A New Efficient Method for Production of Recombinant Antitumor Cytokine TRAIL and Its Receptor-Selective Variant DR5-B
A. V. Yagolovich1,2, A. A. Artykov1,2, D. A. Dolgikh1,2, M. P. Kirpichnikov1,2, and M. E. Gasparian1,a*
1Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, 117997 Moscow, Russia2Lomonosov Moscow State University, Faculty of Biology, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received December 19, 2018; Revised February 25, 2019; Accepted February 26, 2019
The cytokine TRAIL induces apoptosis in tumor cells of various origin without affecting normal cells. Clinical trials of TRAIL-receptor (DR4 and DR5) agonists (recombinant TRAIL or death receptors antibodies) have largely failed because most human tumors were resistant to them. Currently, a second generation of agents targeted at TRAIL-R with increased efficiency has been developed. To this end, we have developed DR5-B, a variant of TRAIL selectively interacting with DR5. We have developed a new efficient method for production of TRAIL and DR5-B using expression of these proteins in Escherichia coli strain SHuffle B. The proteins were isolated from the cytoplasmic fraction of cells and purified to a high degree of homogeneity using metal-affinity and ion-exchange chromatography. The protein yield was 211 and 173 mg from one liter of cell culture for DR5-B and TRAIL, respectively, which significantly exceeded the results obtained by other methods. DR5-B killed tumor cells of different origin more efficiently and rapidly compared with TRAIL. The resulting preparations can be used for the study of TRAIL signaling pathways and in preclinical and clinical trials as antitumor agents.
KEY WORDS: cytokine TRAIL, mutant variant DR5-B, strain E. coli SHuffle B, cancer therapyDOI: 10.1134/S0006297919060051
Abbreviations: DD, death domain; DR, death receptor; HFF, human foreskin fibroblasts; IPTG, isopropyl-β-D-1-thiogalactopyranoside; MTT, 3-[4,5-dimethylthiazol-2]-2,5-diphenyl tetrazolium bromide; OPG, osteoprotegerin; TRAIL, TNF-related apoptosis-inducing ligand.
The natural cytokine TRAIL (TNF-related apoptosis-inducing ligand) is a
potential agent for the treatment of neoplastic diseases, since it is
able to selectively induce apoptosis in tumor cells without affecting
normal cells [1]. However, clinical trials have
shown its low efficiency due to the fact that many types of tumors are
resistant to TRAIL. One of the mechanisms of resistance is associated
with the presence of five TRAIL receptors, of which only two, DR4 and
DR5 (death receptors), transduce the signal of apoptosis, while DcR1,
DcR2, and OPG (osteoprotegerin) are decoy receptors and do not have a
functional cytoplasmic death domain (DD), which is necessary for the
induction of apoptosis [2]. Based on
over-expression experiments with DcR1 and DcR2, it is assumed that
these receptors inhibit the induction of apoptosis by TRAIL due to
ligand uptake [3, 4]. In
addition, a version was proposed whereby DcR2 inhibits TRAIL-induced
apoptosis, forming ligand-independent inactive complexes with the DR5
receptor, or induces signaling pathways that prevent apoptosis, in
particular, activation of NF-κB and MAPK/ERK [5-7]. The functions of these
receptors may vary in different cell types and remain not fully
explored.
The relative contribution of two separate death receptors to the induction of apoptosis in various types of tumor cells has not yet been clarified. Using receptor-selective mutant TRAIL variants, it was shown that the death of chronic lymphocytic leukemia, acute myeloid leukemia, and pancreatic cancer cells occurs by activating the DR4 receptor, while DR5 is the main source of apoptosis induction in other tumor cells of epithelial origin [8]. This differential pro-apoptotic efficacy of DR4 and DR5, depending on the type of cell/cancer, can be used in therapy.
Escherichia coli strains are predominantly used for preparation of recombinant TRAIL, where it tends to form inclusion bodies [9-11]. The preparation of recombinant proteins from inclusion bodies requires the stages of solubilization and refolding, requiring large amounts of denaturing and reducing reagents, which leads to considerable time and high cost. The expression of soluble TRAIL in E. coli [11, 12] was previously reported, but the descriptions of the methods were incomplete, and the yield of the product was low. To obtain TRAIL in soluble form, expression of fusion proteins including maltose-binding protein (MBP), b'a' disulfide isomerase domain (PDIb'a'), or thioredoxin as carrier proteins were used [13, 14]. This approach makes it easier to obtain the target protein but does not fit well with the requirements for clinical preparations.
Earlier, we obtained a receptor-specific variant of TRAIL, DR5-B, with amino acid substitutions (Y189N, R191K, E193R, H264R, I266L, and D269H). It binds to the DR5 receptor as effectively as TRAIL but does not bind to the DR4 receptor and, in contrast to other receptor-selective mutant variants, does not interact with decoy receptors DcR1, DcR2, and OPG [14]. We have shown that DR5-B induces apoptosis in various tumor cell lines more effectively than wild-type TRAIL [15]. The DR5-B variant was obtained by expression in the E. coli BL21(DE3) strain as a fusion protein with thioredoxin and subsequent purification after cleavage of the fusion protein by enteropeptidase [9, 14]. This method made it possible to produce significant amounts of soluble protein, but for a number of reasons it was not suitable for use in clinical studies. An additional step of enzymatic cleavage of the fusion protein, followed by purification of the target protein, increased the laboriousness of the purification and influenced the yield and cost of the preparation due to the high cost of the protease.
