-beta
conformation recognized by 3D6 following humanization to bapineuzumab,
Alzheimers Res. Ther., 6, 31.
14.Leyhe, T., Andreasen, N., Simeoni, M., Reich, A.,
von Arnim, C. A., Tong, X., Yeo, A., Khan, S., Loercher, A., Chalker,
M., Hottenstein, C., Zetterberg, H., Hilpert, J., and Mistry, P. (2014)
Modulation of β-amyloid by a single dose of GSK933776 in patients
with mild Alzheimer’s disease: a phase I study,
Alzheimer’s Res. Ther., 6, 19.
15.Zhao, J., Nussinov, R., and Ma, B. (2017)
Mechanisms of recognition of amyloid-beta (Abeta) monomer, oligomer,
and fibril by homologous antibodies, J. Biol. Chem.,
292, 18325-18343.
16.Adolfsson, O., Pihlgren, M., Toni, N., Varisco,
Y., Buccarello, A. L., Antoniello, K., Lohmann, S., Piorkowska, K.,
Gafner, V., Atwal, J. K., Maloney, J., Chen, M., Gogineni, A., Weimer,
R. M., Mortensen, D. L., Friesenhahn, M., Ho, C., Paul, R., Pfeifer,
A., Muhs, A., and Watts, R. J. (2012) An effector-reduced
anti-beta-amyloid (Abeta) antibody with unique abeta binding properties
promotes neuroprotection and glial engulfment of Abeta, J.
Neurosci., 32, 9677-9689.
17.Ultsch, M., Li, B., Maurer, T., Mathieu, M.,
Adolfsson, O., Muhs, A., Pfeifer, A., Pihlgren, M., Bainbridge, T. W.,
Reichelt, M., Ernst, J. A., Eigenbrot, C., Fuh, G., Atwal, J. K.,
Watts, R. J., and Wang, W. (2016) Structure of crenezumab complex with
Abeta shows loss of beta-hairpin, Sci. Rep., 6,
39374.
18.La Porte, S. L., Bollini, S. S., Lanz, T. A.,
Abdiche, Y. N., Rusnak, A. S., Ho, W. H., Kobayashi, D., Harrabi, O.,
Pappas, D., Mina, E. W., Milici, A. J., Kawabe, T. T., Bales, K., Lin,
J. C., and Pons, J. (2012) Structural basis of C-terminal beta-amyloid
peptide binding by the antibody ponezumab for the treatment of
Alzheimer’s disease, J. Mol. Biol., 421,
525-536.
19.Bohrmann, B., Baumann, K., Benz, J., Gerber, F.,
Huber, W., Knoflach, F., Messer, J., Oroszlan, K., Rauchenberger, R.,
Richter, W. F., Rothe, C., Urban, M., Bardroff, M., Winter, M.,
Nordstedt, C., and Loetscher, H. (2012) Gantenerumab: a novel human
anti-Abeta antibody demonstrates sustained cerebral amyloid-beta
binding and elicits cell-mediated removal of human amyloid-beta, J.
Alzheimer’s Dis., 28, 49-69.
20.Crespi, G. A., Hermans, S. J., Parker, M. W., and
Miles, L. A. (2015) Molecular basis for mid-region amyloid-beta capture
by leading Alzheimer’s disease immunotherapies, Sci Rep.,
5, 9649.
21.Gu, L., Liu, C., Stroud, J. C., Ngo, S., Jiang,
L., and Guo, Z. (2014) Antiparallel triple-strand architecture for
prefibrillar Abeta42 oligomers, J. Biol. Chem.,
289, 27300-27313.
22.Paravastu, A. K., Leapman, R. D., Yau, W. M., and
Tycko, R. (2008) Molecular structural basis for polymorphism in
Alzheimer’s beta-amyloid fibrils, Proc. Natl. Acad. Sci.
USA, 105, 18349-18354.
23.Colvin, M. T., Silvers, R., Ni, Q. Z., Can, T.
V., Sergeyev, I., Rosay, M., Donovan, K. J., Michael, B., Wall, J.,
Linse, S., and Griffin, R. G. (2016) Atomic resolution structure of
monomorphic Aβ42 amyloid fibrils, J. Am. Chem. Soc.,
138, 9663-9674.
