REVIEW: New Perspective on the Reversibility of ATP Synthesis and Hydrolysis by Fo⋅F1-ATP Synthase (Hydrolase)
A. D. Vinogradova
Lomonosov Moscow State University, School of Biology, Department of Biochemistry, 119234 Moscow, Russia
Received June 19, 2019; Revised July 8, 2019; Accepted July 8, 2019
Fo⋅F1-ATPases of mitochondria, chloroplasts, and microorganisms catalyze transformation of proton motive force (the difference between the electrochemical potentials of hydrogen ion across a coupling membrane) to the free energy of ATP phosphoryl potential. It is often stated that Fo⋅F1-ATPases operate as reversible chemo-mechano-electrical molecular machines that provide either ATP synthesis or hydrolysis depending on particular physiological demands of an organism; the microreversibility principle of the enzyme catalysis is usually taken as a dogma. Since 1980, the author has upheld the view that the mechanisms of ATP synthesis and hydrolysis by the Fo⋅F1 complex are different (Vinogradov, A. D. (2000) J. Exp. Biol., 203, 41-49). In this paper, the author proposes a new model considering the existence in coupling membranes of two non-equilibrium isoforms of Fo⋅F1 unidirectionally catalyzing synthesis and/or hydrolysis of ATP.
KEY WORDS: Fo⋅F1-ATPase, reversibility of enzymatic catalysisDOI: 10.1134/S0006297919110038
Abbreviations: Fo⋅F1, H+-translocating ATPase (synthase); p, proton motive force; Pd-SBP, inside-out Paracoccus denitrificans plasma membrane vesicles; SMP, submitochondrial particles.
I graduated from the Department of Biochemistry at the School of Biology, Moscow State University, in 1964 and have held tenured Professor positions for more than 50 years (since 1967), while being strongly convinced that research activity is a mandatory and honorary duty of any Professor at the University. About a quarter of my research papers published with many co-authors, all graduated from our Department, have been on the studies of mitochondrial ATPase, the key component of oxidative phosphorylation. Oxidative phosphorylation was discovered and qualitatively characterized by V. A. Engelhardt and V. A. Belitzer in the Soviet Union in the 1930s. I am honored to associate myself with the successors of Russian bioenergetics school, in which the establishing role of the Department of Biochemistry is well recognized. It is helpful to look back from time to time (the more often, the better), first, to summarize what has been done and, second, to outline further studies. This paper is not a review in a common sense; rather, it is a historical progress report, hopefully, not the final one. Previously, I have published several reviews on the subject with proper citations of the original studies [1-3]. In this essay dedicated to 80th anniversary of the Department of Biochemistry, I will refer exclusively to my own studies started in 1972; some statements will be made as well-established facts without proper references to the literature.For a large number of problems there will be some animal of choice or a few such animals on which it can be most conveniently studied. I have no doubt that there is quite a number of animals which are similarly “created” for special physiological purposes, but I am afraid that most of them are unknown to the men for whom they were “created”.
“The Krogh Principle”, August Krogh, eminent Danish physiologist, 1921 Nobel Prize winner (Krogh, A. (1929) The progress of physiology, Am. J. Physiol., 90, 243-251)
Figure 1 shows maximally simplified scheme of oxidative phosphorylation. Oxidation by oxygen (or other oxidants in some organisms) of low-molecular-mass components (SH2) formed by decomposition of nutrients is catalyzed by a complex of oxidoreductases, the so-called respiratory chain. This oxidation is coupled to the vectorial translocation of protons from one compartment (in) to another (out) separated by the membrane that limits spontaneous mixing of the contents of these compartments and, most importantly, that is slowly (compared to the rate of vectorial translocation) permeable for protons (hydroxonium cations in water). The difference between the electrochemical potentials of hydrogen ions across the membrane (ΔµH+, or proton motive force, p) thus arises. The transfer of protons along the electrical and pH gradients via the Fo⋅F1 complex embedded in the membrane is coupled to the ATP formation from ADP and Pi (bottom of Fig. 1) at the expense of free energy accumulated as p. The reverse reaction, ATP hydrolysis (top of Fig. 1), is coupled with the formation of p in the absence of respiration.
Fig. 1. Simplified scheme of oxidative phosphorylation. Closed fragment of the inner mitochondrial membrane (SMP) or bacterial cytoplasmic membrane (Pd-SBP) is shown. The surface freely accessible for the surrounding medium faces mitochondrial matrix or bacterial cytoplasm. The proton motive force (p) generator is composed of the respiratory chain components: complex I (NADH:ubiquinone-), complex III (ubiquinol:cytochrome c-), and complex IV (cytochrome c:oxygen-) oxidoreductases. Several other ubiquinone reductases operating as respiratory chain components (electron-transferring flavoprotein dehydrogenase, α-glycerophosphate dehydrogenase, dihydroorotate dehydrogenase) are not shown. Fo⋅F1-ATP synthase (blue) and ATP hydrolase (red) are composed of hydrophilic F1 that performs the catalytic function and Fo providing translocation of protons and acts as a mechanical driving device rotating the γ-subunit of F1. Spontaneous proton translocation by other proteins [nucleotide translocase, uncoupling proteins (UCPs), cation translocases] are shown by double-directed arrows in the right part of the fragment.
