Changes in the Metabolism of Sphingoid Bases in the Brain and Spinal Cord of Transgenic FUS(1-359) Mice, a Model of Amyotrophic Lateral Sclerosis
U. A. Gutner1, M. A. Shupik1, O. A. Maloshitskaya2, S. A. Sokolov2, A. P. Rezvykh3, S. Yu. Funikov3, A. T. Lebedev2, A. A. Ustyugov4, and A. V. Alessenko1,a*
1Emanuel Institute of Biochemical Physics, Russian Academy of Sciences, 119991 Moscow, Russia2Lomonosov Moscow State University, Faculty of Chemistry, 119999 Moscow, Russia
3Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, 119991 Moscow, Russia
4Institute of Physiologically Active Compounds, Russian Academy of Sciences, 142432 Chernogolovka, Moscow Region, Russia
* To whom correspondence should be addressed.
Received March 7, 2019; Revised June 17, 2019; Accepted June 17, 2019
The aim of this study was to evaluate changes in the content of sphingoid bases – sphingosine (SPH), sphinganine, and sphingosine-1-phosphate (SPH-1-P) – and in expression of genes encoding enzymes involved in their metabolism in the brain structures (hippocampus, cortex, and cerebellum) and spinal cord of transgenic FUS(1-359) mice. FUS(1-359) mice are characterized by motor impairments and can be used as a model of amyotrophic lateral sclerosis (ALS). Lipids from the mouse brain structures and spinal cord after 2, 3, and 4 months of disease development were analyzed by chromatography/mass spectrometry, while changes in the expression of the SPHK1, SPHK2, SGPP2, SGPL1, ASAH1, and ASAH2 genes were assayed using RNA sequencing. The levels of SPH and sphinganine (i.e., sphingoid bases with pronounced pro-apoptotic properties) were dramatically increased in the spinal cord at the terminal stage of the disease. The ratio of the anti-apoptotic SPH-1-P to SPH and sphinganine sharply reduced, indicating massive apoptosis of spinal cord cells. Significant changes in the content of SPH and SPH-1-P and in the expression of genes related to their metabolism were found at the terminal ALS stage in the spinal cord. Expression of the SGPL gene (SPH-1-P lyase) was strongly activated, while expression of the SGPP2 (SPH-1-P phosphatase) gene was reduced. Elucidation of mechanisms for the regulation of sphingolipid metabolism in ALS will help to identify molecular targets for the new-generation drugs.
KEY WORDS: amyotrophic lateral sclerosis, sphingosine, sphingosine-1-phosphate, SPHK2, SGPP2, SGPL1, ASAH1, ASAH2DOI: 10.1134/S0006297919100055
Abbreviations: ALS, amyotrophic lateral sclerosis; ASAH1, acidic ceramidase gene; ASAH2, neutral ceramidase gene; PKC, protein kinase C; SGPL1, sphingosine-1-phosphate lyase 1 gene; SGPP2, sphingosine-1-phosphate phosphatase 2 gene; SPH, sphingosine; SPH-1-P, sphingosine-1-phosphate; SPHK1, sphingosine kinase 1 gene; SPHK2, sphingosine kinase 2 gene.
Amyotrophic lateral sclerosis (ALS) is an incurable neurodegenerative
disease characterized by selective degeneration of motor neurons in the
spinal cord, motor cortex, and brain stem. Clinically, the disease is
manifested as the muscular exhaustion, speech and swallowing
impairments, fasciculation, and changes in reflexes and plasticity. The
patients die three to five years after appearance of the first disease
symptoms mainly because of the respiratory paralysis. The etiology of
~90% registered ALS cases is unknown, so they are classified as
sporadic. The remaining 10% are hereditary ALS forms associated mainly
with autosomal dominant mutations in particular genes [1].
Mutations associated with ALS mostly lead to the conformational instability and aggregation of proteins (SOD1 [2], VCP [3], OPTIN [4], UBQLN2 [5]), impairments in the RNA processing and transport (C9ORF72 [6], TDP-43 [7], FUS [8, 9]), and changes in the cytoskeleton dynamics (PFN1 [10], DCTN1 [11], TUBA4A [12]) [13]. This outstanding genetic variability of ALS explains its complexity, when different mechanisms result in similar disease pathogenesis. The key features of ALS development are excitotoxicity, oxidative stress, dysfunction of mitochondria [2], and neuroinflammatory and immune reactions [3, 4]. Recently, apoptosis (programmed cell death) has been intensely studied as one of possible mechanisms responsible for the degeneration of motor neurons in ALS [5].
In this connection, much attention has been drawn to the studies on the disorders of lipid metabolism in ALS development. Lipids play an extremely important role in the central nervous system: they serve as energy sources and major structural components of the cell membranes, as well as participate in the cell–cell communications and signal transmission in apoptosis and differentiation. Disturbances in the lipid metabolism negatively impact both structural and physiological properties of the brain and functions of neurons and neuroglia, including membrane transport and control of enzyme activity [14]. Destruction of cell membranes is a characteristic feature of neurodegeneration that emerges in chronic diseases of the central nervous system [15].
Both sporadic and hereditary forms of ALS are accompanied by lipid metabolism disorders, the most frequent of which is hyperlipidemia (a type of dyslipidemia) [16, 17]. One of the characteristic ALS symptoms observed in ~66% patients is weight loss due to the hypermetabolism directly associated with lipid metabolism [16].
Lipids perform the regulatory role by acting as secondary messengers in the inflammation processes during ALS development which are accompanied by the activation of microglia, loss of neuromuscular junctions, and subsequent degeneration of motor neurons. At the same time, the level of neurotoxic molecules (e.g., cytokines) synthesized with an active involvement of lipid messengers increases [17].
Sphingolipids occupy a special place among lipids, since they are the most important source of secondary messengers involved in cell proliferation, differentiation, and apoptosis [18]. Disorders in the sphingolipid metabolism have been found in many hereditary metabolic diseases, such as Fabry, Niemann–Pick and Gaucher lysosomal storage diseases, and in neurodegenerative disorders (Parkinson’s and Alzheimer’s diseases) [19].
The structure of almost all sphingolipids is based on the sphingoid base sphingosine (SPH). Free SPH is produced as a result of ceramide hydrolysis by ceramidase and can be converted back into ceramide by ceramide synthase. Moreover, it can be phosphorylated to sphingosine-1-phosphate (SPH-1-P) by two kinases (Fig. 1).
Fig. 1. Metabolism of sphingoid bases.
