REVIEW: Small Heat Shock Proteins and Human Neurodegenerative Diseases
L. K. Muranova1, A. S. Ryzhavskaya1, M. V. Sudnitsyna1, V. M. Shatov1, and N. B. Gusev1,a*
1Lomonosov Moscow State University, School of Biology, Department of Biochemistry, 119991 Moscow, Russia* To whom correspondence should be addressed.
Received March 30, 2019; Revised May 4, 2019; Accepted June 27, 2019
The review discusses the role of small heat shock proteins (sHsps) in human neurodegenerative disorders, such as Charcot–Marie–Tooth disease (CMT), Parkinson’s and Alzheimer’s diseases, and different forms of tauopathies. The effects of CMT-associated mutations in two small heat shock proteins (HspB1 and HspB8) on the protein stability, oligomeric structure, and chaperone-like activity are described. Mutations in HspB1 shift the equilibrium between different protein oligomeric forms, leading to the alterations in its chaperone-like activity and interaction with protein partners, which can induce damage of the cytoskeleton and neuronal death. Mutations in HspB8 affect its interaction with the adapter protein Bag3, as well as the process of autophagy, also resulting in neuronal death. The impact of sHsps on different forms of amyloidosis is discussed. Experimental studies have shown that sHsps interact with monomers or small oligomers of amyloidogenic proteins, stabilize their structure, prevent their aggregation, and/or promote their specific proteolytic degradation. This effect might be due to the interaction between the β-strands of sHsps and β-strands of target proteins, which prevents aggregation of the latter. In cooperation with the other heat shock proteins, sHsps can promote disassembly of oligomers formed by amyloidogenic proteins. Despite significant achievements, further investigations are required for understanding the role of sHsps in protection against various neurodegenerative diseases.
KEY WORDS: small heat shock proteins, chaperone-like activity, amorphous aggregation, β-amyloids, posttranslational modifications, neurodegenerative diseasesDOI: 10.1134/S000629791911004X
Abbreviations: ACD, α-crystallin domain; CMT, Charcot–Marie–Tooth disease; CTD, C-terminal domain; NTD, N-terminal domain; (s)Hsp, (small) heat shock proteins.
Correct folding of long polypeptide chains synthesized de novo or
renaturation of protein denatured under unfavorable conditions are very
complex processes. The protein homeostasis (proteostasis) in the cell
is maintained by several families of heat shock proteins (Hsps) that
can interact with cell proteins and with each other in order to perform
their function. Human cells contain several Hsp families: HspH
(Hsp110), HspC (Hsp90), HspA (Hsp70), HspD/E (Hsp60/Hsp10), DNAJ
(Hsp40), and HspB (according to the old classification, the number
after Hsp corresponds to the molecular weight of protein monomer) [1, 2]. Each sHsp family is
characterized by specific properties, functions, and intracellular
location. Some Hsps (Hsp110, Hsp90, Hsp70, Hsp60) possess ATPase
activity, whereas other Hsps (DNAJ) regulate the ATPase activity of
their partner (Hsp70) or lack the ATPase activity at all (small Hsps,
sHsps). Efficient folding of polypeptides chains can be achieved only
by coordinated participation of all (or most) of Hsps belonging to
different protein families, each family including several or even tens
of Hsps. For instance, human genome contains 10 genes coding sHsps [3, 4]. sHsp monomers are composed
of 150-250 amino acid residues (a.a.) and have comparatively small
molecular masses [5, 6]. A
characteristic feature of sHsps is the presence of highly conserved
α-crystallin domain (ACD) consisting of 80-100 a.a. organized
into six or seven β-strands (Fig. 1a) [7, 8]. ACD participates in the
formation of sHsp dimers that can contain either identical or different
monomers [9-11]. Both isolated
ACDs and intact sHsps can form amyloid fibrils under specific
conditions in vitro [12, 13]. Interestingly, short ACD fragments that can
prevent aggregation of denatured proteins, i.e., possess the
chaperone-like activity, tend to form amyloid fibrils [14]. In addition to the conserved ACD, sHsps contain
N-terminal (NTD) and C-terminal (CTD) domains that differ
in length and structure (Fig. 1a). sHsps containing
conserved (I/V)P(I/V) tripeptide in the CTD (αA-crystallin
(HspB4), αB-crystallin (HspB5), HspB1) are prone to the formation
of very large oligomers composed of more than 20 monomers, which is due
to the interaction of this conserved tripeptide with the hydrophobic
groove formed by the β4-β8 strands of the neighboring ACD and
leads to the generation of large oligomers composed of several dimers
[15, 16]. sHsps differ in the
length of poorly ordered N-terminal domain (NTD) that might play
an important role in the stabilization of large oligomers and their
interaction with partners and target proteins [11]. This NTD often contains one or several
phosphorylation sites [5, 6],
whose phosphorylation can affect sHsp oligomeric structure [17, 18] and interaction with
partner proteins, e.g., universal 14-3-3 adapter protein [19].
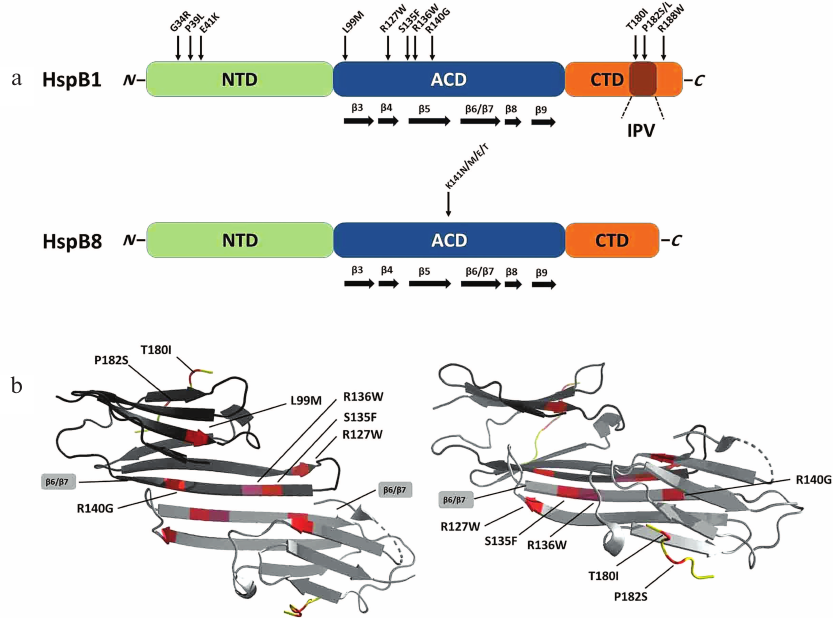
Fig. 1. a) Structures of human HspB1 and HspB8. Green, N-terminal domain (NTD); blue, α-crystallin domain (ACD); orange, C-terminal domain (CTD) with conserved IPV tripeptide. Arrows indicate positions of point mutations associated with Charcot–Marie–Tooth (CMT) disease. b) Ribbon model of the HspB1 dimer fragment containing ACD and CTD (constructed based on PDB 4MJH using PyMol program). Position of β6/β7 strands forming the intermonomer interface is indicated. Left panel, top view; right panel, side view; dimers are rotated by 90° relative to each other.
As already mentioned, the main function of sHsps is the maintenance of protein homeostasis. Hsps can perform this function by different mechanisms. Firstly, sHsps bind partially denatured and misfolded proteins and prevent their aggregation [5, 6]. Formation of such complexes not only prevents aggregation of denatured proteins but keeps them in a state maximally suitable for the interaction with ATP-dependent Hsps that can renature these proteins [20]. Secondly, sHsps promote elimination of denatured proteins via degradation in proteasomes [6, 21] or autophagosomes [22, 23]. Finally, in cooperation with Hsp110, Hsp70, and Hsp40, sHsps can participate in disassembling of amyloid aggregates [24]. Therefore, sHsps play an important role in cell protection against accumulation of partially denatured or misfolded proteins.
