Lymphocyte Phosphatase-Associated Phosphoprotein Is a Substrate of Protein Kinase CK2
T. D. Tsoy1,2,a, N. A. Kruglova1,2, and A. V. Filatov1,2,b*
1Institute of Immunology National Research Center, Federal Medical-Biological Agency, 115522 Moscow, Russia2Lomonosov Moscow State University, Faculty of Biology, 119234 Moscow, Russia
* To whom correspondence should be addressed.
Received June 26, 2018; Revision received August 14, 2018
Lymphocyte phosphatase-associated phosphoprotein (LPAP) is a molecular partner of CD45 phosphatase that plays a key role in the regulation of antigen-specific activation of lymphocytes. The functions of LPAP still remain unknown. We believe that studying LPAP phosphorylation pathways could shed light on its functions. In this work, we studied the phosphorylation of LPAP ectopically expressed in non-lymphoid cells in order to determine the effect of LPAP interaction partners on its phosphorylation. We found that phosphorylation at Ser153 and Ser163 in non-hematopoietic HEK293 cells was conserved, while phosphorylation at Ser99 and Ser172 was almost absent. The pattern of LPAP phosphorylation in K562 erythroid and U937 myeloid cells expressing endogenous CD45 protein was similar to that observed in T and B lymphocytes. We demonstrated for the first time that LPAP is a substrate for protein kinase CK2 that phosphorylates it at Ser153, presumably ensuring LPAP resistance to degradation.
KEY WORDS: LPAP, phosphorylation, ectopic expression, casein kinase 2, human lymphocytesDOI: 10.1134/S0006297918110081
Abbreviations: CIP, calf intestinal phosphatase; CK2, casein kinase 2; 2D-DIGE, two-dimensional difference gel electrophoresis; DTT, dithiothreitol; IEF, isoelectric focusing; LPAP, lymphocyte phosphatase-associated phosphoprotein; PAGE, polyacrylamide gel electrophoresis; PMA, phorbol 12-myristate 13-acetate; SDS, sodium dodecyl sulfate; TBB, 4,5,6,7-tetrabromo-2-azabenzimidazole; WB, Western blotting.
Lymphocyte phosphatase-associated phosphoprotein (LPAP) was identified
in 1991 as a protein tightly associated with the CD45 phosphatase [1]. Initially, it was suggested that LPAP is as a
substrate of CD45 or a regulator of its activity. To test this
hypothesis, three research groups independently generated knockout (KO)
mouse lines deficient by the PTPRCAP gene (LPAP-encoding gene);
however, the phenotypes of the KO and wild-type mice almost did not
differ [2-4].
Later, it was shown that LPAP affects B lymphocyte proliferation and survival [5]. Some data indicated that LPAP cooperates with the c-myc gene product during malignant transformation of pre-B cells [6]. Moreover, single nucleotide polymorphism in the PTPRCAP regulatory region was linked to the development of the diffuse-type gastric cancer [7]. Apart from CD45, LPAP interacts with the CD4 co-receptor and Lck kinase [8, 9]. Nevertheless, the function of LPAP still remains unknown.
The phosphorylation status of LPAP depends on the cell activation state. Hence, LPAP phosphorylation may be a key to the function of this protein. Identification of kinases and phosphatases involved in LPAP phosphorylation/dephosphorylation might help to determine which signaling pathways are mediated by LPAP. Previously, we described four phosphorylation sites in LPAP: Ser99, Ser153, Ser163, and Ser172 [10]. Phosphorylation at these residues produces at least six different LPAP phosphorylated forms. Lymphoid cells, including T, B, and NK lymphocytes, display different patterns of LPAP phosphorylation [11], which may be explained by different representation of LPAP interaction partners in these cells.
In this work, we aimed, first, to elucidate if the LPAP phosphorylation pattern observed in lymphocytes was unique for these cells or could also be found in non-lymphoid cells. This comparison might help to identify potential kinases and phosphatases for LPAP. Also, it might help to conclude if these kinases and phosphatases are constitutively active or inducible, as well as if they are ubiquitously expressed or cell-line specific. Secondly, we also attempted to estimate the influence of LPAP protein partners on LPAP phosphorylation in cell lines of different origin. The third objective was to test the hypothesis formulated earlier [10] that LPAP might serve as a substrate for protein kinase CK2 (CK2).
MATERIALS AND METHODS
Cell cultures, antibodies. Human CEM, Jurkat, K562, U937, and HEK293 cells were cultured in DMEM/F12 media supplemented with 10% fetal calf serum, L-glutamine (4 mM), and gentamycin (80 mg/liter) (PanEco, Russia) in a humidified atmosphere with 5% CO2 at 37°C. Antibodies CL7 and LT45 against LPAP and CD45, respectively, and phospho-specific antibodies were obtained by us earlier [10, 11]. 4,5,6,7-Tetra-bromo-2-azabenzimidazole (TBB; Santa Cruz Biotechnology, USA) and CX-4945 (Selleckchem, Germany) were used for the inhibition of CK2.