24.Tycko, R. (2016) Alzheimer’s disease:
structure of aggregates revealed, Nature, 537,
492-493.
25.Walti, M. A., Ravotti, F., Arai, H., Glabe, C.
G., Wall, J. S., Bockmann, A., Guntert, P., Meier, B. H., and Riek, R.
(2016) Atomic-resolution structure of a disease-relevant Aβ(1-42)
amyloid fibril, Proc. Natl. Acad. Sci. USA, 113,
E4976-E4984.
26.Xiao, Y., Ma, B., McElheny, D., Parthasarathy,
S., Long, F., Hoshi, M., Nussinov, R., and Ishii, Y. (2015) Abeta(1-42)
fibril structure illuminates self-recognition and replication of
amyloid in Alzheimer’s disease, Nat. Struct. Mol. Biol.,
22, 499-505.
27.Qi-Shi, D., Neng-Zhong, X., and Ri-Bo, H. (2015)
Recent development of peptide drugs and advance on theory and
methodology of peptide inhibitor design, Med. Chem.,
11, 235-247.
28.Cho, P. Y., Joshi, G., Johnson, J. A., and
Murphy, R. M. (2014) Transthyretin-derived peptides as beta-amyloid
inhibitors, ACS Chem. Neurosci., 5, 542-551.
29.Parthsarathy, V., McClean, P. L., Holscher, C.,
Taylor, M., Tinker, C., Jones, G., Kolosov, O., Salvati, E., Gregori,
M., Masserini, M., and Allsop, D. (2013) A novel retro-inverso peptide
inhibitor reduces amyloid deposition, oxidation and inflammation and
stimulates neurogenesis in the APPswe/PS1DeltaE9 mouse model of
Alzheimer’s disease, PLoS One, 8,
e54769.
30.Wang, Q., Liang, G., Zhang, M., Zhao, J., Patel,
K., Yu, X., Zhao, C., Ding, B., Zhang, G., Zhou, F., and Zheng, J.
(2014) De novo design of self-assembled hexapeptides as
β-amyloid (Aβ) peptide inhibitors, ACS Chem.
Neurosci., 5, 972-981.
31.Gervais, F., Paquette, J., Morissette, C.,
Krzywkowski, P., Yu, M., Azzi, M., Lacombe, D., Kong, X., Aman, A.,
Laurin, J., Szarek, W. A., and Tremblay, P. (2007) Targeting soluble
Aβ peptide with Tramiprosate for the treatment of brain
amyloidosis, Neurobiol. Aging, 28, 537-547.
32.Giulian, D., Haverkamp, L. J., Yu, J., Karshin,
W., Tom, D., Li, J., Kazanskaia, A., Kirkpatrick, J., and Roher, A. E.
(1998) The HHQK domain of beta-amyloid provides a structural basis for
the immunopathology of Alzheimer’s disease, J. Biol.
Chem., 273, 29719-29726.
33.Gauthier, S., Aisen, P. S., Ferris, S. H.,
Saumier, D., Duong, A., Haine, D., Garceau, D., Suhy, J., Oh, J., Lau,
W., and Sampalis, J. (2009) Effect of tramiprosate in patients with
mild-to-moderate Alzheimer’s disease: exploratory analyses of the
MRI sub-group of the Alphase study, J. Nutr. Health Aging,
13, 550-557.
34.Gauthier, S., Albert, M., Fox, N., Goedert, M.,
Kivipelto, M., Mestre-Ferrandiz, J., and Middleton, L. T. (2016) Why
has therapy development for dementia failed in the last two decades?
Alzheimer’s Dementia, 12, 60-64.
35.Karran, E., Mercken, M., and De Strooper, B.
(2011) The amyloid cascade hypothesis for Alzheimer’s disease: an
appraisal for the development of therapeutics, Nat. Rev. Drug
Discov., 10, 698-712.
36.Schenk, D., Basi, G. S., and Pangalos, M. N.
(2012) Treatment strategies targeting amyloid β-protein, Cold
Spring Harb. Perspect. Med., 2, a006387.