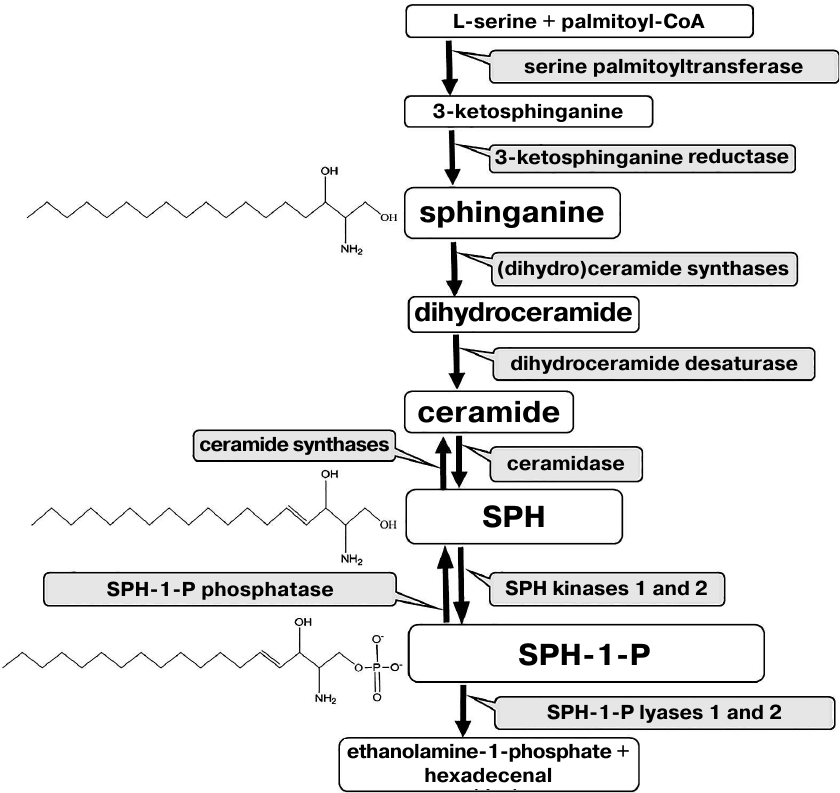
SPH influences cell growth, differentiation, and death [20, 21], affects platelet aggregation [22], inhibits blood coagulation [23], and stimulates mitogenesis [24]; it also has the antitumor [25] and antimicrobial [26] activities. An especially important feature of SPH is its ability to induce apoptosis [20, 27-32]. The mechanism of SPH-mediated apoptosis involves multiple molecular events. It has been shown that in the presence of SPH, apoptosis develops through the caspase-dependent pathway. SPH can induce Bid cleavage, cytochrome c release from the mitochondria, as well as activation of downstream effector caspases 3 or 7 and PARP cleavage. The role of SPH in mitochondrial apoptosis is determined by its effect on the Bcl-2 and Bax proteins: it downregulates Bcl-2 expression and induces Bax cleavage [27].
Similar to ceramide, SPH inhibits protein kinase C (PKC), which is the most important lipid-dependent enzyme determining the cell survival. This effect is associated with the SPH structure that includes primary amino group and long fatty acid residue. The extent of PKC inhibition depends on the presence in the incubation medium of phosphatidylserine, diacylglycerol, Ca2+, lysophosphatidylcholine, phorbol esters, and fatty acids (PKC activators) [33]. Diacylglycerol in the membrane facilitates PKC translocation from the cytosol to the plasma membrane and increases its affinity for Ca2+/calmodulin, thus inducing activation of the enzyme at low cytoplasmic Ca2+ concentrations. Phorbol esters mimic the action of diacylglycerol. The presence of phospholipids and fatty acids is necessary for the PKC activity.
In addition to the effect on PKC, SPH activates a number of other protein kinases, such as casein kinase II [34], mitogen-activated protein kinases [35], sphingosine-dependent kinase (its activity is specifically regulated by SPH and dimethylsphingosine) [36], c-Jun N-terminal kinase [37], protein kinase FA/GSK-3α involved in the heat shock signaling [38], etc.
SPH also influences many other enzymes in the cell. It activates GTP cyclohydrolase [39], adenylate cyclase [40], phospholipase D [41], and phospholipase Cδ [42]. SPH inhibits NADPH oxidase [43], tissue growth factor [44], calmodulin-dependent kinase [45], tyrosine kinase activity of the insulin receptor [46], phosphatidic acid phosphohydrolase [47], and CTP:phosphocholine cytidyltransferase [48], as well as regulates activity of DAG kinase [49]. SPH affects phosphorylation of various receptor proteins [44, 50, 51] and induces Ca2+ mobilization from the intracellular depots [52, 53].
The pro-apoptotic action of SPH is associated with its ability to interact with DNA and influence the activities of replication and transcription enzymes [54], as well as DNA-binding properties of some regulatory proteins including transcription factors and topoisomerases [55, 56]. Some properties of SPH are also characteristic for another sphingoid base – SPH precursor dihydrosphingosine (sphinganine) (Fig. 1).
On the contrary, SPH-1-P has the anti-apoptotic properties and promotes cell proliferation. A special role in these processes belongs to the SPH kinase that decreases the content of SPH in the cell, thereby preventing cell death [20, 27].
Due to their pronounced pro-apoptotic properties, SPH and sphinganine can be directly involved in the death of the central nervous system cells in the course of ALS. However, up to now, there have been no studies on the changes in the levels of sphingoid bases during ALS development in humans and animal models. Alterations in the SPH-1-P content in the course of ALS are also poorly investigated. Therefore, we have paid special attention to the content of SPH and other sphingoid bases in the brain structures and spinal cord and their possible involvement in the neuron death in ALS.
The purpose of the study was to evaluate the changes in the content of sphingoid bases (SPH, sphinganine, and SPH-1-P) and in the expression of genes encoding enzymes involved in their metabolism in the brain structures (hippocampus, cortex, and cerebellum) and spinal cord (lumbar region) in FUS(1-359) transgenic mice that were used as an ALS model.
MATERIALS AND METHODS
Animals. Transgenic FUS(1-359) mice (Bioresource Collection of the Institute of Physiologically Active Compounds, Russian Academy of Sciences) were used as an ALS model. The mice overexpressed the human FUS protein lacking the nuclear location signal under the neuron-specific promoter Thy-1 and displayed characteristic symptoms of ALS. The FUS(1-359) mice have been maintained in a hemizygous state on the CD-1 genetic background according to the previously described protocol [57]. The mice accumulated protein inclusions in the cytoplasm of nerve cells, which led to the damage of axons of motor neurons accompanied by neuroinflammatory response and decrease in the number of motor neurons. The FUS(1-359) mice are characterized by a relatively early manifestation of the disease (2.5-4.5 months) and rapid lethal outcome within several days after symptom manifestation.
Brain structures (hippocampus, cerebellum, and cortex) and spinal cord (lumbar region) were isolated from the mice at the age of 2, 3, and 4 months, which corresponded to the pre-symptomatic, early symptomatic, and symptomatic stages of the disease, respectively. Littermates of the same age but without the transgenic cassette were used as controls.
Lipids were extracted by the method of Bligh and Dyer [58].
Analysis of sphingoid bases (SPH and sphinganine) and SPH-1-P by liquid chromatography/mass spectrometry [59]. Isolated lipids were evaporated under nitrogen flow (PE-8920; Ekroskhim, Russia), dissolved in a chloroform–methanol mixture (2 : 1, v/v) at a concentration of 6.8 mg/ml, and analyzed by high-performance liquid chromatography/mass spectrometry using a TSQ Endura device (Thermo Scientific, USA) equipped with an EclipsePlusC8 column (Agilent, USA) in the multiple reaction monitoring (MRM) regime. MRM transitions for the sphingoid bases were as follows. SPH: MН+ → (M-2Н2O)+, m/z 300 → m/z 264; sphinganine: MН+ → (M-2Н2O)+, m/z 302 → m/z 266; SPH-1-P: MН+ → (M-Н3PO4)+, m/z 381 → m/z 264.