Despite the multilevel protection of cells against proteostasis dysregulation, impairments in the protein folding control can cause certain neurodegenerative diseases. Such impairments can result from mutations in Hsps or accumulation of extremely large amounts of misfolded proteins, the renaturation or elimination of which would be beyond of the capability of the proteostasis-controlling system.
In the first part of our review, we discuss the effects of mutations in sHsps on congenital neuropathies, such as Charcot–Marie–Tooth disease (CMT) and distal hereditary motor neuropathy (dHMN). In the second part of the review, we summarize the data on the role of sHsps in preventing the accumulation of amyloids of different nature in the cells.
MUTATIONS IN sHsps AND TYPE II CHARCOT–MARIE–TOOTH
DISEASE
Inherited neuropathies are commonly occurring and heterogeneous disorders. A neuropathy is classified as CMT if both sensor and motor neurons are damaged or as dHMN if only motor neurons are damaged [25]. Hence, dHMN can be considered as a particular case of CMT. Symptoms and molecular basis of CMT can be very different, thus complicating diagnosis of different forms of this disease [26]. In the simplest case, CMT is classified into two types. Type I CMT is characterized by the myelin sheath damage accompanied by reduced nerve conduction velocity. In type II CMT, the nerve conduction velocity is not changed, but the axon itself is damaged. Type II CMT is observed in 40% CMT patients; about 10% of these patients carry mutations in genes encoding three sHsps – HspB1, HspB3, and HspB8 [27]. At present, more than 30 mutations have been detected in the HspB1 gene, one mutation in the HspB3 gene, and nine mutations in the HspB8 gene [28, 29]. To understand molecular mechanisms underlying the CMT pathology, it is essential to analyze changes induced in the protein structure by these mutations.
In HspB1, mutations associated with CMT has been localized to all three domains of this protein (Fig. 1a). Three point mutations, G34R, P39L, and E41K, in the NTD, lead to the increase in the size of protein oligomers and decrease in the protein thermal stability [18]. Both wild-type HspB1 and mutant proteins are phosphorylated by MAPKAP kinase 2. However, in the case of the wild-type protein, phosphorylation results in rapid (and often complete) dissociation of large oligomers, whereas phosphorylation of the mutants leads only to slight changes in the HspB1 quaternary structure [18]. It was found that phosphorylation-induced dissociation of large oligomers plays an important role in the chaperone-like activity of HspB1 [30]. Therefore, mutations in the NTD disturb phosphorylation-dependent regulation of chaperone-like activity of HspB1.
Most CMT-associated mutations are located in the ACD (Fig. 1a). This domain and especially its β6/β7 strands are involved in the formation of subunit–subunit contacts in large oligomers of sHsps (Fig. 1b) [31]. Therefore, mutations in the ACD could result in significant changes in the HspB1 quaternary structure. Indeed, mutations L99M, R127W, S135F, and R140G cause destabilization of the protein quaternary structure leading to partial dissociation of large HspB1 oligomers at low protein concentration [32-34]. At the same time, at high protein concentration, these mutants tend to form oligomers much larger than the corresponding oligomers formed by the wild-type HspB1, which can be explained by incorrect folding of the protein monomers and exposure of “sticky” regions leading to increased HspB1 aggregation. It should be mentioned that due to the overall destabilization, the L99M, R127W, and S135F mutants easily dissociate even at low phosphorylation levels, i.e., under condition when the wild-type protein remains in the form of large oligomers [32, 34]. In contrast, mutation R136W results in the formation of extremely stable oligomers with the size much larger than that of oligomers formed by the wild-type HspB1. This can be explained by changes in the monomer folding and formation of hydrophobic contacts between F138 residue of one monomer and mutated W136 residue of the neighboring monomer. All analyzed mutants of HspB1 differ from the wild-type protein in their ability to interact with HspB6 and usually demonstrate lower chaperone-like activity toward most model substrates (except insulin) [32-34]. Therefore, mutations in the ACD result in significant changes in the HspB1 quaternary structure, disturb phosphorylation-dependent regulation of protein quaternary structure, and affect HspB1 interaction with protein partners and substrates.
Mutation in the CTD can also be associated with CMT [29]. Mutations T180I, P182S, and R188W are located in close vicinity to the conserved IPV tripeptide (residues 181-183). As already mentioned, this peptide is a fragment of highly flexible CTD that interacts with the ACD domain of the neighboring monomer and stabilize the structure of large HspB1 oligomers [15]. Indeed, mutation P182S decreases protein thermal stability and leads to the formation of very large polydisperse HspB1 aggregates [35]. Mutation R188W is also accompanied by an increase in the HspB1 oligomer size, although it has no significant effect on the protein thermal stability. Mutations P182S and R188W considerably decrease the chaperone-like activity of HspB1 in vitro [35]. These effects can be explained by the fact that the CTD plays an important role in the interaction of sHsps with protein substrates [36].
Summing up, each mutation in HspB1 leads to different alterations in its properties (Fig. 2). Nevertheless, there are some common changes in the structure and properties of HspB1 mutants associated with the CMT disease. Firstly, these mutations affect oligomeric state or stability of HspB1 oligomers. Secondly, they disturb phosphorylation-dependent regulation of protein oligomerization. Thirdly, these mutations affect HspB1 interactions with protein partners and substrates and, as a rule, are accompanied by a decrease in its chaperone-like activity. It is possible that the key factor in the effect of these mutations is the disturbance of proper assembly of HspB1 oligomeric complexes, since reversible association and dissociation of subunits is a prerequisite of normal HspB1 functioning [37].
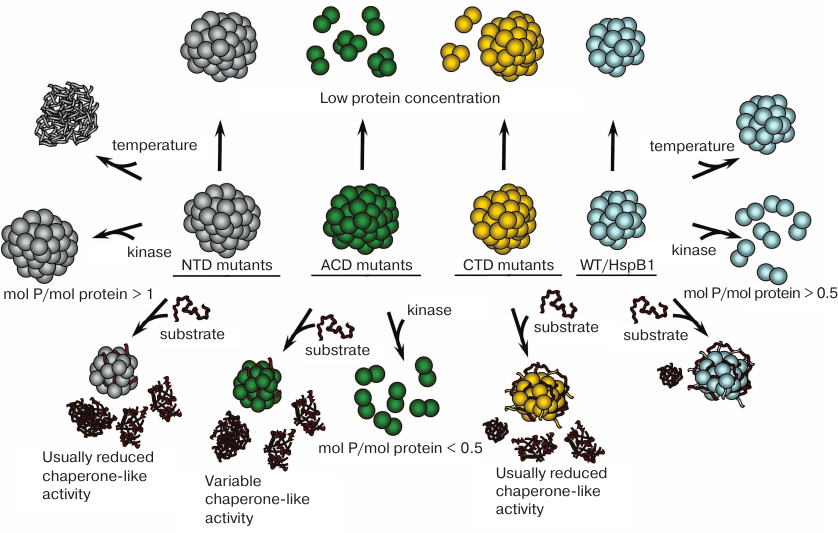
Fig. 2. Changes induced in the structure and properties of CMT-associated HspB1 mutants. Wild-type HspB1 (right column) forms oligomers stable to dissociation, possesses high chaperone-like activity, and is resistant to heat-induced aggregation. Large HspB1 oligomers dissociate to smaller oligomers when phosphorylated at a ratio more than 0.5 mol phosphate per mol protein. Mutants with amino acid substitutions in the NTD (left column) form oligomers stable to dissociation and exhibit lower thermal stability and, as a rule, decreased chaperone-like activity. Oligomers of these mutants do not dissociate after phosphorylation at a ratio of 1 mol phosphate per mol protein and higher. Mutants with amino acid substitutions in the ACD (second from the left column) form large oligomers prone to dissociation at low protein concentration, usually possess decreased chaperone-like activity, and dissociate to smaller oligomers after phosphorylation at a ratio less than 0.5 mol phosphate per mol protein (except R136W mutant). Mutants with amino acid substitutions in the CTD (second from the right column) form large oligomers prone to dissociation at low protein concentration and usually have lower chaperone-like activity.