37.Shankar, G., Li, S., Mehta, T., Garcia-Munoz, A.,
Shepardson, N., Smith, I., Brett, F., Farrell, M., Rowan, M., Lemere,
C., Regan, C., Walsh, D., Sabatini, B., and Selkoe, D. (2008)
Amyloid-beta protein dimers isolated directly from Alzheimer’s
brains impair synaptic plasticity and memory, Nat. Med.,
14, 837-842.
38.Villemagne, V. L., Perez, K. A., Pike, K. E.,
Kok, W. M., Rowe, C. C., White, A. R., Bourgeat, P., Salvado, O., Bedo,
J., Hutton, C. A., Faux, N. G., Masters, C. L., and Barnham, K. J.
(2010) Blood-borne amyloid-β dimer correlates with clinical
markers of Alzheimer’s disease, J. Neurosci.,
30, 6315-6322.
39.Friedlich, A. L., Lee, J.-Y., van Groen, T.,
Cherny, R. A., Volitakis, I., Cole, T. B., Palmiter, R. D., Koh, J.-Y.,
and Bush, A. I. (2004) Neuronal zinc exchange with the blood vessel
wall promotes cerebral amyloid angiopathy in an animal model of
Alzheimer’s disease, J. Neurosci., 24,
3453-3459.
40.Faller, P., and Hureau, C. (2009) Bioinorganic
chemistry of copper and zinc ions coordinated to amyloid-beta peptide,
Dalton Trans., 7, 1080-1094.
41.Kozin, S. A., Zirah, S., Rebuffat, S., Hui Bon
Hoa, G., and Debey, P. (2001) Zinc binding to Alzheimer’s
Aβ(1-16) peptide results in stable soluble complex, Biochem.
Biophys. Res. Commun., 285, 959-964.
42.Kozin, S. A., Mezentsev, Y. V., Kulikova, A. A.,
Indeykina, M. I., Golovin, A. V., Ivanov, A. S., Tsvetkov, P. O., and
Makarov, A. A. (2011) Zinc-induced dimerization of the amyloid-β
metal-binding domain 1-16 is mediated by residues 11-14, Mol.
BioSyst., 7, 1053-1055.
43.Miller, Y., Ma, B., and Nussinov, R. (2010) Zinc
ions promote Alzheimer Abeta aggregation via population shift of
polymorphic states, Proc. Natl. Acad. Sci. USA,
107, 9490-9495.
44.Luhrs, T., Ritter, C., Adrian, M., Riek-Loher,
D., Bohrmann, B., Dobeli, H., Schubert, D., and Riek, R. (2005) 3D
structure of Alzheimer’s amyloid-beta(1-42) fibrils, Proc.
Natl. Acad. Sci. USA, 102, 17342-17347.
45.Dulin, F., Leveille, F., Ortega, J. B., Mornon,
J.-P., Buisson, A., Callebaut, I., and Colloc’h, N. (2008) p3
peptide, a truncated form of Aβ devoid of synaptotoxic effect,
does not assemble into soluble oligomers, FEBS Lett.,
582, 1865-1870.
46.Walsh, D., Klyubin, I., Fadeeva, J., Cullen, W.,
Anwyl, R., Wolfe, M., Rowan, M., and Selkoe, D. (2002) Naturally
secreted oligomers of amyloid beta protein potently inhibit hippocampal
long-term potentiation in vivo, Nature,
416, 535-539.
47.Istrate, A. N., Tsvetkov, P. O., Mantsyzov, A.
B., Kulikova, A. A., Kozin, S. A., Makarov, A. A., and Polshakov, V. I.
(2012) NMR solution structure of rat Aβ(1-16): toward
understanding the mechanism of rats’ resistance to
Alzheimer’s disease, Biophys. J., 102,
136-143.
48.Kozin, S. A., and Makarov, A. A. (2015) New
biomarkers and pharmaceutical targets for diagnostics and therapy of
Alzheimer’s disease (molecular determinants of zinc-dependent
β-amyloid oligomerization), Zh. Nevrol. Psikhiatr. im. S. S.
Korsakova, 115, 5-9.
49.Kulikova, A. A., Makarov, A. A., and Kozin, S. A.
(2015) A role of zinc ions and structural β-amyloid polymorphism
in Alzheimer’s disease onset, Mol. Biol. (Moscow),
49, 249-263.