In this work, we describe a new method for production of recombinant proteins TRAIL and DR5-B that is based on direct expression in the E. coli SHuffle B strain followed by purification of the target protein using affinity and ion-exchange chromatography. The relatively new E. coli strain SHuffle B was developed by New England Biolabs (USA) to improve protein refolding in the cytoplasm and, accordingly, to increase the soluble fraction of proteins upon expression [16]. This technology allows to produce highly purified and homogeneous trimeric preparations with high yields and low levels of endotoxins. The preparation of DR5-B produced by this method demonstrated higher cytotoxic activity compared to TRAIL on tumor cell lines of various origins. High yield and purity of the protein preparation will allow to use the developed technology in clinical studies of DR5-B.
MATERIALS ANS METHODS
Reagents and materials. The cell lines of acute T-lymphoblastic leukemia Jurkat and human foreskin fibroblasts (HFF) were obtained from the Institute of Cytology, Russian Academy of Sciences (St. Petersburg, Russia). Cell lines of colorectal carcinoma HCT116 and Caco-2 were from ATCC (USA). Nutrient media for cell culture (DMEM, EMEM, RPMI 1640), 0.05% trypsin solution with EDTA, phosphate-buffered saline (PBS), and 3-[4,5-dimethylthiazol-2]-2,5-diphenyl tetrazolium bromide (MTT) were from PanEco (Russia); bovine fetal serum was from HyClone (USA); the remaining reagents were from PanReac AppliChem (Germany).
Construction of plasmid vector pET32a/sdr5-b and pET32a/strail for direct expression. Three point mutations (G469C, C471T, A473T) were inserted in the DNA sequence encoding a fused protein Trx/DR5-B by site-directed mutagenesis using oligonucleotide primers gggtaccgacgacgaccatatggtgagagaaagaggtcctc (direct) and agggacctctttctctcaccatatggtcgtcgtcggtaccc (reverse) (Evrogen, Russia) in the plasmid vector pET32a/dr5-b, previously constructed by the authors [14].
Polymerase chain reaction was carried out under the following conditions: 30 s at 95°C, 30 s at 55°C, 14.5 min at 72°C, for 30 amplification cycles. Synthesis of amplified DNA was checked by electrophoresis in 1% agarose gel. The reaction mixture was treated with restriction enzyme DpnI (Thermo Fisher Scientific, USA) for 1 h at 37°C. For DNA production, the reaction mixture was transformed into the bacterial strain E. coli XL-1 Blue (Evrogen), and the cells were grown in LB medium with ampicillin (100 µg/ml) at 37°C for 18 h with stirring at 250 rpm (Thermo Fisher Scientific). Plasmid DNA was isolated from the cell biomass using the GeneJET Plasmid MiniPrep kit (Thermo Fisher Scientific). The presence of mutations was confirmed by automatic sequencing of the DNA (Evrogen).
The resulting plasmid with an additional restriction site for NdeI was digested by XhoI and NdeI restrictases. As a result, four fragments were formed: DNA of the linearized plasmid vector (5445 bp), a fragment of the DR5-B gene (504 bp), a fragment with the thioredoxin gene (345 bp), and a fragment of the tag (129 bp) (since it contained an additional restriction site of NdeI). The fragments were separated on a 1% agarose gel, and the DNA fragments of the linearized plasmid vector and the DR5-B gene were eluted from the gel using a QIAQuick Gel Extraction kit (Qiagen, USA) and ligated using T4 DNA ligase at 10°C for 16 h. The reaction mixture was transformed into E. coli XL-1 Blue strain, and plasmid pET-32a/sdr5-b DNA was isolated. The correct insertion of the gene into the plasmid was confirmed by sequencing of the inserted gene. A similar procedure was performed with the plasmid vector pET-32a/TRAIL designed to express wild-type TRAIL.
Expression and purification of recombinant proteins TRAIL and DR5-B. The TRAIL and DR5-B proteins (a.a. 114-281) were expressed in bacterial strain E. coli SHuffle B (New England Biolabs). The cells were transformed with the plasmid vector pET-32a/sdr5-b and pET-32a/strail and grown with vigorous shaking (250 rpm) at 37°C for 17 h in nutrient medium LB (Luria–Bertani) in the presence of ampicillin (100 μg/ml). The overnight culture was diluted (1 : 100) in liquid TB (Terrific broth) medium with ampicillin (100 μg/ml), grown at 37°C to an optical density of OD600 = 0.6, and the expression of target proteins was induced by inducer of the T7 promoter, isopropyl-β-D-1-thiogalactopyranoside (IPTG). Cell cultures were grown at 28°C for 20 h. The expression level of the target protein was analyzed on a 15% polyacrylamide gel in the presence of SDS. The cells were precipitated by centrifugation at 5000g (Beckman Coulter, USA) at 4°C for 10 min, resuspended in a buffer containing 300 mM NaCl, 50 mM Tris-HCl (pH 8.0), and disrupted in a French Press (Spectronic Instruments, Inc., USA) under a pressure of 2000 psi. The disrupted cells were precipitated by centrifugation at 75,000g (Beckman Coulter) for 30 min. The soluble cell fraction was supplemented with 0.05% (v/v) Triton X-100 and applied to a chromatographic column with Ni-NTA agarose sorbent (Qiagen, USA). The target proteins were eluted in buffer containing 300 mM NaCl, 50 mM NaH2PO4 and 250 mM imidazole (pH 8.0). The purified preparation was dialyzed against buffer containing 45 mM NaCl, 50 mM Tris-HCl, 5 mM β-mercaptoethanol (pH 7.5) at room temperature for 18 h. Then 0.05% (v/v) Triton X-100 was added to the protein solution, and purification was carried out on SP Sepharose sorbent (GE Healthcare, Sweden) in a 0-500 mM NaCl linear gradient using an ӒKTA-FPLC unit (GE Healthcare, Sweden). Protein preparations were dialyzed against buffer containing 150 mM NaCl (pH 7.5) for 18-20 h at 4°C and sterilized by filtration through a sterile syringe filter (Millipore, USA) with 0.2 μm pores with a polyvinyl difluoride membrane (PVDF) and kept at 4°C. Samples after each purification step were analyzed on 15% polyacrylamide gel by staining with Coomassie Brilliant Blue R250 or silver nitrate [16, 17]. During purification, the protein concentration was determined by the Bradford method [18]. The concentration of purified preparations was determined spectrophotometrically on an Ultraspec 2100 instrument (Bio-Rad, USA) at a wavelength of 280 nm using molar extinction coefficients of 25,520 M–1·cm–1 for TRAIL and 24,180 M–1·cm–1 for DR5-B, as determined by the amino acid sequence using the ProtParam program on the ExPASy server (www.expasy.org).