Calibration curves were plotted using SPH, deuterosphingosine, sphinganine, and sphingosine phosphate C17 (Avanti, USA) (MRM transition: 366 → 250). For each lipid, we calculated the ratio of the area under the chromatographic peak to the area under the chromatographic peak of the standard molecule.
Expression of genes encoding enzymes of sphingolipid metabolism was analyzed using RNA sequencing (RNA-Seq). mRNA libraries for sequencing were generated using a kit for isolating poly(A)+ fraction of mRNA (NEB, USA) and NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB) as recommended by the manufacturer [60]. Poly(A)-enriched RNA fraction was sequenced on an Illumina NextSeq 500 platform (Illumina, USA) at the Center of Collective Use (Institute of Molecular Biology, Russian Academy of Sciences) (http://www.eimb.ru/rus/ckp/ccu_genome_c.php). Data processing was performed using the PPLine software [61], including trimming of short readings based on sequencing quality and length with Trimmomatic program [62], alignment of readings to the reference genome of the GRCm38 version with the STAR program [63], and counting of the readings aligned to the genes with the htseq-count program [64].
Statistical data processing and analysis of differential gene expression were carried out with the edgeR package [65] used in the statistical data processing medium of the R language [66]. The significance of differences in the gene expression was estimated by pairwise comparison of experimental groups using standard Fischer F-test and Benjamini–Hochberg procedure for multiple testing correction [67]. The data were visualized using the ggplot2 package [68].
RESULTS
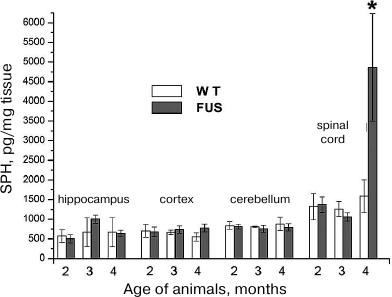
SPH content in the hippocampus, cerebellum, cortex, and spinal cord during the ALS development. The content of SPH capable of inducing apoptosis in the CNS cells was determined in mice at the age of 2, 3, and 4 months. The amount of SPH was determined in lipid specimens and calculated in pg/mg tissue and pg/mg phosphate. Virtually no changes were found in the SPH content calculated per tissue weight in the cerebellum, cortex, and hippocampus in comparison to the control; at the same time, the amount of SPH in the spinal cord at the terminal ALS stage (4 months) increased 4-fold (Fig. 2), which was accompanied by the massive cell death in this CNS region likely caused by the increase in the SPH content. Analysis of changes in SPH amount per mg phosphate (data not presented) revealed its slight increase in the hippocampus at the age of 3 months and in the cortex at the age of 4 months, whereas no changes were observed in the cerebellum. On the contrary, in the spinal cord, the amount of SPH per mg phosphate increased dramatically.
Fig. 2. Changes in the SPH content in the brain structures and spinal cord during ALS development in mice.
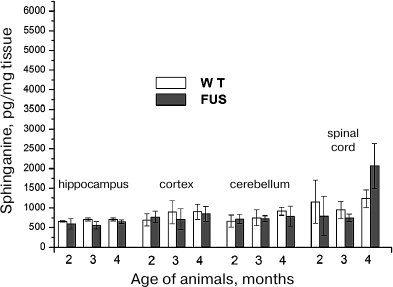
Sphinganine content in the hippocampus, cerebellum, cortex, and spinal cord during ALS development. Analysis of changes in the sphinganine content under the same conditions revealed similar pattern as for SPH. The amount of sphinganine in the spinal cord increased at the age of 4 months, but this increase was less pronounced (2-fold) than in the case of SPH (Fig. 3). In the brain structures, the content of sphinganine (calculated per either mg tissue or mg phosphate) remained virtually the same as in the control during the entire period of observation.
Fig. 3. Changes in the sphinganine content in the brain structures and spinal cord during ALS development in mice.
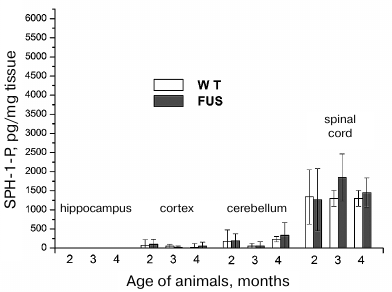
SPH-1-P content in the hippocampus, cerebellum, cortex, and spinal cord during ALS development. SPH-1-P is a product of SPH phosphorylation. Unlike ceramide and SPH, it has the anti-apoptotic properties and participates in the regulation of cell proliferation [20]. Changes in the SPH-1-P level and its ratio to SPH and sphinganine can affect survival of CNS cells. The content of SPH-1-P in the brain structures was low and virtually did not change in the course of disease development (Fig. 4). In the spinal cord, the content of SPH-1-P was considerably higher than in the brain structures and remained constant within the first 4 months. After 4 months, a slight tendency for the increase in the SPH-1-P content was observed. On the contrary, in the control animals, there was a tendency for the decrease in the SPH-1-P content with age, which was likely related to the exhaustion of protective anti-apoptotic reserves of the body. The increase in the SPH-1-P content in the animals with ALS can be explained by the elevation of the amount of content of SPH from which SPH-1-P is synthesized. Moreover, the SPH-1-P ratio to SPH sharply fell in these animals, i.e., the balance between proliferation and apoptosis in the spinal cord cells was disrupted (Figs. 2 and 4).
Fig. 4. Changes in the SPH-1-P content in the brain structures and spinal cord during ALS development in mice.
Expression of genes involved in the metabolism of SPH, sphinganine, and SPH-1-P in the spinal cord during ALS development. Since initially high levels of SPH, sphinganine, and SPH-1-P and changes in the SPH and sphinganine content were observed in the spinal cord only, we analyzed expression of genes encoding enzymes directly involved in the metabolism of these compounds in this CNS region. Expression of the following enzymes was evaluated: 3-ketosphinganine reductase (3-KSR), ceramidase, SPH kinase, SPH-1-P phosphatase, and SPH-1-P lyase (Fig. 1). 3-KSR converts 3-ketosphinganine into sphinganine; dihydroceramide synthase converts sphinganine into dihydroceramide, which is metabolized into ceramide. Ceramidases convert ceramide into SPH (the only SPH source in the cells). By now, five ceramidases have been identified [19]. Acidic ceramidase (ASAH1) and neutral ceramidase (ASAH2) localize to the lysosomal compartments and plasma membrane, respectively, while three alkaline ceramidases have been found in the Golgi apparatus and plasma membrane. SPH kinase phosphorylates SPH with the formation of the anti-apoptotic SPH-1-P. SPH-1-P, in its turn, can be converted back into SPH by SPH-1-P phosphatase or completely removed from the metabolic cycle by SPH-1-P lyase with the generation of ethanolamine phosphate and hexadecenal (Fig. 1). Therefore, SPH content in the cell can be change through several pathways.
