REVIEW: Biochemical Mechanisms and Translational Relevance of Hippocampal Vulnerability to Distant Focal Brain Injury: The Price of Stress Response
N. V. Gulyaeva1,2,a
1Institute of Higher Nervous Activity and Neurophysiology, Russian Academy of Sciences, 117485 Moscow, Russia2Moscow Research and Clinical Center for Neuropsychiatry, Healthcare Department of Moscow, 115419 Moscow, Russia
Received June 7, 2019; Revised August 24, 2019; Accepted August 24, 2019
Focal brain injuries (in particular, stroke and traumatic brain injury) induce with high probability the development of delayed (months, years) cognitive and depressive disturbances which are frequently comorbid. The association of these complications with hippocampal alterations (in spite of the lack of a primary injury of this structure), as well as the lack of a clear dependence between the probability of depression and dementia development and primary damage severity and localization served as the basis for a new hypothesis on the distant hippocampal damage as a key link in the pathogenesis of cognitive and psychiatric disturbances. According to this hypothesis, the excess of corticosteroids secreted after a focal brain damage, in particular in patients with abnormal stress-response due to hypothalamic-pituitary-adrenal axis (HPAA) dysfunction, interacts with corticosteroid receptors in the hippocampus inducing signaling pathways which stimulate neuroinflammation and subsequent events including disturbances in neurogenesis and hippocampal neurodegeneration. In this article, the molecular and cellular mechanisms associated with the regulatory role of the HPAA and multiple functions of brain corticosteroid receptors in the hippocampus are analyzed. Functional and structural damage to the hippocampus, a brain region selectively vulnerable to external factors and responding to them by increased cytokine secretion, forms the basis for cognitive function disturbances and psychopathology development. This concept is confirmed by our own experimental data, results of other groups and by prospective clinical studies of post-stroke complications. Clinically relevant biochemical approaches to predict the risks and probability of post-stroke/post-trauma cognitive and depressive disturbances are suggested using the evaluation of biochemical markers of patients’ individual stress-response. Pathogenetically justified ways for preventing these consequences of focal brain damage are proposed by targeting key molecular mechanisms underlying hippocampal dysfunction.
KEY WORDS: hippocampus, stress, stress response, hypothalamic-pituitary-adrenal axis, corticosteroids, cortisol, corticosterone, glucocorticoid receptor, mineralocorticoid receptor, cytokines, neuroinflammation, neurogenesis, BDNF, interleukins, focal brain injury, stroke, traumatic brain injury, depression, cognitive disturbances, dementiaDOI: 10.1134/S0006297919110087
Abbreviations: ACTH, adrenocorticotropic hormone; Aβ, amyloid β; AD, Alzheimer’s disease; BDNF, brain derived neurotrophic factor; CNS, central nervous system; CRH, corticotropin-releasing hormone; CS, cortisosteroid; GR, glucocorticoid receptor; HPAA, hypothalamic-pituitary-adrenocortical axis; 11HSD, 11β-hydroxysteroid dehydrogenase; IL, interleukin; MCI, mild cognitive impairment; MR, mineralocorticoid receptor; PSD, poststroke depression; TNF-α, tumor necrosis factor-α; TrkB, tropomyosin receptor kinase B.The complicated structure of the hippocampus (different subfields, septo-temporal gradient) and its connections with other essential parts of the brain provide its key position in realizing different forms of behavioral plasticity and response to environmental factors. The hippocampus, an important brain structure for working and spatial memory as well as for emotional behaviors in animals and humans, is a very plastic brain structure, this property being quite predictable in the case of a key mnemonic structure. That, however, is only one side of the coin. Another side, obviously representing the price paid for the plasticity ensuring key involvement in memory and emotions, is selective vulnerability of the hippocampus to numerous stress factors, ischemia, seizures, head trauma, aging, etc. [1]. Considering this, it is not surprising that the hippocampus is a target brain area for the actions of “stress hormones”, primarily corticosteroids (CSs). Together with neurotransmitters, CSs regulate numerous hippocampal functions during development and adulthood, while malfunctions in CS control form the pathogenetic basis for many cerebral diseases, both neurological and psychiatric ones.
CSs are a class of steroid hormones serving as key stress response hormones facilitating stress coping. Acting through specific intracellular receptors in the brain and periphery, CSs regulate behavior, as well as metabolic, cardiovascular, immune and neuroendocrine activities. Their actions are mediated by the glucocorticoid (GR) and mineralocorticoid receptors (MR), members of the nuclear receptor superfamily which, once bound to their ligands, act as transcription factors that can directly modulate gene expression. Interacting with other transcription factors, they also can indirectly regulate the activity of many genes. Recently, membrane-bound GR and MR have been described [2] mediating rapid non-genomic signaling and activating signal transduction pathways, such as protein kinase cascades, to modulate other transcription factors and activate or repress various target genes. These numerous mechanisms mediate CS-dependent regulation of various important functions in a large variety of cells in the CNS and peripheral organs.
A proper response to stressors is critical for survival and adaptive behavior. In mammals, the stress response is primarily mediated by secretion of CSs via the hypothalamic-pituitary-adrenocortical axis (HPAA) as well as sympathetic nervous system response and release of catecholamines through adrenergic neurotransmission. Activation of these pathways results both in a quick adaptive response and subsequent lasting multi-level changes in the brain underlying long-term memories and, thus, forming adaptive experience. Normal HPAA activity (rhythmic and episodic release of adrenal CSs) is essential for maintaining homeostasis. Negative feedback by CSs involves multiple mechanisms limiting HPAA activation and potentially deleterious excessive CS production [3] (Fig. 1a). Adequate CS secretion is tightly regulated by a complex neural circuitry controlling rapid feedback mechanisms. These mechanisms involve non-genomic actions of CSs mediating the immediate inhibition of hypothalamic corticotrophin-releasing hormone (CRH) and pituitary adrenocorticotropic hormone (ACTH) secretion, whereas intermediate and delayed mechanisms mediated by genomic actions involve the modulation of limbic circuitry and peripheral metabolic messengers. Malfunction of this feedback mechanism (e.g., as a consequence of severe and/or chronic stress) results in chronically increased CS levels which have deleterious effect on the hippocampus, a structure with high GR and MR density and, for that reason, highly sensitive to CSs. Chronic CS exposure evokes neuronal cell damage and dendritic atrophy, reduces hippocampal neurogenesis and impairs synaptic plasticity.
