REVIEW: Immunogenetic Factors of Neurodegenerative Diseases: The Role of HLA Class II
M. P. Aliseychik1,2,3,a*, T. V. Andreeva1,2, and E. I. Rogaev1,2,3,b,c*
1Vavilov Institute of General Genetics, Russian Academy of Sciences, Department of Genomics and Human Genetics, 119991 Moscow, Russia2Center for Genetics and Genetic Technologies, Department of Biology, Lomonosov Moscow State University, 119192 Moscow, Russia
3Department of Psychiatry, University of Massachusetts Medical School, Worcester, MA 01655, USA
* To whom correspondence should be addressed.
Received May 28, 2018; Revision received June 21, 2018
An increase in the life expectancy during the last decades in most world countries has resulted in the growing number of people suffering from neurodegenerative disorders, including Alzheimer’s disease, Parkinson’s disease, frontotemporal dementia, and others. Familial forms of neurodegenerative diseases account for 5-10% of all cases and are caused by mutations in specific genes often resulting in pathological protein deposition. The risk factors for neurodegeneration include trauma, lifestyle, and allelic variants of disease-associated genes with incomplete penetrance. Many of these gene variants are located in immunity-related loci, particularly in the human leukocyte antigen locus (HLA class II) coding for proteins of the major histocompatibility complex class II (MHCII). HLA class II plays a key role in the antigen presentation and is expressed in microglial cells. Microglia is a component of innate immunity. On the one hand, microglial cells phagocytize pathological protein deposits; on the other hand, they produce proinflammatory factors accelerating neuronal death. The involvement of adaptive immunity mechanisms (antigen presentation, T cell response, antibody production) in the development of neurodegenerative diseases remains unclear and requires further research, including more detailed studies of the role of identified HLA class II genetic variants.
KEY WORDS: human leukocyte antigen, major histocompatibility complex class II, neurodegeneration, Alzheimer’s disease, Parkinson’s disease, genome-wide association studyDOI: 10.1134/S0006297918090122
Abbreviations: AD, Alzheimer’s disease; ALS, amyotrophic lateral sclerosis; ApoE, apolipoprotein E; CNS, central nervous system; GWAS, genome-wide association study; HLA, human leucocyte antigen; IL, interleukin; IFNγ, interferon γ; MHC, major histocompatibility complex; MHCI(II), major histocompatibility complex class I(II); PD, Parkinson’s disease; TNF, tumor necrosis factor.
An increase in the life expectancy leads to the growing number of
elderly people worldwide. According to 2017 data, 21% of the Russian
Federation population are individuals over 60 years old. Recent
forecasts predict that this category will include 36 million people by
2100 [1]. The increase in the life expectancy is
accompanied by the rise in the number of people suffering from
age-related diseases that can have a significant impact on the quality
of life. Different neurological disorders including Alzheimer’s
(AD) and Parkinson’s (PD) diseases, frontotemporal and vascular
dementias, amyotrophic lateral sclerosis (ALS), and Huntington’s
disease account for 10% of years of life lost (YLL) due to premature
death and disability-adjusted life years (DALY). The data from 2016
show that neurological disorders represent 25% causes of death among
people over 70 years old. Among those, stroke is the most common
pathology (67.3%) followed by AD and other dementias (20.3%). PD
occupies the sevenths place (1.2%) after meningitis, brain cancer,
encephalitis, and epilepsy. In Russia, 4.97% individuals over 70 years
of age die from AD and other types of dementia; in the USA, this number
is even higher – 12.79% [2]. At this moment,
there is no cure for the neurodegenerative diseases that involve slowly
progressing degeneration of the central nervous system (CNS), and the
nature of these disorders mostly remains obscure.
A number of genes have been identified whose mutations cause the development of neurodegenerative disorders. For example, mutations in the APP (amyloid precursor protein), PSEN1 (presenilin 1), and PSEN2 (presenilin 2) genes are associated with the early-onset familial AD (~5% AD cases) [3-5]. However, no specific genes or mutations resulting in sporadic AD (remaining ~95% cases) have been found so far. The risk factors for sporadic AD include genomic variants, trauma, environmental factors, diet, and lifestyle. Pathology-associated low-penetrance genomic variants could be revealed by genome-wide association studies (GWAS) involving large groups of patients and healthy individuals. Identified loci or gene variants should be then validated by analysis of independently assessed cohorts, clarification of the polymorphism location, and biological interpretation of the obtained data.
HLA LOCUS
Human leukocyte antigen (HLA) or the major histocompatibility complex (MHC) locus is located on chromosome 6. It comprises ~0.13% of the genome and contains ~150 protein-encoding genes predominately associated with the immune system. The MHC locus includes three gene classes. MHC class III contains genes for various ligands, receptors, cytokines, and other immune molecules. The highly polymorphic HLA-A, HLA-B, HLA-C, HLA-DR, HLA-DQ, and HLA-DP genes coding proteins involved in antigen presentation by the MHC are located in the HLA class I and II loci. HLA-B is the most polymorphic gene of the MHC class I locus (MHCI); 1077 allele variants of this gene have been described so far. HLA-DRB1 is the most polymorphic gene of the MHC class II locus (MHCII) and has 669 allele variants [6]. The diversity of the HLAI and II genes is essential for efficient immune defense against pathogens at the population level, since a broad variety of antigen-presenting molecules makes possible presentation of a broad array of peptides and, hence, ensures initiation of efficient immune response against a large number of pathogens [7].
In the CNS, MHCI is expressed mostly by the microglia and endothelial cells; low MHCI expression is detected in the astrocytes, oligodendrocytes, and neurons [8, 9]. Neuronal expression of MHCI is typical for the hippocampus, but can be also observed in the cerebral cortex, substantia nigra, and olfactory bulbs [10]. The main source of MHCII in the CNS is microglia; however, low-level MHCII expression is found in astrocytes and endothelial cells [8].
According to the results of multiple studies, HLA polymorphisms and haplotypes are associated mainly with autoimmune pathologies (multiple sclerosis, rheumatoid arthritis, psoriasis, celiac disease, chronic lymphocytic thyroiditis, etc. [6]), but also with psychiatric diseases (e.g., schizophrenia) [11-13] and various neurodegenerative disorders [14-17].
HLA CLASS II AND ALZHEIMER’S DISEASE
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by accumulation of amyloid plaques in the extracellular space and formation of intracellular neurofibrillary tangles consisting of hyperphosphorylated τ protein. Such pathological protein aggregation results in the neuronal damage and brain atrophy causing cognitive dysfunctions and memory loss [18]. Short-term memory loss is one of the most common early AD symptoms [19]; however, the first pathological changes, such as decrease in the β-amyloid level, increase in the τ protein content in the cerebrospinal liquid, and brain atrophy, can be detected 15-20 years prior to the manifestation of the first AD symptoms [20].
Missense mutations in the APP, PSEN1, and PSEN2 genes associated with development of the early-onset familial AD are highly penetrant [3-5]. Mutations in PSEN1 are the most frequent cause of the early-onset autosomal dominant forms of AD. Homologous PSEN1 and PSEN2 genes encode presenilin proteins that act as intramembrane aspartate proteases [21]. The proteolytic processing of APP by presenilins in the membrane and by β-secretase at the outer membrane leads to the production of β-amyloid peptides – major components of amyloid plaques. Interestingly enough, no AD-associated mutations have been found for the β-secretase-encoding BACE gene [22]. Among the factors that may impact the risk of AD development is the level of presenilin gene expression. Thus, it was demonstrated that the transcriptional activity of PSEN2 can be modulated by various physiological and genetic factors [23, 24]. It has been also proven that AD development is associated with the ε4 allele of the apolipoprotein E gene (APOE). This allele is the most common risk factor in the population of European and Asian descent [25, 26]. For example, it was shown that the risk of AD development in elderly people in Russia is 3 times higher for the heterozygous carriers of the APOE ε4 allele and 8 to 10 times higher for the homozygotes carriers this allele [27]. Significantly weaker genetic association of the PICALM, SORL1, PLD3, CLU, CR1, TREM2, CD33, and ABCA7 polymorphic variants with AD has been reported, but not always validated [28-31].