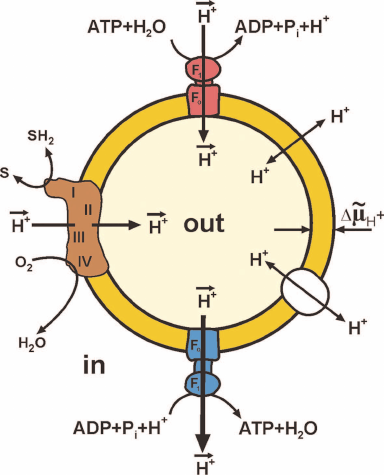
Under physiological conditions, oxygen consumption coupled with ATP formation in cells and tissues is regulated by a number of factors. The simplest and well-known one is the respiratory control. This phenomenon had been originally predicted and experimentally observed by V. A. Belitzer and then confirmed and studied further a decade later by two independent American groups of B. Chance and H. Lardy who used intact isolated mitochondria in their experiments. The authors observed the following phenomena. If no substrates for ATP synthesis (ADP and Pi) were present, oxidation of respiratory substrates (oxygen consumption) was slow and limited by dissipation of p via slow leakage of protons due to specifically catalyzed and spontaneous permeation of protons. Addition of ADP and Pi either strongly or slightly (depending on particular mitochondrial preparation) stimulated respiration which continued until added ADP and Pi transformed to ATP and p became equilibrated by the phosphoryl potential ATP/ADP⋅Pi. Studying parameters of ATP synthase or hydrolase activities of Fo⋅F1 in intact mitochondria is practically impossible, because the overall process involves several other enzymes, such as specific translocases of Pi, adenine nucleotides, and respiratory substrates (Krebs cycle intermediates), each of them depending on p. Figure 1 shows simplified operation of Fo⋅F1 in closed inside-out submitochondrial or bacterial plasma membranes. The outer side of the membrane corresponds to the mitochondrial matrix (or bacterial intracellular) side and the inner side of the vesicle corresponds to the side facing the surrounding medium. The active sites of the respiratory chain components and nucleotide-binding sites of F1 are freely accessible for externally added substrates; these vesicles catalyze the respiration-dependent ATP synthesis. Unfortunately, respiration of most inside-out mitochondrial or plasma membrane vesicles is only insignificantly controlled by the Fo⋅F1 activity, and ATP synthesis in these particles can be measured using ATP traps (glucose plus hexokinase) or luciferin-luciferase ATP-detecting system. It should be mentioned here that the overwhelming majority of conclusions on the mechanism of ATP synthesis are based on the studies of ATP hydrolase activity catalyzed by either soluble F1 or membrane bound Fo⋅F1 or, most recently, on the studies of “visually” detected mechano-chemistry of individual enzyme molecules. Apparently, those conclusions are justified only if the principle of enzyme microreversibility is accepted. Another concern is whether the term equilibrium can be used for the description of respiratory control. Generation and utilization of p are always stationary and never equilibrium processes. The thermodynamic notion equilibrium is often erroneously overused in bioenergetics literature.
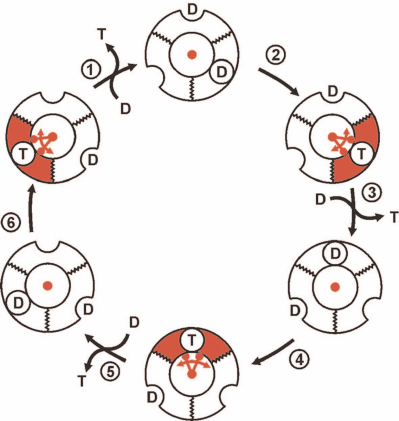
The conventionally accepted view on the events at the catalytically competent nucleotide-binding sites of trimeric α-β-subunit pairs in F1 during p-dependent ATP synthesis is outlined in Fig. 2. It should be noted again that this and numerous analogous schemes of different degree of elaboration are based on the experimental data (kinetics, isotope exchange, structural data) obtained in the preparations unable to synthesize ATP. The scheme shown in Fig. 2 describes sequential formation of three ATP molecules from ADP plus Pi. The mechanism, which is now called rotary alternating site binding-change mechanism, was originally proposed by K. Repke and R. Shon in 1973 and formulated in the generally accepted form by P. Boyer. The three principal aspects of this mechanism are evident:
1) ATP synthesis (formation of phosphoanhydride bond between β- and γ-phosphoryl groups of ATP) proceeds spontaneously and is driven by the high affinity of the product (ATP) to its specific site;
2) free energy of p is required to push out generated ATP from the enzyme active site to the surrounding medium; this is mechanically performed by the F1 γ-subunit located inside the three-lobe “orange” of F1 rotated by the flow of protons through Fo;
3) the stoichiometry is strictly fixed: three ATP molecules per complete (360°) turn of γ-subunit. This is hard to explain when the number of c-subunits in the proton-conductive Fo of Fo⋅F1 is not a multiple of three. The system operating with a variable generated ATP/vectorially translocated proton stoichiometry would be physiologically rational and act as an important regulatory element.