Plasmids and cell transfection. Plasmids coding for wild-type and mutant LPAP were generated by us earlier [11]. The PTPRCAP gene was cloned into the pCMVpA expression vector and the pUCHR IRES GFP lentiviral vector (Addgene, USA). For transfection, HEK293 cells were seeded in 6-well plates at 5·105 cells per well in 2.5 ml of the medium. The next day, the cells were transfected with the corresponding plasmids (4 μg per well) using Lipofectamine 2000 (Invitrogen, USA). In the case of transient transfection with the pCMVpA expression vector, the cells were collected for analysis 48 h after transfection. To generate lentiviral particles, HEK293 cells were transfected with three plasmids: pUCHR LPAP IRES GFP (2.2 μg), packaging plasmid pCMV-Δ8.2R (1.45 μg), and pCMV VSVG (0.45 μg) coding for the vesicular stomatitis Indiana virus (VSV) G protein (all plasmids were from Addgene). Forty-eight hours later, the supernatant with the lentiviruses was collected, filtered, and added to the target cells for stable transfection. LPAP expression was analyzed 4 days after transfection by immunofluorescence and Western blotting.
Immunoprecipitation. Cells (1·107) were lysed in 250 μl of ice-cold buffer containing 1% Triton X-100, 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 5 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 10 mM NaF, and 1 mM Na3VO4 for 30 min at 4°C (all reagents were purchased from Merck, USA). After 30-min incubation on ice, cell debris was pelleted by centrifugation at 20,000g for 25 min at 4°C. The lysates were cleared twice at 4°C by 1-h incubation with 50 μl of 50% (v/v) suspension of BrCN-Sepharose (Thermo Fisher Scientific, USA) coupled to normal mouse IgG. Immunoprecipitation was carried out with the CL7 antibody immobilized on AffiGel-Hz support (Bio-Rad, USA). The immunoprecipitated complexes were washed three times and the bound proteins were eluted by incubation with the SDS-PAGE sample buffer (62.5 mM Tris-HCl (pH 6.8), 10% glycerol, 2% SDS, 5% 2-mercaptoethanol, 0.05% Bromophenol blue) at 80°C for 5 min.
One-dimensional and two-dimensional electrophoresis. Eluted samples were fractionated by Laemmli’s SDS-PAGE in 12 or 18% polyacrylamide gels under reducing conditions. Pre-stained protein markers were from Thermo Fisher Scientific. Phosphorylated proteins were detected in the gel with the Pro-Q Diamond stain (Invitrogen) and visualized with a Molecular Imager FX Pro fluorescent scanner (Bio-Rad). For 2-dimensional difference gel electrophoresis (2D-DIGE), samples labeled with Cy3 and Cy5 were mixed and then fractionated by isoelectric focusing (IEF) on Immobiline DryStrip pH 4-7 strips (GE Healthcare, USA) with Ettan IPGphor 3 (GE Healthcare). The strips were then equilibrated in the reducing solution (50 mM Tris-HCl (pH 6.8), 6 M urea, 30% glycerol, 2% SDS, 1% dithiothreitol (DTT)) and fractionated by 18% SDS-PAGE. Proteins in the gels were visualized with an Amersham Imager 600RGB (GE Healthcare).
Western blotting. After SDS-PAGE, proteins were transferred to PVDF membranes by the semi-dry method. The membranes were blocked with 5% (w/v) fat-free milk in PBS containing 0.1% Tween 20 and incubated for 1 h with primary antibodies. Antibody CL7 conjugated to horseradish peroxidase was used for detection of total LPAP. Polyclonal phospho-specific antibodies (designated as pSer99, pSer153, and pSer163) were used for the detection of LPAP phosphorylated at Ser99, Ser153, and pSer163, respectively. The membranes were washed and incubated with goat antibodies against mouse IgG conjugated with horseradish peroxidase (GE Healthcare). The blots were then visualized with a ChemiDoc XRS System (Bio-Rad) using chemiluminescence reagents (Millipore, USA).
LPAP phosphorylation and dephosphorylation in vitro. LPAP was immunoprecipitated using AffiGel-CL7 and transferred into 50 μl of buffer containing 50 mM Tris-HCl (pH 7.6), 100 mM NaCl, 10 mM MgCl2, and 1 mM DTT. For dephosphorylation, 10 units of calf intestinal phosphatase (CIP; SibEnzyme, Russia) were added, and the reaction mixture was incubated for 1 h at 37°C. The beads were washed with 50 mM Tris-HCl (pH 7.6) and transferred into 50 μl of the CK2 buffer (50 mM Tris-HCl, 10 mM MgCl2, 0.1 mM EDTA, 2 mM DTT, 0.01% Brij-35, pH 7.5). Phosphorylation was carried out by adding 500 units of recombinant CK2 (New England Biolabs, USA) and 200 μM ATP. The beads were incubated for 1 h at 30°C and then washed with 50 mM Tris-HCl (pH 7.6). The proteins were eluted with the sample buffer and analyzed by SDS-PAGE.