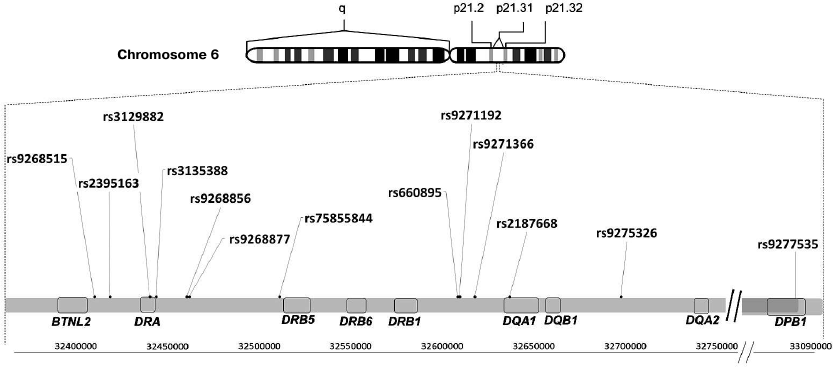
A large study was published in 2013 that was devoted to meta-analysis of GWAS for the late-onset AD forms using four previous GWAS data sets consisting of 17,008 Alzheimer's disease cases and 37,154 controls. The identified genetic associations were validated in an independent set of 8572 Alzheimer's disease cases and 11,312 controls. A total of 19 loci newly associated with AD were identified; the strongest association among those was revealed for the rs9271192 polymorphism (Fig. 1) in the HLA–DRB5–DRB1 region on chromosome 6, where the MHCII genes are located. However, the authors themselves admitted that due to the complex organization of this locus it was extremely difficult to identify which genes were causal [14].
Fig. 1. Location of polymorphisms associated with neurodegenerative diseases in the HLA class II locus. A fragment of the locus 21 (p21.31) on the human chromosome 6 short shoulder (p) is shown with positions of HLA class II genes and polymorphisms associated with neurodegenerative diseases.
The data described above [14] were obtained in individuals of the European descent. In order to test the association of the identified rs9271192 polymorphism with the risk of the late-onset AD in different ethnical groups, this polymorphic marker was analyzed in 982 AD patients and 1344 healthy controls from the Northern China. It was demonstrated that the risk of AD development was 2 times higher in the homozygous carriers of the C allele in comparison to the non-carriers. When these data were stratified by the apolipoprotein E (APOE) ε4 status, the observed association was confined to APOE ε4 non-carriers, which indicates that the nucleotide substitution in the HLA–DRB5–DRB1 region may be an independent AD risk factor [32].
Analysis of brain cortical samples showed that DNA methylation in the HLA–DRA–DRB5 locus could likely affect the AD development. Three CpG sites in this locus were found to be associated with β-amyloid accumulation, and 9 CpG sites – with the density of neurofibrillary tangles [33].
MHC class II (MHCII) is a heterodimer consisting of two types of polypeptide chains (α and β) expressed mainly on the surface of the antigen-presenting cells such as macrophages and dendritic cells. In the CNS, MHCII is predominantly expressed by the microglia [34]. Multiple attempts to evaluate MHCII expression in the brain of AD patients in comparison to the healthy controls produced a large body of controversial data. Hopperton et al. analyzed all papers published by the time of their own study in which they assessed MHCII expression in the post-mortem brain samples of AD patients vs. the control group [35]. The authors found that 36 studies reported upregulated MHCII expression in AD patients (at least in one part of the brain), while seven papers found no difference in the MHCII expression between AD patients and healthy subjects. Forty-one out of the 43 cited studies assessed MHCII expression immunohistochemically; in two papers, quantitative PCR (polymerase chain reaction) or Western blotting were used [35]. Thus, the controversy of the data produced in different studies is likely due to the differences in the brain regions investigated, examination techniques, and the number of samples analyzed.
Meta-analysis of transcriptome data from the brain cortex samples of AD patients and healthy individuals demonstrated upregulation of the HLA-DRA gene expression in AD patients [36] and association of the rs9271192 polymorphism with elevated HLA-DRB1 expression in the cerebral cortex [37].
So, HLA class II is likely associated with the AD development, as confirmed by genetic, epigenetic, and protein expression studies. However, the links between the found polymorphisms, methylation sites, and qualitative and quantitative characteristics of the MHCII protein complex in certain brain regions still have to be established.
HLA CLASS II AND PARKINSON’S DISEASE
Parkinson’s disease is the second most common neurodegenerative disorder. Its clinical symptoms include muscle rigidity, tremor, and bradykinesia [38, 39] resulting from the degeneration of dopaminergic nigrostriatal pathways. This degeneration might be caused by the cytoplasmic accumulation in dopaminergic neurons of protein aggregates (Lewy bodies) consisting predominately of the synaptic protein α-synuclein [38, 40].
PD hereditary forms (5-10% of all cases) are caused by mutations in the SNCA, VPS35, LRRK2, PINK1, PARK2, and PARK7 genes. It is commonly believed that in the rest of cases, PD development is contributed by various factors including polymorphism of low-penetrance genes, such as GBA, MAPT, GAK, BST1, LAMP3, and SYT11 [41, 42].
Unlike AD, whose genetic association with a single polymorphic marker in the MHC has been demonstrated, PD is associated with a group of single-nucleotide polymorphisms in the HLA class II locus: rs3129882, rs75855844, rs9268515, rs2395163, rs660895, and rs4248166 [43] (Fig. 1). However, it is impossible to establish which particular variant associates with the disease because of extensive polymorphism in the HLA locus, uneven allele distribution in populations, and relatively small distances between the polymorphisms. In particular, Hamza et al. proved genetic association of the rs3129882 polymorphism in the HLA–DRA non-coding region with the risk of PD development by studying 2000 PD patients and 1986 healthy controls [15]. At the same time, meta-analysis of GWAS data published by the International Genomic Consortium revealed association between PD and the rs75855844 polymorphism in the HLA-DRB5 gene. The discovery phase of this study included 5333 case and 12,019 control samples; 7053 case and 9007 control samples were analyzed in the replication stage [44]. The results of new meta-analysis of GWAS data were published in 2012 [45]. In this study, the authors conducted detailed investigation of 51 polymorphisms in the HLA–DR region and showed that only the HLA–DRB1 rs660895 polymorphism passed multiple comparison tests and represents a protective factor in PD development [45]. Interestingly, the same polymorphism associates with the risk of rheumatoid arthritis [46], which, in its turn, negatively correlates with PD [47].
Single-nucleotide substitutions in the coding regions of the HLA-DP, HLA-DQ, and HLA-DR genes affect the ability of the MHCII molecule to form stable complex with peptides, which directly impacts the efficiency of antigen presentation [48]. Hence, in order to elucidate the functional role of revealed genetic associations, it is important to understand if they are related to particular allele variants or regulatory sites of the HLA-genes. Genetic studies (2000 patients and 1986 controls in the main reference group; 843 patients and 856 controls in the replicate group) showed that the C*03:04 and DRB1*04:04 alleles are associated with the risk of PD development independently on the closely located polymorphisms. At the same time, the rs3129882 polymorphism and closely located rs9268515 and rs2395163 polymorphisms are associated with PD independently on the HLA alleles [43]. Hence, it can be suggested that both structural and regulator elements of the HLA II locus are involved into PD pathogenesis.
Expression of the HLA class II genes is regulated via formation of three-dimensional structures. Thus, the non-coding regions of the HLA class II genes contain insulator sequences that interact with gene promoters and activate them. This interaction proceeds with the involvement of the CCCTC-binding factor and class II transactivator and results in the formation of loops [49]. Because such regulation system can supposedly coordinate the expression of several genes; nucleotide substitutions in the regulatory elements could result in the systemic effects. It is known, for example, that rs3129882 and rs2395163 are associated with the changes in the expression of the HLA-DR and HLA-DQ genes in various tissues [43]. Further investigation of epigenetic regulation of the HLA locus is necessary in order to answer the question which particular polymorphic variants in the non-coding regions of the HLA class II genes affect pathogenesis of neurodegenerative diseases.
HLA CLASS II AND OTHER NEURODEGENERATIVE DISEASES
Frontotemporal dementia is a common neurodegenerative disease characterized by accumulation of protein aggregates in the brain tissue leading to the neuronal cell death, gliosis, and vascular damage. More than 90% cases of frontotemporal dementia are due to accumulation of MAPT (τ protein), TARDBP (TAR DNA-binding protein), and FUS (fused in sarcoma) in the frontal and temporal lobes of the cerebral cortex. The patients can simultaneously exhibit symptoms of several neurodegenerative diseases. For example, 20% patients with frontotemporal dementia exhibit symptoms of early parkinsonism [50].
Genetic factors contribute significantly to the development of frontotemporal dementia: 30-40% patients with this diagnosis have relatives with neurodegenerative disorders [51, 52]. The majority of described mutations leading to the development of familial frontotemporal have been found in the MAPT gene encoding the tau protein, as well as in the C9orf72 and GRN genes [50, 51]. However, there are many cases, for which no mutations in the known genes have been identified.