Fig. 2. The rotary alternating site binding-change mechanism of ATP synthesis by Fo⋅F1. F1 is shown as a trimer of α-β-subunit pairs and single γ-subunit (shown in red) located inside of the trimer. The binding sites for the substrate (ADP + Pi, designated as D) or product (T) located at the α-β surface exist in two states: opened, shown as a half-circle on the surface of flat ring, and closed (with tightly bound D and T), shown as a circle on the surface of flat ring. D binds to a single substrate binding site of F1 already containing D and T. T is pushed out by the γ-subunit rotation which is driven by proton flow through Fo. The trimer (F1) is fixed, so its rotation is prevented. One-third turning of γ-subunit (120°) results in the ATP release to the surrounding medium and binding of the second ADP molecular at the open site. A single act of ATP release (step 3) regenerates free F1 which differs from its original form only in the position of γ-subunit. Two following 120° turns result in the release of two ATP molecules and regeneration of the original state of F1.
ATP HYDROLASE ACTIVITY OF Fo⋅F1
Membrane-bound complex Fo⋅F1 of SMP or soluble F1 catalyze hydrolysis of ATP and other purine nucleotides. The reaction of ATP hydrolysis in water at pH 8.0 is described by Eq. (1):
It should be emphasized that proton in the right part of Eq. (1) (the so-called stoichiometric proton) is not vectorially translocated proton shown in Fig. 1. The rate of this reaction can be easily measured in weakly buffered medium by either glass pH-electrode or proper pH indicator (Phenol red is convenient to use). At the very beginning of our studies on the ATPase activity of partially purified F1 solubilized from the acetone powder of bovine heart mitochondria, several specific kinetic properties have been noticed [4, 5] which initiated further examination. Shortly, we have realized that studies on the function of F1 in oxidative phosphorylation are relevant if performed on the complete Fo⋅F1 complex operating in the membranes capable of generation and utilization of p. For this, we have developed a procedure for the preparation of coupled inside-out submitochondrial particles (SMP) capable of all the activities described in Fig. 1. Starting from 1979, all our studies have been done in SMP. The specific features of ATP hydrolysis originally observed were as follows:
1) ATP hydrolysis was not exclusively Mg2+-specific; the enzyme catalyzed the reaction, although at lower rates, with other divalent cations, with the highest activity observed for Co2+;
2) the initial reaction rate rapidly (on the minute scale) declined, which could be explained by neither enzyme irreversible inactivation, nor the decrease in the substrate (ATP) or increase in the product (ADP and Pi) concentrations;
3) the enzymatic activity of both F1 and Fo⋅F1 was significantly stimulated by the standard protonophoric uncoupler 2,4-dinitrophenol. This phenomenon observed for the soluble F1 [5] at that time (early 1970s) seemed to strongly contradict the electrochemical mechanism of oxidative phosphorylation by P. Mitchell;
4) the initial hydrolysis rate showed simple hyperbolic dependence on ATP concentration.
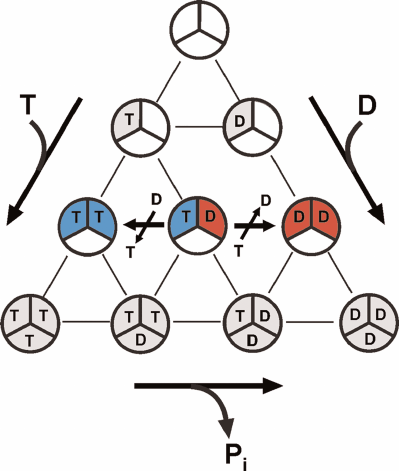
The latter merits short discussion in the light of the alternating site binding-change mechanism and numerous published reports on abnormal (non-Michaelis) dependence of F1- and Fo⋅F1-ATPase activities on ATP concentration. It has been well established that all F1-ATPases are trimers of α-β-pairs containing three rapidly exchangeable catalytically competent nucleotide-binding sites. The possibilities of nucleotide binding to these sites are demonstrated in Fig. 3. The alternating site binding-change catalysis, as any catalytic mechanism, must start and end with the same particular form of the enzyme. The initial and regenerated form of the enzyme operating according to the alternating site binding-change mechanism is F1, which has a single active nucleotide-binding site filled by either ATP (for hydrolytic) or ADP (synthetic) activities and two other sites permanently filled with ATP and ADP, respectively (compare Figs. 2 and 3). The single catalytic site model corresponds to simple hyperbolic dependence of the initial rate on the substrate concentration. The 3α⋅3β hexamer (free enzyme) can form from nucleotide-free subunits or subunits that had already bound nucleotides before the formation of active enzyme. The diagram in Fig. 3 shows all possible variants of ATP and ADP binding to the F1 catalytic sites only. The number of possible different forms of F1 is 10 for the symmetrical trimer. Apparently, the number of various forms greatly increases if we account for the binding of a single Pi molecule to the ADP-filled sites or binding of one to three Pi molecules to the nucleotide-free sites. This simple consideration argues for greater care in assigning the role of catalytic intermediate to any particular crystallographically solved atomic structure of F1 or Fo⋅F1. Interestingly, the catalytic parameters of Fo⋅F1 from Paracoccus denitrificans membranes determined at the ATP and ADP concentrations close to the physiological ones and greatly exceeding Km and Ki, are only slightly different from those usually measured (~1 mM ATP and ~0.1 mM ADP). This suggests that the additional binding of ADP and ATP to the nucleotide-binding sites does not affect the overall catalysis. On the other hand, the steady-state ATP hydrolysis by P. denitrificans Fo⋅F1 requires the presence of ~10–5 M ADP; an increase in the activity of the ATP-regenerating system (phosphoenolpyruvate plus pyruvate kinase) results in the reaction inhibition [6]. Catalytically significant binding of nucleotides appears to be species-specific, as no inhibition of ATP hydrolysis catalyzed by bovine heart Fo⋅F1 by extra activity of the ATP-regenerating system was observed [6]. These observations clearly demonstrate a complexity of ATPase kinetics and call for greater care in the interpretation of experimental results.
