Genetic Association between Alzheimer’s Disease Risk Variant of the PICALM Gene and Auditory Event-Related Potentials in Aging
N. V. Ponomareva1,2,a*, T. V. Andreeva2,3, M. A. Protasova2, Yu. V. Filippova1, E. P. Kolesnikova1, V. F. Fokin1, S. N. Illarioshkin1, and E. I. Rogaev2,3,4,b*
1Research Center for Neurology, 125367 Moscow, Russia2Vavilov Institute of General Genetics, Russian Academy of Sciences, 119991 Moscow, Russia
3Lomonosov Moscow State University, Department of Biology, Center of Genetics and Genetic Technologies, 119991 Moscow, Russia
4Brudnick Neuropsychiatric Research Institute, Department of Psychiatry, University of Massachusetts Medical School, Worcester, USA
* To whom correspondence should be addressed.
Received May 21, 2018; Revision received June 26, 2018
Aging and genetic predisposition are major risk factors in age-related neurodegenerative disorders. The most common neurodegenerative disorder is Alzheimer’s disease (AD). Genome-wide association studies (GWAS) have identified statistically significant association of the PICALM rs3851179 polymorphism with AD. The PICALM G allele increases the risk of AD, while the A allele has a protective effect. We examined the association of the PICALM rs3851179 polymorphism with parameters of the P3 component of auditory event-related potentials (ERPs) in 87 non-demented volunteers (age, 19-77 years) subdivided into two cohorts younger and older than 50 years of age. We found statistically significant association between the AD risk variant PICALM GG and increase in the P3 latency in subjects over 50 years old. The age-dependent increase in the P3 latency was more pronounced in the PICALM GG carriers than in the carriers of the PICALM AA and PICALM AG genotypes. The observed PICALM-associated changes in the neurophysiological processes indicate a decline in the information processing speed with aging due, probably, to neuronal dysfunction and subclinical neurodegeneration of the neuronal networks in the hippocampus and the frontal and parietal cortical areas. Such changes were less pronounced in the carriers of the PICALM gene A allele, which might explain the protective effect of this allele in the cognitive decline and AD development.
KEY WORDS: PICALM genotype, neurodegeneration, aging, genetic predisposition, Alzheimer’s disease, event-related potentials, P300DOI: 10.1134/S0006297918090092
Abbreviations: AD, Alzheimer’s disease; EEG, electroencephalogram; ERPs, event-related potentials; LP, latency period; PD, Parkinson’s disease.
Aging and genetic predisposition are the greatest known risk factors in
the development of age-related neurodegenerative disorders, the most
common of which are Alzheimer’s disease (AD) and
Parkinson’s disease (PD).
The development of AD, especially of the early-onset familial AD, is related to mutations in the genes encoding presenilin 1 (PSEN1) [1, 2], presenilin 2 (PSEN2) [2-4], and amyloid precursor protein (APP) [5]. Polymorphism of the apolipoprotein E gene (ApoE) located on chromosome 19 is the most common AD risk factor. The ApoE ε4 allele markedly elevates the risk of AD; however, it is not necessary or sufficient for AD development [6, 7].
Recently, a remarkable progress in identifying new genes with lower impact and penetrance but still associated with increased risk of AD development, has been achieved by using genome-wide association studies (GWAS). Thus, it was found that the PICALM rs3851179 polymorphism is associated with the AD development [8, 9]. PICALM (chromosome 11q14) has been described as one of the six most common risk genes in the AlzGene susceptibility gene database (http://www.AlzGene.org). The PICALM allele G increases the risk of AD development, while the A allele decreases this risk [10, 11]. It was found that epistatic and additive interaction between genes identified in GWAS (including PICALM and ApoE genotypes) might be involved in AD development [12, 13].
PICALM encodes phosphatidylinositol-binding clathrin assembly protein involved in clathrin-mediated endocytosis (CME) [14]. CME is a component of some metabolic pathways playing a crucial role in AD pathogenesis, such as formation and clearance of β-amyloid and tau protein clearance by autophagy [15-17]. PICALM modulates neuronal trafficking, including the transport of synaptic vesicle protein VAMP2 required for the neurotransmitter release from presynaptic terminals [18]. It was found that PICALM expression depends on the PICALM rs3851179 polymorphism [19].
The PICALM rs3851179 polymorphism is associated with the hippocampus size and the entorhinal cortex thickness in healthy individuals, patients with mild cognitive impairment (MCI), and AD patients [20, 21].
Earlier, we investigated the functional role of the PICALM rs3851179 polymorphism using the resting-state quantitative electroencephalography (qEEG) and revealed association between the studied PICALM polymorphism and age-related changes in the β EEG activity [22].
Other sensitive tools that can be used for assessing functional changes in the brain in normal and pathological aging are cognitive components of event-related potentials (ERPs) that reflect brain activity directly involved in decision making, memory, and attention. ERPs generation is a result of summation of excitatory and inhibitory postsynaptic potentials (EPSPs and IPSPs, respectively) mainly in the pyramidal neurons of the frontal, parietal, and temporal cortical structures and the hippocampus [23-27]. Cognitive ERPs have been used to examine neurophysiological mechanisms underlying cognitive disorders in aging and neurodegenerative diseases (e.g., MCI, AD, and PD) [26, 28, 29]. This method surpasses functional magnetic resonance imaging in temporal resolution (but not in spatial resolution).