50.Barykin, E. P., Mitkevich, V. A., Kozin, S. A.,
and Makarov, A. A. (2017) Amyloid beta modification: a key to the
sporadic Alzheimer’s disease? Front. Genet.,
8, 58.
51.Kozin, S. A., Mitkevich, V. A., and Makarov, A.
A. (2016) Amyloid-β containing isoaspartate 7 as potential
biomarker and drug target in Alzheimer’s disease, Mendeleev
Commun., 26, 269-275.
52.Kulikova, A. A., Cheglakov, I. B., Kukharsky, M.
S., Ovchinnikov, R. K., Kozin, S. A., and Makarov, A. A. (2016)
Intracerebral injection of metal-binding domain of Abeta comprising the
isomerized Asp7 increases the amyloid burden in transgenic mice,
Neurotox. Res., 29, 551-557.
53.Mattson, M. P. (1995) Untangling the
pathophysio-chemistry of [beta]-amyloid, Nat. Struct. Mol.
Biol., 2, 926-928.
54.Mattson, M. P. (2004) Pathways towards and away
from Alzheimer’s disease, Nature, 430,
631-639.
55.Murray, B., Sharma, B., and Belfort, G. (2017)
N-terminal hypothesis for Alzheimer’s disease, ACS Chem.
Neurosci., 8, 432-434.
56.Baker, H. F., Ridley, R. M., Duchen, L. W., Crow,
T. J., and Bruton, C. J. (1994) Induction of beta (A4)-amyloid in
primates by injection of Alzheimer’s disease brain homogenate.
Comparison with transmission of spongiform encephalopathy, Mol.
Neurobiol., 8, 25-39.
57.Ridley, R. M., Baker, H. F., Windle, C. P., and
Cummings, R. M. (2006) Very long term studies of the seeding of
beta-amyloidosis in primates, J. Neural. Transm.,
113, 1243-1251.
58.Langer, F., Eisele, Y. S., Fritschi, S. K.,
Staufenbiel, M., Walker, L. C., and Jucker, M. (2011) Soluble Abeta
seeds are potent inducers of cerebral beta-amyloid deposition, J.
Neurosci., 31, 14488-14495.
59.Morales, R., Duran-Aniotz, C., Castilla, J.,
Estrada, L. D., and Soto, C. (2012) De novo induction of
amyloid-[beta] deposition in vivo, Mol. Psychiatry,
17, 1347-1353.
60.Rosen, R. F., Fritz, J. J., Dooyema, J., Cintron,
A. F., Hamaguchi, T., Lah, J. J., LeVine, H., 3rd, Jucker, M., and
Walker, L. C. (2012) Exogenous seeding of cerebral beta-amyloid
deposition in betaAPP-transgenic rats, J. Neurochem.,
120, 660-666.
61.Watts, J. C., Giles, K., Grillo, S. K., Lemus,
A., DeArmond, S. J., and Prusiner, S. B. (2011) Bioluminescence imaging
of Aβ deposition in bigenic mouse models of Alzheimer’s
disease, Proc. Natl. Acad. Sci. USA, 108,
2528-2533.
62.Eisele, Y. S., Bolmont, T., Heikenwalder, M.,
Langer, F., Jacobson, L. H., Yan, Z. X., Roth, K., Aguzzi, A.,
Staufenbiel, M., Walker, L. C., and Jucker, M. (2009) Induction of
cerebral beta-amyloidosis: intracerebral versus systemic Abeta
inoculation, Proc. Natl. Acad. Sci. USA, 106,
12926-12931.
63.Eisele, Y. S., Obermuller, U., Heilbronner, G.,
Baumann, F., Kaeser, S. A., Wolburg, H., Walker, L. C., Staufenbiel,
M., Heikenwalder, M., and Jucker, M. (2010) Peripherally applied
Abeta-containing inoculates induce cerebral beta-amyloidosis,
Science, 330, 980-982.
64.Meyer-Luehmann, M., Coomaraswamy, J., Bolmont,
T., Kaeser, S., Schaefer, C., Kilger, E., Neuenschwander, A.,
Abramowski, D., Frey, P., Jaton, A. L., Vigouret, J. M., Paganetti, P.,
Walsh, D. M., Mathews, P. M., Ghiso, J., Staufenbiel, M., Walker, L.