Fig. 3. Multiple variants of ATP and ADP binding at potentially active substrate (product)-binding sites of F1. Ten different complexes can be formed: one free of bound nucleotide and nine with differently filled binding sites. “Empty” F1 and all ADP-containing are potentially capable of binding 1 to 3 molecules of Pi, so the number of different complexes becomes significantly larger than 10. The variants of free enzyme regenerated in the mechanism of ATP synthesis or hydrolysis depicted in Fig. 2 are shown in blue and red, respectively.
DEPENDENCE OF ATP HYDROLYSIS ON Mg2+
Most known ATPases use the nucleotide–divalent cation complexes as their substrates. This is also true for F1-type ATPases. However, it appears that Mg2+ have some additional, poorly understood role in their hydrolytic activity. ATP is known to form complexes with Mg2+, and F1-ATPase activity decreases if ATP concentration exceeds the one needed for the ATP4-⋅Mg2+ complex formation. The decline in the enzyme activity corresponds quantitatively to the decrease in free Mg2+ [4, 7]. Kinetically stable ATP⋅lanthanide complexes are not hydrolyzed by F1-ATPase in the absence of Mg2+, and the ATPase activity, although not high (about 10% of the activity observed in the presence of optimal ATP⋅Mg2+ concentration), is induced by adding Mg2+ [7]. The existence of pH-dependent Mg2+-specific binding site with Kd of ~10–5 M is also evident from the data on the ADP-dependent transformation of Fo⋅F1 to its inactive form [8-10]. The participation of Mg2+-specific site in the mechanism of F1-catalyzed ATP hydrolysis and, possibly, in proton translocation seems to be important and somehow forgotten problem.
PROTEIN INHIBITOR (IF1) OF F1-ATPase
Soluble F1 and various SMP might either contain or lack, depending on particular preparative procedure, small water-soluble protein that binds to F1 tightly enough to consider it as a specific subunit of the enzyme. The presence of IF1 in some preparation impedes and often distorts the results of kinetic studies of ATP hydrolysis. Both IF1 and IF1-free SMP are available, and the parameters of Fo⋅F1 interaction with IF1 have been studied [11-13]. This small protein slowly, compared with the catalytic turnover of F1, inhibits ATP hydrolysis. The parameters of inhibition depend on ATP, i.e., on the steady-state amount of some catalytic intermediate(s). The inhibition rate is strongly pH-dependent due to the protonation of IF1 histidine residue responsible for the protein conformational transition to its active inhibitory form [11, 12]. IF1 does not inhibit the ATP synthase activity of Fo⋅F1 [13]. The physiological significance of the ATPase activity inhibition by IF1 is poorly understood. One interesting possibility merits a short discussion.
The release of “stoichiometric” proton [see Eq. (1)] in the ATP hydrolase reaction results in the acidification of surrounding medium. Physiological concentration of ATP in the mitochondrial matrix is 5-10 mM. ATP hydrolysis (acidification) and synthesis (alkalization) may serve as an Fo⋅F1-controlled pH buffer system of the mitochondrial matrix. Strong pH dependence of IF1 within the physiologically relevant pH range and pH dependence of Fo⋅F1-ATPase activity itself (see below) allow to consider a combination of ATP/ADP, IF1, and Fo⋅F1 as a sensitive pH buffer of the mitochondrial matrix.