The most studied and informative parameter is the ERPs positive P3 component (P300) elicited ~300 ms after the onset of a rare stimulus in the cognitive task involving identification of rare “target” stimuli within a series of frequently presented “standard” stimuli (the so-called oddball paradigm).
Multiple studies (including intracranial ones) demonstrated that the P3 component is mainly elicited by the neuronal networks of the frontal, temporal, and parietal cortical areas and the hippocampus, the frontal cortex being responsible predominantly for attention, while temporal and parietal areas – for memory functions [26, 27].
The P3 latency gradually increases with normal aging, and it is significantly elevated in neurodegenerative diseases accompanied by cognitive impairment, especially in AD [23, 26, 28, 30]. The amplitude of P3 decreases in AD (the data on P3 amplitude in normal aging are controversial) [28, 31].
Several studies have demonstrated an association between the genetic risk factors for AD and the parameters of cognitive ERPs. Analysis of cognitive ERPs in a small cohort of 30 elderly females including 10 carriers of the ApoE E4+ genotype found that the latency of the P3 component of auditory ERPs in the examined subjects increased, while the neuropsychological parameters of memory were comparable between ApoE E4+ carriers and non-carriers [32]. It was also shown that the P3 latency was increased in clinically asymptomatic relatives of AD patients [33]. Similarly, elevated latency of the late components of visual ERPs was found in clinically asymptomatic relatives of AD patients [34].
The impact of PICALM polymorphism on cognitive ERPs in normal aging and neurodegenerative diseases remains unexplored.
Our study was aimed at investigating the association between the PICALM rs3851179 polymorphism and the P3 component of auditory ERPs and how it might be affected by aging.
MATERIALS AND METHODS
Subjects. A total of 87 non-demented volunteers, 43 males and 44 females, aged 19-77 (mean age, 48.0 ± 1.7 years) were examined after undergoing neurological and psychometric examination. The exclusion criteria were as follows: neurological or mental disorders including cardiovascular diseases, psychiatric diseases, epilepsy, as well as psychiatric or neurological disorders in subject’s history.
All subjects were divided into groups according to the PICALM rs3851179 polymorphism. The PICALM AA&AG group consisted of homozygous PICALM AA or heterozygous PICALM AG carriers; the PICALM GG group included homozygous PICALM GG carriers. Each group was further subdivided into subgroups with subjects younger and older than 50 years of age.
All subjects were genotyped for ApoE.
Cognitive auditory ERPs recordings. Cognitive auditory ERPs were recorded with a computerized Neuro-KM system (Statokin, Russia) using the monopolar recording in the parietal (P3, P4), frontal (F3, F4) and central (Cz) areas according to the International 10-20 System. Auditory ERPs were recorded according to a standard auditory discrimination protocol using target and non-target stimuli (oddball paradigm). The target stimuli were recognized as 2000-Hz clicks among frequent 1000-Hz standard stimuli. The subject was required to distinguish between the two tones by mentally counting the target tones and not responding to the standard. Binaural stimuli were applied for 50 ms; the stimulus intensity was 80-90 dB (in accordance with the auditory threshold); the frequency was 1 Hz. The stimuli were applied in a pseudo-random order at a 7 : 3 target/non-target ratio. Sweeps to targets were visually inspected for artefacts before being accepted into the average. The averaging number for target stimuli was 40-46. Control experiments demonstrated that the averaging number within the studied range did not influence the parameters of cognitive ERPs. The pre-stimulus interval was 100 ms; the epoch length was 600 ms. We measured the N2–P3 interpeak amplitude (µV). The P3 latency was defined relative to stimulus onset.
Genotyping. DNA was isolated from peripheral blood mononuclear cells by a standard phenol-chloroform extraction method with a Qiagen kit (Qiagen, The Netherlands). PICALM genotyping was performed by PCR followed by restriction fragment length polymorphism (RFLP) analysis as described in [22]. ApoE genotyping was performed by PCR as described in [12].
Statistical analysis. The differences in the P3 latency and amplitude of cognitive ERPs were assessed by ANOVA using repeated measurements in the general linear model (GLM) with the (i) between-subject factors Genotype (PICALM AA&AG vs PICALM GG) and Age (younger vs older than 50 years of age) and (ii) within-subject factors Recording Area (frontal F, parietal P, and central C) and Asymmetry (right and left hemispheres). PICALM AA and PICALM AG genotype carriers were grouped together, because the PICALM AA group contained a relatively small number of subjects (14 individuals), and ANOVA did not reveal significant differences between the ERP parameters in the PICALM AA and PICALM AG carriers. The data were checked for sphericity because of the introduction of within-subject factors into the model. In case the sphericity was violated, the Greenhouse–Geisser correction was applied. The ApoE genotype was included as a fixed factor into the GLM for statistical processing of the results.
The Duncan’s test was used in post-hoc comparisons at p < 0.05.
The dependence of P3 amplitude and latency on age was estimated using Pearson correlation coefficient.
The distribution of polymorphism variants was tested for its correspondence to the Hardy–Weinberg equation by using the χ2-test.