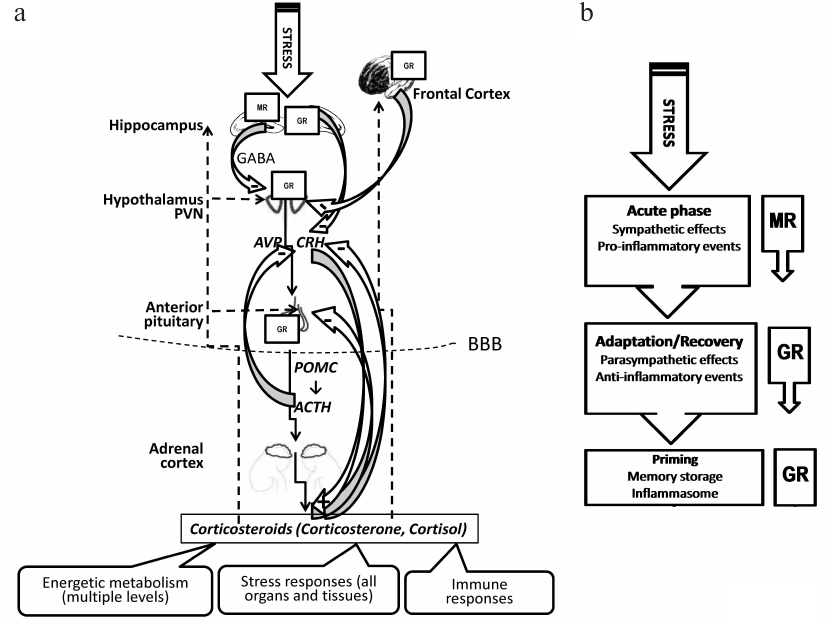
Fig. 1. HPAA regulation, CSs and the involvement of CS receptors in the stress response. a) Organization of stress response in the HPAA [3, 14, 62, 63, 93]. Within this main neuroendocrine axis, a stressor induces release of the adrenocorticotropic hormone (ACTH) secretagogues corticotropin releasing hormone (CRH) and arginine vasopressin (AVP) from the neurosecretory neurons of the medial parvocellular paraventricular nucleus (PVN) of the hypothalamus in the hypophysial portal circulation at the level of the median eminence. The hypophyseal portal vessels transport peptides to the anterior pituitary to enable access to corticotrophs. These secretagogues promote the cleavage of pro-opiomelanocortin (POMC) to ACTH and ACTH release from the pituitary into the systemic circulation, inducing synthesis and release of CSs, cortisol (in some species, e.g., man) or corticosterone (in others, e.g., rodents) at the adrenal cortex. Basal CS release varies in a diurnal pattern, and release increases several-fold after exposure to a stressor. CSs enter the blood circulation and act peripherally and centrally, accessing cognate receptors in virtually every organ and tissue, including the brain (CSs readily pass the blood-brain-barrier, BBB). Cellular effects of CSs are mediated via glucocorticoid receptors (GRs) and mineralocorticoid receptors (MRs). GRs are expressed in most brain structures and regions involved in the HPAA, while the hippocampus is also rich in MR. Regulatory control over the HPAA is mediated via multi-level feedback loops from HPAA structures and from the brain structures, including the hippocampus and frontal cortex. In the hypothalamus, the inhibitory GABAergic input to the PVN neurons is enhanced by excitatory hippocampal output, gamma-aminobutyric acid (GABA) exerting an inhibitory effect on corticotroph secretion. Circulating CSs affect the hypothalamus inhibiting further secretion of CRH, and the pituitary inhibiting the secretion of ACTH. Circulating levels of ACTH also affect the hypothalamus inhibiting secretion of CRH. These negative feedback loops result in a significant decrease of circulating levels of CSs after the end of the stressor. Stress suppresses hippocampal MR expression, and then reduces the hippocampal GR expression, inducing hypersecretion of CRH and AVP, and, ultimately, HPAA axis hyperactivity. b) CS receptors and the neuroendocrine stress response [2, 6, 8, 12, 96, 105]. Different phases in the stress reaction, recovery from stress and adaptation are associated with different pattern of CS secretion and activation of MR and GR. The binding of CS to MR and GR proceeds in the brain in three stages (see main text for details).
Recently, we have suggested a new hypothesis on the principal involvement of stress response mechanisms (including HPAA malfunctioning, interaction of released CSs with hippocampal CS receptors and subsequent inflammatory events) in the remote hippocampal damage underlying delayed cognitive disturbances/dementia and depressive disorders induced by focal brain lesions (e.g., post-stroke and post-traumatic) [4]. Here, we will critically review the literature data and the results of our studies to specify and solidify the molecular part of this concept. We will provide data and logical foundations substantiating that the phenomenon of distant hippocampal vulnerability is closely related to HPAA functioning and, therefore, to the individual stress-response. Key biochemical mechanisms of the CS-dependent stress response will be discussed as well as translational implications.
CS RECEPTORS, HPAA AND STRESS RESPONSE
CSs entering the brain bind to two intracellular receptor types that regulate transcription of CS-responsive genes: the high affinity MR and the GR with approximately 10-fold lower affinity and also exert rapid, non-genomic effects on the excitability and activation of neurons in (amongst others) the hippocampus, hypothalamus, amygdala and prefrontal cortex thus affecting cognition, adaptive behavior and neuroendocrine output within minutes [8, 9]. GR and MR showing a high degree of colocalization in the hippocampus, predominantly reside in the cytoplasm without ligand and are translocated into the nucleus upon ligand binding to act as transcriptional factors. Thus, their subcellular localizations are an important component of their biological activity [10]. It has been hypothesized that MR operates in pro-active mode to prevent homeostatic disturbance, while additional GR activation promotes in reactive fashion recovery after stress. The translational implication is that an imbalance in MR and GR underlies behavioral deficits and neuroendocrine disturbances increasing vulnerability for stress-related brain disorders. MRs respond to low concentrations of the steroid, while higher concentrations are needed for additional activation of GRs. MR occupation in hippocampal neurons was suggested to be relevant for stability of ongoing transmission, for basal activity and sensitivity of the stress response system, for behavioral reactivity and response selection. Additional transient GR activation suppresses excitability, facilitates recovery from the stress response, and promotes information storage.
Thus, the balance of MR- and GR-mediated effects appears critical for the long-term control exerted by CSs over specific aspects of neuronal activity, stress responsiveness, and behavioral adaptation [11]. These suggestions have been experimentally confirmed in further studies which have shown that resilience depends on balanced MR- and GR-mediated actions; brain MR promotes appraisal processes, choice of coping style, learning and memory retrieval; brain GR promotes recovery, rational decisions and contextual memory, while transcription factors and co-regulators confer specificity to MR- and GR-mediated actions [12]. Indeed, CSs exercise fast non-genomic actions on neurons in the hippocampal CA1 region depending on classical MR which are accessible from the outside of the plasma membrane and displaying an order of magnitude lower affinity for corticosterone than the nuclear version involved in neuroprotection. Consequently, this “membrane” receptor could play an important role while CS levels are high, i.e., during the initial phase of the stress response. De Kloet et al. [2] showed that during this phase CSs promote hippocampal excitability amplifying the effect of other stress hormones. These permissive non-genomic effects may contribute to fast behavioral effects and encoding of stress-related information. Notably, CSs have been shown to modulate hippocampal synapses. Dendritic spines are postsynaptic structures of synapses and are essential for synaptic plasticity and cognition. Rapid non-genomic spine modulation was described after applying corticosterone (an acute stress model of the hippocampus), these modulations presumably being mediated by MR [13]. The fast effects of MR are complemented by slower GR-mediated effects which facilitate suppression of temporary raised excitability, recovery from the stressful experience and storage of information for future use (contextualization, rationalization and memory storage of the experience) [12] (Fig. 1b).
These sequential phases in cognitive performance depend on synaptic metaplasticity regulated by coordinate MR- and GR activation including recruitment of co-regulators and transcription factors as determinants of context-dependent specificity in steroid action; they can be modulated by genetic variation and (early) experience. Interestingly, inflammatory responses to damage (see next chapter) seem to be governed by a similarly balanced MR/GR-mediated action as the initiating, terminating and priming mechanisms involved in stress-adaptation [12].
Thus, the difference of GR and MR in distribution and affinity as well as mediating both rapid non-genomic (“membrane” CS receptors) and slow gene-mediated neuronal actions (intracellular CS receptors) provide for a fine and well-regulated association of natural (e.g., stress-induced) shifts in CS level with an intricate pattern of time- and region-dependent changes in neuronal activity and, consequently, with distinct behavioral, in particular, cognitive and emotional patterns. This “one hormone/two receptors system” works in balance, modulating a large spectrum of actions in the CNS.
MRs seem to play a crucial role in the maintenance of the circadian ACTH and cortisol rhythm, through the modulation of CRH and arginine vasopressin release [14]. They bind both aldosterone and glucocorticoids, the latter having much higher affinity for MR than for the closely related GR. Brain MRs have two faces: “Salt” and “Stress”. “Salt” refers to the regulation of salt appetite, and reciprocal arousal, motivation and reward, by a network of aldosterone-selective MR-expressing neurons projecting from the nucleus tractus solitarii and circumventricular organs, while “Stress” is about the limbic-forebrain nuclear and membrane MRs, which act as a switch in the selection of the best stress coping mechanisms [15]. Owing to minimal aldosterone transfer across the blood brain barrier and the absence of the neuronal enzyme 11β-hydroxysteroid dehydrogenase (11HSD) type 2 (an intracellular “gate-keeper”), neuronal MR is fully occupied even at low physiological CS levels and constitutes a key factor in the arising of higher cognitive functions such as memorization, learning and mood [16]. Activation of the limbic MR promotes selective attention, memory retrieval and the appraisal process, while driving emotional expressions of fear and aggression. Subsequently, rising CS concentrations activate GRs in limbic-forebrain circuitry underlying executive functions and memory storage, which contribute to balance with MR-mediated actions to homeostasis, excitability and behavioral adaptation [15]. Thus, key modulators of MR function include GR, which may affect MR function by formation of heterodimers and by differential genomic and non-genomic responses on gene expression, and 11HSDs, determining the availability of intracellular active CSs [17].