Protein content in polyacrylamide gels was evaluated by densitometry using the appropriate ImageJ software option (http://rsbweb.nih.gov/ij/, National Institutes of Health, USA-NIH, Bethesda). This program calculates the intensity of gray in a selected area of a black and white image of a polyacrylamide gel, which allows comparing the protein content of the gel.
Purification of the preparations from endotoxins. DR5-B and TRAIL were further purified on Pierce High Capacity Endotoxin Removal Resin affinity sorbent (Thermo Fisher Scientific). Purified protein preparations were incubated with 10 ml of sorbent for 2 h at room temperature with slow stirring. The mixture of proteins with sorbent was applied to a column and the protein fraction was collected. Endotoxin content was measured using ToxinSensorTM Chromogenic LAL Endotoxin Assay Kit (GenScript, USA).
Analytical ultracentrifugation. To determine the homogeneity of the purified DR5-B and TRAIL preparations, analytical ultracentrifugation was performed in a Beckman Model E centrifuge (Beckman Coulter) at protein concentration of 1 mg/ml in standard tubes of 0.4 ml at 60,000 rpm. The sedimentation coefficient s20,w was calculated using the SEDFIT software.
Biological activity of the preparations. Cell lines of colorectal carcinoma HCT116, Caco-2, and human foreskin fibroblasts (HFF) were cultured in DMEM nutrient medium containing 10% (v/v) bovine fetal serum. Jurkat T-lymphoblastic leukemia cells were cultured in RPMI 1640 culture medium containing 10% bovine fetal serum. All the cell lines were cultured at 37°C and 5% CO2. The cells were seeded in 96-well plates (1·105 cells per well for HCT116, Caco-2 and HFF and 5·105 cells for Jurkat) and incubated with TRAIL or DR5-B for 24 h. To determine cell viability, MTT was added at a final concentration of 0.5 mg/ml and incubated for 3 h at 37°C. The plates were centrifuged at 3000 rpm (ELMI, Latvia) for 5 min, the supernatant was removed, dimethyl sulfoxide (DMSO) was added to each well, and the optical density was measured at a wavelength of 450 nm subtracting the background at 655 nm using an iMark™ Microplate Absorbance Reader (Bio-Rad).
RESULTS
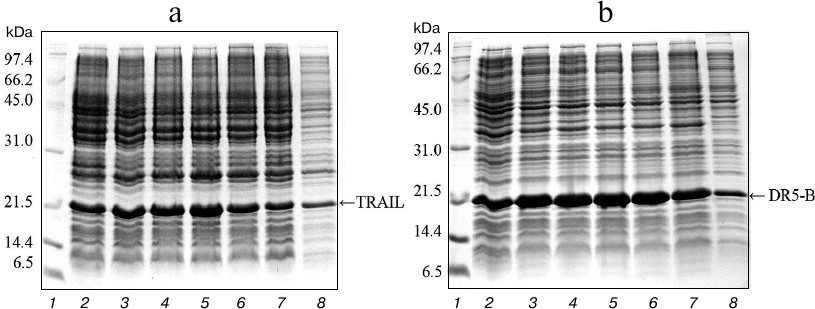
Expression in E. coli SHuffle B and purification of TRAIL and DR5-B proteins. For direct expression of TRAIL and DR5-B proteins, the prepared plasmid vectors pET-32a/sdr5-b and pET-32a/strail were transformed into the high-performance E. coli SHuffle B strain. Large amounts of active proteins with properly formed disulfide bonds are produced in the cytoplasm of this E. coli strain due to the reduced activity of gor and trxB reductases, along with increased expression of DsbC disulfide bond isomerase [19]. The cells were cultured with different concentrations of T7 promoter inducer IPTG, and the content of the target protein was analyzed in 15% polyacrylamide gel (Fig. 1). The expression of TRAIL and DR5-B was also observed in the absence of IPTG, but it was significantly increased in the presence of the inducer. In further experiments for protein purification, cells were grown in the presence of 0.1 mM IPTG. The expression level of DR5-B was higher compared to TRAIL and accounted for 25% of total protein. Evaluation of protein content in polyacrylamide gels by densitometric analysis using the appropriate ImageJ software option (http://rsbweb.nih.gov/ij/) showed that the expression level for 0.1 mM IPTG induction was approximately 676 mg of TRAIL and 782 mg of DR5-B in 1 liter of cell culture, and 45-50% of the proteins were in soluble form (Fig. 1, lane 8).
Fig. 1. Expression of TRAIL (a) and DR5-B (b) in E. coli strain SHuffle B upon induction of the T7 promoter by IPTG at various concentrations. Samples containing 10 μl of culture were centrifuged at 5000g for 5 min, dissolved in sample buffer, and analyzed with 15% Tris-glycine SDS-PAGE. Lanes: 1) molecular weight markers; 2) cell cultures grown without inducer; 3-7) cell cultures grown in the presence of 0.02, 0.05, 0.1, 0.25 and 0.5 mM IPTG, respectively; 8) 10 μg of protein from the soluble fraction of cytoplasmic proteins when expression was induced by 0.1 mM IPTG.