RESULTS
Demographic parameters of the subjects enrolled in the study are shown in Table 1. No significant age and sex differences between the PICALM GG and PICALM AA&AG carriers were found in the cohorts younger and older than 50 years of age, as well as in the total sample (p > 0.05). The frequencies of the G and A alleles were 60.3 and 39.7%, respectively. The observed genotype distribution did not differ from the one expected according to the Hardy–Weinberg equation (χ2 = 0.02; p > 0.99).
Table 1. Demographic characteristics of
healthy subjects with different PICALM genotypes
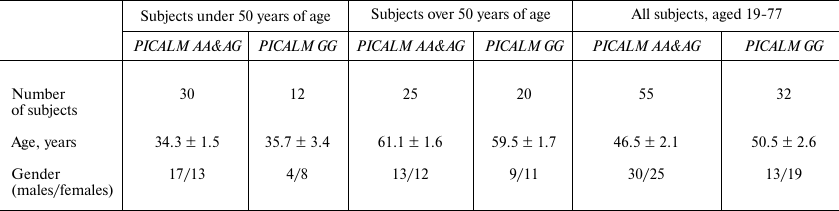
Note: Data are presented as mean ± S.E.
Statistical analysis revealed the dependence of the P3 latency on the PICALM Genotype (F[1.81] = 4.1; p < 0.05). As demonstrated by post-hoc comparison, the P3 latency in the PICALM GG group was longer than in the PICALM AA&AG group in the frontal (p < 0.01), parietal (p < 0.02), and central (p < 0.02) brain areas (Table 2).
Table 2. P3 latency (ms) in healthy subjects
with different PICALM genotypes
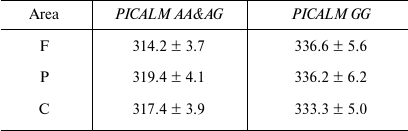
Notes: Data are presented as mean ± S.E; F, frontal area; P,
parietal area; C, central area.
The P3 latency also depended on Age (F[1.81] = 13.1; p < 0.001) (Fig. 1). Thus, in individuals over 50 years old, the P3 latency was greater in the PICALM GG group than in the PICALM AA&AG group (post-hoc comparison) in the frontal (p = 0.02), parietal (p = 0.03), and vertex (p = 0.05) areas. However, in individuals younger than 50 years of age, this difference was not significant (Fig. 1).
Fig. 1. Latency (latent period, LP) of the P3 component of auditory ERPs in healthy subjects under (1) and over (2) 50 years of age carrying different PICALM genotypes. F, P and C are frontal, parietal and central brain areas, respectively; * p < 0.05, significant difference between PICALM GG and PICALM AA&AG genotype carriers; # p < 0.05, significant difference between subjects younger and older than 50 years of age.
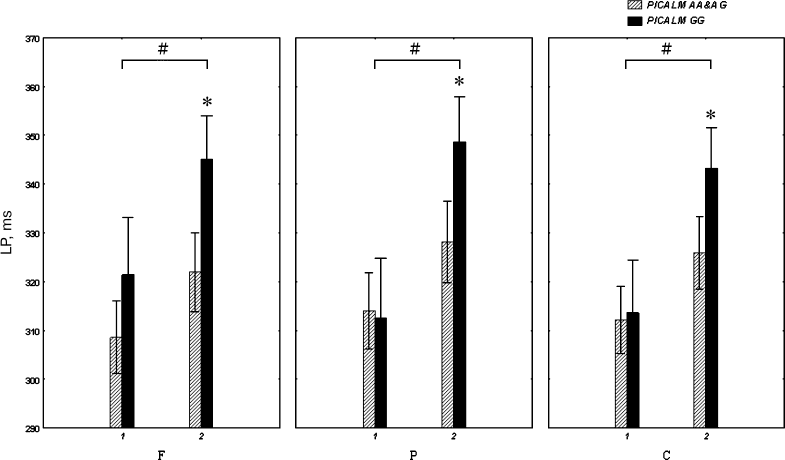
The P3 latency was longer in PICALM GG carriers older vs. younger than 50 years of age in the frontal (p = 0.02), parietal (p < 0.001) and central (p = 0.006) brain areas. The PICALM AA&AG group also showed an increase in the P3 latency with age, although the difference between the subjects younger vs. older than 50 years was not significant (Fig. 1).
The ANOVA did not reveal any effect of between-subject factors (Genotype and Age) on P3 amplitude (Fig. 2).
Fig. 2. Amplitude of the P3 component of auditory ERPs in healthy subjects under (1) and over (2) 50 years of age carrying different PICALM genotypes.
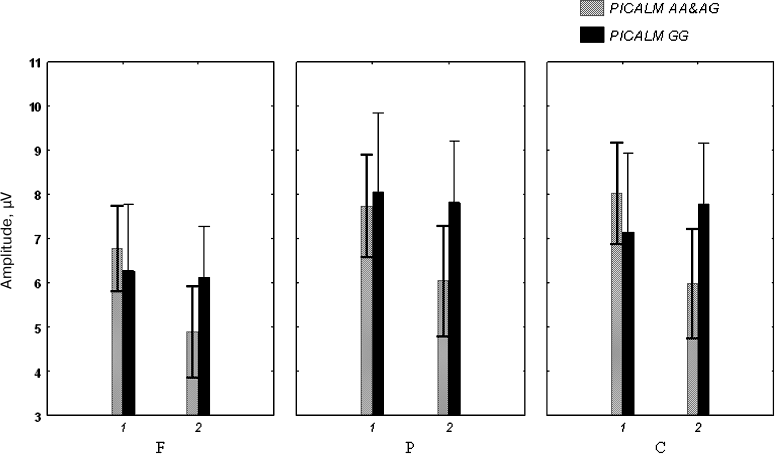
No interaction between the PICALM Genotype and the within-subject factors was observed.