According to the GWAS data (3526 patients with frontotemporal dementia and 9402 healthy controls of the European descent), the polymorphisms rs9268877 and rs9268856 located in the non-coding region between the HLA-DRA and HLA-DRB5 genes in the HLA locus are associated with the risk of frontotemporal dementia (Fig. 1) [16]. It was found that the allele A of the polymorphic marker rs9268877 is associated with the higher risk of the frontotemporal dementia development (OR = 1.2), while the allele A of the polymorphic marker rs9268856, on the contrary, has a protective effect (OR = 0.8) [16]. Due to the fact that the distance between the polymorphisms is less than 1500 bp, it is difficult to assess whether these factors are independent. Investigation of the population of the South-Western China revealed association of the polymorphic variant rs9268856, but not rs9268877, with the increased risk of other neurodegenerative disease – amyotrophic lateral sclerosis (ALS). It was also found that the AA genotype of rs9268856 was associated with a 1.5-fold lower life expectancy in the ALS patients [17].
Multiple sclerosis (MS) is a chronic disease of the nervous system, which is different from the age-related neurodegenerative diseases, as it is mainly an autoimmune disorder that affects brain and spinal cord. Inflammation in the CNS is accompanied by the lymphocytic infiltration causing damage to the axonal myelin. With time, microglia activation is initiated that accompanies extended neurodegeneration [53]. Some HLA class II alleles, such as HLA-DRB1*15:01, HLA-DRB1*13:03, HLA-DRB1*03:01, HLA-DRB1*08:01, and HLA-DQB1*03:02, are currently considered risk factors in MS development; the majority of them are allele variants of the HLA-DRB1 gene. Thus, the risk of MS development in the carriers of the HLA-DRB1*15:01 allele is 8.3 times higher than in the non-carriers [54]. The protective alleles of the HLA class I have been also described for MS, including HLA-A*02:01, HLA-B*44:02, HLA-B*38:01, and HLA-B*55:01 [54]. It was suggested that their association with a decreased MS risk can be explained by less efficient autoantigen presentation. For example, investigation of the crystal structure the HLA-DRB1*15:01 complex with myelin basic protein demonstrated that alanine in the polymorphic position DRβ 71 creates a space for the aromatic side chain of myelin basic protein, which is one of the major autoantigens against which T cell response is developed [55]. Interestingly, a characteristic epitope of β-amyloid was described for HLA-DRB1*15:01 allele variant; presentation of this epitope initiates strong immune response in the transgenic mouse model of AD [56].
INNATE IMMUNE RESPONSE AND NEURODEGENERATION
Neurodegeneration always involves immune system and, as a consequence, is accompanied with inflammation (Fig. 2). In particular, AD patients have a decreased content of lymphocytes in blood [64] and altered cytokine levels in the blood serum [65]. An increase in the number of γδT cells in the blood and changes in the levels of interleukin 2 (IL-2), interleukin 6 (IL-6), and tumor necrosis factor (TNF) were observed in PD patients [66]. Immune response genes (CLU, CR1, TREM2 [67], CD33, ABCA7) are found among genetic loci with AD-associated polymorphisms. Expression of most of these genes is upregulated in the cerebral cortex of AD patients [28, 29]. Polymorphisms associated with the risk of PD development were identified in the immune response genes BTNL2, BST1, and LAMP3 [68].
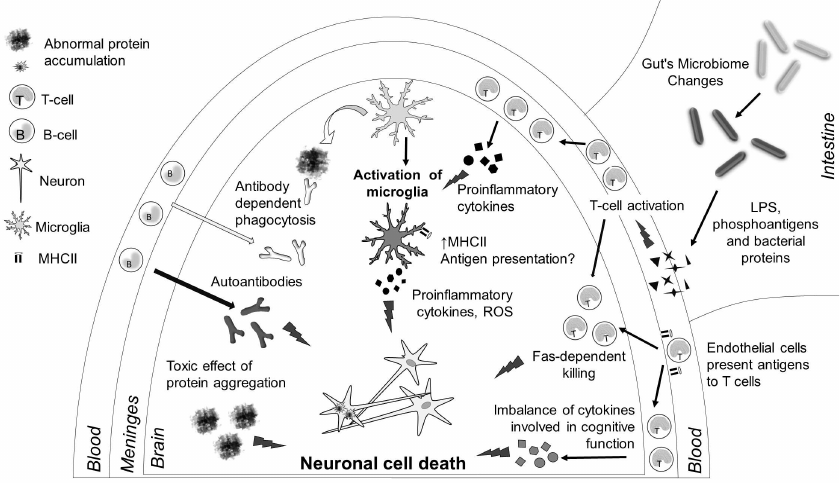
Fig. 2. Immune processes in neurodegenerative diseases. Neuronal death in the brain is the key to pathogenesis of neurodegenerative diseases, and it is caused by several reasons. Aggregates of protein molecules (β-amyloid, α-synuclein, τ protein, etc.) are toxic for the neurons. Microglia cells can phagocytize these aggregates; antibodies promote phagocytosis. However, due to the activation of the innate immunity receptors in neurodegenerative diseases, microglial cells are activated and start producing proinflammatory cytokines, reactive oxygen species (ROS), and reactive nitrogen species, thereby causing neuronal death. which is the second reason for cellular neuron death. Moreover, activation of microglial cells upregulates expression of MHC class II on their surface. Cytokines produced by meningeal T cells can facilitate microglia activation; in some cases, T cells can infiltrate brain tissues and cause apoptosis of neurons via the FAS-dependent pathway. Changes in the gut microflora result in the increase in the amounts of bacterial antigens, liposaccharide (LPS) molecules, and phosphoantigens, which can facilitate activation of the immune system in general and affect the inflammatory processes in the CNS. Autoantibodies produced by B cells as a result of adaptive immune response to neuronal antigens could be an additional factor in neurodegeneration. Finally, changes in the expression of cytokines by meningeal T cells can directly affect cognitive functions.
It has been emphasized more and more often that unlike “classic” neuroinflammatory diseases (MS and encephalitis), pathogenesis of neurodegenerative diseases involves innate immune response [69-71], in which microglia plays the most important role.
Microglial cells are resident macrophages of the CNS. Inflammatory signals, pathogens, Toll-like receptor ligands, and aberrant protein aggregation (β-amyloid, α-synuclein) can activate the microglia leading to the synthesis of proinflammatory cytokines (IL-1, IL-6, TNF) and chemokines, production of reactive oxygen species, and induction of phagocytosis.
The role of microglia in AD pathogenesis is not completely understood. On the one hand, accumulation of β-amyloid in the extracellular space causes continuous activation of the microglia. Production of cytokines by the activated microglia results in progressively increasing number of activated microglial cells by the positive feedback mechanism. Inflammation impairs the function of the nervous system cells and causes eventual death of neurons. After prolonged activation, microglial cells themselves acquire the exhausted phenotype specific of chronic inflammation [69]. On the other hand, microglial cells phagocytize cell debris and β-amyloid and are involved in the processes of tissue repair and isolation of amyloid plaques thus protecting neurons from their toxic effect [72]. Numerous AD-associated genes (CD33, TREM2, PLCG2) are actively expressed in the microglial cells and participate in the processes of their activation [35]. Being the antigen-presenting cells, microglial cells are the main source of MHCII in the nervous tissue [73].
PD development is accompanied with an increase in the activated microglia markers [74]. Conglomerates of microglial cells have been found in the vicinity of α-synuclein deposits. α-Synuclein activates microglia via interaction with the Toll-like receptors. This process is accompanied by the synthesis of proinflammatory cytokines, such as IL-1β, TNF, and IL-6. It is generally accepted that developing inflammation is the main cause of neurodegeneration, although as in the case of AD, microglia is capable of phagocytizing toxic protein deposits, which might slightly slow down PD development [74, 75].
An increase in the number of microglial cells, microglia activation, and disruption of its functions have been also demonstrated in other neurodegenerative pathologies, such as ALS and frontotemporal dementia [76].
ADAPTIVE IMMUNE RESPONSE AND NEURODEGENERATION
Based on the generally accepted opinion that the main immune component in the pathogenesis of neurodegenerative diseases is innate immunity response mediated by the microglia [69-71], it is difficult to interpret the revealed genetic association of the HLA class II genes with the majority of neurodegenerative disorders (table). It is likely that changes in the structural and regulatory regions of the HLA class II genes affect the processes of antigen presentation, and, therefore, modulate adaptive immunity. It was found that polymorphisms in the antigen presentation-related genes other than HLA class II are associated with the development of neurodegenerative diseases. For example, the rs241448 polymorphism associated with the risk of AD development in the APOE ε4 carriers is located in the TAP2 gene, the protein product of which is required for antigen processing [77].