RESULTS
Phosphorylation of LPAP ectopically expressed in non-lymphoid cell lines. Endogenous LPAP from lymphoid cells displayed a characteristic pattern when resolved in 18% PAGE. Apart from the major band at 32 kDa, there was a satellite band migrating at 28 kDa that disappeared after CIP treatment. Similar electrophoretic pattern was also observed for LPAP from the T lymphoblastoid cell line CEM (Fig. 1a). As demonstrated earlier, the satellite band is LPAP phosphorylated at Ser172. HEK293 cells are commonly used for ectopic expression of proteins due to their high transfection efficiency. In our study, we used HEK293 cells because they do not belong to hematopoietic lineage and do not express LPAP and its binding partners CD45, CD4, and Lck.
Fig. 1. Phosphorylation of LPAP ectopically expressed in non-hematopoietic HEK293 cells. a) Comparison of electrophoretic mobility of LPAP from CEM and HEK293 cells; b, c) phosphorylation of point and truncated LPAP mutants, respectively; wild-type (wt) LPAP is shown for comparison; d) 2D-DIGE of S99A and S153A LPAP mutants. LPAP dephosphorylated with CIP was labelled with Cy3 (lower panel); untreated LPAP was labelled with Cy5 (middle panel). Proteins were separated by SDS-PAGE in 18% (a, b, d) or 12% (c) gels. The gels were visualized with a fluorescent imager (d) or stained with the phospho-specific Pro-Q Diamond dye (b, lower panel, and c); proteins were transferred to the PVDF-membrane and probed with mAb CL7 against total LPAP (a and b, upper panel).
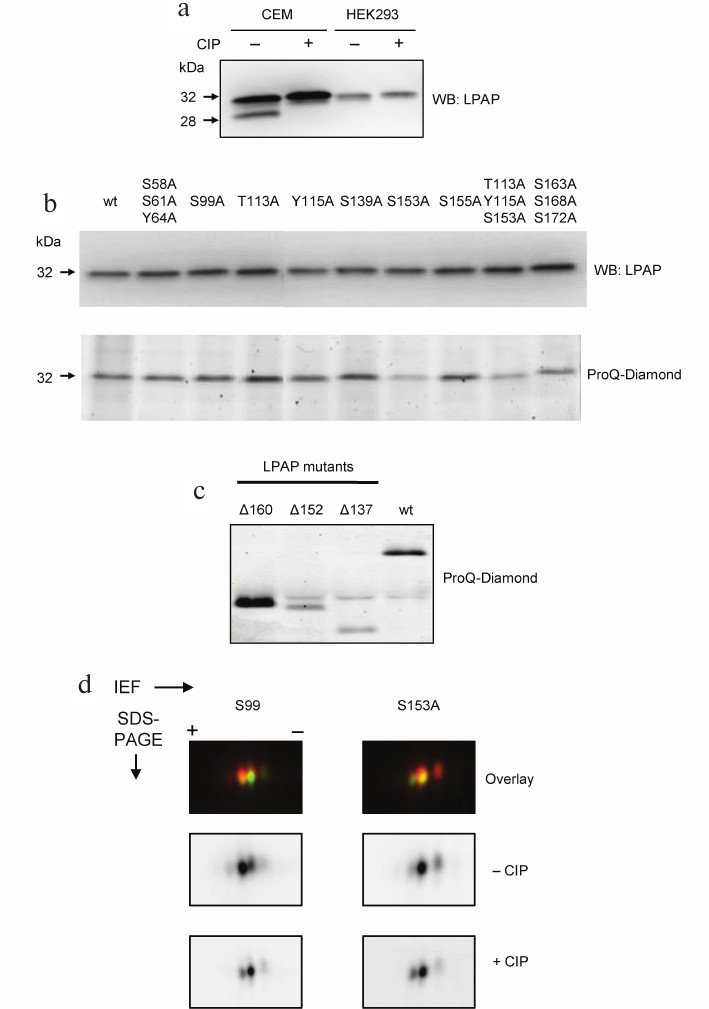
LPAP was successfully expressed in HEK293 cells after both transient and stable transfection. When resolved by SDS-PAGE, it migrated as a single band at 32 kDa without any additional band in the low-molecular-weight region of the gel (Fig. 1a). Therefore, we concluded that LPAP expressed in HEK293 is not phosphorylated at Ser172. However, it did not rule out the possibility that HEK293 cells contain other LPAP phosphorylated forms that do not differ in their electrophoretic mobility from the non-phosphorylated LPAP. To test this, we generated a series of LPAP point mutants in which Ser, Thr, and Tyr residues predicted to be phosphorylated were replaced with Ala. Some mutants contained two or three substitutions. Phosphorylation of LPAP mutants was analyzed by SDS-PAGE and gel staining with Pro-Q Diamond that binds only to phosphorylated proteins. Figure 1b (lower panel) shows that all LPAP point mutants produced the same signal except the mutants containing S153A. The signal intensity for S153A and T113A/Y115A/S153A was approximately two times lower than for the other mutants and wild-type (wt) LPAP. The S163A/S168A/S172A mutant also produced slightly lower signal. All lanes had the same protein load, as evidenced from Fig. 1b, upper panel.