C., and Jucker, M. (2006) Exogenous induction of cerebral
beta-amyloidogenesis is governed by agent and host, Science,
313, 1781-1784.
65.Stohr, J., Watts, J. C., Mensinger, Z. L.,
Oehler, A., Grillo, S. K., DeArmond, S. J., Prusiner, S. B., and Giles,
K. (2012) Purified and synthetic Alzheimer’s amyloid beta
(Aβ) prions, Proc. Natl. Acad. Sci. USA, 109,
11025-11030.
66.Kumar, S., Rezaei-Ghaleh, N., Terwel, D., Thal,
D. R., Richard, M., Hoch, M., Mc Donald, J. M., Wullner, U., Glebov,
K., Heneka, M. T., Walsh, D. M., Zweckstetter, M., and Walter, J.
(2011) Extracellular phosphorylation of the amyloid beta-peptide
promotes formation of toxic aggregates during the pathogenesis of
Alzheimer’s disease, EMBO J., 30,
2255-2265.
67.Nussbaum, J. M., Schilling, S., Cynis, H., Silva,
A., Swanson, E., Wangsanut, T., Tayler, K., Wiltgen, B., Hatami, A.,
Ronicke, R., Reymann, K., Hutter-Paier, B., Alexandru, A., Jagla, W.,
Graubner, S., Glabe, C. G., Demuth, H. U., and Bloom, G. S. (2012)
Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated
amyloid-beta, Nature, 485, 651-655.
68.Tsvetkov, P. O., Popov, I. A., Nikolaev, E. N.,
Archakov, A. I., Makarov, A. A., and Kozin, S. A. (2008) Isomerization
of the Asp7 residue results in zinc-induced oligomerization of
Alzheimer’s disease amyloid β(1-16) peptide,
Chembiochem, 9, 1564-1567.
69.Indeykina, M. I., Popov, I. A., Kozin, S. A.,
Kononikhin, A. S., Kharybin, O. N., Tsvetkov, P. O., Makarov, A. A.,
and Nikolaev, E. N. (2011) Capabilities of MS for analytical
quantitative determination of the ratio of alpha- and betaAsp7 isoforms
of the amyloid-beta peptide in binary mixtures, Anal. Chem.,
83, 3205-3210.
70.Pekov, S., Indeykina, M., Popov, I., Kononikhin,
A., Bocharov, K., Kozin, S. A., Makarov, A. A., and Nikolaev, E. (2017)
Application of MALDI-TOF/TOF-MS for relative quantitation of α-
and β-Asp7 isoforms of amyloid-β peptide, Eur. J. Mass
Spectrom., 24, 141-144.
71.Zakharova, N. V., Shornikova, A. Y., Bugrova, A.
E., Baybakova, V. V., Indeykina, M. I., Kononikhin, A. S., Popov, I.
A., Kechko, O. I., Makarov, A. A., and Nikolaev, E. N. (2017)
Evaluation of plasma peptides extraction methods by high-resolution
mass spectrometry, Eur. J. Mass Spectrom., 23,
209-212.
72.Kostyukevich, Y., Kononikhin, A., Popov, I.,
Indeykina, M., Kozin, S. A., Makarov, A. A., and Nikolaev, E. (2015)
Supermetallization of peptides and proteins during electrospray
ionization, J. Mass Spectrom., 50, 1079-1087.
73.Mekmouche, Y., Coppel, Y., Hochgrafe, K.,
Guilloreau, L., Talmard, C., Mazarguil, H., and Faller, P. (2005)
Characterization of the ZnII binding to the peptide amyloid-beta1-16
linked to Alzheimer’s disease, Chembiochem,
6, 1663-1671.
74.Zirah, S., Rebuffat, S., Kozin, S. A., Debey, P.,
Fournier, F., Lesage, D., and Tabet, J.-C. (2003) Zinc binding
properties of the amyloid fragment Aβ(1-16) studied by
electrospray-ionization mass spectrometry, Int. J. Mass
Spectrom., 228, 999-1016.