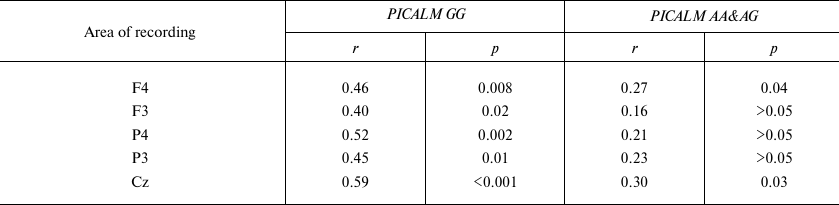
Correlation analysis revealed that the P3 latency in PICALM GG carriers significantly increased with age in all the examined areas. No significant correlation between the P3 latency and age was found in the parietal area in the PICALM AA&AG group, while in the frontal and central areas, this correlation was less significant than in PICALM GG group (Table 3 and Fig. 3).
Fig. 3. Correlation between age and P3 latency (LP) in the vertex (Cz) area in healthy subjects with different PICALM genotypes. Dashed lines, 95% confidence interval.
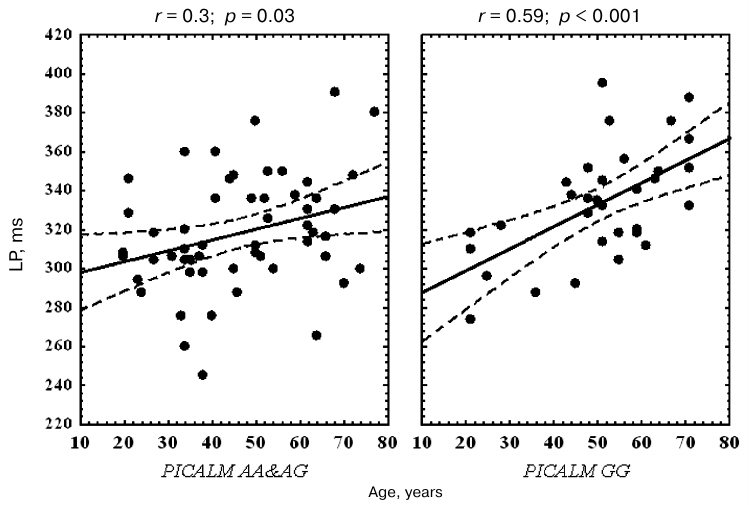
Table 3. Correlation between age and P3
latency in healthy subjects with different PICALM genotypes
DISCUSSION
In this study, we revealed an association between the PICALM rs3851179 polymorphism and the latency of the P3 component of auditory ERPs in adult non-demented volunteers. Significantly increased P3 latency in the carriers of AD risk PICALM GG vs. the carriers of the protective PICALM AA & PICALM AG genotypes was observed in subjects over 50 years old in all examined brain regions (frontal, parietal, and central areas). The correlation between age and increase in the P3 latency was more pronounced in the carriers of the AD risk PICALM GG genotype vs. the PICALM AA & PICALM AG carriers.
More pronounced increase in the P3 latency in the carriers of the PICALM GG AD risk genotype indicates that information processing is slowed down in the hippocampus and parietal and frontal cortical structures of these subjects due probably to early neurodegeneration resulting in the reduced size of the hippocampus and decreased thickness of the entorhinal cortex [26, 27]. These changes were less pronounced in the carriers of the protective A allele. Our results corroborate the data published by Biffi et al. [20] on the association between the PICALM polymorphisms and reduced size of the hippocampus and decreased thickness of the entorhinal cortex. An increase in the P3 latency underlies memory impairments and attention deficit and correlates with neuropsychological measures of these cognitive functions [23-26, 31].
The impact of the PICALM genotype on the neuronal dysfunction and neurodegeneration manifested as altered cognitive ERPs can be caused by the effect of PICALM genotype on the generation and clearance of β-amyloid, as well as clearance of tau protein [14-17]. Acording to the current dominant model of AD pathogenesis, generation and accumulation of neurotoxic β-amyloid in the brain is a pivotal pathogenic event that triggers release and intracellular accumulation of hyperphosphorylated tau protein, impaired mitochondrial function, excitotoxicity, oxidative stress, and inflammatory reactions eventually resulting in neurodegeneration [35].
PICALM-linked processes are also involved in neurotransmission [18]. Previously, we found that the PICALM GG genotype is associated with an increase in the EEG beta activity in aging [22]. The EEG β activity is related to glutamate-mediated neurotransmission. Glutamate-associated excitotoxicity may promote neurodegeneration in carriers of the PICALM GG genotype.