Results obtained with various genetically modified mouse lines confirm the importance of the GR in regulation of the HPAA: interference with GR activity stimulates, whereas increased GR protein levels inhibit, the HPAA. Genetic downregulation of GR protein levels and inactivation of the GR gene in the brain reduce anxiety-related behavior, revealing a central role of GR in emotional behavior. In the GR mutant animals, cellular properties of hippocampal CA1 neurons are changed, and hippocampal-dependent explicit memory is affected. Comparing MR and GR mutant animals suggests the requirement of MR but not GR for dentate gyrus granule cell maintenance [18]. Nevertheless, GR is believed to be the main mediator of the stress response in the proliferation, differentiation, migration, and functional integration of newborn neurons in the hippocampus. GR expression directly regulates the excitation-inhibition balance, key for proper adult neurogenesis. An excitation-inhibition disbalance may underlie aberrant functional integration of newborn neurons associated with psychiatric and paroxysmal brain disorders [19].
The GR is encoded by the NR3C1 gene. CS signaling follows several consecutive steps leading to target gene transactivation, including ligand binding, nuclear translocation of ligand-activated GR complexes, DNA binding, co-activator interaction and recruitment of functional transcriptional machinery. Any step may be impaired and may account for altered CS signaling. In particular, glucocorticoid resistance syndrome may result in a reduced level of functional GR, a decreased hormone affinity and binding, a defect in nuclear GR translocation, a decrease or lack of DNA binding and/or post-transcriptional GR modifications. To date, 26 loss-of-function NR3C1 mutations have been reported in the context of different disorders, the clinical signs being generally associated with hypercortisolism without negative regulatory feedback loop on the HPAA. Some GR polymorphisms (ER22/23EK, GR-9β) have been linked to glucocorticoid resistance and a healthier metabolic profile whereas some others seemed to be associated with CS hypersensitivity (N363S, BclI) [20]. Recently, genomic loci at which either MR or GR bind selectively were identified, and members of the NeuroD transcription factor family were found to be specifically associated with MR-bound DNA in the hippocampus. Using forebrain-specific MR knockout mice, it has been shown that NeuroD acts in a permissive way to enhance MR-mediated transcription, rather than competition for DNA binding, as a mechanism of MR- over GR-specific binding [21].
Hippocampal voltage gated Ca2+ channels are among the most prominent targets of CSs. Occupation of GR maintains steady electrical activity in hippocampal neurons. When the levels of CSs are low, L-type Ca2+ currents of CA1 hippocampal cells are small; when CS levels rise, the amplitude of L-type Ca2+ currents is enhanced through a process requiring DNA binding of GR homodimers. Chronic Ca2+ overload may increase the vulnerability of limbic cells to additional challenges, e.g., during epileptic or ischemic episodes [22]. GRs appear to have the capacity to substantially alter mitochondrial transcript abundance [23]. The regulation of mitochondrial transcripts by stress and CSs will likely prove functionally relevant in many stress-sensitive tissues including the brain.
Epigenetic modifications are induced by a range of stressors, both physical and psychological, and they are critical in explaining how environmental factors, which have no effect on the DNA sequence, can have such profound, long-lasting influences on both physiology and behavior. A novel, rapid non-genomic mechanism in which CSs via GR facilitate signaling of the ERK-MAPK pathway to the downstream nuclear kinases MSK1 and Elk-1 was revealed in dentate gyrus granule neurons. Activation of this signaling pathway results in serine 10 (S10) phosphorylation and lysine 14 (K14) acetylation at histone H3 (H3S10p-K14ac), leading to the induction of the immediate-early genes c-Fos and Egr-1 [24, 25]. CSs, released as part of the stress response and acting via GRs, enhance signaling through the ERK1/2/MSK1-Elk-1 pathway and thereby increase the impact on epigenetic and gene expression mechanisms. An important role of GABA was found in controlling the epigenetic and gene transcriptional responses to psychological stress [24].
Thus, CSs exert their effects on the brain through genomic mechanisms that involve both GRs and MRs directly binding to DNA, as well as by non-genomic and epigenetic mechanisms. CSs synergize both genomically and non-genomically with neurotransmitters, neurotrophic factors and other stress mediators (Fig. 2) to shape an organism’s present and future responses to a stressful environment.
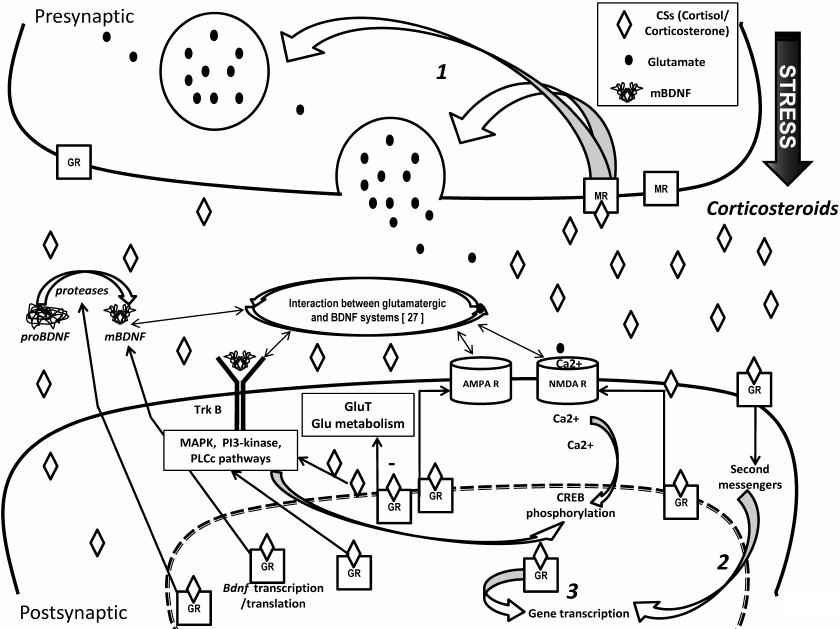
Fig. 2. Mechanisms of synaptic effects of CS and interaction with other systems (glutamatergic synapse as an example) [15, 27, 29, 31]. The interaction of CS with MR and GR underlies interaction and coordination of CS/HPAA with all major systems relevant for neuronal plasticity. CSs can bind, with different affinities, to GRs and MRs existing in both cytoplasmic/nuclear and membrane- bound forms, mediating both delayed and rapid effects, respectively. The effects can result from non-genomic mechanisms (1, mediated by membrane receptors), indirect genomic mechanisms (2, mediated by membrane receptors and second messengers) and genomic mechanisms (3, mediated by cytoplasmic receptors moving to the nucleus and acting as transcription factors). In the genomic mechanism, CSs pass the plasma membrane, enter into the cytosol and bind to GRs, thereby inducing homodimerization (GR–CS complex). Rapid effects of CSs include upregulation of presynaptic glutamate synthesis via presynaptic membrane MR, while later postsynaptic cytosolic GRs regulate ionotropic N-methyl-D-aspartate receptors (NMDAR) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR) increasing their insertion into the post-synaptic membrane as well as glucose (Glu) metabolism and transport reducing plasma membrane glucose transporters (GluT). CSs regulate brain-derived neurotrophic factor (BDNF) synthesis, processing, trafficking, secretion and signaling (see main text for details). The multiple interaction of BDNF/TrkB system with glutamate system (see [27] for details) together with the involvement of CSs in control of both enables to balance and adjust the processes ensuring neuroplasticity.
The key role of neurotrophic factors, in particular brain derived neurotrophic factor (BDNF), in neuroplasticity requires a fine-tuned interaction of the BDNF system with the CS system co-existing throughout the CNS. CS-signaling mediates the regulation of stress response to maintain homeostasis, while neurotrophin signaling plays a primary role in neuronal outgrowth and is crucial for axonal guidance and synaptic integrity. In the hippocampus, high expression of GRs and MRs as well as BDNF and its receptor, tropomyosin-related kinase receptor B (TrkB), have been reported providing for an effective crosstalk between the systems [26]. Interacting long-term effects of stress (including early life stress) on CSs and neurotrophin signaling pathways are manifested in stress-susceptible regions of the hippocampus. Recently, we have analyzed multifaceted interactions between BDNF and glutamate systems [27], here we will discuss how BDNF/TrkB are coordinated with CSs and their receptors in the glutamate synapse. Indeed, the synergy between TrkB and GR signaling is a key factor determining cellular responses to stress [28]. CSs alter expression and signaling of BDNF (Fig. 2). Since BDNF, an essential facilitator of neuronal plasticity, is known to promote brain plasticity, enhance cell survival, increase hippocampal neurogenesis and cellular excitability, it has been hypothesized that specific adverse effects of CSs may be mediated by attenuating BDNF expression and signaling. Suri and Vaidya summarized the data on the influence of CSs on hippocampal BDNF system at multiple levels, spanning from the well-documented CS-induced changes in BDNF mRNA to studies examining alterations in BDNF receptor-mediated signaling [29].