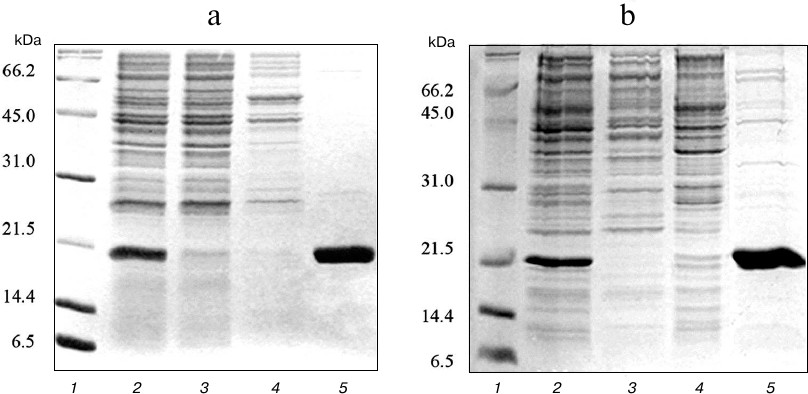
At the first stage, the expressed proteins were purified from soluble fraction of the cell lysate using metal-affinity chromatography on Ni-NTA sorbent. Due to the presence of several histidine residues in the amino acid sequence, TRAIL strongly binds to the Ni-NTA sorbent even in the absence of polyhistidine tag. As a result, TRAIL and DR5-B recombinant proteins were efficiently purified, and the purity of the preparations was approximately 85% (Fig. 2).
Fig. 2. Purification of TRAIL (a) and DR5-B (b) proteins by metal-affinity chromatography on Ni-NTA agarose. Samples containing 15 μg of protein were analyzed with 15% SDS-PAGE. Lanes: 1) molecular weight markers; 2) soluble fraction of cytoplasmic proteins; 3) fraction of proteins not bound to Ni-NTA agarose; 4) fraction of proteins washed with buffer containing 20 mM imidazole; 5) fraction of proteins washed with buffer containing 250 mM imidazole.
To obtain highly purified protein preparations, a second purification step was carried out using cation-exchange chromatography on SP Sepharose sorbent, which can bind 50-100 mg of protein per ml of sorbent. Proteins purified on Ni-NTA agarose were dialyzed against buffer containing 45 mM NaCl, 50 mM Tris-HCl, and 5 mM β-mercaptoethanol and applied to a column with 10 ml of SP Sepharose. Proteins were eluted with a linear NaCl gradient of 0-500 mM at salt concentrations of 150-250 mM. The resulting protein solutions contained practically no impurities of non-target polypeptides (Fig. 3). The developed technology produced highly purified proteins TRAIL and DR5-B in the amount of 173 and 211 mg from one liter of cell culture, respectively. The yields of the TRAIL and DR5-B protein preparations during purification are shown in Table 1.
Fig. 3. Purification of DR5-B protein using ion-exchange chromatography on SP Sepharose sorbent. a) Samples containing 10-15 μg of protein were analyzed on 15% SDS-PAGE. Lanes: 1) molecular weight markers; 2) protein fraction after purification in the first stage on Ni-NTA agarose; 3) fraction of proteins not bound with SP Sepharose; 4) fraction of proteins released from the column at 100 mM NaCl; 5-7) protein fractions after purification on SP Sepharose. b) Analysis of the purity of the preparations with silver nitrate staining of polyacrylamide gels. Lanes: 1) molecular weight markers; 2) TRAIL (500 ng); 3) DR5-B (500 ng).
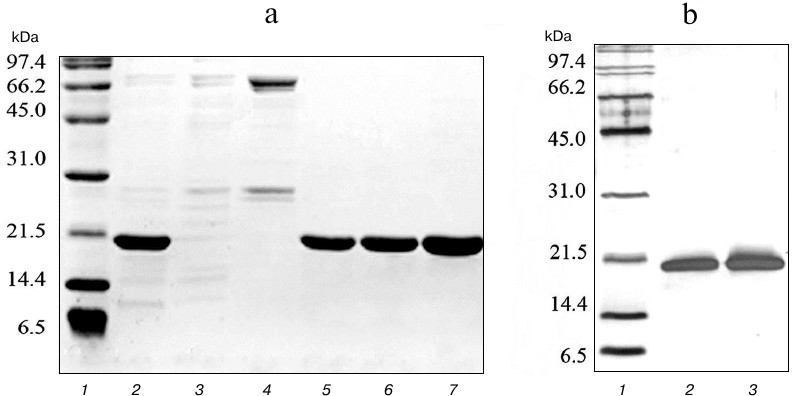
Table 1. Yields of TRAIL and DR5-B proteins
from 1 liter of cell culture during purification

* Amount of protein determined by the Bradford method.
** Amount of protein determined by absorption at 280 nm regarding
the calculated extinction coefficient.
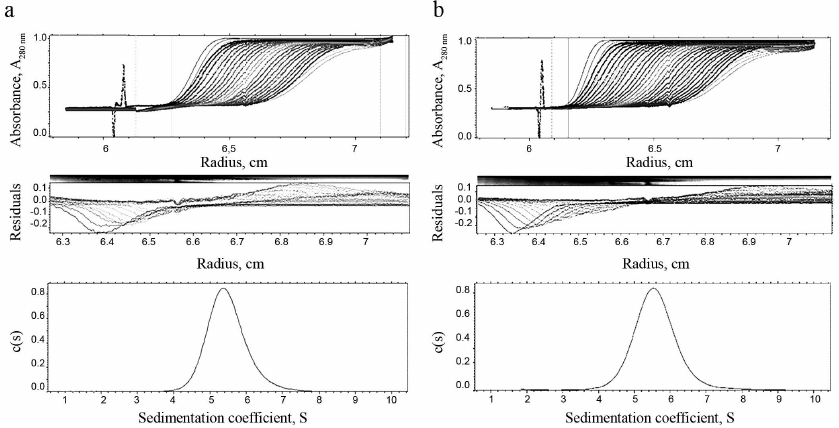
Homogeneity and degree of trimerization of the preparations were investigated by analytical ultracentrifugation (Fig. 4). Sedimentation coefficients (s20,w) 5.49 ± 0.03 and 5.61 ± 0.03 were determined, which corresponds to molecular masses (Mw) 59,105 and 59,156 Da for TRAIL and DR5-B, respectively. The molecular weights of the monomers TRAIL and DR5-B, calculated on the basis of their amino acid sequences, are 19,624.04 and 19,616.11 Da, respectively, so that the molecular weights of both preparations, determined by analytical ultracentrifugation, correspond to their trimers. The shape of the curves in Fig. 4 suggests that the preparations are highly homogeneous and consist of trimeric molecules.