75.Zirah, S., Kozin, S. A., Mazur, A. K., Blond, A.,
Cheminant, M., Segalas-Milazzo, I., Debey, P., and Rebuffat, S. (2006)
Structural changes of region 1-16 of the Alzheimer disease amyloid
β-peptide upon zinc binding and in vitro aging, J. Biol.
Chem., 281, 2151-2161.
76.Tsvetkov, P. O., Kulikova, A. A., Golovin, A. V.,
Tkachev, Y. V., Archakov, A. I., Kozin, S. A., and Makarov, A. A.
(2010) Minimal Zn2+ binding site of amyloid-β,
Biophys. J., 99, L84-L86.
77.Kozin, S. A., Kulikova, A. A., Istrate, A. N.,
Tsvetkov, P. O., Zhokhov, S. S., Mezentsev, Y. V., Kechko, O. I.,
Ivanov, A. S., Polshakov, V. I., and Makarov, A. A. (2015) The English
(H6R) familial Alzheimer’s disease mutation facilitates
zinc-induced dimerization of the amyloid-β metal-binding domain,
Metallomics, 7, 422-425.
78.Kulikova, A. A., Tsvetkov, P. O., Indeykina, M.
I., Popov, I. A., Zhokhov, S. S., Golovin, A. V., Polshakov, V. I.,
Kozin, S. A., Nudler, E., and Makarov, A. A. (2014) Phosphorylation of
Ser8 promotes zinc-induced dimerization of the amyloid-β
metal-binding domain, Mol. BioSyst., 10,
2590-2596.
79.Liu, S.-T., Howlett, G., and Barrow, C. J. (1999)
Histidine-13 is a crucial residue in the zinc ion-induced aggregation
of the Aβ peptide of Alzheimer’s disease,
Biochemistry, 38, 9373-9378.
80.Nisbet, R. M., Nuttall, S. D., Robert, R., Caine,
J. M., Dolezal, O., Hattarki, M., Pearce, L. A., Davydova, N., Masters,
C. L., Varghese, J. N., and Streltsov, V. A. (2013) Structural studies
of the tethered N-terminus of the Alzheimer’s disease
amyloid-β peptide, Proteins, 81,
1748-1758.
81.Adzhubei, A. A., Anashkina, A. A., and Makarov,
A. A. (2017) Left-handed polyproline-II helix revisited: proteins
causing proteopathies, J. Biomol. Struct. Dyn.,
35, 2701-2713.
82.Adzhubei, A. A., Sternberg, M. J. E., and
Makarov, A. A. (2013) Polyproline-II helix in proteins: structure and
function, J. Mol. Biol., 425, 2100-2132.
83.Istrate, A. N., Kozin, S. A., Zhokhov, S. S.,
Mantsyzov, A. B., Kechko, O. I., Pastore, A., Makarov, A. A., and
Polshakov, V. I. (2016) Interplay of histidine residues of the
Alzheimer’s disease Aβ peptide governs its Zn-induced
oligomerization, Sci. Rep., 6, 21734.
84.Polshakov, V. I., Mantsyzov, A. B., Kozin, S. A.,
Adzhubei, A. A., Zhokhov, S. S., van Beek, W., Kulikova, A. A.,
Indeykina, M. I., Mitkevich, V. A., and Makarov, A. A. (2017) A
binuclear zinc interaction fold discovered in the homodimer of
Alzheimer’s amyloid-beta fragment with taiwanese mutation D7H,
Angew. Chem. Int. Ed. Engl., 56, 11734-11739.
85.Mezentsev, Y. V., Medvedev, A. E., Kechko, O. I.,
Makarov, A. A., Ivanov, A. S., Mantsyzov, A. B., and Kozin, S. A.
(2016) Zinc-induced heterodimer formation between metal-binding domains
of intact and naturally modified amyloid-beta species: implication to
amyloid seeding in Alzheimer’s disease? J. Biomol. Struct.
Dyn., 34, 2317-2326.
86.Jucker, M., and Walker, L. C. (2013)
Self-propagation of pathogenic protein aggregates in neurodegenerative
diseases, Nature, 501, 45-51.