Changes in BDNF levels are of vital functional significance. By counteracting the adverse effects of excessive stress-induced CS signaling, BDNF has been implicated as a resilience factor to psychopathology caused by chronic stress [30]. Chronic restraint stress leads to increases in BDNF mRNA and protein in some regions of the brain, e.g., the basal lateral amygdala, but decreases in other regions such as the CA3 region of the hippocampus, and dendritic spine density increases or decreases in line with these changes in BDNF [31]. In view of the powerful influence that BDNF has on dendritic spine growth, these observations suggest that the fundamental reason for the direction and extent of changes in dendritic spine density in a particular region of the brain under stress is due to the local changes in BDNF, the most likely cause of these changes being stress-initiated release of CSs readily entering neurons and altering BDNF gene expression. Differences in the distribution of GR and MR and of their downstream actions may underlie opposite effects on BDNF gene expression. Epigenetic control of BDNF transcription (differences in the extent of methylation and acetylation) in specific parts of the brain following stress may be another alternative [31]. Notably, positive effects of GR activation on memory consolidation critically engage the BDNF pathway. Finsterwald et al. [32] have proposed the hypothesis that mild stress promotes the formation of strong long-term memories because the activation of hippocampal GRs after learning is coupled to the recruitment of the growth and pro-survival BDNF/cAMP response element-binding protein (CREB) pathway, which is well-known to be a general mechanism required for long-term memory formation.
The above discourse is mostly related to a glutamatergic synapse which has been supposed three decades ago to be tightly related to both CSs and hippocampal damage [33]. At present, there is overwhelming evidence for multiple effects of stress on excitatory transmission and synaptic plasticity in the hippocampus, interactions between stress and hippocampal glutamatergic neurons playing a critical role in the cognitive and emotional consequences of aversive stimuli. Mounting evidence suggests that acute and chronic stress, through the release of CSs, induce changes in glutamate neurotransmission in the hippocampus and prefrontal cortex, thereby influencing key aspects of cognitive processing [34]. Dysfunction of glutamatergic neurotransmission is increasingly considered to be a core feature of stress-related mental illnesses. As well as the BDNF system, the excitatory neurotransmitter glutamate is principally involved in phenomena of cellular and synaptic plasticity and the memory phenomenon. The connections between the two systems are numerous and bidirectional, providing for mutual regulation of the glutamatergic and BDNF systems. The available data suggest that it is the complex and well-coordinated nature of these connections that secures optimal synaptic and cellular plasticity in the normal brain. Both systems are associated with the pathogenesis of depression, and the disturbance of tight and well-balanced associations between them results in unfavorable changes in neuronal plasticity underlying depressive disorders and other mood diseases [27]. Recent studies have shed light on the mechanisms by which stress and CSs affect glutamate transmission, including effects on glutamate release, glutamate receptors, glutamate clearance and metabolism (see [34] for review). Indeed, stress impacts on excitatory synapses are mediated by a complex set of neurohormones and neurotransmitters, among which adrenal CSs play a crucial role. Chaouloff and Groc [35] reviewed the tonic and intrinsic effects of CSs on hippocampal excitatory transmission, glutamate receptor trafficking and expression, and synaptic plasticity, paying attention to their temporality (rapid and transient effects followed by slow and persistent genomic effects). Many effects of CSs on the glutamatergic system are mediated by their binding to cytosolic MRs and GRs, further translocation to the nucleus and regulating the transcription of target genes, while rapid effects are mediated by membrane CS receptors (Fig. 2). This important “regulatory triangle” combining (i) HPAA/CSs/CS receptors, (ii) BDNF system and (iii) glutamatergic system represents the heart of the intricate multi-level regulation of signaling and metabolic processes in the hippocampus underlying the ability to adapt to life challenges and support the neuroplasticity equilibrium, while disturbances in this equilibrium result in various cerebral pathologies.
CSs, NEUROINFLAMMATION AND THE HIPPOCAMPUS
It should be noted that CSs have been long ago considered anti-inflammatory and protective agents due to their ability to inhibit gene expression of pro-inflammatory mediators and other possible damaging molecules. Nonetheless, recent studies have uncovered situations in which these hormones can act as pro-inflammatory agents depending on the dose, chronicity of exposure, and the structure/ organ analyzed. In the hippocampus, the excess of CSs may stimulate pro-inflammatory genes ]37]. Considering the accepted notion that high levels of cortisol have anti-inflammatory properties, this association is particularly enigmatic [38] (Fig. 3). Phenomenologically, excessive CS accumulation in the brain appears to be closely associated with pro-inflammatory events, and this relation has been revealed in different rodent models of depression [39]. Stress induces secretion of cytokines, while cytokines may induce hormonal changes similar to those observed following exposure to stress. Extended stress responses and overproduction of cytokines impair neuronal plasticity and increase HPAA activity, sensitizing its response to cytokines and stress. Stress situation and induced CRH release evoke a pro-inflammatory response in the brain, characterized by a complex release of several inflammatory mediators including cytokines, prostanoids, nitric oxide (NO) and transcription factors, all of them participating in multiple interactions between neuroendocrine and neuroimmune systems [40].
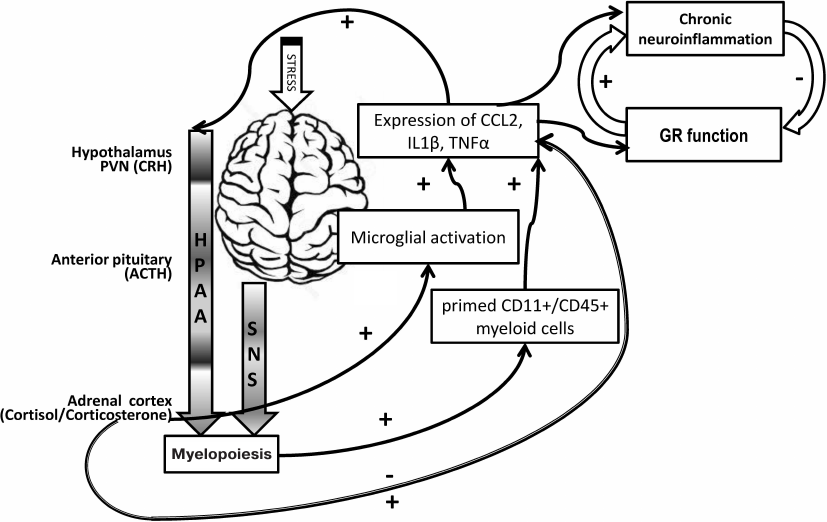
Fig. 3. Corticosteroids and neuroinflammation: stress exposure induces the release of HPAA hormones and upregulates cytokine expression [3, 40, 41, 70, 73]. Stress activates the HPAA and the sympathetic nervous system (SNS). CSs induce activation of microglia, activated microglia releasing pro-inflammatory cytokines including IL-1β and C-C Motif Chemokine Ligand 2 (CCL2). In turn, these cytokine responses contribute to the development of a reactive endothelium in the regional neurovasculature. Both CSs and SNS stimulate the production of primed CD11+/CD45+ myeloid cells (MCs) in the bone marrow. Release of MCs into circulation results in trafficking of these cells to the reactive neurovasculature which is followed by adhesion and diapedesis into the brain. Cytokines cause hypercortisolemia via dysregulation of the HPAA directly by activating it and indirectly by modifying GR sensitivity to CS leading to CS hypersecretion. Cytokines directly stimulate activation of the HPAA via actions both intrinsic and extrinsic to the axis and increase HPAA sensitivity to the following stress challenges. There are myriads of mechanisms mediating deleterious effects of chronically elevated cytokine level in the brain tissue, many of them associated with brain CS receptors. Usually, a viscous cycle occurs (top right): chronic neuroinflammation inhibits GR function, which in turn exacerbates pro-inflammatory cytokine activity and aggravates chronic neuroinflammation.