It is important to answer the question which cellular processes are negatively affected by HspB1 mutations. As already mentioned, mutations in HspB1 are associated with the axonal form of CMT that affects neuronal axons [27]. Therefore, it was suggested that in the case of HspB1 mutants, CMT is caused mainly by the axonal damage [38, 39]. HspB1 might directly or indirectly affect the stability of the cytoskeleton formed by microtubules and intermediate filaments (neurofilaments), which are the major components of neuronal cytoskeleton. Indeed, it was shown that HspB1 interacts with tubulin, thus increasing the stability of microtubules [40]. It is believed that mutations in the ACD promote HspB1 affinity to tubulin and stabilize microtubules [41, 42]. Under normal conditions, microtubules are dynamic structures that constantly undergo reversible polymerization/depolymerization [43]. To compensate for the stabilization of microtubules caused by HspB1 mutations, cells upregulate the activity of histone deacetylase 6 (HDAC6), an enzyme that deacetylates tubulin, thereby inducing microtubule depolymerization and causing damage of the axonal cytoskeleton [43]. In this respect, it should be mentioned that recently developed highly specific inhibitors of histone deacetylase are considered as promising drugs for the treatment of type II CMT [44].
The second important component of cytoskeleton that can be affected by HspB1 is intermediate filaments (neurofilaments). HspB1 mutations S135F and P182L are associated with the neurofilament network damage and can lead to cell death [45, 46]. Mutations R127W, S135F, and P182L are accompanied by an increase in the extent of neurofilament phosphorylation by cdk5 protein kinase and also result in the cytoskeletal damage [47].
Experiments on transgenic mice expressing human HspB1 mutants S135F and R136W correlate with the data obtained on cell cultures. The animals demonstrated symptoms characteristic for CMT, such as locomotion impairments, axonal damage, increased level of neurofilament phosphorylation, and decreased level of tubulin acetylation [48, 49], although less pronounced than in CMT patients. There are no doubts that comprehensive understanding of molecular mechanisms underlying the association of HspB1 mutations with the CMT development requires further clinical and experimental studies.
Mutations in another Hsp, HspB8 (Hsp22), can also be associated with the CMT [29]. As in HspB1, these mutations can be located in NTD, ACD, or CTD of HspB8. However, the mutation hotspot is Lys141 residue that can be replaced by Asn, Met, Glu, or Thr. This residue is homologous to Arg140 in HspB1, Arg116 in HspB4, and Arg120 in HspB5. It is located at the interface of two monomers and participates in the stabilization of the contact between the monomers by forming a salt bridge with negatively charged residue of the neighboring monomer [31]. Unlike HspB1, HspB8 forms only small oligomers that presumably exist as an equilibrium mixture of dimers with monomers [50, 51]. Probably due to this fact, mutation K141E does not affect the quaternary structure of HspB8. However, it destabilizes the structure of HspB8 and makes it more susceptible to limited proteolysis [52]. Depending on the nature of protein substrate, the K141E mutant possesses either equal or slightly lower chaperone-like activity than the wild-type HspB8 [52]. Some experiments indicated that K141 substitution decreases HspB8 affinity to the adapter protein Bag3 [53, 54], whereas other studies demonstrated that this mutation, on the contrary, increases HspB8 affinity to Bag3 [28]. Bag3 forms heterooligomeric complex with HspB8, heat shock protein Hsc70, and chaperone-interacting ubiquitin ligase (CHIP) that catalyzes ubiquitination of denatured proteins followed by proteolytic degradation in autophagosomes [23, 29]. Mutation-induced changes in the interaction between HspB8 and Bag3 can disturb the process of proteolytic degradation of denatured proteins and lead to various neurodegenerative diseases. Recently published data indicate that certain HspB1 mutations can also affect normal processes of autophagy and phagophore formation [55].
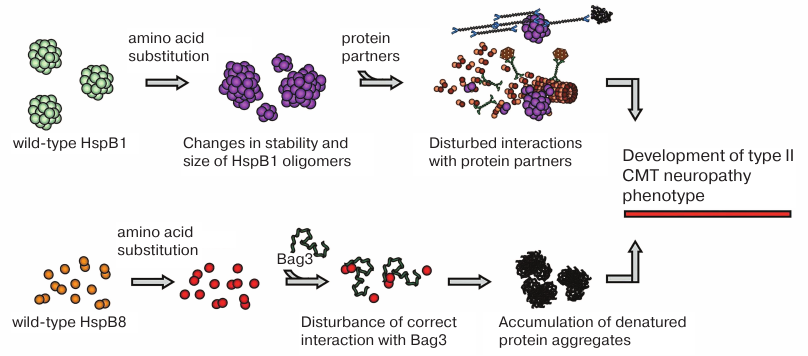
To conclude this part of the review, mutations in sHsps are associated with the axonal form of CMT. Mutations in HspB1 shift the equilibrium between different oligomeric forms, thereby affecting HspB1 interaction with target proteins, in particular, cytoskeletal components (Fig. 3). Cytoskeleton damage can result in neuronal death. Mutations in HspB8 alter its interaction with Bag3, leading to the impairments in the chaperone-assisted ubiquitination and autophagy. This results in the accumulation of denatured proteins followed by cell death (Fig. 3). In other words, mutations in sHsps disturb proteostasis.
Fig. 3. Probable mechanisms underlying effects of CMT-associated mutations. Mutations in HspB1 are often associated with changes in the stability of protein oligomers or in the regulation of HspB1 oligomeric state, which disturbs HspB1 interaction with protein targets and partners resulting in cytoskeletal damage and other impairments. Mutations in HspB8 affect its interaction with the adapter protein Bag3, affecting autophagy and leading to the accumulation of denatured protein aggregates.
Let us address another problem, namely, how sHsps prevent accumulation of protein aggregates and amyloid fibrils formed by denatured proteins and proteins prone to amyloidosis.
sHsps AND AMYLOIDOSIS
Many proteins contain in their structure long stretches of amino acid sequence that can form β-strands. In addition, certain point mutations can increase the probability of β-strand formation from α-helices or random coils. When such regions are brought in close vicinity to each other and are present at high concentrations, they can interact with the formation of prefibrils due to lateral aggregation. The prefibrils can then transform into fibrils and inclusion bodies. Among proteins prone to amyloid formation are α-synuclein, amyloid peptide Aβ1-40, prions, tau protein, and many others.
The effect of sHsps on synuclein aggregation has been comprehensively studied. Synuclein, a comparatively small protein of 140 a.a., belongs to intrinsically disordered proteins. During interaction with the membrane or formation of tetramers, most of the synuclein sequence forms ordered α-helical regions. In addition, synuclein can exist as unordered monomers or misfolded monomers prone to aggregation [56]. The probability of misfolding is increased by the action of stress factors and mutations A53T, A30P, and E46K [57]. Destabilized misfolded monomers form β-amyloid prefibrils that are then transformed into fibrils or Lewy bodies detected in neurons of Parkinson’s disease patients [57].
Formation of synuclein aggregates (Lewy bodies) is often accompanied by upregulation of HspB1 and HspB5 expression [58]. Detailed studies on the impact of sHsps on synuclein aggregation have led to the conclusion that HspB1 and HspB5 do not form tight complexes with synuclein monomers; however, they can stabilize the structure of the monomer and prevent its transition to the form prone to oligomerization and aggregation. These effects were observed for both intact sHsps and their isolated ACDs [59]. HspB1 and HspB5 also interact with small synuclein aggregates and even with amyloid fibrils, preventing their dissociation and induction of secondary nucleation [60]. It was shown that HspB1 can be located on the surface of synuclein fibrils, thereby decreasing their hydrophobicity and preventing their aggregation and elongation [61].
Interestingly, isolated ACD efficiently stabilizes the structure of monomeric synuclein but is unable to interact with synuclein fibrils and cannot prevent their aggregation and elongation [61]. It was suggested that the sites responsible for the prevention of amorphous aggregation of model protein substrates and for the inhibition of amyloid formation are located in different parts of the sHsp molecule. The sites responsible for the prevention of amorphous aggregation are located in the NTD, whereas the sites responsible for the prevention of amyloid peptide Aβ1-40 aggregation are located in the central ACD [62]. This is possible because of the interaction of ACD containing six or seven β-strands with β-strands of the protein target. In this respect, it should be mentioned that under in vitro conditions, crystallin itself can form functionally active β-amyloids with the chaperone-like activity comparable to that of intact protein [12, 13]. Moreover, recently published data indicate that β-amyloid can be accumulated in cataract eye lens [63].