Polymorphic markers in HLA class II locus associated with
neurodegenerative diseases
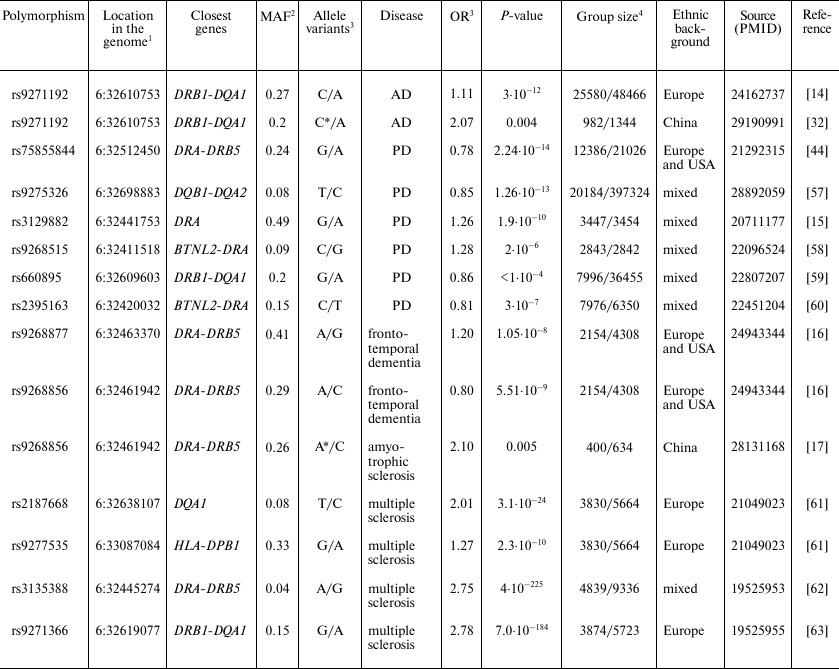
1 Human references genome GRCh38.p7 was used.
2 MAF, minor allele frequency in population according to the
data of 1000 genomes project (www.internationalgenome.org).
3 OR, Odds ratio.
4 Group of patients with diagnosed disease/control group.
* Association with homozygous genotype for the indicated allele was
analyzed.
It is possible that neurodegeneration induces adaptive immune response. Indeed, T cells from the blood of AD patients demonstrate increased reactivity towards β-amyloid. Antibodies against β-amyloid were found in the blood of aged individuals, and the titer of these antibodies was higher in AD patients [65]. The data obtained in Rag-5xfAD mice lacking T, B, and NK cells and exhibiting pathological changes similar to those observed in AD patients indicate that antibodies produced by B cells are required for efficient phagocytosis of β-amyloid by the microglia [78]. Hence, adaptive immunity can play a protective role in AD and other neurodegenerative diseases.
On the other hand, Th1 and Th17 lymphocytes can migrate into the brain in response to proinflammatory signals generated by the microglia and to aggravate inflammation by producing interferon γ (IFNγ) [79]. The study of post-mortem samples from PD patients demonstrated that both CD4+ and CD8+ T lymphocytes are capable of brain infiltration. It was established using the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of PD that CD4+ cells cause the death of dopaminergic neurons, most likely, via interaction of the FAS receptor with its ligand (FASL) [80]. There is an opinion that antigen presentation in the complexes with MHCI and MHCII by brain endothelial cells regulates selective permeability of the blood-brain barrier for T lymphocytes [81].
Neuronal death accompanying all neurodegenerative diseases can also induce the B cell response, in particular, production of autoantibodies. For example, immunoglobulins isolated from the serum of ALS patients induce apoptosis in a cell line of motoneurons [82]. Antibodies against β-amyloid and α-synuclein have been found in the serum of healthy people; however, the titers of these antibodies are often higher in AD and PD patients, respectively [83, 84].
Activated microglia expressing MHCII can present antigens, thus inducing adaptive immune response. Disruption of the blood-brain barrier has been demonstrated for the majority of neurodegenerative diseases. This disruption can lead to the changes in the transport of compounds and cause infiltration of the peripheral blood cells into the brain [85]. However, lymphocyte infiltration is not a typical feature of all neurodegenerative diseases; it is manifested rather locally.
CONCLUSIONS AND DIRECTIONS FOR FUTURE RESEARCH
Based on the information presented above, we can postulate the following three statements on the relationship between the immune system and neurodegenerative diseases. First, genetic association between polymorphisms in the immune response genes (including the HLA class II locus) and neurodegenerative diseases have been clearly demonstrated. Second, pathogenesis of neurodegenerative diseases (especially during disease progression) is determined by the innate immunity response mediated mostly by the microglia. Third, adaptive immunity exerts dual action on the neurodegeneration process: the antibody-mediated response promotes phagocytosis of aberrant protein deposits, while the T cell immune response facilitates neuronal death (although does not represent its main cause). At present, there are no sufficient data to establish the links between these statements. In particular, the role of antigen presentation in pathogenesis of neurodegenerative diseases is poorly understood. Which cells participate in this process? Which antigens are presented? Where does it all happen?
The fact that polymorphisms in the HLA class II locus are the risk factors of neurodegenerative diseases suggests that the process of antigen presentation can be essential during the disease onset, but then becomes secondary at the later stages of disease pathogenesis. As mentioned above, AD development starts 15-20 years before the disease can be diagnosed [20]; therefore, the majority of studies are conducted at the disease progression rather than prodromal stage. Transgenic models of AD are based on the reproduction of hereditary AD forms achieved by introduction of AD-related high-penetrance mutations into the genome. Hence, novel approaches must be developed to investigate the role of immune system in the early stages of neurodegenerative disorders.
It is possible that in certain cases, initial immune response is induced not so much by autoantigens, but by viral or bacterial proteins. There is no consensus so far on the viral theory of neurodegenerative diseases. However, some studies report the link between the level of antibodies against the herpes simplex virus [86, 87] or cytomegalovirus [88] and the risk of AD development. Viral infections might facilitate induction of neuroinflammation and represent one of the factors in the development of neurodegenerative diseases.
A large body of data has emerged recently implying that MHCI expressed in neurons plays a non-immune function. It is likely involved in synaptic plasticity, formation and elimination of synapses [89-91]. It cannot be ruled out that in addition to its important role in immune response, MHCII contributes to the functioning of the nervous system via yet unknown mechanisms. For this reason, the data on the role of MHCII in neurogenesis are extremely important. MHCII is expressed in neuronal stem cells during brain embryonic development [92]. Adult neurogenesis is associated with the increase in the MHCII content at the surface of microglia in the neurogenic niches in response to IFNγ and IL-4 produced by T lymphocytes of the brain meningeal membranes [93]. Meningeal immunity can affect cognitive functions through expression of cytokines. For example, IL-4 produced by meningeal T lymphocytes participates in the formation and maintenance of spatial memories [94], while IFNγ is involved in the implementation of social behavioral programs [95]. Neurodegeneration processes are often linked to neurogenesis disruption [96]; that is why detailed elucidation of the molecular mechanisms of functioning of neuronal stem cells is required for understanding the pathogenesis of neurodegenerative diseases.
Finally, it is known that gut microbiota affects the development of pathological processes in neurodegenerative diseases. The treatment of transgenic mouse AD and PD models with antibiotics alleviates the disease symptoms [97-99]. It is possible that changes in the bacterial and viral microflora result in impaired immune tolerance of the entire body (including the CNS) and initiation of autoimmune and neuroinflammatory processes. The cells of innate and adaptive immune systems activated by bacterial molecules (protein antigens, LPS, phosphoantigens, etc.) can migrate from the gut to the brain meninges, where they produce proinflammatory factors, thereby promoting neuroinflammation and neurodegeneration. Hence, HLA class II polymorphisms associated with the development of neurodegenerative diseases could affect presentation of not only neuronal antigens, but also viral or bacterial antigens and, therefore, facilitate inflammation caused by viruses and microbes.
Funding
This work was supported by the Russian Science Foundation (project no. 14-44-00077) and by the NIH grant (no. R01AG054712).
REFERENCES
1.World Population Prospects: the 2017 revision.
Vol. II: Demographic Profiles (2017) United Nations, N. Y.
2.GBD 2015 Neurological Disorders Collaborator Group,
Feigin, V. L., Abajobir, A. A., Abate, K. H., Abd-Allah, F., Abdulle,
A. M., Abera, S. F., Abyu, G. Y., Ahmed, M. B., Aichour, A. N.,
Aichour, I., Aichour, M. T. E., Akinyemi, R. O., Alabed, S.,
Al-Raddadi, R., Alvis-Guzman, N., Amare, A. T., et al. (2017) Global,
regional, and national burden of neurological disorders during
1990-2015: a systematic analysis for the global burden of disease study
2015, Lancet Neurol., 16, 877-897.
3.Rogaev, E. I., Sherrington, R., Rogaeva, E. A.,
Levesque, G., Ikeda, M., Liang, Y., Chi, H., Lin, C., Holman, K.,
Tsuda, T., Mar, L., Sorbi, S., Nacmias, B., Piacentini, S., Amaducci,
L., Chumakov, I., Cohen, D., Lannfelt, L., Fraser, P. E., Rommens, J.