Catecholamines and CSs play critical roles in the regulation of brain cytokines after stress exposure. The neuroendocrine responses to psychological stressors affect the immediate and long-term regulation of inflammatory cytokines IL-1β, TNF-α, and IL-6 within the hippocampus, hypothalamus, or prefrontal cortex, this regulation changing across time with repeated stress exposure (see [41] for review). Central catecholamines induce the secretion of IL-1β from microglia, IL-1β being a key factor in the further activation of microglia and recruitment of monocytes into the brain. Normally, elevated CSs inhibit the production of brain cytokines by suppressing noradrenergic locus coeruleus neurons and inhibiting the NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells) signaling pathway. However, in repeated stress, CSs and peripheral catecholamines facilitate inflammatory responses to future stimuli by stimulating monocytes to leave the bone marrow, downregulating inhibitory receptors on microglia, and priming neuroimmune inflammatory responses mediated by peripheral monocytes or macrophages [42] (Fig. 3).
The hippocampus is selectively vulnerable to neuroinflammation, and this may be one of the bases for the involvement of this structure in the pathogenesis of many mental and neurological diseases (see [4] for review). Stress challenges produce time-dependent and stressor-specific patterns of cytokine/chemokine expression in the brain and inflammation-related genes are up-regulated and exhibit unique expression profiles in males and females depending upon individual, cooperative or antagonistic interactions between steroid hormone receptors (estrogen receptors and GR). Accumulating evidence suggests that impaired balance and crosstalk between GR and NFκB, an effector of the immune axes, may play a key role in mediating the harmful effects of chronic stress on mood and behavior [43]. According to the “MR/GR balance hypothesis” [44], inflammatory responses to damage seem to be governed by a balanced MR/GR-mediated action as the initiating, terminating and priming mechanisms involved in stress/adaptation (Fig. 1b). MRs are associated with pro-inflammatory bias in the hippocampus of spontaneously hypertensive SHR rats. In these rats, MR/GR imbalance has been demonstrated: MR expression is increased and MR activation by endogenous corticosterone induces a pro-inflammatory cascade contributing to microglia activation, overexpression of pro-inflammatory mediators, down-regulation of anti-inflammatory factors, ultimately resulting in hippocampal tissue damage [45]. Administration of IL-1β produces a long-lasting increase in corticosterone; IL-1β also influences MR function in the hippocampus and causes a shift in the MR/GR balance, which may underlie prolonged activation of the HPAA during an immune response [11].
It has been suggested that CSs induce synthesis and release of high mobility group box-1 (HMGB1), an endogenous danger signal or alarmin, from microglia, which signals through TLR2/TLR4, thereby priming the NLRP3 inflammasome. When CS levels reach a critical threshold, they may thus function as an alarmin by inducing HMGB1, thereby preparing an organism’s innate immune system (NLRP3 inflammasome priming) for subsequent immune challenges. This may confer a significant survival advantage by enhancing the central innate immune and sickness response to immune challenges [46].
Within the brain, stress elevates both norepinephrine and CSs, and both affect several genomic and signaling cascades responsible for modulating memory strength. Glia are one of the main targets for stress-induced changes, astrocytes, microglia, and oligodendrocytes all having unique contributions to learning and memory. Furthermore, these three types of glia express receptors for both CSs and norepinephrine and are hence immediate targets of stress hormone actions. It is becoming increasingly clear that inflammatory cytokines and immunomodulatory molecules released by glia during stress may promote many of the behavioral effects of acute and chronic stress [47]. Microglia are the predominant immune cells of the CNS playing key physiological roles in maintaining homeostasis, particularly in response to stress, and mediating synaptic plasticity, learning and memory. Many of these effects are believed to be driven by stress-linked signaling molecules, first of all CSs. The majority of studies have demonstrated that stress induces significant structural remodeling of microglia and can augment the release of pro-inflammatory mediators [39]. CSs increase ramification of hippocampal microglia and may modulate age-associated changes in microglial morphology [48]. Stress-induced microglial alterations, rather than being epiphenomena, have behavioral implications, involving microglia in directly regulating definite aspects of cognitive function and emotional regulation. Since neuroinflammation is regarded as a key mechanism of neuronal cell damage and neurodegeneration, specifying molecular microglial-dependent mechanisms of neuroinflammation control by CSs and their receptors is of primary importance for preventing brain diseases.
CSs, COGNITIVE FUNCTIONS AND THE HIPPOCAMPUS
Patients with AD, Parkinson’s or Huntington’s disease show chronically high cortisol levels suggesting changes occurring in HPAA control. In experimental models of these diseases, chronic stress or CS treatment was found to exacerbate both the behavioral symptoms and neurodegenerative processes [52]. Clinical studies demonstrate that, in AD, elevated cortisol is associated with poorer overall cognitive functioning, as well as with poorer episodic memory, executive functioning, language, spatial memory, processing speed, and social cognition [53]. Similarly, chronically elevated corticosterone or CS administration induces cognitive impairment and abnormal (depressive-like, anxious) behavior in animals. Moreover, in cognitively healthy subjects, higher cortisol levels have been associated with an increased risk of cognitive decline and AD, while patients with dementia and mild cognitive impairment (MCI) have been found to have higher cerebrospinal fluid cortisol levels than cognitively healthy controls (see [53] for review). Elevated cerebrospinal fluid cortisol may also be associated with a more rapid cognitive decline in MCI.
Basal forebrain cholinergic neurons, believed to mediate cognitive function, are implicated in the etiology of stress, cognitive aging, AD, and other neurodegenerative diseases. These neurons are involved in stress response and cognition and project to different cortical sites and limbic structures, including the hippocampus, primary CS target tissue associated with cognitive function together with HPAA response. There is the molecular interactive link between the CSs/MR/GR and cholinergic system that contributes to basal forebrain cholinergic neuronal degeneration in stress-induced acceleration of cognitive decline in aging and AD [54].
Certainly, high cortisol may stimulate different signal mechanisms potentially contributing to neurodegeneration. High cortisol may exert neurotoxic effects on the hippocampus and promote oxidative stress and amyloid β (Aβ) peptide toxicity. AD is the most common form of dementia, characterized by the pathological accumulation of two proteins, the Aβ and hyperphosphorylated forms of the microtubule-associated protein tau in the neurons of the hippocampus and prefrontal cortex. Aβ, a peptide released by synapses in physiological conditions, is pathologically accumulated in brain structures involved in memory processing and represents a key toxic hallmark of AD. The oligomeric form of Aβ is believed to affect synapse function and there is accumulating evidence that GRs participate in oligomeric Aβ generation, indicating a tight functional interplay between Aβ and GR activities at excitatory synapses [55]. Although clinical studies have so far failed to establish a direct causative link between CSs and AD pathogenesis, evidence from pre-clinical studies has shown that increased CS levels accelerate the formation of Aβ in AD animal models by promoting the amyloidogenic pathway in parallel reducing Aβ clearance, through transcriptional mechanisms involving the GR. Conversely, effects of stress on tau phosphorylation seem to be mainly mediated by the CRH receptor (CRFR1) and independent from stress-induced CS elevation [51].
Chronic stress is an environmental risk factor modulating microglial function, microglia being causally linked to Aβ accumulation, tau pathology, neurodegeneration, and synaptic loss in AD, though also plays beneficial roles, particularly in the phagocytic elimination of Aβ [56]. Possible other mechanisms underlying the involvement of CS excess in AD also include their interactions with inflammatory mediators, neurotransmitters, and growth factors.
The fact that HPAA disturbances are associated with memory impairments, and hypercortisolemic conditions with atrophy of the hippocampus have been confirmed by many studies. However, recent discoveries support a more complicated picture of HPAA function and pathology in acquiring, retrieving, and consolidating new memories. These findings include: the existence of an “inverted U-shaped relationship” between stimulation of brain GR and memory performance; different responses of distinct areas of the hippocampus to CS stimulation; potential reversibility of hippocampal atrophy in some conditions, although whether such atrophy is a cause or effect of these pathological conditions remains obscure [57].