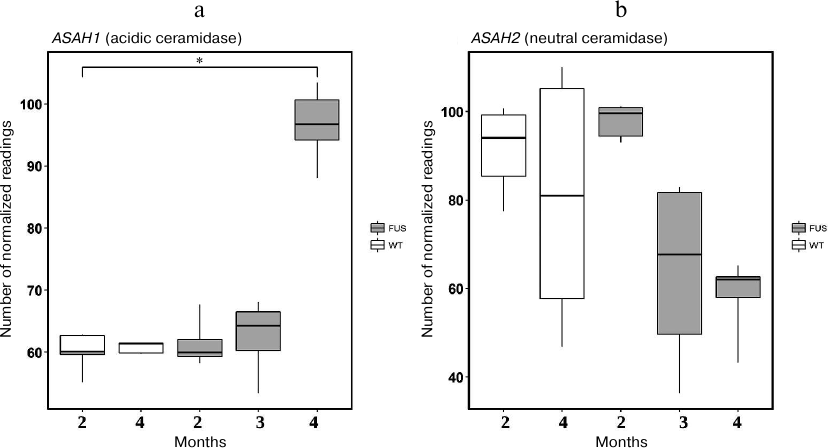
First, we evaluated expression of the 3-KSR gene involved in sphinganine synthesis in the spinal cord of transgenic (FUS) and non-transgenic wild type (WT) mice. In 4-month-old mice, the level of 3-KSR mRNA in transgenic mice was slightly increased as compared to the control WT animals (Fig. 5). Next, we investigated expression of four genes encoding ceramidases and found that expression of the ASAH1 (acidic ceramidase) gene was upregulated, whereas the expression of the ASAH2 (neutral ceramidase) gene was significantly downregulated during progression of the FUS-mediated proteinopathy (p < 0.05, Fischer test) (Fig. 6). The content of mRNAs for alkaline ceramidases (ACER2 and ACER3) remained unchanged, and no expression of the ACER1 mRNA was observed in the spinal cord (data not presented).
Fig. 5. The level of 3-KSR mRNA in the spinal cord of transgenic (FUS) and control wild-type (WT) mice in the course of development of the FUS-mediated proteinopathy. The data are obtained by RNA-Seq analysis and presented as a number of mapped reads per million of sequenced reads (CPM, counts per million).
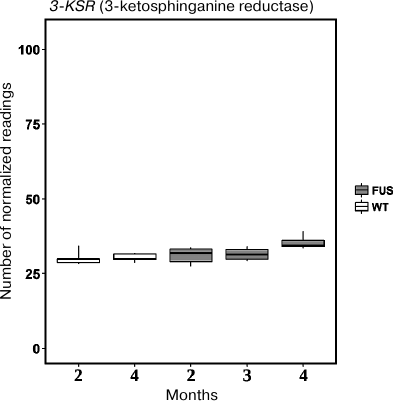
Fig. 6. The levels of mRNAs for acidic (ASAH1) (a) and neutral (ASAH2) (b) ceramidases in the spinal cord of transgenic (FUS) and control wild-type (WT) mice; * p < 0.05 (Fischer test).
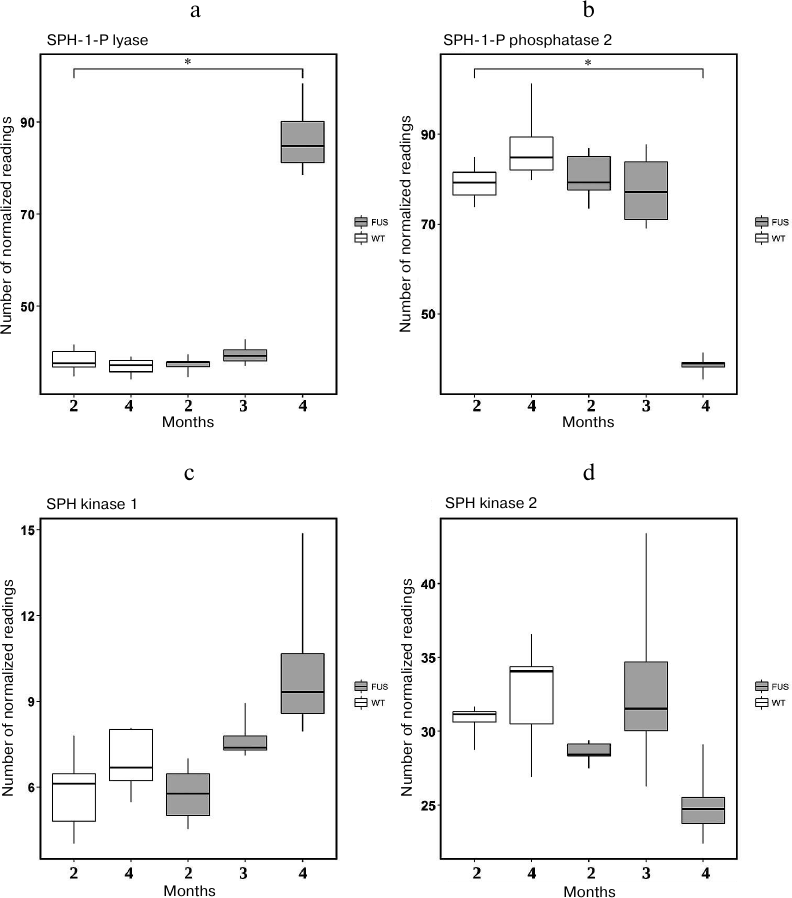
Analyzing changes in the expression of genes encoding SPH-1-P lyase (Sgpl) (Fig. 7a), SPH-1-P phosphatase 2 (Sgpp2) (Fig. 7b ), SPH-1-P kinase 1 (Sphk1) (Fig. 7c), and SPH-1-P kinase 2 (Sphk2) (Fig. 7d), we suggested that the SPH-1-P degradation by SPH-1-P lyase to the final products ethanolamine phosphate and hexadecenal was dramatically activated, because expression of the gene encoding SPH-1-P lyase was sharply increased. At that, expression of genes encoding phosphatase 2 and kinases 1 and 2 decreases (Fig. 7), which could result in decrease in the content of these enzymes. Based on these data, we concluded that despite significant increase in the content of SPH and sphinganine, SPH-1-P metabolism was shifted towards its degradation (Fig. 7). Moreover, the ratio of SPH-1-P to sphingoid bases considerably decreased, thus creating conditions for active apoptosis.
Fig. 7. Expression of genes encoding enzymes involved in metabolism of SPH and SPH-1-P in transgenic (FUS) and control wild-type (WT) mice. a) SPH-1-P lyase; b) SPH-1-P phosphatase; c, d) SPH kinases 1 and 2, respectively; * p < 0.05 (Fischer’s test).
DISCUSSION
The mechanisms of motor neuron degeneration in ALS are poorly understood and the reasons for the ALS onset remain unclear. So far, there are no efficient approaches for the ALS treatment. However, the latest studies have established that degeneration of motor neurons in ALS occurs via apoptotic death [5]. Apoptosis is regulated by various interconnected pathways that eventually result in the programmed cell death. In addition to the genetic regulation, apoptosis is controlled by free radicals, death receptors, caspases, proapoptotic proteins of the Bcl-2 family, inhibitors of apoptosis proteins (IAPs), tumor suppressor protein p53, tumor necrosis factor-α, and many other apoptosis-associated molecules. Apoptosis can be initiated by the damage to DNA, mitochondria, and lysosomal membranes. Some of the apoptosis initiation mechanisms have been observed in the animal ALS models [5, 69-71].