M., and George-Hyslop, P. H. S. (1995) Familial Alzheimer’s
disease in kindreds with missense mutations in a gene on chromosome 1
related to the Alzheimer’s disease type 3 gene, Nature,
376, 775-778.
4.Goate, A., Chartier-Harlin, M.-C., Mullan, M.,
Brown, J., Crawford, F., Fidani, L., Giuffra, L., Haynes, A., Irving,
N., James, L., Mant, R., Newton, P., Rooke, K., Roques, P., Talbot, C.,
Pericak-Vance, M., Roses, A., Williamson, R., Rossor, M., Owen, M., and
Hardy, J. (1991) Segregation of a missense mutation in the amyloid
precursor protein gene with familial Alzheimer’s disease,
Nature, 349, 704-706.
5.Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva,
E. A., Levesque, G., Ikeda, M., Chi, H., Lin, C., Li, G., Holman, K.,
Tsuda, T., Mar, L., Foncin, J.-F., Bruni, A. C., Montesi, M. P., Sorbi,
S., Rainero, I., Pinessi, L., Nee, L., Chumakov, I., Pollen, D.,
Brookes, A., Sanseau, P., Polinsky, R. J., Wasco, W., Da Silva, H. A.
R., Haines, J. L., Pericak-Vance, M. A., Tanzi, R. E., Roses, A. D.,
Fraser, P. E., Rommens, J. M., and St. George-Hyslop, P. H. (1995)
Cloning of a gene bearing missense mutations in early-onset familial
Alzheimer’s disease, Nature, 375, 754-760.
6.Shiina, T., Hosomichi, K., Inoko, H., and Kulski,
J. K. (2009) The HLA genomic loci map: expression, interaction,
diversity and disease, J. Hum. Genet., 54, 15-39.
7.Parham, P., and Ohta, T. (1996) Population biology
of antigen presentation by MHC class I molecules, Science,
272, 67-74.
8.Chastain, E. M. L., Duncan, D. S., Rodgers, J. M.,
and Miller, S. D. (2011) The role of antigen presenting cells in
multiple sclerosis, Biochim. Biophys. Acta, 1812,
265-274.
9.Hoftberger, R., Aboul-Enein, F., Brueck, W.,
Lucchinetti, C., Rodriguez, M., Schmidbauer, M., Jellinger, K., and
Lassmann, H. (2004) Expression of major histocompatibility complex
class l molecules on the different cell types in multiple sclerosis
lesions, Brain Pathol., 14, 43-50.
10.Mokhtari, R., and Lachman, H. M. (2016) The major
histocompatibility complex (MHC) in schizophrenia: a review, J.
Clin. Cell. Immunol., 7, 479.
11.Schwab, S. G., Hallmayer, J., Albus, M., Lerer,
B., Eckstein, G. N., Borrmann, M., Segman, R. H., Hanses, C., Freymann,
J., Yakir, A., Trixler, M., Falkai, P., Rietschel, M., Maier, W., and
Wildenauer, D. B. (2000) A genome-wide autosomal screen for
schizophrenia susceptibility loci in 71 families with affected
siblings: support for loci on chromosome 10p and 6, Mol.
Psychiatry, 5, 638-649.
12.Wright, P., Donaldson, P. T., Underhill, J. A.,
Choudhuri, K., Doherty, D. G., and Murray, R. M. (1996) Genetic
association of the HLA DRB1 gene locus on chromosome 6p21.3 with
schizophrenia, Am. J. Psychiatry, 153, 1530-1533.
13.Sekar, A., Bialas, A. R., de Rivera, H., Davis,
A., Hammond, T. R., Kamitaki, N., Tooley, K., Presumey, J., Baum, M.,
Van Doren, V., Genovese, G., Rose, S. A., Handsaker, R. E., Daly, M.
J., Carroll, M. C., Stevens, B., McCarroll, S. A., and McCarroll, S. A.
(2016) Schizophrenia risk from complex variation of complement
component 4, Nature, 530, 177-183.
14.Lambert, J. C., Ibrahim-Verbaas, C. A., Harold,
D., Naj, A. C., Sims, R., Bellenguez, C., Jun, G., DeStefano, A. L.,
Bis, J. C., Beecham, G. W., Grenier-Boley, B., Russo, G.,
Thornton-Wells, T. A., Jones, N., Smith, A. V., et al. (2013)
Meta-analysis of 74,046 individuals identifies 11 new susceptibility
loci for Alzheimer’s disease, Nat. Genet., 45,
1452-1458.
15.Hamza, T. H., Zabetian, C. P., Tenesa, A.,
Laederach, A., Montimurro, J., Yearout, D., Kay, D. M., Doheny, K. F.,
Paschall, J., Pugh, E., Kusel, V. I., Collura, R., Roberts, J.,
Griffith, A., Samii, A., Scott, W. K., Nutt, J., Factor, S. A., and
Payami, H. (2010) Common genetic variation in the HLA region is
associated with late-onset sporadic Parkinson’s disease, Nat.
Genet., 42, 781-785.
16.Ferrari, R., Hernandez, D. G., Nalls, M. A.,
Rohrer, J. D., Ramasamy, A., Kwok, J. B. J., Dobson-Stone, C., Brooks,
W. S., Schofield, P. R., Halliday, G. M., Hodges, J. R., Piguet, O.,
Bartley, L., Thompson, E., Haan, E., et al. (2014) Frontotemporal
dementia and its subtypes: a genome-wide association study, Lancet
Neurol., 13, 686-699.
17.Yang, X., Zheng, J., Tian, S., Chen, Y., An, R.,
Zhao, Q., and Xu, Y. (2017) HLA-DRA/HLA-DRB5 polymorphism affects risk
of sporadic ALS and survival in a southwest Chinese cohort, J.
Neurol. Sci., 373, 124-128.
18.De Strooper, B., and Karran, E. (2016) The
cellular phase of Alzheimer’s disease, Cell, 164,
603-615.
19.Verheijen, J., and Sleegers, K. (2018)
Understanding alzheimer disease at the interface between genetics and
transcriptomics, Trends Genet., 34, 434-447.
20.Bateman, R. J., Xiong, C., Benzinger, T. L. S.,
Fagan, A. M., Goate, A., Fox, N. C., Marcus, D. S., Cairns, N. J., Xie,
X., Blazey, T. M., Holtzman, D. M., Santacruz, A., Buckles, V., Oliver,
A., Moulder, K., Aisen, P. S., Ghetti, B., Klunk, W. E., McDade, E.,
Martins, R. N., Masters, C. L., Mayeux, R., Ringman, J. M., Rossor, M.
N., Schofield, P. R., Sperling, R. A., Salloway, S., Morris, J. C., and
Dominantly Inherited Alzheimer Network, for the D. I. A. (2012)
Clinical and biomarker changes in dominantly inherited
Alzheimer’s disease, N. Engl. J. Med., 367,
795-804.
21.Grigorenko, A. P., Moliaka, Y. K., Plotnikova, O.
V., Smirnov, A., Nikishina, V. A., Goltsov, A. Y., Gusev, F., Andreeva,
T. V., Nelson, O., Bezprozvanny, I., and Rogaev, E. I. (2017)
Mutational re-modeling of di-aspartyl intramembrane proteases:
uncoupling physiologically-relevant activities from those associated
with Alzheimer’s disease, Oncotarget, 8,
82006-82026.
22.Nicolaou, M., Song, Y. Q., Sato, C. A.,
Orlacchio, A., Kawarai, T., Medeiros, H., Liang, Y., Sorbi, S.,
Richard, E., Rogaev, E. I., Moliaka, Y., Bruni, A. C., Jorge, R.,
Percy, M., Duara, R., Farrer, L. A., St. George-Hyslop, P., and
Rogaeva, E. A. (2001) Mutations in the open reading frame of the
beta-site APP cleaving enzyme (BACE) locus are not a common cause of
Alzheimer’s disease, Neurogenetics, 3, 203-206.
23.Lukiw, W. J., Gordon, W. C., Rogaev, E. I.,
Thompson, H., and Bazan, N. G. (2001) Presenilin-2 (PS2) expression
up-regulation in a model of retinopathy of prematurity and
pathoangiogenesis, Neuroreport, 12, 53-57.
24.Riazanskaia, N., Lukiw, W. J., Grigorenko, A.,
Korovaitseva, G., Dvoryanchikov, G., Moliaka, Y., Nicolaou, M., Farrer,
L., Bazan, N. G., and Rogaev, E. (2002) Regulatory region variability
in the human presenilin-2 (PSEN2) gene: potential contribution to the
gene activity and risk for AD, Mol. Psychiatry, 7,
891-898.