Importantly, the dentate gyrus of the hippocampus maintains production of new neurons throughout life. Adult neurogenesis is closely associated with hippocampal function, including learning and memory, anxiety regulation and feedback of the stress response. Altered neurogenesis is suggested to be involved in the onset of brain diseases, particularly mental disorders and neurodegenerative diseases. Stress affects all range of hippocampal neurogenesis, including the production, migration and survival of new neurons, and CSs have been implicated in stress-induced impairment of adult neurogenesis. It is generally assumed that CSs have a negative impact on both embryonic and adult neural stem/progenitor cell proliferation, this phenomenon being related to the pathophysiology of brain diseases, such as depression and autism spectrum disorders, as well as impairments of learning and memory [58, 59]. However, this view is rather skewed since, depending on the stress nature and severity as well as on the stress response of the organism, increases in CS levels are sometimes associated with enhanced adult neurogenesis in the dentate gyrus, though in other situations they are suppressive. Although the effects of acute and mild stress on adult neurogenesis are generally brief and can be quickly overcome, chronic exposure and more severe forms of stress can induce longer lasting reductions in neurogenesis [60]. In these circumstances, the factors that buffer against the suppressive influence of elevated CSs remain unclear and under debate [61]. There is evidence for both direct and indirect effects of CSs on neural stem/progenitor cell proliferation and adult neurogenesis and a hypothesis has been formulated that CS rhythmicity and oscillations originating from the activity of the HPAA, may be crucial for the normal neurogenesis in the hippocampus [59].
It is the hippocampal sensitivity to stress which is believed to explain the negative impact of stress and related stress hormones on animal and human cognitive function. However, in the last two decades, new data were gathered showing that stress impacts on many mnemonic cortical and subcortical brain structures other than the hippocampus [62], these data remaining out of the scope of this article. Indeed, most CS effects on the brain are exerted through MRs and GRs, inducing intricate and often opposite actions on the cerebral structures implicated in various cognitive functions.
CSs, DEPRESSION, NEUROINFLAMMATION AND THE HIPPOCAMPUS
Hyperactivity of the HPAA as one of the fundamental biological mechanisms underlying major depression results from diminished feedback inhibition of CS-induced reduction of HPAA signaling and increased CRH secretion from the paraventricular nucleus and extra-hypothalamic neurons, while the hippocampus normally inhibits stress-induced HPAA activation [63] (Fig. 1a). A growing number of studies indicate that abnormal function of MR and GR is a crucial component of the pathophysiology of depression. During chronic stress-induced inhibition of systemic feedback, cytosolic GR levels are significantly modified in the hippocampus and prefrontal cortex, both structures known to be deeply involved in the pathogenesis of depression [40]. Long-term disturbances in the release pattern of CSs and in the responsiveness of their receptors give rise to structural and functional changes in neuronal properties which may contribute to the expression of a psychopathology [64].
Depression is highly prevalent in infectious, autoimmune and neurodegenerative diseases and at the same time, depressed patients show impaired immune function [65]. It was initially thought that the hypercortisolemia caused a suppression of immune function, but it is now apparent that chronic stress and depression induce a hypoactivity of the GRs on immune cells and in limbic regions of the brain. Depression is now thought to be associated with activation of some links of cellular immunity resulting in the hypersecretion of pro-inflammatory cytokines and the hyperactivity of the HPAA, such immune activation inducing “stress-like” behavioral and neurochemical changes [66]. Increased inflammation and hyperactivity of HPAA, two hallmarks in major depression, are often associated: development of depression is believed to be related with disturbance of the allostasis and inflammatory activation of the immune system. This results in a chronic increase in the CS and pro-inflammatory cytokine levels, inducing an allostatic load and, sequentially, neurodegeneration, irreversible cognitive impairment and permanent disability [67]. Enhanced susceptibility to inflammatory and autoimmune disease in depression is related to impairments in HPAA activity which may be associated to hypocortisolism, or to glucocorticoid resistance resulting from impairments in local factors affecting CS availability and function, including the GR [68].
Both inflammation and the activation of the HPAA by stress are normal components of the stress response, but when stress is prolonged and hypercortisolemia persists, the peripheral macrophages become activated together with the central microglia, the neuronal networks are damaged and become dysfunctional (Fig. 3) [69]. Since communication occurs between the endocrine, immune systems and CNS, an activation of the inflammatory responses can affect neuroendocrine processes, and vice versa.
Physical, psychological or combined-stress conditions evoke a pro-inflammatory response in the brain and other systems, characterized by a complex release of several inflammatory mediators. Pro-inflammatory cytokines, including IL-1β, IL-6 and TNF-α are implicated in the etiologies of clinical depression and anxiety disorders. CSs inhibit (via GR), while catecholamines stimulate (via β-adrenergic receptors) IL-1β production [40]. Inflammatory cytokines contribute to depression via action on three major pathways in the brain: the neuroendocrine system; neurotransmitter depletion; and neuroprogression pathways. Cytokines cause hypercortisolemia by dysregulation of the HPAA directly by activating it and indirectly by modifying GR sensitivity to cortisol leading to cortisol hypersecretion (Fig. 3). Cytokines deplete central synaptic serotonin levels by reducing its synthesis and increasing its reuptake. They may also deplete neurotrophic factors, in particular BDNF, which are believed to play a neuroprotective role against depression. Cytokines activate cellular cascades that cause excitotoxicity and apoptosis and inhibit neurogenesis in the hippocampus [70].
Notably, depression is comorbid with many other conditions associated with neurodegeneration. The simple model that CSs induce neurodegeneration does not seem accurate, but rather that elevated cytokines, in the context of glucocorticoid resistance, are probably the offenders and chronic inflammatory changes in this situation may represent a common feature responsible for the enhanced vulnerability of depressed patients to develop neurodegenerative changes later in life [65]. Patients with depression exhibit all cardinal features of inflammation, including increased circulating levels of inflammatory inducers, activated sensors, and inflammatory mediators targeting all tissues. Pro-inflammatory cytokines modulate mood behavior and cognition by reducing brain monoamine levels, activating neuroendocrine responses, promoting excitotoxicity (increased glutamate levels), activating the tryptophan-kynurenine pathway resulting in the synthesis of the neurotoxic NMDA receptor agonist quinolinic acid and 3-hydroxykynurenine, thereby enhancing oxidative stress, contributing to neurodegeneration and impairing brain plasticity [69]. It is believed now that a vicious circle exists closely intertwining neuropsychiatric disorders and inflammation which are powering each other in a bidirectional loop. Depression facilitates inflammatory reactions and inflammation promotes depression and other neuropsychiatric disorders. Potential triggers of chronic inflammation include changes in neuroendocrine regulation, metabolism, diet/microbiota, and negative health behaviors, including early-life [71]. The complexity of mechanisms underlying depression makes it difficult to create a general concept of pathogenesis of depression, and the current theories on CS hypersecretion and serotonergic dysfunctions do not provide sufficient explanations for the nature of the disease. Since inflammatory and neurodegenerative (I&ND) processes play an important role in depression and enhanced neurodegeneration in depression may at least partly be caused by inflammatory processes, the I&ND hypothesis of depression has been generated, an extension of a cytokine hypothesis considering that external, e.g., psychosocial stressors, and internal stressors, e.g., organic inflammatory disorders or conditions, may trigger depression via inflammatory processes [72].
Hippocampal GRs and the HPAA have close interactions with pro-inflammatory cytokines and neuroinflammation. Elevated pro-inflammatory cytokine levels and GR functional resistance are among the most widely investigated factors in the pathophysiology of depression. These two major components create a vicious cycle (Fig. 3). Chronic neuroinflammation inhibits GR function, which in turn exacerbates pro-inflammatory cytokine activity and aggravates chronic neuroinflammation. On the other hand, neuroinflammation causes an imbalance between oxidative stress and the anti-oxidant system, which is also associated with depression. Although cytokines and GRs, important players in depression, are essential components of a whole system of inflammatory and endocrine interactions, rather than play independent parts [73, 74].