The results obtained for mutant LPAPs were corroborated by the data on LPAP truncated forms. As is shown in Fig. 1c, full-length LPAP (wt) produced a strong signal with Pro-Q Diamond. The signal intensity for the Δ160 mutant lacking the C-terminal sequence (a.a. 160-206) was even higher than for wt LPAP. However, when larger molecules fragments were removed, as in the mutants Δ153 (deleted a.a. 153-206) and Δ138 (deleted a.a. 138-206), the intensity of the Pro-Q Diamond signal strongly decreased. It should be noted that the sequence between Asp152 and Val160 contains only one phosphorylation site, Ser153. Therefore, LPAP phosphorylated at Ser153 is the main phosphorylated form of ectopically expressed LPAP in HEK293 cells.
2D-DIGE confirmed that LPAP expressed in HEK293 cells was represented by two forms. On 2D gels, LPAP migrated as two spots with relatively similar intensity (Fig. 1d). The spot in the acidic area of the gel disappeared after CIP treatment and, therefore, corresponded to the phosphorylated LPAP. By contrast, the intensity of the spot in the basic area increased after CIP treatment. This spot corresponded to the nonphosphorylated LPAP. Moreover, the spot in the acidic area was absent in the S153A mutant but not in the other mutants, for example S99A.
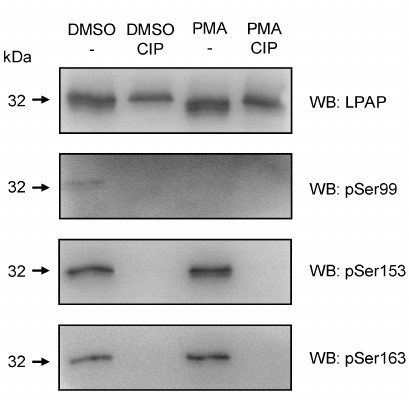
Finally, different phosphorylated forms of LPAP were detected by Western blotting using phospho-specific antibodies. The most intense signal was generated by antibodies against pSer153 (Fig. 2). The level of Ser153 phosphorylation did not change after cell activation. The content of pSer163-LPAP was low but increased after cell activation with phorbol 12-myristate 13-acetate (PMA). Antibodies against the pSer99 detected a very faint band that disappeared after cell activation with PMA.
Fig. 2. Phosphorylation of LPAP ectopically expressed in non-hematopoietic HEK293 cells detected by phospho-specific antibodies. LPAP was purified from the cells treated with dimethyl sulfoxide (DMSO) (vehicle) or activated with PMA. Control samples were incubated with CIP. The samples were resolved by 18% SDS-PAGE, transferred to PVDF membrane, and probed with mAb CL7 against total LPAP or with phospho-specific antibodies.
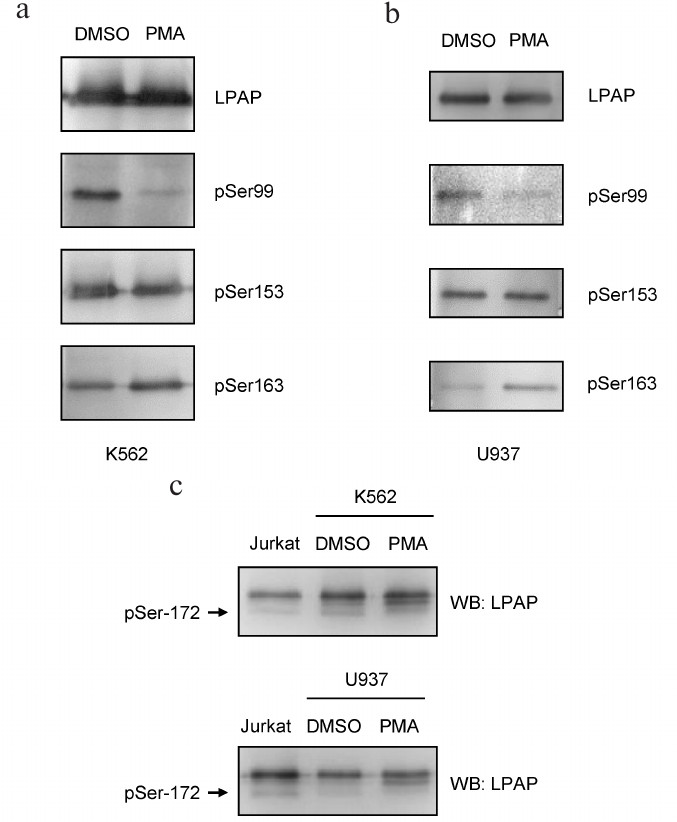
Phosphorylation of LPAP ectopically expressed in erythroid and myeloid cells. To express LPAP in hematopoietic cells, we chose non-lymphoid K562 and U937 cell lines. Since chemical transfection of these cells is inefficient, we used stable transfection with the lentiviral vectors. Similar to HEK293 cells, pSer153-LPAP was constitutively present in K562 and U937 cells (Fig. 3). pSer163-LPAP was also detected in HEK293 cells; its level increased after cell activation with PMA. pSer99-LPAP was clearly detectable in resting cells; however, its amount strongly decreased after cell activation. To detect pSer172-LPAP, we used SDS-PAGE because this phosphorylated form could be separated from other phospho-species due to higher electrophoretic mobility in 18% gel. The signal of pSer172-LPAP was strong in K562 cells and very weak in U937 cells and disappeared completely after cell activation with PMA.