25.Saunders, A. M., Strittmatter, W. J., Schmechel,
D., George-Hyslop, P. H., Pericak-Vance, M. A., Joo, S. H., Rosi, B.
L., Gusella, J. F., Crapper-MacLachlan, D. R., and Alberts, M. J.
(1993) Association of apolipoprotein E allele epsilon 4 with late-onset
familial and sporadic Alzheimer’s disease, Neurology,
43, 1467-1472.
26.Bertram, L., McQueen, M. B., Mullin, K., Blacker,
D., and Tanzi, R. E. (2007) Systematic meta-analyses of Alzheimer
disease genetic association studies: the AlzGene database, Nat.
Genet., 39, 17-23.
27.Korovaitseva, G. I., Shcherbatykh, T. V.,
Selezneva, N. V., Gavrilova, S. I., Golimbet, V. E., Voskresenskaia, N.
I., and Rogaev, E. I. (2001) Genetic association between the
apolipoprotein E (ApoE) gene alleles and various forms of
Alzheimer’s disease, Genetika, 37, 529-535.
28.Karch, C. M., Cruchaga, C., and Goate, A. M.
(2014) Alzheimer’s disease genetics: from the bench to the
clinic, Neuron, 83, 11-26.
29.Van Cauwenberghe, C., Van Broeckhoven, C., and
Sleegers, K. (2016) The genetic landscape of Alzheimer disease:
clinical implications and perspectives, Genet. Med., 18,
421-430.
30.Papassotiropoulos, A., Lambert, J. C., Wavrant-De
Vrieze, F., Wollmer, M. A., von der Kammer, H., Streffer, J. R.,
Maddalena, A., Huynh, K. D., Wolleb, S., Lutjohann, D., Schneider, B.,
Thal, D. R., Grimaldi, L. M. E., Tsolaki, M., Kapaki, E., Ravid, R.,
Konietzko, U., Hegi, T., Pasch, T., Jung, H., Braak, H., Amouyel, P.,
Rogaev, E. I., Hardy, J., Hock, C., and Nitsch, R. M. (2005)
Cholesterol 25-hydroxylase on chromosome 10q is a susceptibility gene
for sporadic Alzheimer’s disease, Neurodegener. Dis.,
2, 233-241.
31.Golenkina, S. A., Gol’tsov, A. I.,
Kuznetsova, I. L., Grigorenko, A. P., Andreeva, T. V., Reshetov, D. A.,
Kunizheva, S. S., Shagam, L. I., Morozova, I. I., Goldenkova-Pavlova,
I. V., Shimshilashvili, K., Viacheslavova, A. O., Faskhutdinova, G.,
Gareeva, A. E., Zainullina, A. G., Khusnutdinova, E. K., Puzyrev, V.
P., Stepanov, V. A., Kolotvin, A. V., Samokhodskaia, L. M., Selezneva,
N. D., Gavrilova, S. I., and Rogaev, E. I. (2010) Analysis of clusterin
gene (CLU/APOJ) polymorphism in Alzheimer’s disease
patients and in normal cohorts from Russian populations, Mol. Biol.
(Moscow), 44, 620-626.
32.Lu, R. C., Yang, W., Tan, L., Sun, F. R., Tan, M.
S., Zhang, W., Wang, H. F., and Tan, L. (2017) Association of HLA-DRB1
polymorphism with Alzheimer’s disease: a replication and
meta-analysis, Oncotarget, 8, 93219-93226.
33.Yu, L., Chibnik, L. B., Srivastava, G. P.,
Pochet, N., Yang, J., Xu, J., Kozubek, J., Obholzer, N., Leurgans, S.
E., Schneider, J. A., Meissner, A., De Jager, P. L., and Bennett, D. A.
(2015) Association of brain DNA methylation in SORL1,
ABCA7, HLA-DRB5, SLC24A4, and BIN1 with
pathological diagnosis of Alzheimer’s disease, JAMA
Neurol., 72, 15-24.
34.Lee, Y. B., Nagai, A., and Kim, S. U. (2002)
Cytokines, chemokines, and cytokine receptors in human microglia, J.
Neurosci. Res., 69, 94-103.
35.Hopperton, K. E., Mohammad, D., Trepanier, M. O.,
Giuliano, V., and Bazinet, R. P. (2018) Markers of microglia in
post-mortem brain samples from patients with Alzheimer’s disease:
a systematic review, Mol. Psychiatry, 23, 177-198.
36.Yokoyama, J. S., Wang, Y., Schork, A. J.,
Thompson, W. K., Karch, C. M., Cruchaga, C., McEvoy, L. K., Witoelar,
A., Chen, C. H., Holland, D., Brewer, J. B., Franke, A., Dillon, W. P.,
Wilson, D. M., Mukherjee, P., Hess, C. P., Miller, Z., Bonham, L. W.,
Shen, J., Rabinovici, G. D., Rosen, H. J., Miller, B. L., Hyman, B. T.,
Schellenberg, G. D., Karlsen, T. H., Andreassen, O. A., Dale, A. M.,
and Desikan, R. S. (2016) Association between genetic traits for
immune-mediated diseases and Alzheimer’s disease, JAMA
Neurol., 73, 691-697.
37.Allen, M., Kachadoorian, M., Carrasquillo, M. M.,
Karhade, A., Manly, L., Burgess, J. D., Wang, C., Serie, D., Wang, X.,
Siuda, J., Zou, F., Chai, H. S., Younkin, C., Crook, J., Medway, C.,
Nguyen, T., Ma, L., Malphrus, K., Lincoln, S., Petersen, R. C.,
Graff-Radford, N. R., Asmann, Y. W., Dickson, D. W., Younkin, S. G.,
and Ertekin-Taner, N. (2015) Late-onset Alzheimer’s disease risk
variants mark brain regulatory loci, Neurol. Genet., 1,
e15.
38.Dexter, D., and Jenner, P. (2013) Parkinson
disease: from pathology to molecular disease mechanisms, Free Radic.
Biol. Med., 62, 132-144.
39.Jenner, P., and Olanow, C. W. (2006) The
pathogenesis of cell death in Parkinson’s disease,
Neurology, 66, 10.
40.Spillantini, M. G., Schmidt, M. L., Lee, V.
M.-Y., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997)
α-Synuclein in Lewy bodies, Nature, 388,
839-840.
41.Van der Brug, M. P., Singleton, A., Gasser, T.,
and Lewis, P. A. (2015) Parkinson’s disease: from human genetics
to clinical trials, Sci. Transl. Med., 7, 205ps20.
42.Lill, C. M. (2016) Genetics of Parkinson’s
disease, Mol. Cell. Probes, 30, 386-396.
43.Wissemann, W. T., Hill-Burns, E. M., Zabetian, C.
P., Factor, S. A., Patsopoulos, N., Hoglund, B., Holcomb, C., Donahue,
R. J., Thomson, G., Erlich, H., and Payami, H. (2013) Association of
Parkinson disease with structural and regulatory variants in the HLA
region, Am. J. Hum. Genet., 93, 984-993.
44.International Parkinson Disease Genomics
Consortium, Nalls, M. A., Plagnol, V., Hernandez, D. G., Sharma, M.,
Sheerin, U.-M., Saad, M., Simon-Sanchez, J., Schulte, C., Lesage, S.,
Sveinbjornsdottir, S., Stefansson, K., Martinez, M., Hardy, J.,
Heutink, P., Brice, A., Gasser, T., Singleton, A. B., and Wood, N. W.
(2011) Imputation of sequence variants for identification of genetic
risks for Parkinson’s disease: a meta-analysis of genome-wide
association studies, Lancet, 377, 641-649.
45.Ahmed, I., Tamouza, R., Delord, M.,
Krishnamoorthy, R., Tzourio, C., Mulot, C., Nacfer, M., Lambert, J.-C.,
Beaune, P., Laurent-Puig, P., Loriot, M.-A., Charron, D., and Elbaz, A.
(2012) Association between Parkinson’s disease and the HLA-DRB1
locus, Mov. Disord., 27, 1104-1110.
46.Rai, E., and Wakeland, E. K. (2011) Genetic
predisposition to autoimmunity – what have we learned? Semin.
Immunol., 23, 67-83.
47.Rugbjerg, K., Friis, S., Ritz, B., Schernhammer,
E. S., Korbo, L., and Olsen, J. H. (2009) Autoimmune disease and risk
for Parkinson disease: a population-based case-control study,
Neurology, 73, 1462-1468.
48.Hammer, J., Valsasnini, P., Tolba, K., Bolin, D.,
Higelin, J., Takacs, B., and Sinigaglia, F. (1993) Promiscuous and
allele-specific anchors in HLA-DR-binding peptides, Cell,
74, 197-203.