The PICALM rs3851179 polymorphism was also shown to be associated with PD development [36], although some studies failed to find this association [37]. The most damaged cells in PD are dopaminergic neurons of substantia nigra. α-Synuclein plays a crucial role in PD pathogenesis, affects clathrin-mediated endocytosis of NMDA receptors and influences NMDA-dependent death of dopaminergic neurons [38].
The latency of cognitive ERPs is increased in both non-demented and demented PD patients, but this increase is more pronounced in the demented PD patents [39]. PD is also characterized by cognitive impairments, although less pronounced than in AD [40]. It cannot be ruled out that age-associated changes in cognitive ERPs found in our study are caused by some PD-related factors; however, the significance of such factors in the modulation of cognitive ERPs requires further investigation.
Overall, our results confirm that investigation of the role of genetic factors in the development of functional alterations in aging brain is relevant to unveil the mechanisms underlying such alterations. This approach could potentially allow to develop personalized strategies of treatment and efficient prevention of neurodegenerative disorders [2, 7, 40-44].
In conclusion, we found that the latency of P3 (P300) component of auditory ERPs was increased in healthy carriers of the AD risk PICALM GG genotype vs. carriers of the PICALM AA and PICALM AG genotypes, with the effect being more pronounced in subjects older than 50 years of age. The processing of information in the PICALM GG carriers progressively slows down with aging due probably to neuronal dysfunction and subclinical neurodegeneration in the neuronal networks of the frontal and parietal cortical structures and the hippocampus. In the PICALM allele A carriers, these changes were less pronounced, which may explain protective effects of this allele on the rate of cognitive decline in aging and, probably, in AD development.
Funding
This study was supported by the Russian Science Foundation (project no. 14-44-00077; genotyping) and the National Institutes of Health, USA (grant R01AG054712 to E.R.; statistical analysis).
Conflict of Interests
The authors declare no financial or other conflicts of interest.
Ethical Committee Approval
All procedures with volunteers were conducted in accordance with the ethical standards of the National Committee for Research Ethics and the Declaration of Helsinki 1964, as well as its subsequent revisions and comparable ethical standards.
All subjects provided a written informed consent to participate in the study performed in accordance with the protocol approved by the Ethics Committee at the Research Center of Neurology and Vavilov Institute of General Genetics, Russian Academy of Sciences.
REFERENCES
1.Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva,
E. A., Levesque, G., Ikeda, M., Chi, H., Lin, C., Li, G., Holman, K.,
Tsuda, T., Mar, L., Foncin, J. F., Bruni, A. C., Montesi, M. P., Sorbi,
S., Rainero, I., Pinessi, L., Nee, L., Chumakov, I., Pollen, D.,
Brookes, A., Sanseau, P., Polinsky, R. J., Wasco, W., Da Silva, H. A.,
Haines, J. L., Perkicak-Vance, M. A., Tanzi, R. E., Roses, A. D.,
Fraser, P. E., Rommens, J. M., and St. George-Hyslop, P. H. (1995)
Cloning of a gene bearing missense mutations in early-onset familial
Alzheimer’s disease, Nature, 375, 754-760.
2.Rogaev, E., Sherrington, R., Rogaeva, E., Levesque,
G., Ikeda, M., Liang, Y., Chi, H., Lin, C., Holman, K., and Tsuda, T.
(1995) Familial Alzheimer’s disease in kindreds with missense
mutations in a gene on chromosome 1 related to the Alzheimer’s
disease type 3 gene, Nature, 376, 775-778.
3.Levy-Lahad, E., Wasco, W., Poorkaj, P., Romano, D.
M., Oshima, J., Pettingell, W. H., Yu, C. E., Jondro, P. D., Schmidt,
S. D., Wang, K., Crowley, A. C., Fu, Y. F., Guenette, S. Y., Galas, D.,
Nemens, E., Wijsman, E. M., Bird, Th. D., Schellenberg, G. D., and
Tanzi, R. E. (1995) Candidate gene for the chromosome 1 familial
Alzheimer’s disease locus, Science, 269,
973-977.
4.Rogaev, E. I., Sherrington, R., Wu, C., Levesque,
G., Liang, Y., Rogaeva, E. A., Ikeda, M., Holman, K., Lin, C., Lukiw,
W. J., de Jong, P. J., Fraser, P. E., Rommens, J. M., and St.
George-Hyslop, P. (1997) Analysis of the 5′-sequence, genomic
structure, and alternative splicing of the presenilin-1 gene (PSEN1)
associated with early onset Alzheimer disease, Genomics, 40,
415-424.
5.Goate, A., Chartier-Harlin, M. C., Mullan, M.,
Brown, J., Crawford, F., Fidani, L., Giuffra, L., Haynes, A., Irving,
N., James, L., Mant, R., Newton, P., Rooke, K., Roques, P., Talbot, C.,
Pericak-Vance, M., Roses, A., Williamson, R., Rossor, M., Owen, M., and
Hardy, J. (1991) Segregation of a missense mutation in the amyloid
β-protein precursor gene with familial Alzheimer’s disease,
Nature, 349, 704-706.
6.Saunders, A. M., Strittmatter, W. J., Schmechel,
D., George-Hyslop, P. H., Pericak-Vance, M. A., Joo, S. H., Rosi, B.