Fig. 3. Phosphorylation of LPAP ectopically expressed in erythroid K562 cells and myeloid U937 cells. a, b) 12% SDS-PAGE; c) 18% SDS-PAGE; the first lane is loaded with LPAP from Jurkat cells as a control for Ser172 phosphorylation. LPAP was purified from the cells treated with DMSO (vehicle) or cells activated with PMA. Western blotting (WB) was performed with mAb CL7 against total LPAP or phospho-specific antibodies.
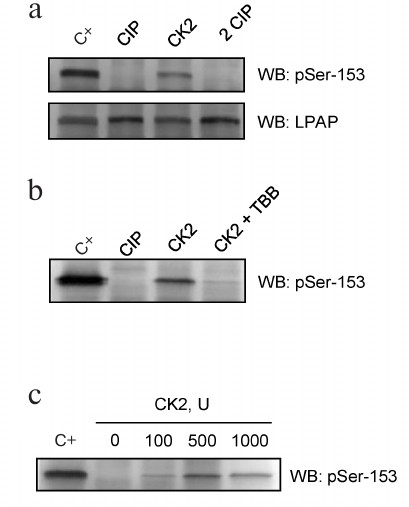
Protein kinase CK2 phosphorylates LPAP at Ser153. Our experiments demonstrated that LPAP was phosphorylated at Ser153 in cells of different origin independently of the cell activation state. Previously, we hypothesized that this residue is phosphorylated by CK2 [10]. First, the ability of CK2 to phosphorylate LPAP was shown in vitro. LPAP was purified from Jurkat cells (Fig. 4a; C+) and dephosphorylated by treatment with CIP. Incubation with the recombinant CK2 and ATP restored the LPAP phosphorylation status, while subsequent treatment with CIP resulted in LPAP dephosphorylation (Fig. 4a; 2 CIP). The level of LPAP phosphorylation at Ser153 was analyzed by Western blotting using phospho-specific antibodies. Blot staining with mAb CL7 confirmed that the amount of LPAP did not change after the treatments (Fig. 4a; lower panel). When the reaction was performed in the presence of 1 μM TBB (CK2 inhibitor), no Ser153 phosphorylation was detected (Fig. 4b). The effect of CK2 was dose-dependent with the maximum phosphorylation observed when 500 units of the enzymes were added to the reaction mixture (Fig. 4c). This concentration of CK2 led to LPAP phosphorylation that was very close to the phosphorylation level of LPAP isolated from cells.
Fig. 4. LPAP phosphorylation in vitro by recombinant CK2. a) LPAP purified from Jurkat cells (C+) was subjected to dephosphorylation by CIP, then phosphorylated by recombinant CK2, and dephosphorylated again by CIP (2 CIP); b) samples in the lanes C+, CIP, and CK2 were prepared similarly to the samples in panel (a); the sample in the CK2+TBB lane was phosphorylated in the presence of TBB (1 μM); c) LPAP phosphorylation with different amounts of CK2. LPAP was purified from Jurkat cells and subjected to 12% SDS-PAGE followed by Western blotting (WB) with phospho-specific antibodies against pSer153 or mAb CL7 against total LPAP.