Exposure to chronic stress early in life as well as in adulthood has been shown to reduce the expression of GR (in particular through epigenetic mechanisms) and to up-regulate the expression of the co-chaperone gene FKBP5, which restrains GR activity by limiting the translocation of the receptor complex to the nucleus. Another mechanism that contributes to changes in GR responsiveness is the state of receptor phosphorylation that controls activation, subcellular localization and transcriptional activity. GR phosphorylation represents an important mechanism for the cross-talk between neurotrophic signaling and GR-dependent transcription, bridging two important players for mood disorders. One gene that lies downstream from GR and may contribute to stress-related changes is serum glucocorticoid kinase-1 (SGK1); its expression is significantly increased after exposure to chronic stress in rodents as well as in the blood of drug-free depressed patients. SGK1 up-regulation may ultimately reduce hippocampal neurogenesis and contribute to the structural abnormalities that have been reported to occur in depressed patients [75]. Stress dynamically regulates co-expression networks of GR-dependent major depression risk genes, changes in the correlation structure of functional genetic variants forming a tight CS-responsive co-expression network possibly contributing to early-life adversity-associated disease risk. A subset of genetic variants may contribute to the risk for depression by altering circuit-level effects of early and adult social experiences on the network formation and structure [76].
Allostatic overload, which can occur during chronic stress, can reshape the HPAA through epigenetic modification of genes in the hippocampus, hypothalamus and other stress-responsive brain regions [77]. Epigenetic processes may play an important role in the etiology of stress-related mental disorders such as major depressive and anxiety disorders. Psychologically stressful events evoke a long-term impact on behavior through changes in hippocampal function brought about by distinct glutamatergic and CS-driven changes in epigenetic regulation of gene transcription, which are modulated by local GABAergic interneurons and limbic afferent inputs [24].
The influence of CSs on hippocampus-dependent learning and memory processes is now indisputable, though interpretations from recent studies suggest that the idea that CS-induced hippocampal cell death accounts fully for the associated cognitive and emotional deficits is only partially correct [78]. Changes in neurogenesis are believed to play an important role in affective diseases, in particular, depression. Indeed, a specific sub-population of hippocampal neurons, the granule cells of the dentate gyrus, a constantly functioning neurogenic niche, is more sensitive to excess of CSs, though the mechanisms of this unique vulnerability remain obscure. Thus, CSs, through MR and GR, influence not only cell death, but also cell birth and differentiation, MR occupation being essential for their survival. Using the model of chronic unpredictable stress, it was shown that MR-dependent neurogenesis was closely related with anxiety-like behavior [79]. While excessive occupation of GR can induce loss of neurons in the absence of MR activation, additional MR occupation usually results in less severe and potentially reversible consequences involving only dendritic atrophy and loss of synaptic contacts. GR-mediated serious injury in the dentate gyrus subfield might be the cause of hippocampal volume loss in depression [80].
DEPRESSION, DEMENTIA AND DAMAGED HIPPOCAMPUS: A 2-IN-1
CONSTRUCTION
The hippocampal area of the adult brain contains neural stem cells or more committed neural progenitor cells, which retain throughout the human life the ability of self-renewal and to differentiate into multiple neural cell lineages, such as neurons, astrocytes, and oligodendrocytes. Importantly, these characteristic cells contribute significantly to the above-indicated functions of the hippocampus, while various stressors and CSs influence proliferation, differentiation, and fate of these cells [83]. Disordered CSs in depression may inhibit neurogenesis, but the contribution of diminished neurogenesis to the onset or progression of AD is still debated. However, both CSs and cytokines also reduce BDNF, implicated in hippocampal neurogenesis, depression and dementia this reinforcing the notion that those cases of depression with disordered CSs may be a risk for AD.
Chronic inflammation considered to be central to the pathogenesis of major depression could predispose depressed patients to neurodegenerative changes in later life. Indeed, there is now clinical evidence that depression is a common antecedent of AD and may be an early manifestation of dementia before the cognitive declines becomes apparent [81]. Cytokines, including IL-1β, IL-6 and TNF-α, are increased in the blood in some cases of depression, and increased cytokines are a known risk for later AD. Inflammatory and CS-mediated changes occur in both depression and AD, both cytokines and CSs potentially having pro-inflammatory actions in the brain [81, 82]. It is hypothesized that the progress from depression to dementia could result from the activation of macrophages in the blood, and microglia in the brain, that release pro-inflammatory cytokines. These cytokines stimulate a cascade of inflammatory changes (such as an increase in prostaglandin E2, nitric oxide in addition to more pro-inflammatory cytokines) and a hypersecretion of cortisol. The latter inhibits protein synthesis thereby reducing the synthesis of neurotrophic factors and preventing repair of damaged neuronal networks. Neurotoxic end products of the tryptophan-kynurenine pathway, such as quinolinic acid, accumulate in astrocytes and neurons in both depression and dementia. Thus, increased neurodegeneration, reduced neuroprotection and neuronal repair are common pathological features of major depression and dementia explaining why major depression is a frequent prelude to dementia [84].
All this reasoning is associated with a search for common links between molecular mechanisms of dementia and depression as well as possible links between CS disturbances and chronic low grade inflammation with changes in brain structure that could precipitate neurodegenerative changes associated with AD and other dementias as well as in major depression. Quite right in general, these assumptions do not contain a core statement: the structure of the hippocampus makes this brain region tightly involved in both emotional behavior and cognition. The hippocampus is not a homogeneous brain area, and it is the complex organization of this structure that underlies its relevance and functional pleiotropism. To keep the story simple, the plain paradigm “cognition controlled by the dorsal (posterior) hippocampus vs. emotions controlled by the ventral (anterior) hippocampus” attributes cognitive disturbances/dementia mainly to the dorsal hippocampus, while the development of emotional disturbances, including depressive behavior, is, at least at the start, associated with the ventral hippocampus (see [4] for review). In other words, the hippocampus works as a 2-in-1 system, controlling cognition and emotions in the healthy brain, while hippocampal damage may underlie both cognitive and emotional disturbances. This simple view may be a basis for both understanding the comorbidity of depression and dementia and exploring causative links between two pathologies and mechanisms of their association. The hypothesis which we have suggested [4] is a logical consequence of this view: dysfunctional stress response mechanisms (including interaction of released CSs with hippocampal receptors and subsequent inflammatory events) are involved in the remote hippocampal damage underlying delayed dementia and depression induced by focal brain damage (e.g., post-stroke or post-traumatic).
ABNORMAL STRESS RESPONSE AND STRESS PERIODS IN PATIENTS AFTER
FOCAL BRAIN LESIONS
Poststroke depression (PSD) is the most prevalent psychiatric disorder after stroke, which is independently correlated with negative clinical outcome. The identification of specific biomarkers could help to increase the sensitivity of PSD diagnosis and elucidate its pathophysiological mechanisms. According to a recent meta-analysis data, the following molecular candidates differentiate PSD patients from non-depressed stroke subjects: hypercortisolemia and blunted cortisol awakening response, increased early markers of inflammation (high-sensitivity C-reactive protein, ferritin, neopterin, and glutamate), serum pro-inflammatory cytokines (TNF-α, IL-1β, IL-6, IL-18, IFN-γ), as well as decreased pro-inflammatory/anti-inflammatory ratios (TNF-α/IL-10, IL-1β/IL-10, IL-6/IL-10, IL-18/IL-10, IFN-γ/IL-10); serum BDNF concentrations; BDNF met/met genotypes; higher BDNF promoter methylation status [84]. Importantly, symptoms of depression after a stroke or a transient ischemic attack increased the risk of cognitive impairment and functional deterioration at 2-year follow-up [91]. In the situation, when the hippocampus is chronically overloaded with CSs, it may become primed or even damaged before focal brain injury. The TABASCO study demonstrated the predictive role of hippocampal mean diffusivity, suggesting that these changes may precede and contribute to volumetric and connectivity changes in the hippocampi, potentially serving as a marker for early identification of patients at risk of developing cognitive impairment or dementia [92]. All these data confirm the importance of abnormal stress-response due to HPAA dysfunction in the development of hippocampal damage-mediated cognitive and emotional post-stroke disturbances.
Chronic stress-induced activation of the HPAA takes many forms (chronic basal hypersecretion, sensitized stress responses, and even adrenal exhaustion), with manifestation dependent upon factors such as stressor chronicity, intensity, frequency, and modality. Importantly, individual response to acute or chronic stress is determined by numerous factors, including genetics, early life experience, environmental conditions, sex and age. The context in which stressors occur will determine whether individual acute or chronic stress responses are adaptive or maladaptive (pathological) [93]. Stress-induced excess of CS secretion is the main factor contributing to remote hippocampal damage underlying post-stroke and post-trauma cognitive and depressive disturbances. During the post-injury period two major diverse sequences of pathological events occur; one associated with the direct damage to the brain tissue, the second associated with stress and subsequent HPAA activation, excessive CS release and stimulation of hippocampal GR and MR inducing remote functional and structural changes in the hippocampus (neuroinflammation, disturbances in neurogenesis, synaptic plasticity, neurodegeneration, etc.) [4].