Fig. 4. Results of analytical ultracentrifugation of TRAIL (a) and DR5-B (b) preparations. The preparations were centrifuged at 60,000 rpm in standard 0.4-ml cells at a concentration of 1 mg/ml. Protein sedimentation profiles were recorded at the absorbance of 280 nm. The c(s) parameter is the coefficient of continuous sedimentation distribution determined using the SEDFIT program (http://www.analyticalultracentrifugation.com).
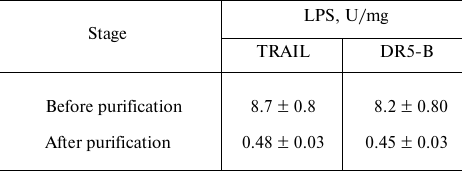
Purification of TRAIL and DR5-B preparations from endotoxins. The content of endotoxins in the purified preparations was evaluated using the ToxinSensorTM Chromogenic LAL Endotoxin Assay Kit (USA) according to the manufacturer’s instructions. When the purification of proteins was carried out without detergent Triton X-100, the level of endotoxins was more than 35 U/mg (data not shown), which is quite low for recombinant proteins purified from E. coli strains. The relatively low endotoxin content is probably due to the use of cation-exchange chromatography [20]. The effect of detergent Triton X-100 during purification on the content of endotoxins in the preparation was investigated. At low concentration of Triton X-100 (0.05%), the level of endotoxins decreased 5-fold and amounted to 8 ± 0.8 U/mg (Table 2). This content of endotoxins allows the use of the drug in biological and preclinical studies, but it exceeds the maximum allowable dose for use in clinical trials (≤5 U/kg per hour). Therefore, the preparations were further purified on Pierce High Capacity Endotoxin Removal Resin affinity sorbent with covalently bound ε-poly-L-lysine to remove endotoxins. The total content of endotoxins in the purified preparations of TRAIL and DR5-B did not exceed 0.5 U/mg (Table 2). It should be noted that during the purification of the drugs from endotoxins, the loss of proteins was insignificant (1-2%).
Table 2. Content of lipopolysaccharides
(LPS) in samples of TRAIL and DR5-B preparations before and after
purification on the affinity sorbent for endotoxin removal
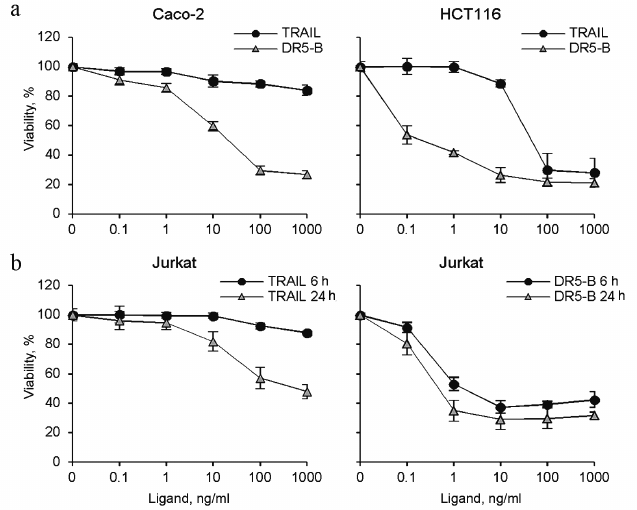
Cytotoxic activity of TRAIL and DR5-B preparations. Cytotoxic activity of the TRAIL and DR5-B preparations was studied on the cell lines of colorectal carcinoma HCT116 and Caco-2, as well as on T-lymphoblastic leukemia Jurkat (Fig. 5). DR5-B protein effectively killed HCT116 and Jurkat cells at concentrations of 0.1-1 ng/ml, whereas TRAIL exhibited cytotoxic activity only at concentrations above 10 ng/ml. In addition, DR5-B killed Jurkat cells much faster than TRAIL (Fig. 5b). DR5-B during 6 h killed only 10% fewer cells in comparison with 24-h incubation, whereas TRAIL was ineffective at 6-h incubation. Caco-2 cells were almost resistant to TRAIL, whereas DR5-B killed 80% of these cells. Earlier, using Colo205 cell line, it was shown that the receptor-selective variant TRAIL-D269H/E195R accelerated activation of the DR5 receptor due to bypass of the ligand-induced heterooligomerization of the DR5 receptor with decoy receptors resulting in increased apoptosis [21]. However, in Jurkat and Caco-2 cells, the level of surface expression of decoy receptors is extremely low [15, 22] and insignificant for inhibition of TRAIL mediated apoptosis. We suggest that the degree of oligomerization of DR5 receptor upon binding to DR5-B is higher compared to TRAIL, which provides an increase in the apoptotic signal. It was previously shown that when TRAIL binds to pre-dimerized DR5, the receptors are clustered in a highly organized network, which is required to induce effective apoptosis [23]. Interestingly, a dimeric ligand of DR5 receptor caused apoptosis in BJAB cells but did not kill Jurkat and HCT116 cells due to the difference in the level of DR5 oligomerization on the cell surface before addition of the ligand [24].