87.Hosoda, R., Saido, T. C., Otvos, L. J., Arai, T.,
Mann, D. M. A., Lee, V. M.-Y., Trojanowski, J. Q., and Iwatsubo, T.
(1998) Quantification of modified amyloid [beta] peptides in Alzheimer
disease and Down syndrome brains, J. Neuropathol. Exp. Neurol.,
57, 1089-1095.
88.Roher, A. E., Lowenson, J. D., Clarke, S.,
Wolkow, C., Wang, R., Cotter, R. J., Reardon, I. M., Zurcher-Neely, H.
A., Heinrikson, R. L., Ball, M. J., and Greenberg, B. D. (1993)
Structural alterations in the peptide backbone of beta-amyloid core
protein may account for its deposition and stability in
Alzheimer’s disease, J. Biol. Chem., 268,
3072-3083.
89.Shimizu, T., Matsuoka, Y., and Shirasawa, T.
(2005) Biological significance of isoaspartate and its repair system,
Biol. Pharm. Bull., 28, 1590-1596.
90.Jucker, M., and Walker, L. C. (2011) Pathogenic
protein seeding in Alzheimer disease and other neurodegenerative
disorders, Ann. Neurol., 70, 532-540.
91.Kozin, S. A., Cheglakov, I. B., Ovsepyan, A. A.,
Telegin, G. B., Tsvetkov, P. O., Lisitsa, A. V., and Makarov, A. A.
(2013) Peripherally applied synthetic peptide isoAsp7-Aβ(1-42)
triggers cerebral β-amyloidosis, Neurotox. Res.,
24, 370-376.
92.Borchelt, D. R., Ratovitski, T., van Lare, J.,
Lee, M. K., Gonzales, V., Jenkins, N. A., Copeland, N. G., Price, D.
L., and Sisodia, S. S. (1997) Accelerated amyloid deposition in the
brains of transgenic mice coexpressing mutant presenilin 1 and amyloid
precursor proteins, Neuron, 19, 939-945.
93.Garcia-Alloza, M., Robbins, E. M., Zhang-Nunes,
S. X., Purcell, S. M., Betensky, R. A., Raju, S., Prada, C., Greenberg,
S. M., Bacskai, B. J., and Frosch, M. P. (2006) Characterization of
amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer
disease, Neurobiol. Dis., 24, 516-524.
94.Mitkevich, V. A., Petrushanko, I. Y., Yegorov, Y.
E., Simonenko, O. V., Vishnyakova, K. S., Kulikova, A. A., Tsvetkov, P.
O., Makarov, A. A., and Kozin, S. A. (2013) Isomerization of Asp7 leads
to increased toxic effect of amyloid-β42 on human neuronal cells,
Cell Death Dis., 4, e939.
95.Yurinskaya, M. M., Mitkevich, V. A., Kozin, S.
A., Evgen’ev, M. B., Makarov, A. A., and Vinokurov, M. G. (2015)
HSP70 protects human neuroblastoma cells from apoptosis and oxidative
stress induced by amyloid peptide isoAsp7-Abeta(1-42), Cell Death
Dis., 6, e1977.
96.Moreth, J., Mavoungou, C., and Schindowski, K.
(2013) Passive anti-amyloid immunotherapy in Alzheimer’s disease:
what are the most promising targets? Immun. Ageing,
10, 18.
97.Busche, M. A., Grienberger, C., Keskin, A. D.,
Song, B., Neumann, U., Staufenbiel, M., Forstl, H., and Konnerth, A.
(2015) Decreased amyloid-beta and increased neuronal hyperactivity by
immunotherapy in Alzheimer’s models, Nat. Neurosci.,
18, 1725-1727.
98.Lawrence, J. L. M., Tong, M., Alfulaij, N.,
Sherrin, T., Contarino, M., White, M. M., Bellinger, F. P., Todorovic,
C., and Nichols, R. A. (2014) Regulation of presynaptic
Ca2+, synaptic plasticity and contextual fear conditioning
by a N-terminal β-amyloid fragment, J. Neurosci.,
34, 14210-14218.
99.Mediannikov, O., and Morozov, A. (2014) Peptide
compound useful for inhibiting amyloid plaque formation, France Patent
2,966,827 (PCT/FR2011/052477, WO2012056157A1, EP2632938A1,
JP2013542217A, US20130252901A1, RU2013106757/04(010044),
CA2808196A1).