ADP(Mg2+)-INHIBITED FORM OF Fo⋅F1
When discussing kinetics of any enzymatic reaction and developing the models for the reaction mechanism, the steady-state approximation is usually assumed for the initial reaction rates. The “initial rates” of ATP hydrolysis catalyzed by Fo⋅F1 depleted of IF1 depend on the “history” of particular enzyme preparation. A decrease in the substrate (ATP) concentration can proceed either at a constant rate, with the lag-phase, or with initial burst of activity. This behavior discovered many years ago and commonly called ADP(Mg2+)-inhibition is due to the specific binding of ADP in the presence of Mg2+ at the F1 active site located in one of the α-β-subunit pairs, most likely in the β-subunit. Preincubation of F1 or Fo⋅F1 with very low ADP or ATP concentrations (almost equal to the F1 concentration) in the presence of Mg2+ results in complete inhibition of the ATP hydrolase activity [14, 15]. Full but slow activation (on the minute scale) of ATPase could be induced by Pi [16], EDTA (binding of Mg2+) [9], incubation of the inhibited enzyme with phosphoenolpyruvate plus pyruvate kinase [17], sulfite anion [18], and addition of substrate amounts of ATP [19]. Energization of the membrane results in rapid activation of the ATPase activity of the ADP(Mg2+)-inhibited enzyme [20]. The absence of F1-ATPase activity in many prokaryotes is likely due, at least partially, to natural “preincubation” with ADP. Interestingly, the enzyme preincubated with ADP at the concentrations significantly higher than those needed for the “stoichiometric” inhibition is activated substantially faster than the enzyme inhibited with low ADP concentrations [21]. A number of features of F1- or Fo⋅F1-ATPase, such as activation by anions and specific inhibition by azide [18], stimulation by 2,4-dinitrophenol [5], differences in the interaction with other than ATP purine nucleotides [18], effects of Mg2+ [22], specific activation by the detergent LDAO, and the absence or very low ATPase activity of prokaryotic membranes can be satisfactory explained by the kinetic schemes published in a series of our works. Potentially important role in the regulation of ATPase of the enzyme ε-subunit is also widely discussed, although the data on its effects are somehow contradictory. In fact, our analysis of ADP(Mg2+)-inhibition and, particularly, its absence in the ATP synthesis by coupled membrane preparations, have lead us to the proposal, which has been gradually accepted by other researchers that the mechanisms of the reactions of ATP synthesis and hydrolysis catalyzed by Fo⋅F1-ATPases are different and those molecular machines do not obey the principles of microreversibility (detailed equilibrium). This hypothesis has been formulated many years ago in a general form: there is an equilibrium controlled by a number of effectors between the two forms of Fo·F1:
each form catalyzing either hydrolysis (F1Н) or synthesis (F1S) of ATP [22, 23]. This suggestion explained the unidirectional effects of IF1, antibiotic aurovertin, azide, Pi, and others. It has happened very difficult to prove or disprove this hypothesis because of ambiguous interpretation of results usually obtained on notoriously heterogeneous preparations.
CHANGING THE STUDY OBJECT: Paracoccus denitrificans
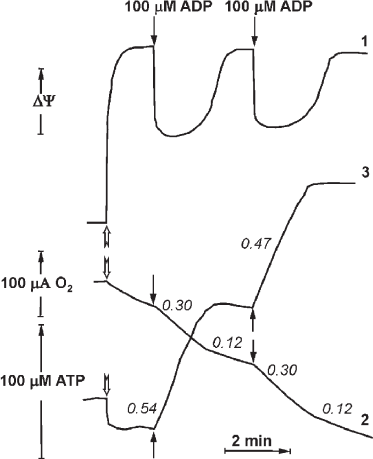
In 1970, P. John and W. Hamilton reported the respiratory control in the inside-out plasma membranes (Pd-SBP) of the soil bacterium P. denitrificans. This phenomenon appeared to be quantitatively and quantitatively similar to the respiratory control observed in intact mammalian mitochondria. Synchronous recording of oxygen consumption, membrane potential (Δψ), and ATP synthesis in the plasma membrane vesicles of P. denitrificans is shown in Fig. 4 (reproduced from [24]). Shortly, another surprising observation was reported by the Oxford group that coupled Pd-SBP catalyzed oxidative phosphorylation (ATP synthesis) but did not show the ATPase activity. Following the “Krogh principle” (see above), we have started studies of the enzyme activity in coupled membranes from P. denitrificans. We believe that despite well-documented differences in some catalytic properties of Fo⋅F1 in various microorganisms, the basic functional principles of F1-type ATPases are the same. We suspected that the absence of ATP hydrolase activity in Pd-SBP was due to the above-described ADP(Mg2+) induced inhibition. Indeed, we were able to show that Pd-Fo⋅F1 catalyzed both synthesis and hydrolysis of ATP at comparable rates if the membranes were pre-energized by coupled oxidation of succinate or NADH [25, 26]. We also developed a special experimental setup to detect active ATP hydrolysis that included membranes oxidizing succinate or NADH and ATPase initiated by simultaneously added malonate (for instant inhibition of succinate oxidation) and ATP. Once induced, ATP hydrolysis proceeds until p is supported by H+ translocation via the activated Fo⋅F1 complex. The reaction is thus energetically self-supported; in other words, a portion of free energy derived from ATP hydrolysis is used to keep Fo⋅F1 in the catalytically active state. This operation mechanism is not a standard catalytic cycle but should rather be considered as machine functioning. This arrangement is physiologically warranted: the enzyme should “know” that the hydrolysis of ATP formed by other metabolic pathways (Entner–Doudoroff pathway of anaerobic glucose catabolism in P. denitrificans) is used to maintain vitally required membrane potential, i.e., ATP is not hydrolyzed uselessly. The ATPase activity declines when the membrane potential drops below permissible level. As noted above and shown in Fig. 4, the respiration rate decreases when added ADP is phosphorylated to ATP, which can be explained by “equilibration” of p and phosphoryl potential (ATP/ADP ratio). However, the ATP/ADP ratio in this controlled state depends on neither Pi concentration, nor the amount of added ATP. These data suggest that the controlled state is determined kinetically rather than thermodynamically [24].