Fig. 5. Cytotoxic activity of recombinant protein preparations TRAIL and DR5-B on various tumor cell lines. Cell viability was determined using the MTT test. a) HCT116 and Caco-2 colorectal carcinoma cells were incubated with the drugs for 24 h. b) Jurkat acute T-lymphoblastic leukemia cells were incubated with the cytokines for 6 and 24 h. Values are presented as mean ± standard deviation (n = 3).
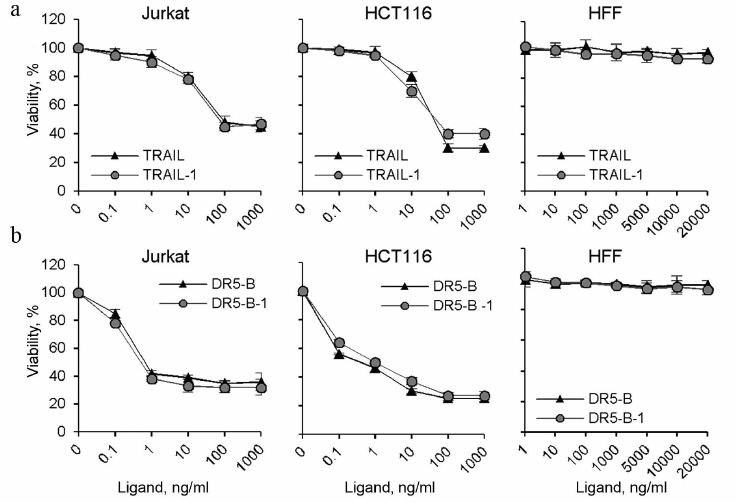
It should be noted that the DR5-B and TRAIL preparations produced in this work using direct expression and the preparations produced by expression as a fusion protein with thioredoxin (TRAIL-1 and DR5-B-1) [15] showed the same cytotoxic activity on HCT116 and Jurkat cells (Fig. 6). Neither the TRAIL nor DR5-B produced as described showed cytotoxicity untransformed human foreskin fibroblasts (HFF) (Fig. 6), indicating that the developed new method for producing TRAIL and DR5-B did not affect specificity of their action.
Fig. 6. Comparative analysis of cytotoxic activity of TRAIL and DR5-B preparations produced by direct expression in the E. coli SHuffle B strain and preparations produced after expression of fusion proteins with thioredoxin in the strain BL23(DE3) (TRAIL-1 and DR5-B-1). HCT116, Jurkat, and HFF cells were incubated with TRAIL (a) and DR5-B (b) preparations for 24 h, and cell viability was determined using the MTT test. Values are presented as mean ± standard deviation (n = 3).
DISCUSSION
Expression of hybrid recombinant proteins fused to a domain of a “carrying” protein is a convenient way to produce proteins for molecular biological studies. For example, a method for cytoplasmic expression of TRAIL was developed earlier using a fusion protein with maltose-binding protein (MBP). For purification of the target protein, the fusion protein was cleaved by tobacco engraving virus protease [13]. The yield of protein reported in this work was 36 mg from one liter of cell culture. In another method for cytoplasmic expression of TRAIL, it was fused with Fh8-ΔI-CM tag, where Fh8 is a low molecular weight 8 kDa polypeptide antigen of Fasciola hepatica, which improves the solubility of the protein, and ΔI-CM is a calmodulin-binding polypeptide that undergoes self-cleavage at low pH values [25]. The protein yield in this study was 77.5 mg of protein from one liter of cell culture with a purity of 89%. Earlier in our laboratory, we developed a method to produce TRAIL variants by expressing the hybrid proteins fused with thioredoxin in E. coli BL21(DE3) strain, where the solubility of the fusion protein was 20% of the total expression [9, 14].
However, therapeutic proteins obtained by the hybrid method have a number of limitations on use as pharmaceuticals. Hybrid expression involves the use of specific proteases for cleaving the “carrier” protein. Despite the fact that these proteases cleave fusion proteins at a specific site located between the carrier and target protein, the cleavage produces traces of nonspecific products that are difficult to remove. In addition, the purification process of the target protein includes the additional step of the removal of the proteases. Also, the use of proteases significantly increases the cost of the final product due to the high labor costs and high cost of the enzyme, which makes it difficult to scale up the production of a drug for clinical trials.
Since the antitumor cytokine TRAIL is a potential agent for the treatment of neoplastic diseases, the task was set to develop a new method for producing this cytokine with a high yield and low cost. To this end, a new strategy was developed for the expression and purification of TRAIL. The protein was for the first time expressed in the E. coli strain SHuffle B without carrier proteins and tags and in soluble form. This facilitated the purification procedure and improved the yield of the target protein. During the two stages of purification (affinity and ion-exchange chromatography), 173 and 211 mg of TRAIL and DR5-B preparations were obtained, respectively, with a purity of 98%. The higher yield of DR5-B might be related to higher solubility of the protein, due to the replacement of hydrophobic and uncharged amino acid residues with hydrophilic and charged residues (such as Y189N, Q193R, H264R) [14].
Due to the use of detergent Triton X-100 in low concentrations (0.05%) at the purification stages, the level of bacterial endotoxins in the preparations was reduced 3-4-fold (from 35 to 8.2 U/mg) [26]. Since therapeutic doses of TRAIL preparations are quite high (5-10 mg/kg of weight), the level of endotoxins in TRAIL or DR5-B should not exceed 0.5 U/kg [27]. The preparations were additionally purified from endotoxins on a special endotoxin-binding sorbent, reducing the level to an acceptable concentration (less than 0.5 U/ml).