Degeneration of motor neurons caused by mutant superoxide dismutase-1 (mSOD1) was modeled in mSOD1 transgenic mice. During disease progression, these animals demonstrated mitochondria-dependent apoptosis associated with the proapoptotic Bax protein translocation the from the cytosol to the mitochondria, cytochrome c release from the mitochondria to the cytosol, and caspase-9 activation in the spinal cord [71].
Despite numerous studies on apoptosis, the role of sphingolipids in its development and regulation has been poorly studied, which might be due to the diversity of sphingolipid molecules, their complex metabolism, and multi-level control of their functions, as well as involvement of sphingolipids in virtually all physiological and pathophysiological processes in the cell. On the contrary, the role of ceramides and sphingoid bases in the induction of apoptosis has been well understood. SPH can induce apoptosis by all the above-mentioned mechanisms. In particular, it causes DNA degradation [54], transmits signals from the tumor necrosis factor-α [28-30], induces oxidative stress, and activates caspases [31, 32] and other apoptosis signaling molecules. At the same, the role of sphingoid bases in the death of motor neurons in ALS remains unknown.
Our results demonstrate that ALS development is associated with the dysregulation of the metabolism of sphingoid bases, including SPH, sphinganine, and SPH-1-P, mainly in the spinal cord, whereas in the brain structures, the content of these compounds remains low and does not change with the disease progression. This correlates well with the fact that mostly motor neurons are damaged in ALS. The ratio of the anti-apoptotic SPH-1-P to the pro-apoptotic SPH and sphinganine in the affected tissues is sharply decreased, which indicates pronounced activation of cell death in the spinal cord structures, but not in the brain.
We also observed that ALS development was accompanied by dysregulation of the expression of genes encoding enzymes of sphingolipid metabolism mainly in the spinal cord. Out of four ceramidase genes studied, expression of Asah1 mRNA (acidic ceramidase) was upregulated, whereas expression of the Asah2 mRNA (plasma membrane neutral ceramidase) was significantly downregulated in the course of FUS-mediated proteinopathy progression (Fig. 6). Acidic ceramidase of lysosomes (ASAH1) hydrolyzes C10-C14 ceramides with saturated bonds or C18:1 and C18:2 ceramides with unsaturated bonds. ASAH2 hydrolyzes C16-C22 and C26-C36 ceramides. Therefore, SPH is generated by the lysosomal ceramidase from ceramides with shorter fatty acids. The changes in the activity of lysosomal ceramidase can indicate development of the lysosomal apoptosis. The levels of mRNAs for alkaline ceramidases (Acer2 and Acer3) remain unchanged, whereas no expression of the Acer1 gene was observed in the spinal cord. These ceramidases are located in the endoplasmic reticulum and Golgi complex, i.e., do not participate in the generation of SPH from ceramides. The sharp increase in the expression of SPH-1-P lyase at the ALS terminal stage demonstrates exhaustion of the anti-apoptotic reserves of motor neurons and rapid apoptosis development. The observed upregulation of the SPH kinase gene expression on the 2nd and 3rd months of ALS development can be explained by the protective properties of the spinal cord motor neurons. However, these properties are negated by the rapid cell death at the terminal stage of ALS development at 4 months.
The use of mass spectrometry allowed us to study the differences in the content of three most important sphingoid bases (which are difficult to analyze by other methods) and to demonstrate for the first time the involvement of sphingoid bases in the death of spinal cord neurons during ALS development. Moreover, these data were obtained using the ALS animal model that closely mimics the sporadic form of ALS in humans. Deeper understanding of biological pathways regulating metabolism of different sphingolipids during ALS development can lead to the identification of novel targets for pharmaceutical preparations, e.g., enzymes participating in the metabolism of sphingoid bases involved in ALS pathogenesis, such as acidic ceramidase responsible for the SPH generation. An example of such promising pharmaceutical preparation for the ALS treatment is the SPH synthetic analog Fingolimod, which is phosphorylated in the body and displays the effect similar to that of SPH-1-P, the anti-apoptotic metabolite of sphingolipids [72]. Comprehensive studies of the changes in the metabolism of sphingoid bases in ALS will facilitate the understanding of pathological aspects of this disease and promote the development of new drugs for the treatment of this neurodegenerative pathology.
Funding. This work was supported by the Russian Science Foundation (project no. 19-13-00378; investigation of neurodegenerative processes in FUS(1-359) mice) and State Task no. 44.4 (State Registration 01201253310) for the Institute of Biochemical Physics (studies of lipid properties in neuropathologies). Transgenic animals were provided by the Program of support of bioresource collections of the Institute of Physiologically Active Compounds (IPAC), Russian Academy of Sciences (animal maintenance).
Acknowledgements. The study was performed using equipment of the Center of Collective Use of IPAC within the framework of the program of the Presidium of the Russian Academy of Sciences on priority directions no. 18 “Innovative developments in biomedicine”. The data of high-performance sequencing were analyzed at the Genome Center of Collective Use, Institute of Molecular Biology (http://www.eimb.ru/rus/ckp/ccu_genome_c.php).
Conflict of interest. The authors declare no conflict of interest.
Compliance with ethical norms. All international, national, and/or institutional guidelines for the care and use of animals have been observed.
REFERENCES
1.Van Den Bosch, L. (2011) Genetic rodent models of
amyotrophic lateral sclerosis, J. Biomed. Biotechnol.,
2011, 348765, doi: 10.1155/2011/348765.
2.Robberecht, W., Sapp, P., Viaene, M. K., Rosen, D.,
McKenna-Yasek, D., Haines, J., Horvitz, R., Theys, P., and Brown, R.,
Jr. (1994) Cu/Zn superoxide dismutase activity in familial and sporadic
amyotrophic lateral sclerosis, J. Neurochem., 62,
384-387, doi: 10.1046/j.1471-4159.1994.62010384.x.
3.Koppers, M., van Blitterswijk, M. M., Vlam, L.,
Rowicka, P. A., van Vught, P. W., Groen, E. J., Spliet, W. G.,
Engelen-Lee, J., Schelhaas, H. J., de Visser, M., van der Kooi, A. J.,
van der Pol, W. L., Pasterkamp, R. J., Veldink, J. H., and van den
Berg, L. H. (2012) VCP mutations in familial and sporadic amyotrophic
lateral sclerosis, Neurobiol. Aging, 33,
837.e7-13, doi: 10.1016/j.neurobiolaging.2011.10.006.
4.Daoud, H., Suhail, H., Szuto, A., Camu, W.,
Salachas, F., Meininger, V., Bouchard, J. P., Dupre, N., Dion, P. A.,
and Rouleau, G. A. (2012) UBQLN2 mutations are rare in French and
French-Canadian amyotrophic lateral sclerosis, Neurobiol. Aging,
33, 2230-2233, doi:
10.1016/j.neurobiolaging.2012.03.015.
5.Sathasivam, S., Ince, P. G., and Shaw, P. J. (2001)
Apoptosis in amyotrophic lateral sclerosis: a review of the evidence,
Neuropathol. Appl. Neurobiol., 27, 257-274, doi:
10.1046/j.0305-1846.2001.00332.x.
6.DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B.