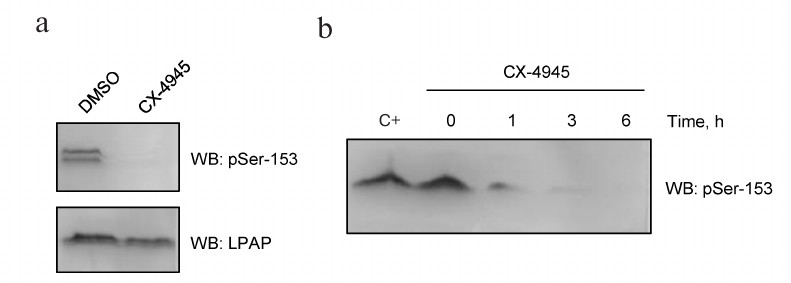
In order to block CK2 activity in vivo, we used its more potent inhibitor CX-4945 (Silmitasertib). CX-4945 has a high affinity to the ATP-binding domain of CK2 and blocks the kinase catalytic subunit. The inhibitory concentration (IC50) of CX-4945 in Jurkat cells is as low as 0.1 μM [12]. When Jurkat cells were incubated with 10 μM CX-4945 for 4 h, antibodies against pSer153 were no longer able to stain LPAP (Fig. 5a, upper panel). Western blotting of cell lysates revealed a non-specific band above LPAP. Control staining with mAb CL7 showed that the total protein load was the same (Fig. 5a, lower panel). The effect of CX-4945 was already detectable after 1 h of incubation (Fig. 5b); after 6 h, Ser153 was completely dephosphorylated.
Fig. 5. CX-4945 inhibitor blocks LPAP phosphorylation at Ser153. a) Jurkat cells treated with 10 µM CX-4945 or DMSO (vehicle control); b) LPAP phosphorylation at different inhibition times. C+, without inhibitor. Cell lysates were fractionated by 12% SDS-PAGE and analyzed by Western blotting (WB) with phospho-specific antibodies against pSer153 or mAb CL7 against total LPAP.
DISCUSSION
Transfection of HEK293 with LPAP-bearing genetic constructs demonstrated that LPAP could be expressed at high levels in non-lymphoid cells lacking CD45. This contradicts the earlier observation that LPAP degrades rapidly in the absence of CD45 [1]. It is possible that such unusual stability of LPAP can be explained by its specific phosphorylation in non-lymphoid cells. The most pronounced characteristic of LPAP expressed in HEK293 cells is the total absence of its phosphorylation at Ser172. Moreover, the level of LPAP phosphorylation at Ser99 is also very low, which might be explained by the fact that kinases responsible for LPAP phosphorylation at Ser99 and Ser172 are not expressed in HEK293. We also cannot exclude the possibility that in order to be phosphorylated at these residues, LPAP has to form a complex with CD45 and/or CD4 that are present only in lymphoid cells. It should be noted that LPAP expressed in the non-lymphoid HEK293 cells was nevertheless phosphorylated at Ser153 and Ser163.
Then, we transfected erythroid cells K562 and myeloid cells U937 with the PTPRCAP gene so that the conditions of LPAP ectopic expression were closer to those in T and B cells. The choice of these cell lines was dictated by the fact that K562 and U937 cells do not express endogenous LPAP but have high levels of CD45. Moreover, U937 cells express the CD4 co-receptor. Therefore, LPAP expression in K562 and U937 allowed us to reconstitute, at least partially, the molecular complex observed in T and B lymphocytes. Under these conditions, LPAP phosphorylation at Ser99 and Ser172 was restored. After cell activation, this phosphorylation disappeared, similarly to what happens in T and B cells.
Phosphorylation at Ser163 was detected in all the cell lines tested. However, its dependence on the cell activation state was different in different cells. Thus, the extent of Ser163 phosphorylation was strongly regulated by the cell activation in U937 cells but remained virtually independent on the cell activation state in K562 cells. Analysis of LPAP phosphorylation in various cell lines failed to find a correlation between the LPAP phosphorylation pattern and expression of different CD45 isoforms.
The kinases responsible for LPAP phosphorylation at Ser99, Ser163, and Ser172 are still under debate. These residues are not located in the well-known consensus motifs recognized by particular kinases. By contrast, Ser153 is positioned in the acidic motif specific for CK2. We demonstrated that CK2 phosphorylates immunopurified LPAP in vitro. This result was corroborated by the fact that CX-4945, a specific inhibitor of CK2 [12], blocked Ser153 phosphorylation in vivo. CK2 is a widely expressed kinase with a constitutive activity [13]. In accordance with these data, we observed that the majority of LPAP molecules were phosphorylated at Ser153 in all cell types regardless of the cell activation state.
CK2 phosphorylates Ser/Thr residues in the consensus motifs that can have up to five acidic amino acids. The minimal consensus for CK2 is S/T–X–X–D/E [14, 15]. Ser153 is located in the S–D–T–E sequence that fits into this consensus. The activity of CK2 towards Ser153 can be enhanced by aspartic and glutamic acid residues located at positions –1 and +5 [14].
CK2 mediates a variety of cellular processes, including cell proliferation, apoptosis, transcription, and translation [16]. Among CK2 activities, we would like to highlight its role in the PI3K/Akt-pathway, in which CK2 controls Akt phosphorylation at Ser129 [17]. In lymphocytes, CK2 performs specific functions, e.g., regulates CD4+ T cell differentiation towards TH17 or Treg [18].
CK2 phosphorylation can control the stability of the corresponding substrates. Some proteins, such as transcription factor ATF4 [19], suppressor PML [20], and VHL protein [21], are directed to degradation after CK2 phosphorylation. Other proteins, including Bid [22], transcriptions factors max [23] and myc [24], PTEN phosphatase [25], connexin 45.6 [26], and presenilin [27] become more stable after phosphorylation by CK2.