According to the prevailing concept, sHsps, including HspB1 and HspB5, prevent aggregation of synuclein by stabilizing its monomers and/or suppressing fibril formation. Unexpectedly, it was found that overexpression of HspB5 in human glioblastoma cells promotes accumulation of synuclein aggregates in astrocytes [64], which may be due to the competition between overexpressed HspB5 and HspB8 for the interaction with Bag3 and inhibition of autophagy.
Apart from synuclein, sHsps (HspB1, HspB5, HspB6, HspB8) can interact with Aβ-amyloid peptides. Different sHsps were found to accumulate in senile plaques formed mostly by amyloid peptides in the cells of Alzheimer’s disease patients [65, 66]. sHsps (HspB1, HspB5, HspB6, HspB8) detected in these aggregates can be covalently linked to the amyloid peptide by transglutaminase [65]. sHsps not only co-localize with amyloid peptide aggregates but can also prevent their formation [67-69]. It is suggested that depending on the nature of amyloid peptide (Aβ1-42 or D-Aβ1-40), sHsps can affect interaction of its monomers (or small peptide oligomers) with the outer cell membrane, while HspB5 can prevents transition of protofibrils into mature fibrils [69]. Addition of amyloid peptide to the culture of cortical rat astrocytes was accompanied by the HspB1 release and binding of the added peptide [68]. HspB6 was found to protect neuroblastoma SH-SY5Y cells from the accumulation of Aβ-amyloid peptide aggregates [70]. HspB6 interacts with the peptide site responsible for its polymerization and aggregation. Phosphorylation of HspB6 promotes its interaction with the low-molecular-weight forms of amyloid peptide and increases its efficiency in preventing amyloidosis. Even small N-terminal peptide of HspB6 (25 a.a.) phosphorylated at Ser residue prevents aggregation of amyloid peptide fibrils [70].
Tau is another aggregation-prone protein that forms neurofibrillary tangles in the cells of Alzheimer’s disease patients. Tau is a multifunctional intrinsically disordered protein that stabilizes microtubules [71] and can be phosphorylated by many protein kinases. Tau hyperphosphorylation decreases its interaction with tubulin and increases the probability of tau aggregation with the formation of inclusion bodies, which results in the development of various tauopathies [72, 73].
HspB1 predominantly interacts with hyperphosphorylated tau protein, thereby decreasing the amount of protein available for aggregation. Moreover, HspB1 increases the rate of dephosphorylation of paired helical filaments formed by the hyperphosphorylated tau [74]. It is believed that HspB1 recognizes the phosphorylation sites in tau structure, thus preventing its aggregation and promoting proteolytic degradation of this protein [75]. Experimental data indicate that hyperphosphorylation leads to further tau destabilization. This highly destabilized protein tends to aggregate, while HspB1 inhibits this process. In parallel, destabilized tau protein can undergo renaturation or proteolytic degradation, while HspB1 promotes both these processes [76]. The peptide corresponding to a.a. 244-369 of tau protein tends to form fibrils; HspB1 transiently interacts with this peptide and decreases the rate of fibril formation [77]. It was suggested that the VQI sequence twice repeated in the 244-369 a.a. peptide interacts with the hydrophobic groove formed by the ACD β4-β8 strands in HspB1, i.e., the sites occupied by the CTD (I/V)X(I/V) peptide in the absence of substrates [77, 78].
sHsps can affect tau aggregation both directly and indirectly. For instance, it was found that the adapter protein 14-3-3 transiently and weakly interacts with the non-phosphorylated tau; phosphorylation of the latter strongly increases the binding affinity of 14-3-3 protein [79-82]. Depending on the conditions and location of phosphorylation sites, 14-3-3 can either promote further tau phosphorylation and aggregation and/or stabilize tau aggregates, i.e., prevent their disassembly and emergence of especially deleterious small oligomers serving as new oligomerization seeds [83]. Phosphorylated HspB6 forms tight complexes with 14-3-3 [84] and, therefore, can efficiently compete with tau for the interaction with 14-3-3. Hence, phosphorylated HspB6 can indirectly modulate the effect of 14-3-3 on tau aggregation.
Experimental data on the impact of sHsps on prion aggregation are controversial. Introduction of the scrapie-inducing prion (scrapie 263 agent) into the hamster brain upregulated HspB5 synthesis; however, the authors failed to demonstrate HspB5 co-localization with PrPSc aggregates. The brain levels of HspB5 are significantly increased in various prion diseases, although it is unlikely that this increase affects pathogenesis of prion infections [85]. At the same time, yeast Hsp26 and Hsp42 were found to prevent prionogenesis of the yeast prion Sup35. Hsp42 suppressed the growth of fibrils from the ends, whereas Hsp26 inhibited self-association of prion fibrils. Moreover, by cooperating with Hsp40, Hsp70, and Hsp104, sHsps can destabilize prion fibrils and promote their disassembly [86].
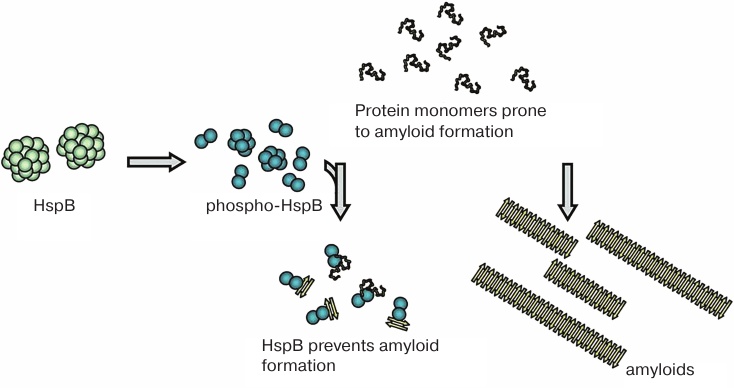
In conclusion, sHsps predominantly interact with monomers (or small oligomers) of intrinsically disordered proteins prone to amyloid formation. By binding to these proteins, sHsps stabilize their structure, prevent their aggregation, and/or facilitate their proteolytic degradation (Fig. 4). It is highly probable that this type of interaction occurs with the participation of amyloidogenic β4-β8 strands of the ACDs of sHsps [62]. The binding of sHsps results in the formation of mixed structures, in which β-strands of sHsps interact with β-strands of amyloidogenic protein monomers. The similarity between the structures formed by amyloidogenic proteins and sHsps is supported by the fact that both sHsps (HspB5) and amyloids of tau protein interact with α7 nicotine acetylcholine receptors, inducing signal transmission through Stat3, activation of autophagy, and suppression of secretion of proinflammatory interleukins [87, 88]. It was hypothesized that in large sHsps oligomers, the β4-β8 strands responsible for the interaction with amyloidogenic proteins are occupied with the CTD tripeptide (I/V)P(I/V). Because of this, large sHsp oligomers interact poorly with amyloidogenic proteins. Stress factors and associated phosphorylation cause large sHsp oligomers to dissociate to small oligomers. This results in the exposure of hydrophobic β4-β8 strands that become available for the interaction with amyloidogenic proteins. After this, sHsps can efficiently prevent aggregation of protein substrates [89].
Fig. 4. Small oligomers formed upon sHsp phosphorylation at different sites can prevent amorphous aggregation of partially denatured proteins and accumulation of amyloid fibrils by binding monomers (or small oligomers) of amyloidogenic proteins.
sHsps are important components of a complex chaperone system that ensure correct protein folding and prevent accumulation of partially denatured proteins in the cell. Certain sHsps (such as HspB1, HspB4, HspB5) exist in a form of labile large oligomers that are in the equilibrium with small oligomers. Mutations can affect the equilibrium between different oligomeric forms, thermal stability, chaperone-like activity, and interactions of sHsps with protein partners. Mutations in HspB8 can influence its interaction with the adapter protein Bag3 and autophagy regulation, ensuring selective proteolysis of misfolded proteins. Therefore, mutations in sHsps can lead to neurodegenerative disorders, such as CMT. Under certain conditions, ACD β-strands can interact with β-strands of amyloidogenic proteins and stabilize the structure of the latter, prevent their aggregation, and/or promote their proteolytic degradation. Hence, sHsps can prevent or delay the development of neurodegenerative disorders, such as Parkinson’s and Alzheimer’s diseases, different forms of tauopathies, and prion diseases.