L., Gusella, J. F., Crapper-MacLachlan, D. R., and Alberts, M. J.
(1993) Association of apolipoprotein E allele epsilon 4 with late-onset
familial and sporadic Alzheimer’s disease, Neurology,
43, 1467-1472.
7.Rogaev, E. I. (1999) Genetic factors and a
polygenic model of Alzheimer’s disease, Genetika,
35, 1558-1571.
8.Harold, D., Abraham, R., Hollingworth, P., Sims,
R., Gerrish, A., Hamshere, M. L., Pahwa, J. S., Moskvina, V., Dowzell,
K., Williams, A., Jones, N., Thomas, C., Stretton, A., Morgan, A. R.,
Lovestone, S., Powell, J., Proitsi, P., Lupton, M. K., Brayne, C.,
Rubinsztein, D. C., Gill, M., Lawlor, B., Lynch, A., Morgan, K., Brown,
K. S., Passmore, P. A., Craig, D., McGuinness, B., Todd, S., Holmes,
C., Mann, D., Smith, A. D., Love, S., Kehoe, P. G., Hardy, J., Mead,
S., Fox, N., Rossor, M., Collinge, J., Maier, W., Jessen, F.,
Schurmann, B., Heun, R., van den Bussche, H., Heuser, I., Kornhuber,
J., Wiltfang, J., Dichgans, M., Frolich, L., Hampel, H., Hull, M.,
Rujescu, D., Goate, A. M., Kauwe, J. S., Cruchaga, C., Nowotny, P.,
Morris, J. C., Mayo, K., Sleegers, K., Bettens, K., Engelborghs, S., De
Deyn, P. P., Van Broeckhoven, C., Livingston, G., Bass, N. J., Gurling,
H., McQuillin, A., Gwilliam, R., Deloukas, P., Al-Chalabi, A., Shaw, C.
E., Tsolaki, M., Singleton, A. B., Guerreiro, R., Mühleisen, T.
W., Nothen, M. M., Moebus, S., Jockel, K. H., Klopp, N., Wichmann, H.
E., Carrasquillo, M. M., Pankratz, V. S., Younkin, S. G., Holmans, P.
A., O’Donovan, M., Owen, M. J., and Williams, J. (2009)
Genome-wide association study identifies variants at CLU and
PICALM associated with Alzheimer’s disease, Nat.
Genet., 41, 1088-1093.
9.Lambert, J.-C., Heath, S., Even, G., Campion, D.,
Sleegers, K., Hiltunen, M., Combarros, O., Zelenika, D., Bullido, M.
J., Tavernier, B., Letenneur, L., Bettens, K., Berr, C., Pasquier, F.,
Fievet, N., Barberger-Gateau, P., Engelborghs, S., De Deyn, P., Mateo,
I., Franck, A., Helisalmi, S., Porcellini, E., Hanon, O., de Pancorbo,
M. M., Lendon, C., Dufouil, C., Jaillard, C., Leveillard, T., Alvarez,
V., Bosco, P., Mancuso, M., Panza, F., Nacmias, B., Bossu, P.,
Piccardi, P., Annoni, G., Seripa, D., Galimberti, D., Hannequin, D.,
Licastro, F., Soininen, H., Ritchie, K., Blanche, H., Dartigues, J.-F.,
Tzourio, C., Gut, I., Van Broeckhoven, C., Alperovitch, A., Lathrop,
M., and Amouyel, P. (2009) Genome-wide association study identifies
variants at CLU and CR1 associated with Alzheimer’s
disease, Nat. Genet., 41, 1094-1099.
10.Carrasquillo, M. M., Belbin, O., Hunter, T. A.,
Ma, L., Bisceglio, G. D., Zou, F., Crook, J. E., Pankratz, V. S.,
Dickson, D. W., Graff-Radford, N. R., Petersen, R. C., Morgan, K., and
Younkin, S. G. (2010) Replication of CLU, CR1, and
PICALM associations with Alzheimer’s disease, Arch.
Neurol., 67, 961-964.
11.Wang, Z., Lei, H., Zheng, M., Li, Y., Cui, Y.,
and Hao, F. (2016) Meta-analysis of the association between
Alzheimer’s disease and variants in GAB2, PICALM,
and SORL1, Mol. Neurobiol., 53, 6501-6510.
12.Golenkina, S. A., Gol’tsov, A. I.,
Kuznetsova, I. L., Grigorenko, A. P., Andreeva, T. V., Reshetov, D. A.,
Kunizheva, S. S., Shagam, L. I., Morozova, I. I., Goldenkova-Pavlova,
I. V., Shimshilashvili, K., Viacheslavova, A. O., Faskhutdinova, G.,
Gareeva, A. E., Zainullina, A. G., Khusnutdinova, E. K., Puzyrev, V.
P., Stepanov, V. A., Kolotvin, A. V., Samokhodskaia, L. M., Selezneva,
N. D., Gavrilova, S. I., and Rogaev, E. I. (2010) Analysis of clusterin
gene (CLU/APOJ) polymorphism in Alzheimer’s disease
patients and in normal cohorts from Russian populations, Mol. Biol.
(Moscow), 44, 620-620.