Fig. 4. Synchronous registration of the electrical component Δψ (curve 1, arbitrary units), respiration (2), and ATP synthesis (3) after addition of limited amount of ADP (100 μM) to the suspension of coupled Pd-SBP. The reactions were initiated by adding Pd-SBP (shown by open arrows) to the medium containing 0.25 M sucrose, 1 mM Hepes (pH 7.0), 0.1 mM EDTA, 5 mM MgCl2, 5 mM potassium phosphate, 5 mM semicarbazide, 5 mM ethanol, 450 U alcohol dehydrogenase, and 60 μM NADH (30°C). Electrical potential (1) was registered as Oxonol IV response; respiration (2) was measured with an oxygen-sensitive electrode; ATP synthesis (3) was registered with a pH-sensitive electrode (reproduced from [24] with permission from Elsevier).
KINETICS OF p-DEPENDENT ATP SYNTHESIS
ATP synthesis from ADP and Pi is a classical bi- or three-substrate reaction, if vectorially transported proton is considered as a substrate. The following simplified kinetic scheme of the reaction is:
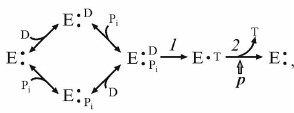 (3)
(3)where D and T are ADP and ATP, respectively.
The dependence of the reaction initial rate on the ADP and Pi concentrations corresponds to random binding kinetics, and steps 1 and 2 in the scheme (3) are separated by the irreversible step (ping-pong kinetics). A decrease in p results in the proportional decrease in the steady-state concentration of the intermediate which produces the product [27]. Note that the same kinetic pattern is expected if random binding of substrates is preceded by the energy-dependent transformation of the enzyme to the form capable of (random) substrate binding.
KINETICS OF ATP HYDROLYSIS: EFFECT OF Pi
It is well known that Pi, a natural substrate of oxidative phosphorylation, does not inhibit ATP hydrolysis by soluble F1 or membrane-bound Fo⋅F1 at the concentrations significantly exceeding its Km in the ATP synthesis. This suggests different pathways of ATP synthesis and hydrolysis, as we have repeatedly emphasized in our publications. The effect of Pi on the ATP hydrolase activity of coupled Pd-Fo⋅F1 was eventually studied. We found that Pi does not inhibit ATP hydrolysis, but is required for ordered sequential product release, and such unusual behavior is due to the ADP(Mg2+)-induced inhibition. The kinetic scheme of the energy-producing coupled ATP hydrolysis [28] is shown below.
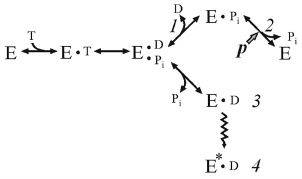 (4)
(4)where E*•D is ADP(Mg2+), inactive form of Fo⋅F1.
Dissociation of the products proceeds in the orderly manner: p prevents release of Pi, and ADP must leave first. If p is absent, Pi has low affinity to the active site and its dissociation leads to the formation of intermediate 3 rapidly transforming to the ADP(Mg2+)-inhibited form. Comparison of schemes (3) and (4) shows that the substrates bind randomly when Fo⋅F1 catalyzes ATP synthesis, whereas the products of hydrolysis (ADP and Pi) must be released sequentially, and the steady-state reaction should include p-dependent Pi binding.
OTHER EFFECTORS OF Fo⋅F1
Low concentrations of the antibiotic venturicidin specifically inhibit Fo⋅F1, thus preventing proton translocation by Fo, similar to the classic inhibitor of mammalian oxidative phosphorylation oligomycin. Oligomycin virtually does not inhibit Fo⋅F1 of prokaryotes. The sensitivity of ATP synthesis and hydrolysis by Pd-Fo⋅F1 to venturicidin is substantially different: the half-inhibitory concentrations for the ATP synthesis and hydrolysis are ~0.3 and 1.0 nmol per mg protein, respectively, for the membranes preincubated under identical conditions. The inhibition is non-competitive with respect to the substrates. Another unidirectional effector, acidification, strongly inhibits ATPase activity at pH 7.0 without affecting ATP synthesis [29]. A decrease in the ATP hydrolysis upon acidification does not reciprocally activate ATP synthesis, as might have been expected if the effect of pH change were due to the shift in the equilibrium depicted in scheme (2).