F., Boxer, A. L., Baker, M., Rutherford, N. J., Nicholson, A. M.,
Finch, N. A., Flynn, H., Adamson, J., Kouri, N., Wojtas, A., Sengdy,
P., Hsiung, G. Y., Karydas, A., Seeley, W. W., Josephs, K. A., Coppola,
G., Geschwind, D. H., Wszolek, Z. K., Feldman, H., Knopman, D. S.,
Petersen, R. C., Miller, B. L., Dickson, D. W., Boylan, K. B.,
Graff-Radford, N. R., and Rademakers, R. (2011) Expanded GGGGCC
hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome
9p-linked FTD and ALS, Neuron, 72, 245-256, doi:
10.1016/j.neuron.2011.09.011.
7.Mackenzie, I. R., Bigio, E. H., Ince, P. G., Geser,
F., Neumann, M., Cairns, N. J., Kwong, L. K., Forman, M. S., Ravits,
J., Stewart, H., Eisen, A., McClusky, L., Kretzschmar, H. A., Monoranu,
C. M., Highley, J. R., Kirby, J., Siddique, T., Shaw, P. J., Lee, V.
M., and Trojanowski, J. Q. (2007) Pathological TDP-43 distinguishes
sporadic amyotrophic lateral sclerosis from amyotrophic lateral
sclerosis with SOD1 mutations, Ann. Neurol., 61, 427-434,
doi: 10.1002/ana.21147.
8.Kwiatkowski, T. J., Bosco, D. A., Leclerc, A. L.,
Tamrazian, E., and Vanderburg, C. R. (2009) Mutations in the
FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral
sclerosis, Science, 323, 1205-1208, doi:
10.1126/science.1166066.
9.Deng, H. X., Zhai, H., Bigio, E. H., Yan, J.,
Fecto, F., Ajroud, K., Mishra, M., Ajroud-Driss, S., Heller, S., Sufit,
R., Siddique, N., Mugnaini, E., and Siddique, T. (2010)
FUS-immunoreactive inclusions are a common feature in sporadic and
nonSOD1 familial amyotrophic lateral sclerosis, Ann. Neurol.,
67, 739-748, doi: 10.1002/ana.22051.
10.Daoud, H., Dobrzeniecka, S., Camu, W., Meininger,
V., Dupre, N., Dion, P. A., and Rouleau, G. A. (2013) Mutation analysis
of PFN1 in familial amyotrophic lateral sclerosis patients,
Neurobiol. Aging, 34, 1311.e1-1311.e2, doi:
10.1016/j.neurobiolaging.2012.09.001.11.
11.Munch, C., Sedlmeier, R., Meyer, T., Homberg, V.,
Sperfeld, A. D., Kurt, A., Prudlo, J., Peraus, G., Hanemann, C. O.,
Stumm, G., and Ludolph, A. C. (2004) Point mutations of the p150
subunit of dynactin (DCTN1) gene in ALS, Neurology,
63, 724-726, doi: 10.1212/01.WNL.0000134608.83927.B1.
12.Rademakers, R., and van Blitterswijk, M. (2014)
Excess of rare damaging TUBA4A variants suggests cytoskeletal defects
in ALS, Neuron, 84, 241-243, doi:
10.1016/j.neuron.2014.10.002.
13.Ghasemi, M., and Brown, R. H. (2017) Genetics of
amyotrophic lateral sclerosis, Cold Spring Harb. Perspect. Med.,
7, a024125.
14.Hussain, G., Anwar, H., Rasul, A., Imran, A.,
Qasim, M., Zafar, S., Imran, M., Kamran, S. K. S., Aziz, N., Razzaq,
A., Ahmad, W., Shabbir, A., Iqbal, J., Baig, S. M., Ali, M., Gonzalez
de Aguilar, J. L., Sun, T., Muhammad, A., and Muhammad Umair, A. (2019)
Lipids as biomarkers of brain disorders, Crit. Rev. Food Sci.
Nut., 7, 1-24, doi:
10.1080/10408398.2018.1529653.
15.Hussain, G., Wang, J., Rasul, A., Anwar, H.,
Imran, A., Qasim, M., Zafar, S., Kamran, S. K. S., Razzaq, A., Aziz,
N., Ahmad, W., Shabbir, A., and Iqbal, J. (2019) Role of cholesterol
and sphingolipids in brain development and neurological diseases,
Lipids Health Dis., 18, 26-38, doi:
10.1186/s12944-019-0965-z.
16.Dupuis, L., Corcia, P., Fergani, A., Gonzalez De
Aguilar, J. L., Bonnefont-Rousselot, D., Bittar, R., Seilhean, D.,
Hauw, J. J., Lacomblez, L., Loeffler, J. P., and Meininger, V. (2008)
Dyslipidemia is a protective factor in amyotrophic lateral sclerosis,
Neurology, 70, 1004-1009, doi:
10.1212/01.wnl.0000285080.70324.27.
17.Chen, X., Yazdani, S., Piehl, F., Magnusson, P.
K. E., and Fang, F. (2018) Polygenic link between blood lipids and
amyotrophic lateral sclerosis, Neurobiol. Aging, 67,
202.e1-202.e6, doi: 10.1016/j.neurobiolaging.2018.03.022.
18.Albi, E., Alessenko, A., and Grosch, S. (2018)
Sphingolipids in inflammation, Mediators Inflamm., 2018,
Article ID 7464702, 3 p., doi: 10.1155/2018/7464702.
19.Alessenko, A. V. (2013) Potential role of
sphingolipids in neuropathogenesis of Alzheimer’s diseases,
Biomed. Khim., 59, 25-50, doi:
10.18097/pbmc20135901025.
20.Maceyka, M., and Spiegel, S. (2014) Sphingolipid
metabolites in inflammatory disease, Nature, 510, 58-67,
doi: 10.1038/nature13475.
21.Okuwa, H., Kanno, T., Fujita, Y., Gotoh, A.,
Tabata, C., Fukuoka, K., Nakano, T., and Nishizaki, T. (2012)
Sphingosine suppresses mesothelioma cell proliferation by inhibiting
PKC-δ and inducing cell cycle arrest at the G(0)/G(1) phase,
Cell Physiol. Biochem., 30, 995-1004, doi:
10.1159/000341476.
22.Hannun, Y. A., Loomis, C. R., Merrill, A. H.,
Jr., and Bell, R. M. (1986) Sphingosine inhibition of protein kinase C
activity and of phorbol dibutyrate binding in vitro and in human
platelets, J. Biol. Chem., 261, 12604-12609.
23.Conkling, P. R., Patton, K. L., Hannun, Y. A.,
Greenberg, C. S., and Weinberg, J. B. (1989) Sphingosine inhibits
monocyte tissue factor-initiated coagulation by altering factor VII
binding, J. Biol. Chem., 264, 18440-18444.
24.Zhang, H., Buckley, N. E., Gibson, K., and
Spiegel, S. (1990) Sphingosine stimulates cellular proliferation via a
protein kinase C-independent pathway, J. Biol. Chem.,
265, 76-81.
25.Hakomori, S.-I. (1992) Functional role of
glycosphingolipids in tumor progression, J. Exp. Med.,
168, 211-222, doi: 10.1620/tjem.168.211.
26.Bibel, D. J., Aly, R., and Shinefield, H. R.
(1995) Topical sphingolipids in antisepsis and antifungal therapy,
Clin. Exp. Dermatol. Clin. Exp. Dermatol., 20,
395-400.