Funding. This study was supported by the Russian Foundation for Basic Research (project 19-04-00038).
Acknowledgements. All authors of this paper are alumni of the Department of Biochemistry, School of Biology, Moscow State University. Our investigation would have been impossible if not based on the knowledge and skills obtained in the course of our study at our department, on traditions laid by the founder of our department Academician Sergei E. Severin. In the year of the 80th anniversary of the Department of Biochemistry, we would like to wish our department great achievements and to voice the hope that in the future, despite all difficulties, the Department of Biochemistry will be able to educate interested and skillful biochemists.
Conflict of interest. The authors declare no conflict of interest in financial or any other area.
Compliance with ethical norms. This article does not contain studies with human participants or animals performed by any of the authors.
REFERENCES
1.Vos, M. J., Hageman, J., Carra, S., and Kampinga,
H. H. (2008) Structural and functional diversities between members of
the human HSPB, HSPH, HSPA, and DNAJ chaperone families,
Biochemistry, 47, 7001-7011; doi: 10.1021/bi800639z.
2.Vilasi, S., Bulone, D., Caruso Bavisotto, C.,
Campanella, C., Marino Gammazza, A., San Biagio, P. L., Cappello, F.,
Conway de Macario, E., and Macario, A. J. L. (2017) Chaperonin of group
I: oligomeric spectrum and biochemical and biological implications,
Front. Mol. Biosci., 4, 99; doi:
10.3389/fmolb.2017.00099.
3.Fontaine, J. M., Rest, J. S., Welsh, M. J., and
Benndorf, R. (2003) The sperm outer dense fiber protein is the 10th
member of the superfamily of mammalian small stress proteins, Cell
Stress Chaperones, 8, 62-69.
4.Kappe, G., Franck, E., Verschuure, P., Boelens, W.
C., Leunissen, J. A., and de Jong, W. W. (2003) The human genome
encodes 10 alpha-crystallin-related small heat shock proteins:
HspB1-10, Cell Stress Chaperones, 8, 53-61.
5.Mymrikov, E. V., Seit-Nebi, A. S., and Gusev, N. B.
(2011) Large potentials of small heat shock proteins, Physiol.
Rev., 91, 1123-1159; doi: 91/4/1123.
6.Bakthisaran, R., Tangirala, R., and Rao, C. M.
(2015) Small heat shock proteins: role in cellular functions and
pathology, Biochim. Biophys. Acta, 1854, 291-319; doi:
10.1016/j.bbapap.2014.12.019.
7.Bagneris, C., Bateman, O. A., Naylor, C. E.,
Cronin, N., Boelens, W. C., Keep, N. H., and Slingsby, C. (2009)
Crystal structures of alpha-crystallin domain dimers of
alphaB-crystallin and Hsp20, J. Mol. Biol., 392,
1242-1252; doi: S0022-2836(09)00936-X.
8.Baranova, E. V., Weeks, S. D., Beelen, S., Bukach,
O. V., Gusev, N. B., and Strelkov, S. V. (2011) Three-dimensional
structure of alpha-crystallin domain dimers of human small heat shock
proteins HSPB1 and HSPB6, J. Mol. Biol., 411, 110-122;
doi: S0022-2836(11)00574-2.
9.Mymrikov, E. V., Seit-Nebi, A. S., and Gusev, N. B.
(2012) Heterooligomeric complexes of human small heat shock proteins,
Cell Stress Chaperones, 17, 157-169; doi:
10.1007/s12192-011-0296-0.
10.Delbecq, S. P., Rosenbaum, J. C., and Klevit, R.
E. (2015) A mechanism of subunit recruitment in human small heat shock
protein oligomers, Biochemistry, 54, 4276-4284; doi:
10.1021/acs.biochem.5b00490.
11.Heirbaut, M., Lermyte, F., Martin, E. M., Beelen,
S., Sobott, F., Strelkov, S. V., and Weeks, S. D. (2017) Specific
sequences in the N-terminal domain of human small heat-shock protein
HSPB6 dictate preferential hetero-oligomerization with the orthologue
HSPB1, J. Biol. Chem., 292, 9944-9957; doi:
10.1074/jbc.M116.773515.
12.Carver, J. A., Ecroyd, H., Truscott, R. J. W.,
Thorn, D. C., and Holt, C. (2018) Proteostasis and the regulation of
intra- and extracellular protein aggregation by ATP-independent
molecular chaperones: lens alpha-crystallins and milk caseins, Acc.
Chem. Res., 51, 745-752; doi:
10.1021/acs.accounts.7b00250.
13.Garvey, M., Ecroyd, H., Ray, N. J., Gerrard, J.
A., and Carver, J. A. (2017) Functional amyloid protection in the eye
lens: retention of alpha-crystallin molecular chaperone activity after
modification into amyloid fibrils, Biomolecules, 7, E67;
doi: 10.3390/biom7030067.
14.Tanaka, N., Tanaka, R., Tokuhara, M., Kunugi, S.,
Lee, Y. F., and Hamada, D. (2008) Amyloid fibril formation and
chaperone-like activity of peptides from alphaA-crystallin,
Biochemistry, 47, 2961-2967; doi: 10.1021/bi701823g.
15.Delbecq, S. P., Jehle, S., and Klevit, R. (2012)
Binding determinants of the small heat shock protein,
alphaB-crystallin: recognition of the 'IxI' motif, EMBO J.,
31, 4587-4594; doi: emboj2012318.
16.Hochberg, G. K., and Benesch, J. L. (2014)
Dynamical structure of alphaB-crystallin, Prog. Biophys. Mol.
Biol., 115, 11-20; doi:
10.1016/j.pbiomolbio.2014.03.003.
17.Jovcevski, B., Kelly, M. A., Rote, A. P., Berg,
T., Gastall, H. Y., Benesch, J. L., Aquilina, J. A., and Ecroyd, H.
(2015) Phosphomimics destabilize Hsp27 oligomeric assemblies and
enhance chaperone activity, Chem. Biol., 22, 186-195;
doi: 10.1016/j.chembiol.2015.01.001.
18.Muranova, L. K., Weeks, S. D., Strelkov, S. V.,
and Gusev, N. B. (2015) Characterization of mutants of human small heat
shock protein HspB1 carrying replacements in the N-terminal domain and
associated with hereditary motor neuron diseases, PLoS One,
10, e0126248; doi: 10.1371/journal.pone.0126248.
19.Sluchanko, N. N., Beelen, S., Kulikova, A. A.,
Weeks, S. D., Antson, A. A., Gusev, N. B., and Strelkov, S. V. (2017)
Structural basis for the interaction of a human small heat shock
protein with the 14-3-3 universal signaling regulator,
Structure, 25, 305-316; doi:
10.1016/j.str.2016.12.005.
20.Zwirowski, S., Klosowska, A., Obuchowski, I.,
Nillegoda, N. B., Pirog, A., Zietkiewicz, S., Bukau, B., Mogk, A., and
Liberek, K. (2017) Hsp70 displaces small heat shock proteins from
aggregates to initiate protein refolding, EMBO J., 36,
783-796; doi: 10.15252/embj.201593378.
21.Ahner, A., Gong, X., Schmidt, B. Z., Peters, K.
W., Rabeh, W. M., Thibodeau, P. H., Lukacs, G. L., and Frizzell, R. A.
(2013) Small heat shock proteins target mutant cystic fibrosis
transmembrane conductance regulator for degradation via a small
ubiquitin-like modifier-dependent pathway, Mol. Biol. Cell,
24, 74-84; doi: 10.1091/mbc.E12-09-0678.