49.Choi, N. M., Majumder, P., and Boss, J. M. (2011)
Regulation of major histocompatibility complex class II genes, Curr.
Opin. Immunol., 23, 81-87.
50.Bang, J., Spina, S., and Miller, B. L. (2015)
Frontotemporal dementia, Lancet, 386, 1672-1682.
51.Blauwendraat, C., Wilke, C., Simon-Sanchez, J.,
Jansen, I. E., Reifschneider, A., Capell, A., Haass, C.,
Castillo-Lizardo, M., Biskup, S., Maetzler, W., Rizzu, P., Heutink, P.,
and Synofzik, M. (2018) The wide genetic landscape of clinical
frontotemporal dementia: systematic combined sequencing of 121
consecutive subjects, Genet. Med., 20, 240-249.
52.Rohrer, J. D., Guerreiro, R., Vandrovcova, J.,
Uphill, J., Reiman, D., Beck, J., Isaacs, A. M., Authier, A., Ferrari,
R., Fox, N. C., Mackenzie, I. R. A., Warren, J. D., de Silva, R.,
Holton, J., Revesz, T., Hardy, J., Mead, S., and Rossor, M. N. (2009)
The heritability and genetics of frontotemporal lobar degeneration,
Neurology, 73, 1451-1456.
53.Compston, A., and Coles, A. (2008) Multiple
sclerosis, Lancet, 372, 1502-1517.
54.Moutsianas, L., Jostins, L., Beecham, A. H.,
Dilthey, A. T., Xifara, D. K., Ban, M., Shah, T. S., Patsopoulos, N.
A., Alfredsson, L., Anderson, C. A., Attfield, K. E., Baranzini, S. E.,
Barrett, J., Binder, T. M. C., Booth, D., Buck, D., Celius, E. G.,
Cotsapas, C., D’Alfonso, S., Dendrou, C. A., Donnelly, P.,
Dubois, B., Fontaine, B., Fugger, L., Goris, A., Gourraud, P.-A.,
Graetz, C., Hemmer, B., Hillert, J., Kockum, I., Leslie, S., Lill, C.
M., Martinelli-Boneschi, F., Oksenberg, J. R., Olsson, T., Oturai, A.,
Saarela, J., Sondergaard, H. B., Spurkland, A., Taylor, B., Winkelmann,
J., Zipp, F., Haines, J. L., Pericak-Vance, M. A., Spencer, C. C. A.,
Stewart, G., Hafler, D. A., Ivinson, A. J., Harbo, H. F., Hauser, S.
L., De Jager, P. L., Compston, A., McCauley, J. L., Sawcer, S., McVean,
G., Sawcer, S., and McVean, G. (2015) Class II HLA interactions
modulate genetic risk for multiple sclerosis, Nat. Genet.,
47, 1107-1113.
55.Smith, K. J., Pyrdol, J., Gauthier, L., Wiley, D.
C., and Wucherpfennig, K. W. (1998) Crystal structure of HLA-DR2
(DRA*0101, DRB1*1501) complexed with a peptide from human myelin basic
protein, J. Exp. Med., 188, 1511-1520.
56.Zota, V., Nemirovsky, A., Baron, R., Fisher, Y.,
Selkoe, D. J., Altmann, D. M., Weiner, H. L., and Monsonego, A. (2009)
HLA-DR alleles in amyloid-peptide autoimmunity: a highly immunogenic
role for the DRB1*1501 allele, J. Immunol., 183,
3522-3530.
57.Chang, D., Nalls, M. A., Hallgrimsdottir, I. B.,
Hunkapiller, J., van der Brug, M., Cai, F., International
Parkinson’s Disease Genomics Consortium; 23andMe Research Team,
Kerchner, G. A., Ayalon, G., Bingol, B., Sheng, M., Hinds, D., Behrens,
T. W., Singleton, A. B., Bhangale, T. R., and Graham, R. R. (2017) A
meta-analysis of genome-wide association studies identifies 17 new
Parkinson’s disease risk loci, Nat. Genet., 49,
1511-1516.
58.Hill-Burns, E. M., Factor, S. A., Zabetian, C.
P., Thomson, G., and Payami, H. (2011) Evidence for more than one
Parkinson’s disease-associated variant within the HLA region,
PLoS One, 6, e27109.
59.Ahmed, I., Tamouza, R., Delord, M.,
Krishnamoorthy, R., Tzourio, C., Mulot, C., Nacfer, M., Lambert, J.-C.,
Beaune, P., Laurent-Puig, P., Loriot, M.-A., Charron, D., and Elbaz, A.
(2012) Association between Parkinson’s disease and the HLA-DRB1
locus, Mov. Disord., 27, 1104-1110.
60.Pankratz, N., Beecham, G. W., DeStefano, A. L.,
Dawson, T. M., Doheny, K. F., Factor, S. A., Hamza, T. H., Hung, A. Y.,
Hyman, B. T., Ivinson, A. J., Krainc, D., Latourelle, J. C., Clark, L.
N., Marder, K., Martin, E. R., Mayeux, R., Ross, O. A., Scherzer, C.
R., Simon, D. K., Tanner, C., Vance, J. M., Wszolek, Z. K., Zabetian,
C. P., Myers, R. H., Payami, H., Scott, W. K., Foroud, T., and PD GWAS
Consortium (2012) Meta-analysis of Parkinson’s disease:
identification of a novel locus, RIT2, Ann. Neurol., 71,
370-384.
61.Field, J., Browning, S. R., Johnson, L. J.,
Danoy, P., Varney, M. D., Tait, B. D., Gandhi, K. S., Charlesworth, J.
C., Heard, R. N., Stewart, G. J., Kilpatrick, T. J., Foote, S. J.,
Bahlo, M., Butzkueven, H., Wiley, J., Booth, D. R., Taylor, B. V.,
Brown, M. A., Rubio, J. P., and Stankovich, J. (2010) A polymorphism in
the HLA-DPB1 gene is associated with susceptibility to multiple
sclerosis, PLoS One, 5, e13454.
62.De Jager, P. L., Jia, X., Wang, J., de Bakker, P.
I. W., Ottoboni, L., Aggarwal, N. T., Piccio, L., Raychaudhuri, S.,
Tran, D., Aubin, C., Briskin, R., Romano, S., International MS Genetics
Consortium, Baranzini, S. E., McCauley, J. L., Pericak-Vance, M. A.,
Haines, J. L., Gibson, R. A., Naeglin, Y., Uitdehaag, B., Matthews, P.
M., Kappos, L., Polman, C., McArdle, W. L., Strachan, D. P., Evans, D.,
Cross, A. H., Daly, M. J., Compston, A., Sawcer, S. J., Weiner, H. L.,
Hauser, S. L., Hafler, D. A., and Oksenberg, J. R. (2009) Meta-analysis
of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new
multiple sclerosis susceptibility loci, Nat. Genet., 41,
776-782.
63.Australia and New Zealand Multiple Sclerosis
Genetics Consortium (ANZgene) (2009) Genome-wide association study
identifies new multiple sclerosis susceptibility loci on chromosomes 12
and 20, Nat. Genet., 41, 824-828.
64.Richartz-Salzburger, E., Batra, A., Stransky, E.,
Laske, C., Kohler, N., Bartels, M., Buchkremer, G., and Schott, K.
(2007) Altered lymphocyte distribution in Alzheimer’s disease,
J. Psychiatr. Res., 41, 174-178.
65.Wyss-Coray, T. (2006) Inflammation in
Alzheimer’s disease: driving force, bystander or beneficial
response? Nat. Med., 12, 1005-1015.
66.Hirsch, E. C., and Hunot, S. (2009)
Neuroinflammation in Parkinson’s disease: a target for
neuroprotection? Lancet Neurol., 8, 382-397.
67.Sims, R., van der Lee, S. J., Naj, A. C.,
Bellenguez, C., Badarinarayan, N., Jakobsdottir, J., Kunkle, B. W.,
Boland, A., Raybould, R., Bis, J. C., Martin, E. R., Grenier-Boley, B.,
Heilmann-Heimbach, S., Chouraki, V., Kuzma, A. B., et al. (2017) Rare
coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated
innate immunity in Alzheimer’s disease, Nat. Genet.,
49, 1373-1384.
68.Simon-Sanchez, J., van Hilten, J. J., van de
Warrenburg, B., Post, B., Berendse, H. W., Arepalli, S., Hernandez, D.
G., de Bie, R. M. A., Velseboer, D., Scheffer, H., Bloem, B., van Dijk,
K. D., Rivadeneira, F., Hofman, A., Uitterlinden, A. G., Rizzu, P.,
Bochdanovits, Z., Singleton, A. B., and Heutink, P. (2011) Genome-wide
association study confirms extant PD risk loci among the Dutch, Eur.