The receptor-selective mutant variant DR5-B killed HCT116 tumor cells more effectively compared to TRAIL, as we showed earlier with preparations produced by expressing fusion proteins with thioredoxin in E. coli BL23(DE3) strain [15, 28]. In addition, DR5-B killed Jurkat cells not only more efficiently but also faster than TRAIL, which is no less important, since TRAIL is quickly washed out of human blood (the half-life is about 30 min) [29]. TRAIL-resistant Caco-2 cells were highly sensitive to DR5-B. We suggest that in these cells with high surface expression of DR5 [22] the ligand-induced oligomerization of this receptor is higher when it binds to DR5-B compared to TRAIL, which contributes to the formation of active death-inducing signal complexes and the amplification of the apoptosis signal.
Thus, the developed technology for production of the antitumor cytokine TRAIL and its mutant variant DR5-B allows to produce relatively inexpensive drugs with high purity that be used for treatment of various oncological diseases.
Funding. This work was financially supported by the Russian Foundation for Basic Research (projects no. 18-34-00812 and no. 16-04-0068) and by the grant of the Presidium of the Russian Academy of Sciences “Molecular and Cell Biology”.
Conflict of interest. The authors declare they have no conflict of interest in financial or any other sphere.
Ethics committee approval. This article does not contain any studies with human participants or animals performed by any of the authors.
REFERENCES
1.Guicciardi, M. E., and Gores, G. J. (2009) Life and
death by death receptors, FASEB J., 23, 1625-1637; doi:
10.1096/fj.08-111005.
2.Pan, G., O’Rourke, K., Chinnaiyan, A. M.,
Gentz, R., Ebner, R., Ni, J., and Dixit, V. M (1997) The receptor
for the cytotoxic ligand TRAIL, Science, 276, 111-113;
doi: 10.1126/science.276.5309.111.
3.Degli-Esposti, M. A., Smolak, P. J., Walczak, H.,
Waugh, J., Huang, C. P., DuBose, R. F., Goodwin, R. G., and Smith, C.
A. (1997) Cloning and characterization of TRAIL-R3, a novel member of
the emerging TRAIL receptor family, J. Exp. Med., 186,
1165-1170; doi: 10.1084/jem.186.7.1165.
4.Degli-Esposti, M. A., Dougall, W. C., Smolak, P.
J., Waugh, J. Y., Smith, C. A., and Goodwin, R. G. (1997) The novel
receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated
apoptosis, yet retains an incomplete death domain, Immunity,
7, 813-820; doi: 10.1016/S1074-7613(00)80399-4.
5.Merino, D., Lalaoui, N., Morizot, A., Schneider,
P., Solary, E., and Micheau, O. (2006) Differential inhibition of
TRAIL-mediated DR5-DISC formation by decoy receptors 1 and 2, Mol.
Cell Biol., 26, 7046-7055; doi: 10.1128/MCB.00520-06.
6.Clancy, L., Mruk, K., Archer, K., Woelfel, M.,
Mongkolsapaya, J., Screaton, G., Lenardo, M. J., and Chan, F. K. (2005)
Preligand assembly domain-mediated ligand-independent association
between TRAIL receptor 4 (TR4) and TR2 regulates TRAIL-induced
apoptosis, Proc. Natl. Acad. Sci. USA, 102, 18099-18104;
doi: 10.1073/pnas.0507329102.
7.Nguyen, P. T., Nguyen, D., Chea, C., Miyauchi, M.,
Fujii, M., and Takata, T. (2018) Interaction between N-cadherin and
decoy receptor-2 regulates apoptosis in head and neck cancer,
Oncotarget, 9, 31516-31530; doi:
10.18632/oncotarget.25846.
8.Van Roosmalen, I. A., Quax, W. J., and Kruyt, F. A.
(2014) Two death-inducing human TRAIL receptors to target in cancer:
similar or distinct regulation and function? Biochem.
Pharmacol., 91, 447-456; doi: 10.1016/j.bcp.2014.08.010.
9.Gasparian, M. E., Ostapchenko, V. G., Yagolovich,
A. V., Tsygannik, I. N., Chernyak, B. V., Dolgikh, D. A., and
Kirpichnikov, M. P. (2007) Overexpression and refolding of
thioredoxin/TRAIL fusion from inclusion bodies and further purification
of TRAIL after cleavage by enteropeptidase, Biotechnol. Lett.,
29, 1567-1573; doi: 10.1007/s10529-007-9446-y.
10.Lin, Z., Lei, H., and Cao, P. (2007) Expression,
purification, and in vitro refolding of soluble tumor necrosis
factor-related apoptosis inducing ligand (TRAIL), Protein Expr.
Purif., 51, 276-282; doi: 10.1016/j.pep.2006.07.026.
11.Wang, D., and Shi, L. (2009) High-level
expression, purification, and in vitro refolding of soluble
tumor necrosis factor-related apoptosis-inducing ligand (TRAIL),
Appl. Biochem. Biotechnol., 157, 1-9; doi:
10.1007/s12010-007-8079-x.
12.Ashkenazi, A., Pai, R. C., Fong, S., Leung, S.,
Lawrence, D. A., Marsters, S. A., Blackie, C., Chang, L., McMurtrey, A.
E., Hebert, A., DeForge, L., Koumenis, I. L., Lewis, D., Harris, L.,
Bussiere, J., Koeppen, H., Shahrokh, Z., and Schwall, R. H. (1999)
Safety and antitumor activity of recombinant soluble Apo2 ligand, J.
Clin. Invest., 104, 155-162; doi: 10.1172/JCI6926.
13.Do, B. H., Nguyen, M. T., Song, J. A., Park, S.,
Yoo, J., and Jang, J. (2017) Soluble prokaryotic expression and
purification of bioactive Tumor Necrosis Factor-Related
Apoptosis-Inducing Ligand, J. Microbiol. Biotechnol., 27,
2156-2164; doi: 10.4014/jmb.1705.05070.