27.Taha, T. A., Mullen, N. D., and Obeid, L. M.
(2006) A house divided: ceramide, sphingosine, and
sphingosine-1-phosphate in programmed cell death, Biochim. Biophys.
Acta, 1758, 2027-2036.
28.Alessenko, A. V., and Khrenov, A. V. (1999) Role
of sphingosine in induced apoptosis, Lipids, 34, S75-S76,
doi: 10.1007/bf02562235.
29.Krown, K. A., Page, M. T., Nguyen, C., Zechner,
D., Gutierrez, V., Comstock, K. L., Gembotski, C. C., Quintana, P. J.,
and Sabbadini, R. A. (1996) Tumor necrosis factor alpha-induced
apoptosis in cardiac myocytes. Involvement of the sphingolipid
signaling cascade in cardiac cell death, J. Clin. Invest.,
98, 2854-2865, doi: 10.1172/jci119114.
30.Sweeney, E. A., Sakakura, C., Shirahama, T.,
Masamune, A., Ohta, H., Hakamori, S., and Igarashi, Y. (1996)
Sphingosine and its methylated derivative N,N-dimethylsphingosine (DMS)
induce apoptosis in a variety of human cancer cell lines, Int. J.
Cancer, 66, 358-366, doi:
10.1002/(sici)1097-0215(19960503)66:3%3C358::aid-ijc16%3E3.0.co;2-7.
31.Cuvillier, O., Nava, V. T., Murthy, S. K.,
Edsall, L. C., Levade, T., Milstien, S., and Spiegel, S. (2001)
Sphingosine generation, cytochrome c release, and activation of
caspase-7 in doxorubicin-induced apoptosis of MCF7 breast
adenocarcinoma cells, Cell Death Differ., 8, 162-171,
doi: 10.1038/sj.cdd.4400793.
32.Hung, W. C., Chang, H. C., and Chuang, L. Y.
(1999) Activation of caspase-3-like proteases in apoptosis induced by
sphingosine and other long-chain bases in Hep3B hepatoma cells,
Biochem. J., 338, 161-166.
33.Smith, E. R., Merrill, A. H., Obeid, L. M., and
Hannun, Y. A. (2000) Effects of sphingosine and other sphingolipids on
protein kinase C, Methods Enzymol., 312, 361-373, doi:
10.1016/s0076-6879(00)12921-0.
34.McDonald, O. B., Hannun, Y. A., Reynolds, C. H.,
and Sahyoun, N. (1991) Activation of casein kinase II by sphingosine,
J. Biol. Chem., 266, 21773-21776, doi:
ncbi.nlm.nih.gov/pubmed/1939200.
35.Coroneos, E., Wang, Y., Panuska, J. R.,
Templeton, D. J., and Kester, M. (1996) Sphingolipid metabolites
differentially regulate extracellular signal-regulated kinase and
stress-activated protein kinase cascades, Biochem. J.,
316, 13-17, doi: 10.1042/bj3160013.
36.Megidish, T., White, T., Takio, K., Titani, K.,
Igarashi, Y., and Hakomori, S. (1995) The signal modulator protein
14-3-3 is a target a sphingosine- or N,N-dimethylsphingosine-depend
kinase in 3T3 (A31) cells, Biochem. Biophys. Res. Commun.,
216, 739-747, doi: 10.1006/bbrc.1995.2684.
37.Pyne, S., and Pyne, N. J. (1996) The differential
regulation of cyclic AMP by sphingomyelin-derived lipids and the
modulation of sphingolipid-stimulated extracellular signal regulated
kinase-2 in airway smooth, Biochem. J., 315,
917-923, doi: 10.1042/bj3150917.
38.Yang, S. D., Chang, H. C., and Lee, S. C. (1996)
Okadaic acid, sphingosine, and phorbol ester reversibly modulate heat
induction on protein kinase FA/GSK-3 alpha in A431 cells, J. Cell.
Biochem., 60, 218-225, doi:
10.1002/(SICI)1097-4644(19960201)60:2%3C218::AID-JCB6%3E3.0.CO;2-#.
39.Anastasiadis, P. Z., Kuhn, D. M., Blitz, J.,
Imerman, B. A., Louie, M. C., and Levine, R. A. (1996) Regulation of
tyrosine hydrolase and tetrahydrobiopterin biosynthetic in PC12 cells
by NGF, EGF and IFN-gamma, Brain Res., 713, 125-133, doi:
10.1016/0006-8993(95)01494-2.
40.Zhou, H., Summers, S. A., Birnbaum, M. J., and
Pittman, R. N. (1998) Inhibition of Akt kinase by cell-permeable
ceramide and its implications for ceramide-induced apoptosis, J.
Biol. Chem., 273, 16568-16575, doi:
10.1074/jbc.273.26.16568.
41.Lavie, Y., and Liscovitch, M. (1990) Activation
of phospholipase D by sphingoid bases in NG108-15 neural derived cells,
J. Biol. Chem., 265, 3868-3872.
42.Matecki, A., Stopa, M., Was, A., and Pawelczyk,
T. (1997) Effect of sphingomyelin and its metabolites on the activity
of human recombinant PLC delta 1, Int. Biochem. Cell. Biol.,
29, 815-828, doi: 10.1016/s1357-2725(97)00014-9.
43.Sasaki, J. I., Yamaguchi, M., Yamane, H.,
Okamura, N., and Ishibashi, S. (1996) Sphingosine inhibition of NADPH
oxidase activation in a cell-free system, J. Biochem. (Tokyo),
120, 705-709, doi: 10.1093/oxfordjournals.jbchem.a021468.
44.Pushkareva, M. Y., Khan, W. A., Alessenko, A. V.,
Sahyoun, N., and Hannun, Y. A. (1992) Sphingosine activation of protein
kinases in Jurkat T cells. In vitro phosphorylation of
endogenous protein substrates and specificity of action, J. Biol.
Chem., 267, 15246-15251.
45.Jefferson, A. B., and Schulman, H. (1988)
Sphingosine inhibits calmodulin-depended enzymes, J. Biol.
Chem., 263, 15241-15244.
46.Arnold, R. S., and Newton, A. C. (1991)
Inhibition of the insulin receptor tyrosine kinase by sphingosine,
Biochemistry, 30, 7747-7754.
47.Mullmann, T. J., Siegel, M. I., Egan, R. W., and
Billah, M. M. (1991) Sphingosine inhibits phosphatidate
phosphohydrolase in human neutrophils by a PKC-independent mechanism,
J. Biol. Chem., 266, 2013-2016.
48.Sohal, P. S., and Corne, R. B. (1990) Sphingosine
inhibits the activity of rat liver CTP-phosphocholine
cytidyltransferase, J. Biol. Chem., 265, 11746-11750.
49.Sakane, S., Takemura, H., Yamada, K., Imoto, K.,
Kaneko, M., and Ohshika, H. (1996) Different effects of sphingosine,
R59022 and anionic amphiphiles on two diacylglycerol kinase isozymes
purified from porcine thymus cytosol, J. Biol. Chem.,
271, 1148-1155, doi: 10.1016/0014-5793(89)81134-2.