13.Naj, A. C., Jun, G., Reitz, C., Kunkle, B. W.,
Perry, W., Park, Y. S., Beecham, G. W., Rajbhandary, R. A.,
Hamilton-Nelson, K. L., Wang, L. S., Kauwe, J. S., Huentelman, M. J.,
Myers, A. J., Bird, T. D., Boeve, B. F., Baldwin, C. T., Jarvik, G. P.,
Crane, P. K., Rogaeva, E., Barmada, M. M., Demirci, F. Y., Cruchaga,
C., Kramer, P., Alzheimer’s Disease Genetics Consortium,
Ertekin-Taner, N., Hardy, J., Graff-Radford, N. R., Green, R. C.,
Larson, E. B., St. George-Hyslop, P., Buxbaum, J. D., Evans, D.,
Schneider, J. A., Lunetta, K. L., Kamboh, M. I., Saykin, A. J., Reiman,
E. M., De Jager, P. L., Bennett, D. A., Morris, J. C., Montine, T. J.,
Goate, A. M., Blacker, D., Tsuang, D. W., Hakonarson, H., Kukull, W.
A., Foroud, T. M., Martin, E. R., Haines, J. L., Mayeux, R., Farrer, L.
A., Schellenberg, G. D., and Pericak-Vance, M. A. (2014) Age-at-onset
in late onset Alzheimer disease is modified by multiple genetic loci,
JAMA Neurol., 71, 1394-1404.
14.Xu, W., Tan, L., and Yu, J.-T. (2015) The role of
PICALM in Alzheimer’s disease, Mol. Neurobiol.,
52, 399-413.
15.Xiao, Q., Gil, S. C., Yan, P., Wang, Y., Han, S.,
Gonzales, E., Perez, R., Cirrito, J. R., and Lee, J. M. (2012) Role of
phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia
(PICALM) in intracellular amyloid precursor protein (APP)
processing and amyloid plaque pathogenesis, J. Biol. Chem.,
287, 21279-21289.
16.Ando, K., Brion, J. P., Stygelbout, V., Suain,
V., Authelet, M., Dedecker, R., Chanut, A., Lacor, P., Lavaur, J.,
Sazdovitch, V., Rogaeva, E., Potier, M. C., and Duyckaerts, C. (2013)
Clathrin adaptor CALM/PICALM is associated with
neurofibrillary tangles and is cleaved in Alzheimer’s brains,
Acta Neuropathol., 125, 861-878.
17.Moreau, K., Fleming, A., Imarisio, S., Lopez
Ramirez, A., Mercer, J. L., Jimenez-Sanchez, M., Bento, C. F., Puri,
C., Zavodszky, E., Siddiqi, F., Lavau, C. P., Betton, M., O’Kane,
C. J., Wechsler, D. S., and Rubinsztein, D. C. (2014) PICALM
modulates autophagy activity and tau accumulation, Nat. Commun.,
5, 4998.
18.Harel, A., Mattson, M. P., and Yao, P. J. (2011)
CALM, a clathrin assembly protein, influences cell surface
GluR2 abundance, Neuromolec. Med., 13, 88-90.
19.Parikh, I., Fardo, D. W., and Estus, S. (2014)
Genetics of PICALM expression and Alzheimer’s disease,
PLoS One, 9, e91242.
20.Biffi, A., Anderson, C. D., Desikan, R. S.,
Sabuncu, M., Cortellini, L., Schmansky, N., Salat, D., and Rosand, J.
(2010) Genetic variation and neuroimaging measures in Alzheimer’s
disease, Arch. Neurol., 67, 677-685.
21.Furney, S. J., Simmons, A., Breen, G., Pedroso,
I., Lunnon, K., Proitsi, P., Hodges, A., Powell, J., Wahlund, L.-O.,
Kloszewska, I., Mecocci, P., Soininen, H., Tsolaki, M., Vellas, B.,
Spenger, C., Lathrop, M., Shen, L., Kim, S., Saykin, A. J., Weiner, M.
W., and Lovestone, S. (2011) Genome-wide association with MRI atrophy
measures as a quantitative trait locus for Alzheimer’s disease,
Mol. Psychiatry, 16, 1130-1138.
22.Ponomareva, N. V., Andreeva, T. V., Protasova, M.
S., Shagam, L. I., Malina, D. D., Goltsov, A. Y., Fokin, V. F.,
Illarioshkin, S. N., and Rogaev, E. I. (2017) Quantitative EEG during
normal aging: association with the Alzheimer’s disease genetic
risk variant in PICALM gene, Neurobiol. Aging, 51,
e1-e8.
23.Gnezditskiy, V. V. (1997) Cerebral
Event-Related Potentials in Clinical Practice [in Russian],
TGRU.
24.Zenkov, L. R., and Ronkin, M. A. (2013)
Functional Diagnostics of Nervous System Diseases. Manual for
Physicians [in Russian], MEDpress-inform.
25.Romanov, A. S., Sharova, E. V., Kuznetsova, O.
A., Oknin, L. V., Volynskiy, P. E., and Shchekut’ev, G. A. (2011)
An opportunity for using wavelet synchronization in assessment of
long-latency components of auditory evoked potentials in healthy
humans, Zh. Vysshei Nerv. Deyat. im. I. P. Pavlova, 61,
112-118.