HYPOTHESIS AND CONCLUSION
ATP synthesis and hydrolysis catalyzed by Fo⋅F1-ATPases are, in my opinion, the most complex reactions for kinetic analysis, because their substrates (products) are vectorially translocating protons. Our attempts to understand their kinetic mechanism have led us to the conclusion that Fo⋅F1 operates differently and unidirectionally. I would like to emphasize that everything discussed above is a very simplified picture. For example, in addition to schemes with the three active substrate (product)-binding sites, some F1-ATPases have the so-called tightly bound non-exchangeable nucleotides. Thus, the ε-subunit binds ATP(ADP), and its conformational change influences ATP hydrolase activity; Pd-Fo⋅F1 contains recently described ζ-subunit functionally equivalent to the mitochondrial IF1.
Analysis of our recent data has led us to the suggestion that coupling membranes bear two non-equilibrium forms of Fo⋅F1: one having the properties best suited for the most efficiently p-regulated ATP synthesis, and the other one – for ATP-dependent generation of p. Hence, the equilibrium arrow in scheme (2) (F1H ↔ F1S) has to be eliminated. The terms equilibrium and isoforms should be clarified. So far, there are no data on the existence of true isoenzymes of Fo⋅F1 encoded by different genes, although this possibility cannot be excluded for some subunits of the Fo⋅F1 complex. The isoforms may be represented by differently post-translationally modified proteins or by different sets of tightly bound annular phospholipids. The existence of isoforms with different number of c-subunit in the proton-translocating Fo seems particularly attractive, as it would result in changes in the overall cell bioenergetics.
A note worth to make concerns equilibrium. Some biochemical processes are known to occur on the microsecond time scale. On the other hand, rearrangements of tertiary and quaternary protein structure might take minutes or hours. The term isoform used here refers to the structures with the lifetime many times exceeding the protein catalytic turnover.
It is difficult not to mention numerous proposed “biomedical” projects, for example, studies of mycobacterial (tuberculosis) or trypanosomal Fo⋅F1 aimed to find their specific inhibitors (financial support!). Different sensitivity of ATP synthase and hydrolase activities of Fo⋅F1 has raised some new thoughts on these important old practical problems.
The existence of Fo⋅F1 isoforms operating unidirectionally is by no means proven. Experiment aimed to examine this, in my opinion, interesting possibility are currently underway in our laboratory.
Funding. This work has been continuously supported by the Russian Foundation for Basic Research, starting from its foundation (including current project 17-04-00706/17).
Acknowledgements. The author is grateful to his colleagues (all graduated from the Department of Biochemistry) who participated in the studies of Fo⋅F1. I thank Dr. V. G. Grivennikova for her great help in preparation of this manuscript and editorial comments.
Conflict of interest. The author declares no conflict of interest in financial or any other sphere.
Ethical approval. All procedures performed in the described studies were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
REFERENCES
1.Vinogradov, A. D. (1984) Catalytic properties of
mitochondrial ATP-synthetase, Biokhimiya, 49,
1220-1238.
2.Vinogradov, A. D. (1999) Mitochondrial ATP
synthase: fifteen years later, Biochemistry (Moscow), 64,
1219-1229.
3.Vinogradov, A. D. (2000) Steady-state and
pre-steady-state kinetics of the mitochondrial
Fo⋅F1-ATPase: is ATP synthase a reversible
molecular machine? J. Exp. Biol., 203, 41-49.
4.Akimenko, V. K., Minkov, I. B., Bakeeva, L. E., and
Vinogradov, A. D. (1972) Isolation and properties of soluble ATPase
from bovine heart mitochondria, Biokhimiya, 37,
348-359.
5.Akimenko, V. K., Minkov, I. B., and Vinogradov, A.
D. (1971) Action of uncouplers on soluble mitochondrial ATPase,
Biokhimiya, 36, 655-658.
6.Zharova, T. V., and Vinogradov, A. D. (2006)
Requirement of medium ADP for the steady-state hydrolysis of ATP by the
proton-translocating Paracoccus denitrificans
Fo·F1-ATP synthase, Biochim. Biophys.
Acta, 1757, 304-310; doi: 10.1016/j.bbabio.2006.03.001.
7.Syroeshkin, A. V., Galkin, M. A., Sedlov, A. V.,
and Vinogradov, A. D. (1999) Kinetic mechanism of
Fo·F1 mitochondrial ATPase:
Mg2+ requirement for Mg·ATP hydrolysis,
Biochemistry (Moscow), 64, 1128-1137.
8.Minkov, I. B., Fitin, A. F., Vasilyeva, E. A., and
Vinogradov, A. D. (1979) Mg2+-induced ADP-dependent
inhibition of the ATPase activity of beef heart mitochondrial coupling
factor F1, Biochem. Biophys. Res. Commun., 89,
1300-1306; doi: 10.1016/0006-291x(79)92150-8.
9.Bulygin, V. V., and Vinogradov, A. D. (1991)
Interaction of Mg2+ with Fo·F1
mitochondrial ATPase as related to its slow active/inactive transition,
Biochem. J., 276, 149-156; doi: 10.1042/bj2760149.
10.Bulygin, V. V., Syroeshkin, A. V., and
Vinogradov, A. D. (1993) Nucleotide/H+-dependent change in
Mg2+ affinity at the ATPase inhibitory site of the
mitochondrial F1·Fo-ATP synthase, FEBS
Lett., 328, 193-196; doi: 10.1016/0014-5793(93)80991-3.