50.Davis, R. J., Girones, N., and Faucher, M. (1988)
Two alternative mechanisms control the interconversion of functional
states of the epidermal growth factor receptor, J. Biol. Chem.,
263, 5373-5379.
51.Fausher, M., Girones, N., Hannun, Y. A., Bell, R.
M., and Davis, R. J. (1988) Regulation of the epidermal growth factor
receptor phosphorylation state by sphingosine in A431 human epidermoid
carcinoma cells, J. Biol. Chem., 263, 5319-5327.
52.Huang, W. C., and Chueh, S. H. (1996) Calcium
mobilization from the intracellular mitochondrial and non-mitochondrial
stores of the rat cerebellum, Brain Res., 718,
151-158, doi: 10.1016/0006-8993(96)00108-4.
53.Czajkowski, R., and Baranska, J. (1999)
Sphingosine and phorbol ester modulate protein kinase C activity and
modify ATP-evoked calcium mobilization in glioma C6 cells, Biochem.
Biophys. Res. Commun., 260, 614-618, doi:
10.1006/bbrc.1999.0946.
54.Tamiya-Koizumi, K., Murate, T., Suzuki, M.,
Simbulan, C. M., Nakagawa, M., Takemura, M., Furuta, K., Izuta, S., and
Yoshida, S. (1997) Inhibition of DNA primase by sphingosine and its
analogues parallels with their growth suppression of cultured human
leukemic cells, Biochem. Mol. Biol. Int., 41, 1179-1189,
doi: 10.1080/15216549700202271.
55.Koiv, A., Palvimo, J., and Kinnunen, P. K. (1995)
Evidence for ternary complex formation by histone H1 and liposomes,
Biochemistry, 34, 8018-8027, doi:
10.1021/bi00025a007.
56.Hwong, C. L., Chen, M. S., and Hwang, J. L.
(1989) Phorbol ester transiently increases topoisomerase I mRNA levels
in human skin fibroblasts, J. Biol. Chem., 264,
14923-14926.
57.Deikin, A. V., Kovrazhkina, E. A., Ovchinnikov,
R. K., Bronovitsky, E. V., Razinskaya, O. D., Smirnov, A. P.,
Ermolkevich, T. G., Elyakov, A. B., Popov, A. N., Fedorov, E. N.,
Lytkina, O. A., Kukharsky, M. S., Tarasova, T. V., Shelkovnikova, T.
A., Ustyugov, A. A., Ninkina, N. N., Goldman, I. L., Sadchikov, E. R.,
Bachurin, S. O., and Skvortsova, V. I. (2014) Model of amyotrophic
lateral sclerosis based on the strain of transgenic mice expressing the
mutant form of human FUS protein, Korsakov Zh. Nevrol.,
8, 63-70.
58.Bligh, T. G., and Dyer, W. J. (1959) A rapid
method of total lipid extraction and purification, Can. J. Biochem.
Physiol., 37, 911-917, doi: 10.1139/o59-099.
59Sullards, M. C. (2000) Analysis of sphingomyelin, glucosylceramide,
ceramide, sphingosine, and sphingosine 1-phosphate by tandem mass
spectrometry, Methods Enzymol., 312, 32-45, doi:
10.1016/S0076-6879(00)12898-8.
60.Funikov, S. Y., Rezvykh, A. P., Mazin, P. V.,
Morozov, A. V., Maltsev, A. V., Chicheva, M. M., Vikhareva, E. A.,
Evgen’ev, M. B., and Ustyugov, A. A. (2018) FUS(1-359) transgenic
mice as a model of ALS: pathophysiological and molecular aspects of the
proteinopathy, Neurogenetics, 19, 189-204, doi:
10.1007/s10048-018-0553-9.
61.Krasnov, G. S., Dmitriev, A. A., Kudryavtseva, A.
V., Shargunov, A. V., Karpov, D. S., Uroshlev, L. A., Melnikova, N. V.,
Blinov, V. M., Poverennaya, E. V., Archacov, A. I., Lisitsa, A. V., and
Ponomarenko, E. A. (2015) PPLine: an automated pipeline for SNP, SAP,
and splice variant detection in the context of proteogenomics, J.
Proteome Res., 14, 3729-3737, doi:
10.1021/acs.jproteome.5b00490.
62.Bolger, A. M., Lohse, M., and Usadel, B. (2014)
Trimmomatic: a flexible trimmer for Illumina sequence data,
Bioinformatics, 30, 2114-2120, doi:
10.1093/bioinformatics/btu170.
63.Dobin, A., Davis, C. A., Schlesinger, F.,
Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M., and
Gingeras, T. R. (2013) STAR: ultrafast universal RNA-seq aligner,
Bioinformatics, 29, 15-21, doi:
10.1093/bioinformatics/bts635.
64.Anders, S., Pyl, P. T., and Huber, W. (2015)
HTSeq – a Python framework to work with high-throughput
sequencing data, Bioinformatics, 31, 166-169, doi:
10.1101/002824.
65.Robinson, M. D., McCarthy, D. J., and Smyth, G.
K. (2010) edgeR: a bioconductor package for differential expression
analysis of digital gene expression data, Bioinformatics,
26, 139-140, doi: 10.1093/bioinformatics/btp616.
66.RC Team (2000) R Language Definition, R
Foundation for Statistical Computing, Vienna, Austria.
67.Thissen, D., Steinberg, L., and Kuang, D. (2002)
Quick and easy implementation of the Benjamini–Hochberg procedure
for controlling the false positive rate in multiple comparisons, J.
Educ. Behav. Stat., 27, 77-83, doi:
10.3102/10769986027001077.
68.Wickham, H. (2011) ggplot2, WIREs Comp.
Stats., 3, 180-185, doi: 10.1002/wics.147.
69.Hensley, K., Floyd, R. A., Gordon, B., Mou, S.,
Pye, Q. N., Stewart, C., West, M., and Williamson, K. (2002) Temporal
patterns of cytokine and apoptosis-related gene expression in spinal
cords of the G93A-SOD1 mouse model of amyotrophic lateral sclerosis,
J. Neurochem., 82, 365-367, doi:
10.1046/j.1471-4159.2002.00968.x.
70.Ekegren, T., Grundstrom, E., Lindholm, D., and
Aquilonius, S. M. (1999) Upregulation of Bax protein and increased DNA
degradation in ALS spinal cord motor neurons, Acta Neurol.
Scand., 100, 317-321, doi:
10.1111/j.1600-0404.1999.tb00403.x.
71.Guegan, C., Vila, M., Rosoklija, G., Hays, A. P.,
and Przedborski, S. (2001) Recruitment of the mitochondrial-dependent
apoptotic pathway in amyotrophic lateral sclerosis, J.
Neurosci., 21, 6569-6576, doi:
10.1523/JNEUROSCI.21-17-06569.2001.
72.Berry, J. D., Paganoni, S., Atassi, N., Macklin,
E. A., Goyal, N., Rivner, M., Simpson, E., Appel, S., Grasso, D. L.,
Mejia, N. I., Mateen, F., Gill, A., Vieira, F., Tassinari, V., and
Perrin, S. (2017) Phase IIa trial of fingolimod for amyotrophic lateral
sclerosis demonstrates acceptable acute safety and tolerability,
Muscle Nerve, 56, 1077-1084,doi:
10.1002/mus.25733.