14.Gasparian, M. E., Chernyak, B. V., Dolgikh, D.
A., Yagolovich, A. V., Popova, E. N., Sycheva, A. M., Moshkovskii, S.
A., and Kirpichnikov, M. P. (2009) Generation of new TRAIL mutants
DR5-A and DR5-B with improved selectivity to death receptor 5,
Apoptosis, 14, 778-787; doi:
10.1007/s10495-009-0349-3.
15.Gasparian, M. E., Bychkov, M. L., Yagolovich, A.
V., Dolgikh, D. A., and Kirpichnikov, M. P. (2015) Mutations enhancing
selectivity of antitumor cytokine TRAIL to DR5 receptor increase its
cytotoxicity against tumor cells, Biochemistry (Moscow),
80, 1080-1091; doi: 10.1134/S0006297915080143.
16.Simpson, R. J. (2010) Rapid Coomassie blue
staining of protein gels, Cold Spring Harb. Protoc.,
2010, pdb.prot5413; doi: 10.1101/pdb.prot5413.
17.Switzer, R. C., 3rd, Merril, C. R., and Shifrin,
S. (1979) A highly sensitive silver stain for detecting proteins and
peptides in polyacrylamide gels, Anal. Biochem., 98,
231-237; doi: 10.1016/0003-2697(79)90732-2.
18.Bradford, M. M. (1976) A rapid and sensitive
method for the quantitation of microgram quantities of protein
utilizing the principle of protein–dye binding, Anal.
Biochem., 72, 248-254; doi:
10.1016/0003-2697(76)90527-3.
19.Lobstein, J., Emrich, C. A., Jeans, C., Faulkner,
M., Riggs, P., and Berkmen, M. (2012) SHuffle, a novel Escherichia
coli protein expression strain capable of correctly folding
disulfide bonded proteins in its cytoplasm, Microb. Cell Fact.,
11, 56; doi: 10.1186/1475-2859-11-56.
20.Reichelt, P., Schwarz, C., and Donzeau, M. (2006)
Single step protocol to purify recombinant proteins with low endotoxin
contents, Protein Expr. Purif., 46, 483-488; doi:
10.1016/j.pep.2005.09.027.
21.Szegezdi, E., van der Sloot, A. M., Mahalingam,
D., O’Leary, L., Cool, R. H., Munoz, I. G., Montoya, G., Quax, W.
J., de Jong, S., Samali, A., and Serrano, L. (2012) Kinetics in signal
transduction pathways involving promiscuous oligomerizing receptors can
be determined by receptor specificity: apoptosis induction by TRAIL,
Mol. Cell. Proteom., 11, M111.013730; doi:
10.1074/mcp.M111.013730.
22.Van Geelen, C. M., de Vries, E. G., Le, T. K.,
van Weeghel, R. P., and de Jong, S. (2003) Differential modulation of
the TRAIL receptors and the CD95 receptor in colon carcinoma cell
lines, Br. J. Cancer, 89, 363-373; doi:
10.1038/sj.bjc.6601065.
23.Valley, C. C., Lewis, A. K., Mudaliar, D. J.,
Perlmutter, J. D., Braun, A. R., Karim, C. B., Thomas, D. D., Brody, J.
R., and Sachs, J. N. (2012) Tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL) induces death receptor 5 networks
that are highly organized, J. Biol. Chem., 287,
21265-21278; doi: 10.1074/jbc.M111.306480.
24.Chekkat, N., Lombardo, C. M., Seguin, C.,
Lechner, M. C., Dufour, F., Nomine, Y., De Giorgi, M., Frisch, B.,
Micheau, O., Guichard, G., Altschuh, D., and Fournel, S. (2018)
Relationship between the agonist activity of synthetic ligands of
TRAIL-R2 and their cell surface binding modes, Oncotarget,
9, 15566-15578; doi: 10.18632/oncotarget.24526.
25.Zhang, M., Wang, Z., Chi, L., Sun, J., and Shen,
Y. (2018) Enhanced production of soluble tumor necrosis factor-related
apoptosis-inducing ligand in Escherichia coli using a novel
self-cleavable tag system Fh8-ΔI-CM, Protein Expr. Purif.,
148, 16-23; doi: 10.1016/j.pep.2018.03.005.
26.Malyala, P., and Singh, M. (2008) Endotoxin
limits in formulations for preclinical research, J. Pharm. Sci.,
97, 2041-2044; doi: 10.1002/jps.21152.
27.Herbst, R. S., Eckhardt, S. G., Kurzrock, R.,
Ebbinghaus, S., O’Dwyer, P. J., Gordon, M. S., Novotny, W.,
Goldwasser, M. A., Tohnya, T. M., Lum, B. L., Ashkenazi, A., Jubb, A.
M., and Mendelson, D. S. (2010) Phase I dose-escalation study of
recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in
patients with advanced cancer, J. Clin. Oncol., 28,
2839-2846; doi: 10.1200/JCO.2009.25.1991.
28.Gasparian, M. E., Bychkov, M. L., Yagolovich, A.
V., Kirpichnikov, M. P., and Dolgikh, D. A. (2017) The effect of
cisplatin on cytotoxicity of anticancer cytokine TRAIL and its
receptor-selective mutant variant DR5-B, Dokl. Biochem.
Biophys., 477, 385-388; doi: 10.1134/S1607672917060114.
29.Soria, J. C., Smit, E., Khayat, D., Besse, B.,
Yang, X., Hsu, C. P., Reese, D., Wiezorek, J., and Blackhall, F. (2010)
Phase 1b study of dulanermin (recombinant human Apo2L/TRAIL) in
combination with paclitaxel, carboplatin, and bevacizumab in patients
with advanced non-squamous non-small-cell lung cancer, J. Clin.
Oncol., 28, 1527-1533; doi:
10.1200/JCO.2009.25.4847.