100.Tsvetkov, P. O., Cheglakov, I. B., Ovsepyan, A.
A., Mediannikov, O. Y., Morozov, A. O., Telegin, G. B., and Kozin, S.
A. (2015) Peripherally applied synthetic tetrapeptides HAEE and RADD
slow down the development of cerebral beta-amyloidosis in AbetaPP/PS1
transgenic mice, J. Alzheimer’s Dis., 46,
849-853.
101.Aisen, P. S., Gauthier, S., Ferris, S. H.,
Saumier, D., Haine, D., Garceau, D., Duong, A., Suhy, J., Oh, J., Lau,
W. C., and Sampalis, J. (2011) Tramiprosate in mild-to-moderate
Alzheimer’s disease – a randomized, double-blind,
placebo-controlled, multi-centre study (the Alphase study), Arch.
Med. Sci., 7, 102-111.
102.Jucker, M., and Walker, L. C. (2015)
Neurodegeneration: amyloid-[beta] pathology induced in humans,
Nature, 525, 193-194.
103.Sacks, C. A., Avorn, J., and Kesselheim, A. S.
(2017) The failure of solanezumab – how the FDA saved taxpayers
billions, N. Engl. J. Med., 376, 1706-1708.
104.Moro, M. L., Phillips, A. S., Gaimster, K.,
Paul, C., Mudher, A., Nicoll, J. A. R., and Boche, D. (2018)
Pyroglutamate and isoaspartate modified amyloid-beta in ageing and
Alzheimer’s disease, Acta Neuropathol. Commun.,
6, 3.
105.Bu, X. L., Xiang, Y., Jin, W. S., Wang, J.,
Shen, L. L., Huang, Z. L., Zhang, K., Liu, Y. H., Zeng, F., Liu, J. H.,
Sun, H. L., Zhuang, Z. Q., Chen, S. H., Yao, X. Q., Giunta, B., Shan,
Y. C., Tan, J., Chen, X. W., Dong, Z. F., Zhou, H. D., Zhou, X. F.,
Song, W., and Wang, Y. J. (2017) Blood-derived amyloid-[beta] protein
induces Alzheimer’s disease pathologies, Mol. Psychiatry,
doi: 10.1038/mp.2017.204.
106.Frederickson, C. J., Koh, J.-Y., and Bush, A.
I. (2005) The neurobiology of zinc in health and disease, Nat. Rev.
Neurosci., 6, 449-462.
107.Lauren, J. (2014) Cellular prion protein as a
therapeutic target in Alzheimer’s disease, J.
Alzheimer’s Dis., 38, 227-244.
108.Lauren, J., Gimbel, D. A., Nygaard, H. B.,
Gilbert, J. W., and Strittmatter, S. M. (2009) Cellular prion protein
mediates impairment of synaptic plasticity by amyloid-beta oligomers,
Nature, 457, 1128-1132.
109.Parri, R. H., and Dineley, T. K. (2010)
Nicotinic acetylcholine receptor interaction with beta-amyloid:
molecular, cellular, and physiological consequences, Curr. Alzheimer
Res., 7, 27-39.
110.Spevacek, A. R., Evans, E. G. B., Miller, J.
L., Meyer, H. C., Pelton, J. G., and Millhauser, G. L. (2013) Zinc
drives a tertiary fold in the prion protein with familial disease
mutation sites at the interface, Structure, 21,
236-246.
111.Watt, N. T., Griffiths, H. H., and Hooper, N.
M. (2013) Neuronal zinc regulation and the prion protein, Prion,
7, 203-208.
112.Watt, N. T., Griffiths, H. H., and Hooper, N.
M. (2014) Lipid rafts: linking prion protein to zinc transport and
amyloid-β toxicity in Alzheimer’s disease, Fron. Cell
Dev. Biol., 2, 41.
113.Zawisza, I., Rozga, M., and Bal, W. (2012)
Affinity of copper and zinc ions to proteins and peptides related to
neurodegenerative conditions (Aβ, APP, α-synuclein, PrP),
Coord. Chem. Rev., 256, 2297-2307.