11.Panchenko, M. V., and Vinogradov, A. D. (1985)
Interaction between the mitochondrial ATP synthetase and ATPase
inhibitor protein, FEBS Lett., 184, 226-230; doi:
10.1016/0014-5793(85)80611-6.
12.Panchenko, M. V., and Vinogradov, A. D. (1989)
Kinetics of the interaction of ATPase of submitochondrial fragments and
a natural protein-inhibitor, Biokhimiya, 54, 569-579.
13.Vasil’eva, E. A., Panchenko, M. V., and
Vinogradov, A. D. (1989) Interaction of ATPase from submitochondrial
fragments and a natural inhibitor protein during
ΔμH+ generation on a membrane, Biokhimiya,
54, 1490-1498.
15.Fitin, A. F., Vasilyeva, E. A., and Vinogradov,
A. D. (1979) An inhibitory high affinity binding site for ADP in the
oligomycin-sensitive ATPase of beef heart submitochondrial particles,
Biochem. Biophys. Res. Commun., 86, 434-439; doi:
10.1016/0006-291x(79)90884-2.
16.Yalamova, M. V., Vasilyeva, E. A., and
Vinogradov, A. D. (1982) Mutually dependent influence of ADP and
Pi on the activity of mitochondrial adenosine
triphosphatase, Biochem. Int., 4, 337-344.
17.Vasilyeva, E. A., Fitin, A. F., Minkov, I. B.,
and Vinogradov, A. D. (1980) Kinetics of interaction of adenosine
diphosphate and adenosine triphosphate with ATPase of bovine heart
submitochondrial particles, Biochem. J., 188,
807-815.
18.Vasilyeva, E. A., Minkov, I. B., Fitin, A. F.,
and Vinogradov, A. D. (1982) Kinetic mechanism of mitochondrial
adenosine triphosphatase. Inhibition by azide and activation by
sulfite, Biochem. J., 202, 15-23; doi:
10.1042/bj2020015.
19.Vasilyeva, E. A., Minkov, I. B., Fitin, A. F.,
and Vinogradov, A. D. (1982) Kinetic mechanism of mitochondrial
adenosine triphosphatase. ADP-specific inhibition as revealed by the
steady-state kinetics, Biochem. J., 202, 9-14; doi:
10.1042/bj2020009.
20.Galkin, M. A., and Vinogradov, A. D. (1999)
Energy-dependent transformation of the catalytic activities of the
mitochondrial Fo·F1-ATP-synthase, FEBS
Lett., 448, 123-126; doi: 10.1016/s0014-5793(99)00347-6.
21.Bulygin, V. V., and Vinogradov, A. D. (1989)
Kinetic evidence of the interaction of three nucleotide-binding centers
of mitochondrial ATP-synthetase, Biokhimiya, 54,
1359-1367.
22.Minkov, I. B., Vasilyeva, E. A., Fitin, A. F.,
and Vinogradov, A. D. (1980) Differential effects of ADP on ATPase and
oxidative phosphorylation in submitochondrial particles, Biochem.
Int., 1, 478-485.
23.Syroeshkin, A. V., Vasilyeva, E. A., and
Vinogradov, A. D. (1995) ATP synthesis catalyzed by the mitochondrial
F1·Fo-ATP-synthase is not a reversal of
its ATPase activity, FEBS Lett., 366, 29-32; doi:
10.1016/0014-5793(95)00487-t.
24.Zharova, T. V., and Vinogradov, A. D. (2014)
ATPase/synthase activity of Paracoccus denitrificans
Fo·F1 as related to the respiratory
control phenomenon, Biochim. Biophys. Acta, 1837,
1322-1329; doi: 10.1016/j.bbabio.2014.04.002.
25.Zharova, T. V., and Vinogradov, A. D. (2003)
Proton-translocating ATP-synthase of Paracoccus denitrificans:
ATP-hydrolytic activity, Biochemistry (Moscow), 68,
1101-1108.
26.Zharova, T. V., and Vinogradov, A. D. (2004)
Energy-dependent transformation of
Fo·F1-ATPase in Paracoccus
denitrificans plasma membranes, J. Biol. Chem., 279,
12319-12324; doi: 10.1074/jbc.M311397200.
27.Kegyarikova, K. A., Zharova, T. V., and
Vinogradov, A. D. (2010) Paracoccus denitrificans
proton-translocating ATPase: kinetics of oxidative phosphorylation,
Biochemistry (Moscow), 75, 1264-1271.
28.Zharova, T. V., and Vinogradov, A. D. (2006)
Energy-linked binding of Pi is required for continuous
steady-state proton-translocating ATP hydrolysis catalyzed by
Fo·F1-ATP-synthase, Biochemistry,
45, 14552-14558; doi: 10.1021/bi061520v.
29.Zharova, T. V., and Vinogradov, A. D. (2017)
Functional heterogeneity of Fo·F1
H+-ATPase/synthase in coupled Paracoccus
denitrificans plasma membranes, Biochim. Biophys. Acta,
1858, 939-944; doi: 10.1016/j.bbabio.2017.08.006.