22.Rusmini, P., Cristofani, R., Galbiati, M.,
Cicardi, M. E., Meroni, M., Ferrari, V., Vezzoli, G., Tedesco, B.,
Messi, E., Piccolella, M., Carra, S., Crippa, V., and Poletti, A.
(2017) The role of the heat shock protein B8 (HSPB8) in motoneuron
diseases, Front. Mol. Neurosci., 10, 176; doi:
10.3389/fnmol.2017.00176.
23.Carra, S., Crippa, V., Rusmini, P., Boncoraglio,
A., Minoia, M., Giorgetti, E., Kampinga, H. H., and Poletti, A. (2012)
Alteration of protein folding and degradation in motor neuron diseases:
implications and protective functions of small heat shock proteins,
Prog. Neurobiol., 97, 83-100; doi:
S0301-0082(11)00176-6.
24.Torrente, M. P., and Shorter, J. (2013) The
metazoan protein disaggregase and amyloid depolymerase system: Hsp110,
Hsp70, Hsp40, and small heat shock proteins, Prion, 7,
457-463.
25.Weis, J., Claeys, K. G., Roos, A., Azzedine, H.,
Katona, I., Schroder, J. M., and Senderek, J. (2017) Towards a
functional pathology of hereditary neuropathies, Acta
Neuropathol., 133, 493-515; doi:
10.1007/s00401-016-1645-y.
26.Saporta, A. S., Sottile, S. L., Miller, L. J.,
Feely, S. M., Siskind, C. E., and Shy, M. E. (2011)
Charcot–Marie–Tooth disease subtypes and genetic testing
strategies, Ann. Neurol., 69, 22-33; doi:
10.1002/ana.22166.
27.Yoshimura, A., Yuan, J. H., Hashiguchi, A., Ando,
M., Higuchi, Y., Nakamura, T., Okamoto, Y., Nakagawa, M., and
Takashima, H. (2018) Genetic profile and onset features of 1005
patients with Charcot–Marie–Tooth disease in Japan, J.
Neurol. Neurosurg. Psychiatry, 90, 195-202; doi:
10.1136/jnnp-2018-318839.
28.Echaniz-Laguna, A., Geuens, T., Petiot, P.,
Pereon, Y., Adriaenssens, E., Haidar, M., Capponi, S., Maisonobe, T.,
Fournier, E., Dubourg, O., Degos, B., Salachas, F., Lenglet, T.,
Eymard, B., Delmont, E., Pouget, J., Juntas Morales, R., Goizet, C.,
Latour, P., Timmerman, V., and Stojkovic, T. (2017) Axonal neuropathies
due to mutations in small heat shock proteins: clinical, genetic, and
functional insights into novel mutations, Hum. Mutat.,
38, 556-568; doi: 10.1002/humu.23189.
29.Adriaenssens, E., Geuens, T., Baets, J.,
Echaniz-Laguna, A., and Timmerman, V. (2017) Novel insights in the
disease biology of mutant small heat shock proteins in neuromuscular
diseases, Brain, 140, 2541-2549; doi:
10.1093/brain/awx187.
30.Jovcevski, B., Kelly, M. A., Aquilina, J. A.,
Benesch, J. L. P., and Ecroyd, H. (2017) Evaluating the effect of
phosphorylation on the structure and dynamics of Hsp27 dimers by means
of ion mobility mass spectrometry, Anal. Chem., 89,
13275-13282; doi: 10.1021/acs.analchem.7b03328.
31.Clark, A. R., Lubsen, N. H., and Slingsby, C.
(2012) sHSP in the eye lens: crystallin mutations, cataract and
proteostasis, Int. J. Biochem. Cell Biol., 44, 1687-1697;
doi: 10.1016/j.biocel.2012.02.015.
32.Nefedova, V. V., Sudnitsyna, M. V., Strelkov, S.
V., and Gusev, N. B. (2013) Structure and properties of G84R and L99M
mutants of human small heat shock protein HspB1 correlating with motor
neuropathy, Arch. Biochem. Biophys., 538, 16-24; doi:
10.1016/j.abb.2013.07.028.
33.Nefedova, V. V., Datskevich, P. N., Sudnitsyna,
M. V., Strelkov, S. V., and Gusev, N. B. (2013) Physico-chemical
properties of R140G and K141Q mutants of human small heat shock protein
HspB1 associated with hereditary peripheral neuropathies,
Biochimie, 95, 1582-1592; doi:
10.1016/j.biochi.2013.04.014.
34.Weeks, S. D., Muranova, L. K., Heirbaut, M.,
Beelen, S., Strelkov, S. V., and Gusev, N. B. (2018) Characterization
of human small heat shock protein HSPB1 alpha-crystallin domain
localized mutants associated with hereditary motor neuron diseases,
Sci. Rep., 8, 688; doi: 10.1038/s41598-017-18874-x.
35.Chalova, A. S., Sudnitsyna, M. V., Strelkov, S.
V., and Gusev, N. B. (2014) Characterization of human small heat shock
protein HspB1 that carries C-terminal domain mutations associated with
hereditary motor neuron diseases, Biochim. Biophys. Acta,
1844, 2116-2126; doi: 10.1016/j.bbapap.2014.09.005.
36.Carver, J. A., Grosas, A. B., Ecroyd, H., and
Quinlan, R. A. (2017) The functional roles of the unstructured N- and
C-terminal regions in alphaB-crystallin and other mammalian small
heat-shock proteins, Cell Stress Chaperones, 22, 627-638;
doi: 10.1007/s12192-017-0789-6.
37.Dahiya, V., and Buchner, J. (2019) Functional
principles and regulation of molecular chaperones, Adv. Protein.
Chem. Struct. Biol., 114, 1-60; doi:
10.1016/bs.apcsb.2018.10.001.
38.Bucci, C., Bakke, O., and Progida, C. (2012)
Charcot–Marie–Tooth disease and intracellular traffic,
Prog. Neurobiol., 99, 191-225; doi:
10.1016/j.pneurobio.2012.03.003.
39.Pareyson, D., Saveri, P., Sagnelli, A., and
Piscosquito, G. (2015) Mitochondrial dynamics and inherited peripheral
nerve diseases, Neurosci. Lett., 596, 66-77; doi:
10.1016/j.neulet.2015.04.001.
40.Almeida-Souza, L., Asselbergh, B., d'Ydewalle,
C., Moonens, K., Goethals, S., de Winter, V., Azmi, A., Irobi, J.,
Timmermans, J. P., Gevaert, K., Remaut, H., Van Den Bosch, L.,
Timmerman, V., and Janssens, S. (2011) Small heat-shock protein HSPB1
mutants stabilize microtubules in Charcot–Marie–Tooth
neuropathy, J. Neurosci., 31, 15320-15328; doi:
31/43/15320.
41.d'Ydewalle, C., Krishnan, J., Chiheb, D. M., Van
Damme, P., Irobi, J., Kozikowski, A. P., Vanden Berghe, P., Timmerman,
V., Robberecht, W., and Van Den Bosch, L. (2011) HDAC6 inhibitors
reverse axonal loss in a mouse model of mutant HSPB1-induced
Charcot–Marie–Tooth disease, Nat. Med., 17,
968-974; doi: 10.1038/nm.2396.
42.Benedetti, S., Previtali, S. C., Coviello, S.,
Scarlato, M., Cerri, F., Di Pierri, E., Piantoni, L., Spiga, I., Fazio,
R., Riva, N., Natali Sora, M. G., Dacci, P., Malaguti, M. C., Munerati,
E., Grimaldi, L. M., Marrosu, M. G., De Pellegrin, M., Ferrari, M.,
Comi, G., Quattrini, A., and Bolino, A. (2010) Analyzing
histopathological features of rare Charcot–Marie–Tooth
neuropathies to unravel their pathogenesis, Arch. Neurol.,
67, 1498-1505; doi: 10.1001/archneurol.2010.303.
43.Almeida-Souza, L., Timmerman, V., and Janssens,
S. (2011) Microtubule dynamics in the peripheral nervous system: a
matter of balance, Bioarchitecture, 1, 267-270; doi:
10.4161/bioa.1.6.19198.