Importantly, Ser153 is preceded by the sequence 148–E–A–R–D–S–154 that to some extent corresponds to the consensus motif D/V–G/A/T/S/N–X–D–Z cleaved by caspase-3 [28]. It is possible that phosphorylation at Ser153 protects LPAP from degradation by caspase-3.
CK2 is one of the most pleiotropic kinases with more than 300 protein substrates [13, 14], the most common ones being transcription factors, nuclear proteins, and signaling molecules. Our work has added a new target, LPAP, to this list.
Funding
This work was supported by the Russian Foundation for Basic Research (projects 18-34-00705 and 17-04-00526).
Conflict of Interests
The authors declare no conflict of interests.
Ethical Approval
This paper does not include description of research on humans or animals.
REFERENCES
1.Schraven, B., Schoenhaut, D., Bruyns, E., Koretzky,
G., Eckerskorn, C., Wallich, R., Kirchgessner, H., Sakorafas, P.,
Labkovsky, B., Ratnofsky, S., and Meuer, S. (1994) LPAP, a novel 32-kDa
phosphoprotein that interacts with CD45 in human lymphocytes, J.
Biol. Chem., 269, 29102-29111.
2.Matsuda, A., Motoya, S., Kimura, S., McInnis, R.,
Maizel, A. L., and Takeda, A. (1998) Disruption of lymphocyte function
and signaling in CD45-associated protein-null mice, J. Exp.
Med., 187, 1863-1870.
3.Kung, C., Okumura, M., Seavitt, J. R., Noll, M. E.,
White, L. S., Pingel, J. T., and Thomas, M. L. (1999) CD45-associated
protein is not essential for the regulation of antigen
receptor-mediated signal transduction, Eur. J. Immunol.,
29, 3951-3955.
4.Ding, I., Bruyns, E., Li, P., Magada, D., Paskind,
M., Rodman, L., Seshadri, T., Alexander, D., Giese, T., and Schraven,
B. (1999) Biochemical and functional analysis of mice deficient in
expression of the CD45-associated phosphoprotein LPAP, Eur. J.
Immunol., 29, 3956-3961.
5.Kleiman, E., Salyakina, D., De Heusch, M., Hoek, K.
L., Llanes, J. M., Castro, I., Wright, J. A., Clark, E. S., Dykxhoorn,
D. M., Capobianco, E., Takeda, A., Renauld, J.-C., and Khan, W. N.
(2015) Distinct transcriptomic features are associated with
transitional and mature B-cell populations in the mouse spleen,
Front. Immunol., 6, 30.
6.Wolf, I., Bouquet, C., and Melchers, F. (2016)
cDNA-library testing identifies transforming genes cooperating with
c-myc in mouse pre-B cells, Eur. J. Immunol., 46,
2555-2565.
7.Ju, H., Lim, B., Kim, M., Kim, Y. S., Kim, W. H.,
Ihm, C., Noh, S.-M., Han, D. S., Yu, H.-J., Choi, B. Y., and Kang, C.
(2009) A regulatory polymorphism at position-309 in PTPRCAP is
associated with susceptibility to diffuse-type gastric cancer and gene
expression, Neoplasia, 11, 1340-1347.
8.Krotov, G. I., Krutikova, M. P., Zgoda, V. G., and
Filatov, A. V. (2007) Profiling of the CD4 receptor complex proteins,
Biochemistry (Moscow), 72, 1216-1224.
9.Leitenberg, D., Falahati, R., Lu, D. D., and
Takeda, A. (2007) CD45-associated protein promotes the response of
primary CD4 T cells to low-potency T-cell receptor (TCR) stimulation
and facilitates CD45 association with CD3/TCR and Lck,
Immunology, 121, 545-554.
10.Kruglova, N. A., Meshkova, T. D., Kopylov, A. T.,
Mazurov, D. V., and Filatov, A. V. (2017) Constitutive and
activation-dependent phosphorylation of lymphocyte
phosphatase-associated phosphoprotein (LPAP), PLoS One,
12, e0182468.
11.Filatov, A., Kruglova, N., Meshkova, T., and
Mazurov, D. (2015) Lymphocyte phosphatase-associated phosphoprotein
proteoforms analyzed using monoclonal antibodies, Clin. Transl.
Immunol., 4, e44.
12.Pierre, F., Chua, P. C., O’Brien, S. E.,
Siddiqui-Jain, A., Bourbon, P., Haddach, M., Michaux, J., Nagasawa, J.,
Schwaebe, M. K., Stefan, E., Vialettes, A., Whitten, J. P., Chen, T.
K., Darjania, L., Stansfield, R., Anderes, K., Bliesath, J., Drygin,
D., Ho, C., Omori, M., Proffitt, C., Streiner, N., Trent, K., Rice, W.