J. Hum. Genet., 19, 655-661.
69.Heppner, F. L., Ransohoff, R. M., and Becher, B.
(2015) Immune attack: the role of inflammation in Alzheimer’s
disease, Nat. Rev. Neurosci., 16, 358-372.
70.Prinz, M., and Priller, J. (2017) The role of
peripheral immune cells in the CNS in steady state and disease, Nat.
Neurosci., 20, 136-144.
71.Ransohoff, R. M. (2016) How neuroinflammation
contributes to neurodegeneration, Science, 353,
777-783.
72.Li, Q., and Barres, B. A. (2017) Microglia and
macrophages in brain homeostasis and disease, Nat. Rev.
Immunol., 18, 225-242.
73.Hayes, G. M., Woodroofe, M. N., and Cuzner, M. L.
(1987) Microglia are the major cell type expressing MHC class II in
human white matter, J. Neurol. Sci., 80, 25-37.
74.Harrison, I. F., Smith, A. D., and Dexter, D. T.
(2018) Pathological histone acetylation in Parkinson’s disease:
neuroprotection and inhibition of microglial activation through SIRT 2
inhibition, Neurosci. Lett., 666, 48-57.
75.Sanchez-Guajardo, V., Tentillier, N., and
Romero-Ramos, M. (2015) The relation between α-synuclein and
microglia in Parkinson’s disease: recent developments,
Neuroscience, 302, 47-58.
76.Radford, R. A., Morsch, M., Rayner, S. L., Cole,
N. J., Pountney, D. L., and Chung, R. S. (2015) The established and
emerging roles of astrocytes and microglia in amyotrophic lateral
sclerosis and frontotemporal dementia, Front. Cell. Neurosci.,
9, 414.
77.Bullido, M. J., Martinez-Garcia, A., Artiga, M.
J., Aldudo, J., Sastre, I., Gil, P., Coria, F., Munoz, D. G.,
Hachinski, V., Frank, A., and Valdivieso, F. (2007) A TAP2 genotype
associated with Alzheimer’s disease in APOE4 carriers,
Neurobiol. Aging, 28, 519-523.
78.Marsh, S. E., Abud, E. M., Lakatos, A.,
Karimzadeh, A., Yeung, S. T., Davtyan, H., Fote, G. M., Lau, L.,
Weinger, J. G., Lane, T. E., Inlay, M. A., Poon, W. W., Blurton-Jones,
M., and Mcewen, B. S. (2016) The adaptive immune system restrains
Alzheimer’s disease pathogenesis by modulating microglial
function, Proc. Natl. Acad. Sci. USA, 1, E1316-1325.
79.Bryson, K. J., and Lynch, M. A. (2016) Linking T
cells to Alzheimer’s disease: from neurodegeneration to
neurorepair, Curr. Opin. Pharmacol., 26, 67-73.
80.Brochard, V., Combadiere, B., Prigent, A.,
Laouar, Y., Perrin, A., Beray-Berthat, V., Bonduelle, O.,
Alvarez-Fischer, D., Callebert, J., Launay, J.-M., Duyckaerts, C.,
Flavell, R. A., Hirsch, E. C., and Hunot, S. (2008) Infiltration of
CD4+ lymphocytes into the brain contributes to neurodegeneration in a
mouse model of Parkinson disease, J. Clin. Invest., 119,
182-192.
81.Lopes Pinheiro, M. A., Kooij, G., Mizee, M. R.,
Kamermans, A., Enzmann, G., Lyck, R., Engelhardt, B., and de Vries, H.
E. (2016) Immune cell trafficking across the barriers of the central
nervous system in multiple sclerosis and stroke, Biochim. Biophys.
Acta, 1862, 461-471.
82.Alexianu, M. E., Mohamed, A. H., Smith, R. G.,
Colom, L. V., and Appel, S. H. (2002) Apoptotic cell death of a hybrid
motoneuron cell line induced by immunoglobulins from patients with
amyotrophic lateral sclerosis, J. Neurochem., 63,
2365-2368.
83.Neff, F., Wei, X., Nolker, C., Bacher, M., Du,
Y., and Dodel, R. (2008) Immunotherapy and naturally occurring
autoantibodies in neurodegenerative disorders, Autoimmun. Rev.,
7, 501-507.
84.Papachroni, K. K., Ninkina, N., Papapanagiotou,
A., Hadjigeorgiou, G. M., Xiromerisiou, G., Papadimitriou, A.,
Kalofoutis, A., and Buchman, V. L. (2006) Autoantibodies to
alpha-synuclein in inherited Parkinson’s disease, J.
Neurochem., 101, 749-756.
85.Sweeney, M. D., Sagare, A. P., and Zlokovic, B.
V. (2018) Blood-brain barrier breakdown in Alzheimer disease and other
neurodegenerative disorders, Nat. Rev. Neurol., 14,
133-150.
86.Carbone, I., Lazzarotto, T., Ianni, M.,
Porcellini, E., Forti, P., Masliah, E., Gabrielli, L., and Licastro, F.
(2014) Herpes virus in Alzheimer’s disease: relation to
progression of the disease, Neurobiol. Aging, 35,
122-129.
87.Letenneur, L., Peres, K., Fleury, H., Garrigue,
I., Barberger-Gateau, P., Helmer, C., Orgogozo, J.-M., Gauthier, S.,
and Dartigues, J.-F. (2008) Seropositivity to herpes simplex virus
antibodies and risk of Alzheimer’s disease: a population-based
cohort study, PLoS One, 3, e3637.
88.Barnes, L. L., Capuano, A. W., Aiello, A. E.,
Turner, A. D., Yolken, R. H., Torrey, E. F., and Bennett, D. A. (2015)
Cytomegalovirus infection and risk of Alzheimer disease in older black
and white individuals, J. Infect. Dis., 211, 230-237.
89.Goddard, C. A., Butts, D. A., and Shatz, C. J.
(2007) Regulation of CNS synapses by neuronal MHC class I, Proc.
Natl. Acad. Sci. USA, 104, 6828-6833.
90.Boulanger, L. M. (2009) Immune proteins in brain
development and synaptic plasticity, Neuron, 64,
93-109.
91.Lee, H., Brott, B. K., Kirkby, L. A, Adelson, J.
D., Cheng, S., Feller, M. B., Datwani, A., and Shatz, C. J. (2014)
Synapse elimination and learning rules co-regulated by MHC class I
H2-Db, Nature, 509, 195-200.
92.Vagaska, B., New, S. E. P., Alvarez-Gonzalez, C.,
D’Acquisto, F., Gomez, S. G., Bulstrode, N. W., Madrigal, A., and
Ferretti, P. (2016) MHC-class-II are expressed in a subpopulation of
human neural stem cells in vitro in an IFNγ-independent
fashion and during development, Sci. Rep., 6, 24251.
93.De Miranda, A. S., Zhang, C.-J., Katsumoto, A.,
and Teixeira, A. L. (2017) Hippocampal adult neurogenesis: does the
immune system matter? J. Neurol. Sci., 372, 482-495.
94.Derecki, N. C., Cardani, A. N., Yang, C. H.,
Quinnies, K. M., Crihfield, A., Lynch, K. R., and Kipnis, J. (2010)
Regulation of learning and memory by meningeal immunity: a key role for
IL-4, J. Exp. Med., 207, 1067-1080.
95.Filiano, A. J., Xu, Y., Tustison, N. J., Marsh,
R. L., Baker, W., Smirnov, I., Overall, C. C., Gadani, S. P., Turner,
S. D., Weng, Z., Peerzade, S. N., Chen, H., Lee, K. S., Scott, M. M.,
Beenhakker, M. P., Litvak, V., and Kipnis, J. (2016) Unexpected role of
interferon-γ in regulating neuronal connectivity and social
behaviour, Nature, 535, 425-429.
96.Winner, B., and Winkler, J. (2015) Adult
neurogenesis in neurodegenerative diseases, Cold Spring Harb.
Perspect. Biol., 7, a021287.
97.Erny, D., and Prinz, M. (2017) Microbiology: gut
microbes augment neurodegeneration, Nature, 544,
304-305.
98.Sharon, G., Sampson, T. R., Geschwind, D. H., and
Mazmanian, S. K. (2016) The central nervous system and the gut
microbiome, Cell, 167, 915-932.
99.Minter, M. R., Zhang, C., Leone, V., Ringus, D.
L., Zhang, X., Oyler-Castrillo, P., Musch, M. W., Liao, F., Ward, J.
F., Holtzman, D. M., Chang, E. B., Tanzi, R. E., and Sisodia, S. S.
(2016) Antibiotic-induced perturbations in gut microbial diversity
influences neuro-inflammation and amyloidosis in a murine model of
Alzheimer’s disease, Sci. Rep., 6, 30028.