44.Benoy, V., Vanden Berghe, P., Jarpe, M., Van
Damme, P., Robberecht, W., and Van Den Bosch, L. (2017) Development of
improved HDAC6 inhibitors as pharmacological therapy for axonal
Charcot–Marie–Tooth disease, Neurotherapeutics,
14, 417-428; doi: 10.1007/s13311-016-0501-z.
45.Zhai, J., Lin, H., Julien, J. P., and Schlaepfer,
W. W. (2007) Disruption of neurofilament network with aggregation of
light neurofilament protein: a common pathway leading to motor neuron
degeneration due to Charcot–Marie–Tooth disease-linked
mutations in NFL and HSPB1, Hum. Mol. Genet., 16,
3103-3116; doi: 10.1093/hmg/ddm272.
46.Ackerley, S., James, P. A., Kalli, A., French,
S., Davies, K. E., and Talbot, K. (2006) A mutation in the small
heat-shock protein HSPB1 leading to distal hereditary motor
neuronopathy disrupts neurofilament assembly and the axonal transport
of specific cellular cargoes, Hum. Mol. Genet., 15,
347-354; doi: 10.1093/hmg/ddi452.
47.Holmgren, A., Bouhy, D., De Winter, V.,
Asselbergh, B., Timmermans, J. P., Irobi, J., and Timmerman, V. (2013)
Charcot–Marie–Tooth causing HSPB1 mutations increase
Cdk5-mediated phosphorylation of neurofilaments, Acta
Neuropathol., 126, 93-108; doi:
10.1007/s00401-013-1133-6.
48.Srivastava, A. K., Renusch, S. R., Naiman, N. E.,
Gu, S., Sneh, A., Arnold, W. D., Sahenk, Z., and Kolb, S. J. (2012)
Mutant HSPB1 overexpression in neurons is sufficient to cause
age-related motor neuronopathy in mice, Neurobiol. Dis.,
47, 163-173; doi: S0969-9961(12)00124-6.
49.Lee, J., Jung, S. C., Joo, J., Choi, Y. R., Moon,
H. W., Kwak, G., Yeo, H. K., Lee, J. S., Ahn, H. J., Jung, N., Hwang,
S., Rheey, J., Woo, S. Y., Kim, J. Y., Hong, Y. B., and Choi, B. O.
(2015) Overexpression of mutant HSP27 causes axonal neuropathy in mice,
J. Biomed. Sci., 22, 43; doi:
10.1186/s12929-015-0154-y.
50.Kim, M. V., Seit-Nebi, A. S., Marston, S. B., and
Gusev, N. B. (2004) Some properties of human small heat shock protein
Hsp22 (H11 or HspB8), Biochem. Biophys. Res. Commun.,
315, 796-801; doi: 10.1016/j.bbrc.2004.01.130.
51.Chowdary, T. K., Raman, B., Ramakrishna, T., and
Rao, C. M. (2004) Mammalian Hsp22 is a heat-inducible small heat-shock
protein with chaperone-like activity, Biochem. J., 381,
379-387; doi: 10.1042/BJ20031958.
52.Kim, M. V., Kasakov, A. S., Seit-Nebi, A. S.,
Marston, S. B., and Gusev, N. B. (2006) Structure and properties of
K141E mutant of small heat shock protein HSP22 (HspB8, H11) that is
expressed in human neuromuscular disorders, Arch. Biochem.
Biophys., 454, 32-41; doi: S0003-9861(06)00267-0.
53.Shemetov, A. A., and Gusev, N. B. (2011)
Biochemical characterization of small heat shock protein HspB8
(Hsp22)–Bag3 interaction, Arch. Biochem. Biophys.,
513, 1-9; doi: S0003-9861(11)00249-9.
54.Carra, S., Boncoraglio, A., Kanon, B., Brunsting,
J. F., Minoia, M., Rana, A., Vos, M. J., Seidel, K., Sibon, O. C., and
Kampinga, H. H. (2010) Identification of the Drosophila ortholog
of HSPB8: implication of HSPB8 loss of function in protein folding
diseases, J. Biol. Chem., 285, 37811-37822; doi:
10.1074/jbc.M110.127498.
55.Haidar, M., Asselbergh, B., Adriaenssens, E., De
Winter, V., Timmermans, J. P., Auer-Grumbach, M., Juneja, M., and
Timmerman, V. (2019) Neuropathy-causing mutations in HSPB1 impair
autophagy by disturbing the formation of SQSTM1/p62 bodies,
Autophagy, 15, 1051-1068; doi:
10.1080/15548627.2019.1569930.
56.Sharma, S. K., and Priya, S. (2017) Expanding
role of molecular chaperones in regulating alpha-synuclein misfolding;
implications in Parkinson’s disease, Cell. Mol. Life Sci.,
74, 617-629; doi: 10.1007/s00018-016-2340-9.
57.Cox, D., Carver, J. A., and Ecroyd, H. (2014)
Preventing alpha-synuclein aggregation: the role of the small
heat-shock molecular chaperone proteins, Biochim. Biophys. Acta,
1842, 1830-1843; doi: 10.1016/j.bbadis.2014.06.024.
58.Leak, R. K. (2014) Heat shock proteins in
neurodegenerative disorders and aging, J. Cell Commun. Signal.,
8, 293-310; doi: 10.1007/s12079-014-0243-9.
59.Cox, D., Selig, E., Griffin, M. D., Carver, J.
A., and Ecroyd, H. (2016) Small heat-shock proteins prevent
alpha-synuclein aggregation via transient interactions and their
efficacy is affected by the rate of aggregation, J. Biol. Chem.,
291, 22618-22629; doi: 10.1074/jbc.M116.739250.
60.Cox, D., and Ecroyd, H. (2017) The small heat
shock proteins alphaB-crystallin (HSPB5) and Hsp27 (HSPB1) inhibit the
intracellular aggregation of alpha-synuclein, Cell Stress
Chaperones, 22, 589-600; doi: 10.1007/s12192-017-0785-x.
61.Cox, D., Whiten, D. R., Brown, J. W. P.,
Horrocks, M. H., San Gil, R., Dobson, C. M., Klenerman, D., van Oijen,
A. M., and Ecroyd, H. (2018) The small heat shock protein Hsp27 binds
alpha-synuclein fibrils, preventing elongation and cytotoxicity, J.
Biol. Chem., 293, 4486-4497; doi:
10.1074/jbc.M117.813865.
62.Mainz, A., Peschek, J., Stavropoulou, M., Back,
K. C., Bardiaux, B., Asami, S., Prade, E., Peters, C., Weinkauf, S.,
Buchner, J., and Reif, B. (2015) The chaperone alphaB-crystallin uses
different interfaces to capture an amorphous and an amyloid client,
Nat. Struct. Mol. Biol., 22, 898-905; doi:
10.1038/nsmb.3108.
63.Alperstein, A. M., Ostrander, J. S., Zhang, T.
O., and Zanni, M. T. (2019) Amyloid found in human cataracts with
two-dimensional infrared spectroscopy, Proc. Natl. Acad. Sci.
USA, 116, 6602-6607; doi:
10.1073/pnas.1821534116.
64.Lu, S. Z., Guo, Y. S., Liang, P. Z., Zhang, S.
Z., Yin, S., Yin, Y. Q., Wang, X. M., Ding, F., Gu, X. S., and Zhou, J.
W. (2019) Suppression of astrocytic autophagy by alphaB-crystallin
contributes to alpha-synuclein inclusion formation, Transl.
Neurodegener., 8, 3; doi: 10.1186/s40035-018-0143-7.
65.Boros, S., Kamps, B., Wunderink, L., de Bruijn,
W., de Jong, W. W., and Boelens, W. C. (2004) Transglutaminase
catalyzes differential crosslinking of small heat shock proteins and
amyloid-beta, FEBS Lett., 576, 57-62; doi:
S0014579304010816.