G., and Ryckman, D. M. (2011) Discovery and SAR of
5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic acid (CX-4945), the first
clinical stage inhibitor of protein kinase CK2 for the treatment of
cancer, J. Med. Chem., 54, 635-654.
13.Franchin, C., Borgo, C., Zaramella, S., Cesaro,
L., Arrigoni, G., Salvi, M., and Pinna, L. (2017) Exploring the CK2
paradox: restless, dangerous, dispensable, Pharmaceuticals,
10, E11.
14.Pinna, L. A. (2002) Protein kinase CK2: a
challenge to canons, J. Cell Sci., 115, 3873-3878.
15.Kuenzel, E., Mulligan, J., and Sommercorn, J.
(1987) Substrate specificity determinants for casein kinase II as
deduced from studies with synthetic peptides, J. Biol. Chem.,
262, 9136-9140.
16.Ahmad, K. A., Wang, G., Unger, G., Slaton, J.,
and Ahmed, K. (2008) Protein kinase CK2 – a key suppressor of
apoptosis, Adv. Enzyme Regul., 48, 179-187.
17.Ruzzene, M., Penzo, D., and Pinna, L. A. (2002)
Protein kinase CK2 inhibitor 4,5,6,7-tetrabromobenzotriazole (TBB)
induces apoptosis and caspase-dependent degradation of haematopoietic
lineage cell-specific protein 1 (HS1) in Jurkat cells,
Biochem. J., 364, 41-47.
18.Ulges, A., Witsch, E. J., Pramanik, G., Klein,
M., Birkner, K., Buhler, U., Wasser, B., Luessi, F., Stergiou, N.,
Dietzen, S., Bruhl, T.-J., Bohn, T., Bundgen, G., Kunz, H., Waisman,
A., Schild, H., Schmitt, E., Zipp, F., and Boppa, T. (2016) Protein
kinase CK2 governs the molecular decision between encephalitogenic TH17
cell and Treg cell development, PNAS, 113,
10145-10150.
19.Ampofo, E., Sokolowsky, T., Gotz, C., and
Montenarh, M. (2013) Functional interaction of protein kinase CK2 and
activating transcription factor 4 (ATF4), a key player in the cellular
stress response, Biochim. Biophys. Acta, 1833,
439-451.
20.Scaglioni, P. P., Yung, T. M., Cai, L. F.,
Erdjument-Bromage, H., Kaufman, A. J., Singh, B., Teruya-Feldstein, J.,
Tempst, P., and Pandolfi, P. P. (2006) A CK2-dependent mechanism for
degradation of the PML tumor suppressor, Cell, 126,
269-283.
21.Ampofo, E., Kietzmann, T., Zimmer, A., Jakupovic,
M., Montenarh, M., and Gotz, C. (2010) Phosphorylation of the von
Hippel–Lindau protein (VHL) by protein kinase CK2 reduces its
protein stability and affects p53 and HIF-1α mediated
transcription, IJBCB, 42, 1729-1735.
22.Desagher, S., Osen-Sand, A., Montessuit, S.,
Magnenat, E., Vilbois, F., Hochmann, A., Journot, L., Antonsson, B.,
and Martinou, J.-C. (2001) Phosphorylation of Bid by casein kinases I
and II regulates its cleavage by caspase 8, Mol. Cell, 8,
601-611.
23.Krippner-Heidenreich, A., Talanian, R. V., Sekul,
R., Kraft, R., Thole, H., Ottleben, H., and Lu, B. (2001) Targeting of
the transcription factor Max during apoptosis:
phosphorylation-regulated cleavage by caspase-5 at an unusual glutamic
acid residue in position P11, Biochem. J., 358,
705-715.
24.Channavajhala, P., and Seldin, D. C. (2002)
Functional interaction of protein kinase CK2 and c-Myc in
lymphomagenesis, Oncogene, 21, 5280-5288.
25.Vazquez, F., Grossman, S. R., Takahashi, Y.,
Rokas, M. V., Nakamura, N., and Sellers, W. R. (2001) Phosphorylation
of the PTEN tail acts as an inhibitory switch by preventing its
recruitment into a protein complex, J. Biol. Chem., 276,
48627-48630.
26.Yin, X., Gu, S., and Jiang, J. X. (2001) The
development-associated cleavage of lens connexin 45.6 by caspase-3-like
protease is regulated by casein kinase II-mediated phosphorylation,
J. Biol. Chem., 276, 34567-34572.
27.Walter, J., Schindzielorz, A., Grunberg, J., and
Haass, C. (1999) Phosphorylation of presenilin-2 regulates its cleavage
by caspases and retards progression of apoptosis, PNAS,
96, 1391-1396.
28.Zhirnov, O. P., and Syrtzev, V. V. (2009)
Influenza virus pathogenicity is determined by caspase cleavage motifs
located in the viral proteins, J. Mol. Genet. Med., 3,
124-132.