26.Polich, J. (2007) Updating P300: an integrative
theory of P3a and P3b, Clin. Neurophysiol., 118,
2128-2148.
27.Joos, K., Gilles, A., Van de Heyning, P., De
Ridder, D., and Vanneste, S. (2014) From sensation to percept: the
neural signature of auditory event-related potentials, Neurosci.
Biobehav. Rev., 42, 148-156.
28.Kropotov, J., Ponomarev, V., Tereshchenko, E. P.,
Muller, A., and Jancke, L. (2016) Effect of aging on ERP components of
cognitive control, Front. Aging Neurosci., 8, 69.
29.Lister, J. J., Harrison Bush, A. L., Andel, R.,
Matthews, C., Morgan, D., and Edwards, J. D. (2016) Cortical auditory
evoked responses of older adults with and without probable mild
cognitive impairment, Clin. Neurophysiol., 127,
1279-1287.
30.Goodin, D. S., Squires, K. C., and Starr, A.
(1978) Long latency event-related components of the auditory evoked
potential in dementia, Brain, 101, 635-648.
31.Braverman, E. R., Blum, K., Hussman, K. L., Han,
D., Dushaj, K., Li, M., Marin, G., Badgaiyan, R. D., Smayda, R., and
Gold, M. S. (2015) Evoked potentials and memory/cognition tests
validate brain atrophy as measured by 3T MRI (NeuroQuant) in
cognitively impaired patients, PLoS One, 10,
e0133609.
32.Irimajiri, R., Golob, E. J., and Starr, A. (2010)
ApoE genotype and abnormal auditory cortical potentials in healthy
older females, Neurobiol. Aging, 31, 1799-1804.
33.Green, J., and Levey, A. I. (1999) Event-related
potential changes in groups at increased risk for Alzheimer’s
disease, Arch. Neurol., 56, 1398-1403.
34.Ponomareva, N. V., Fokin, V. F., Selesneva, N.
D., and Voskresenskaia, N. I. (1998) Possible neurophysiological
markers of genetic predisposition to Alzheimer’s disease,
Dement. Geriatr. Cogn. Disord., 9, 267-273.
35.Hardy, J., and Selkoe, D. J. (2002) The amyloid
hypothesis of Alzheimer’s disease: progress and problems on the
road to therapeutics, Science, 297, 353-356.
36.Santos-Reboucas, C. B., Goncalves, A. P., Dos
Santos, J. M., Abdala, B. B., Motta, L. B., Laks, J., de Borges, M. B.,
de Rosso, A. L. Z., Pereira, J. S., Nicaretta, D. H., and Pimentel, M.
M. G. (2017) rs3851179 Polymorphism at 5′ to the PICALM
gene is associated with Alzheimer’s and Parkinson’s
diseases in Brazilian population, Neuromolec. Med., 19,
293-299.
37.Kalinderi, K., Bostantjopoulou, S., Katsarou, Z.,
Clarimon, J., and Fidani, L. (2012) Lack of association of the
PICALM rs3851179 polymorphism with Parkinson’s disease in
the Greek population, Int. J. Neurosci., 122,
502-605.
38.Cheng, F., Li, X., Li, Y., Wang, C., Wang, T.,
Liu, G., Baskys, A., Ueda, K., Chan, P., and Yu, S. (2011)
α-Synuclein promotes clathrin-mediated NMDA receptor endocytosis
and attenuates NMDA-induced dopaminergic cell death, J.
Neurochem., 119, 815-825.
39.Nojszewska, M., Pilczuk, B., Zakrzewska-Pniewska,
B., and Rowinska-Marcinska, K. (2009) The auditory system involvement
in Parkinson disease: electrophysiological and neuropsychological
correlations, J. Clin. Neurophysiol., 26, 430-437.
40.Illarioshkin, S. N., and Ivanova-Smolenskaya, I.
A. (2011) Trembling Hyperkinesis [in Russian], Atmosfera,
Moscow.
41.Illarioshkin, S. N., Ivanova-Smolenskaia, I. A.,
Markova, E. D., Shadrina, M. I., Kliushnikov, S. A., Zagorovskaia, T.
V., Miklina, N. I., Slominskii, P. A., and Limborskaia, S. A. (2004)
Molecular genetic analysis of hereditary neurodegenerative diseases,
Genetika, 40, 816-826.
42.Ponomareva, N. V., Goltsov, A. Y., Kunijeva, S.
S., Scheglova, N. S., Malina, D. D., Mitrofanov, A. A., Boikova, T. I.,
and Rogaev, E. I. (2012) Age- and genotype-related neurophysiologic
reactivity to oxidative stress in healthy adults, Neurobiol.
Aging, 33, 839.e11-21.
43.Ponomareva, N., Andreeva, T., Protasova, M.,
Shagam, L., Malina, D., Goltsov, A., Fokin, V., Mitrofanov, A., and
Rogaev, E. (2013) Age-dependent effect of Alzheimer’s risk
variant of CLU on EEG alpha rhythm in non-demented adults, Front.
Aging Neurosci., 5, 86.
44.Fokin, V. F., and Ponomareva, N. V. (2003)
Neuronergetics and Brain physiology. [in Russian], Antidor,
Moscow.