66.Wilhelmus, M. M., Otte-Holler, I., Wesseling, P.,
de Waal, R. M., Boelens, W. C., and Verbeek, M. M. (2006) Specific
association of small heat shock proteins with the pathological
hallmarks of Alzheimer’s disease brains, Neuropathol. Appl.
Neurobiol., 32, 119-130; doi:
10.1111/j.1365-2990.2006.00689.x.
67.Zerovnik, E. (2017) Co-chaperoning by
amyloid-forming proteins: cystatins vs. crystallins, Eur. Biophys.
J., 46, 789-793; doi: 10.1007/s00249-017-1214-x.
68.Nafar, F., Williams, J. B., and Mearow, K. M.
(2016) Astrocytes release HspB1 in response to amyloid-beta exposure
in vitro, J. Alzheimers Dis., 49, 251-263; doi:
10.3233/JAD-150317.
69.Wilhelmus, M. M., Boelens, W. C., Otte-Holler,
I., Kamps, B., de Waal, R. M., and Verbeek, M. M. (2006) Small heat
shock proteins inhibit amyloid-beta protein aggregation and
cerebrovascular amyloid-beta protein toxicity, Brain Res.,
1089, 67-78; doi: S0006-8993(06)00762-1.
70.Cameron, R. T., Quinn, S. D., Cairns, L. S.,
MacLeod, R., Samuel, I. D., Smith, B. O., Carlos Penedo, J., and
Baillie, G. S. (2014) The phosphorylation of Hsp20 enhances its
association with amyloid-beta to increase protection against neuronal
cell death, Mol. Cell. Neurosci., 61, 46-55; doi:
10.1016/j.mcn.2014.05.002.
71.Sotiropoulos, I., Galas, M. C., Silva, J. M.,
Skoulakis, E., Wegmann, S., Maina, M. B., Blum, D., Sayas, C. L.,
Mandelkow, E. M., Mandelkow, E., Spillantini, M. G., Sousa, N., Avila,
J., Medina, M., Mudher, A., and Buee, L. (2017) Atypical, non-standard
functions of the microtubule associated tau protein, Acta
Neuropathol. Commun., 5, 91; doi:
10.1186/s40478-017-0489-6.
72.Sierra-Fonseca, J. A., and Gosselink, K. L.
(2018) Tauopathy and neurodegeneration: a role for stress,
Neurobiol. Stress, 9, 105-112; doi:
10.1016/j.ynstr.2018.08.009.
73.Zabik, N. L., Imhof, M. M., and Martic-Milne, S.
(2017) Structural evaluations of tau protein conformation:
methodologies and approaches, Biochem. Cell Biol., 95,
338-349; doi: 10.1139/bcb-2016-0227.
74.Shimura, H., Miura-Shimura, Y., and Kosik, K. S.
(2004) Binding of tau to heat shock protein 27 leads to decreased
concentration of hyperphosphorylated tau and enhanced cell survival,
J. Biol. Chem., 279, 17957-17962; doi:
10.1074/jbc.M400351200.
75.Kumar, P., Jha, N. K., Jha, S. K., Ramani, K.,
and Ambasta, R. K. (2015) Tau phosphorylation, molecular chaperones,
and ubiquitin E3 ligase: clinical relevance in Alzheimer’s
disease, J. Alzheimers Dis., 43, 341-361; doi:
10.3233/JAD-140933.
76.Abisambra, J. F., Blair, L. J., Hill, S. E.,
Jones, J. R., Kraft, C., Rogers, J., Koren, J., 3rd, Jinwal, U. K.,
Lawson, L., Johnson, A. G., Wilcock, D., O'Leary, J. C., Jansen-West,
K., Muschol, M., Golde, T. E., Weeber, E. J., Banko, J., and Dickey, C.
A. (2010) Phosphorylation dynamics regulate Hsp27-mediated rescue of
neuronal plasticity deficits in tau transgenic mice, J.
Neurosci., 30, 15374-15382; doi:
10.1523/JNEUROSCI.3155-10.2010.
77.Baughman, H. E. R., Clouser, A. F., Klevit, R.
E., and Nath, A. (2018) HspB1 and Hsc70 chaperones engage distinct tau
species and have different inhibitory effects on amyloid formation,
J. Biol. Chem., 293, 2687-2700; doi:
10.1074/jbc.M117.803411.
78.Janowska, M. K., Baughman, H. E. R., Woods, C.
N., and Klevit, R. E. (2019) Mechanisms of small heat shock proteins,
Cold Spring Harb. Perspect. Biol., a034025; doi:
10.1101/cshperspect.a034025.
79.Hashiguchi, M., Sobue, K., and Paudel, H. K.
(2000) 14-3-3zeta is an effector of tau protein phosphorylation, J.
Biol. Chem., 275, 25247-25254; doi:
10.1074/jbc.M003738200.
80.Sadik, G., Tanaka, T., Kato, K., Yamamori, H.,
Nessa, B. N., Morihara, T., and Takeda, M. (2009) Phosphorylation of
tau at Ser214 mediates its interaction with 14-3-3 protein:
implications for the mechanism of tau aggregation, J.
Neurochem., 108, 33-43; doi:
10.1111/j.1471-4159.2008.05716.x.
81.Sluchanko, N. N., Seit-Nebi, A. S., and Gusev, N.
B. (2009) Effect of phosphorylation on interaction of human tau protein
with 14-3-3zeta, Biochem. Biophys. Res. Commun., 379,
990-994; doi: S0006-291X(09)00007-2.
82.Sluchanko, N. N., Seit-Nebi, A. S., and Gusev, N.
B. (2009) Phosphorylation of more than one site is required for tight
interaction of human tau protein with 14-3-3zeta, FEBS Lett.,
583, 2739-2742; doi: S0014-5793(09)00593-6.
83.Sluchanko, N. N., and Gusev, N. B. (2011)
Probable participation of 14-3-3 in tau protein oligomerization and
aggregation, J. Alzheimers Dis., 27, 467-476; doi:
10.3233/JAD-2011-110692.
84.Sluchanko, N. N., Sudnitsyna, M. V., Chernik, I.
S., Seit-Nebi, A. S., and Gusev, N. B. (2011) Phosphomimicking
mutations of human 14-3-3zeta affect its interaction with tau protein
and small heat shock protein HspB6, Arch. Biochem. Biophys.,
506, 24-34; doi: S0003-9861(10)00463-7.
85.Wang, K., Zhang, J., Xu, Y., Ren, K., Xie, W. L.,
Yan, Y. E., Zhang, B. Y., Shi, Q., Liu, Y., and Dong, X. P. (2013)
Abnormally upregulated alphaB-crystallin was highly coincidental with
the astrogliosis in the brains of scrapie-infected hamsters and human
patients with prion diseases, J. Mol. Neurosci., 51,
734-748; doi: 10.1007/s12031-013-0057-x.
86.Duennwald, M. L., Echeverria, A., and Shorter, J.
(2012) Small heat shock proteins potentiate amyloid dissolution by
protein disaggregases from yeast and humans, PLoS Biol.,
10, e1001346; doi: 10.1371/journal.pbio.1001346.
87.Rothbard, J. B., Rothbard, J. J., Soares, L.,
Fathman, C. G., and Steinman, L. (2018) Identification of a common
immune regulatory pathway induced by small heat shock proteins, amyloid
fibrils, and nicotine, Proc. Natl. Acad. Sci. USA, 115,
7081-7086; doi: 10.1073/pnas.1804599115.
88.Rothbard, J. B., Kurnellas, M. P., Ousman, S. S.,
Brownell, S., Rothbard, J. J., and Steinman, L. (2018) Small heat shock
proteins, amyloid fibrils, and nicotine stimulate a common immune
suppressive pathway with implications for future therapies, Cold
Spring Harb. Perspect. Med., 9, a034223; doi:
10.1101/cshperspect.a034223.
89.Liu, Z., Wang, C., Li, Y., Zhao, C., Li, T., Li,
D., Zhang, S., and Liu, C. (2018) Mechanistic insights into the switch
of alphaB-crystallin chaperone activity and self-multimerization, J.
Biol. Chem., 293, 14880-14890; doi:
10.1074/jbc.RA118.004034.