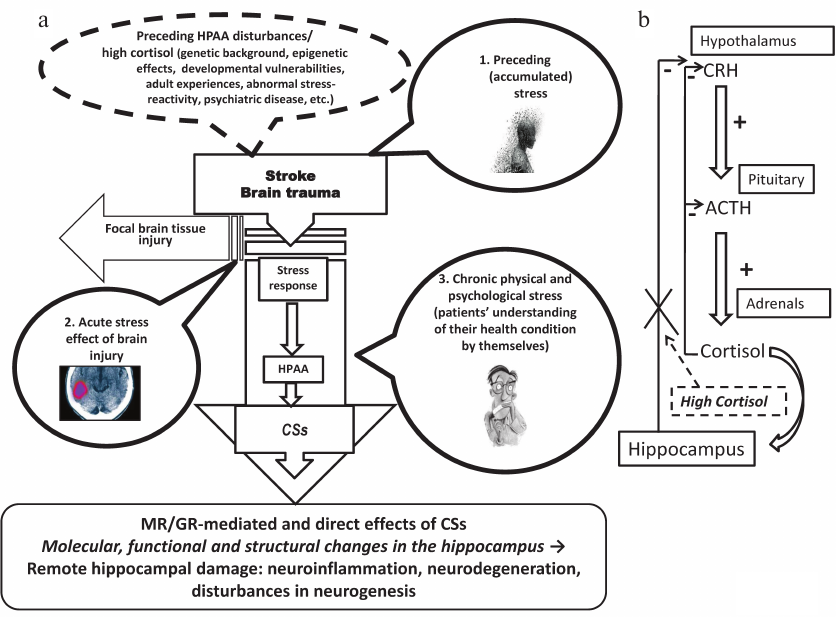
Why are HPAA dysfunction and abnormal stress response so important in both pre- and post-stroke periods (Fig. 4a)? Potential preceding stress associated with prolonged periods of high cortisol may trigger signaling and metabolic changes in the hippocampus which can be worsened during following stress periods. Preceding cortisol load can be easily monitored by hair cortisol assays [94]. Focal brain injury, a stressogenic factor, induces CS augmentation increasing hippocampal GR and MR signaling. The following period of treatment and recovery should be regarded as a time of chronic physical and psychological stress, in particular, induced by patients’ understanding of their health condition by themselves. Taking into account possible preceding HPAA disturbances associated with high cortisol, including abnormal stress-reactivity, psychiatric disease, genetic background, these multiple chronic stress factors affect a patient after a focal brain injury (mainly stroke or trauma) potentially inducing and subsequently aggravating remote hippocampal damage. Normally, hypothalamic CRH activates ACTH release from the pituitary gland and following CS secretion by adrenals, cortisol inhibiting its own secretion via a negative feedback loop (Fig. 1). When cortisol is chronically elevated and the hippocampus becomes functionally damaged, the hippocampus-dependent HPAA slowdown becomes impaired [53] and cortisol level will further increase, inducing additional hippocampal damage. The HPAA malfunctioning vicious circle underlying hippocampal atrophy is depicted in Fig. 4b.
Fig. 4. Periods of stress load after focal brain lesion and remote hippocampal damage. a) Phases of potential stress load before and after focal brain lesion (see main text for details). b) Vicious circle of HPAA dysfunction underlying hippocampal atrophy (see main text for details).
DISTANT HIPPOCAMPAL DAMAGE AFTER FOCAL BRAIN LESION: ARE THERE
WAYS FOR PREVENTION?
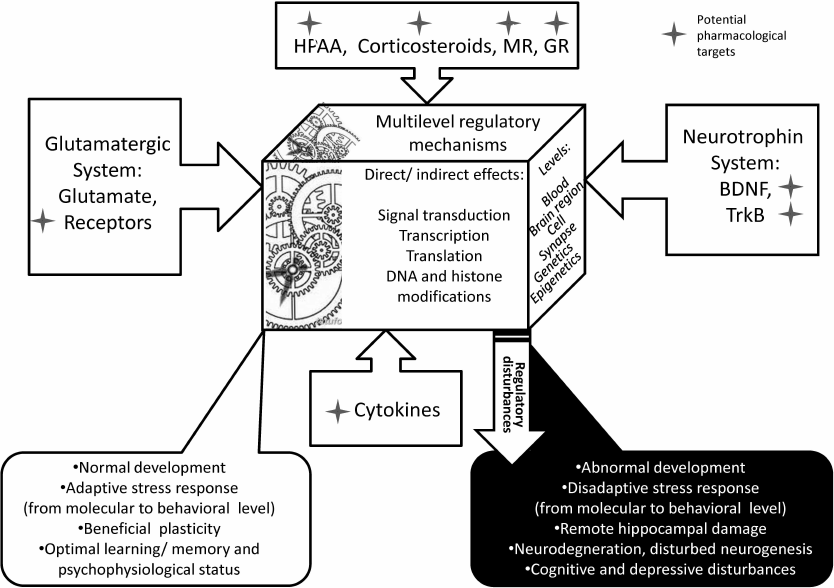
Fig. 5. The “magic mechanical box” of multilevel stress response mechanisms. The ample plastic response of the brain at any level is defined by cross-talks between major systems – HPAA/CS, glutamatergic, neurotrophic, and cytokine – and their finely tuned reciprocal regulation. Normally, the effect of a natural stress factor on any link results in beneficial co-regulatory changes in other processes inducing multilevel adaptive changes. If the stressful challenge is severe and the breakage cannot be easily compensated by adaptive adjustment, the machinery of the box begins malfunctioning inducing pathological alterations, including molecular events resulting in brain cell dysfunction and, ultimately, behavioral disturbances, the hallmarks of neurological and mind diseases. Asterisks designate potential pharmacological targets for preventing or treatment of abnormal functioning of the system.
The simultaneous assessment of neural, endocrine, and immune biomarkers through various modern noninvasive methods is widely discussed [68] and is useful to evaluate HPAA functionality, inflammation and neurotrophic factors. This may help to estimate the risk for further development of cognitive and depressive disturbances. Hair cortisol is regarded as a promising marker of HPAA activity alterations due to stress, somatic and mental health conditions. For instance, levels of hair cortisol were significantly lower in female patients with major depression, while serum cortisol levels were significantly higher in patients, as compared with controls. Decreased hair cortisol found in these patients suggests down regulation of HPAA activity during the preceding month [94]. Thus, assessment of “accumulated” and systemic CSs gives information about HPAA malfunction. Determination of the concentration of chemokines and their receptors is an important indicator of activation of the immune and neuroendocrine systems. The activity of these systems reflects the severity of the disease and provides important information for effective antidepressant treatment [67].
One major principle to access HPAA and stress response mechanisms should be to use a mild challenge. In our previous study, females with major depression manifested an elevated HPAA activity as well as IL-6 and ciliary neurotrophic factor (CNTF) levels at baseline [95]. Besides, specific stress-induced increase in glucose and TNF-α was revealed in this group, which was absent in control subjects (a mild brief cognitive test was used as a challenge). The data confirm the impairments of stress response in major depression and suggest that the reaction of simple metabolic and pro-inflammatory indices to a mild stressogenic challenge may be indicative of a depressive state. Understanding the mechanistic crosstalk between CSs and BDNF holds promise for the identification of potential therapeutic targets [29]. Serum BDNF is also stress-sensitive, characterized by a significant post-stress increase and subsequent decline to recovery. Linz et al. [30] found indications for an antagonistic relationship between BDNF and cortisol; higher BDNF peaks after psychosocial stress were associated with steeper cortisol recovery, while the magnitude of the cortisol stress response was linked to steeper BDNF recovery after stress. Notably, the brain response to stress depends on an individual’s genetic background in interaction with life events [96]. For example, cortisol and emotion/cognition are affected by MR gene haplotypes based on rs5522 and rs2070951. MR haplotype 1 (GA) moderates the effects of (early) life stressors and reproductive cycle. MR haplotype 2 (CA) is a gain of function variant that protects females against depression by association with an optimistic, resilient phenotype [97]. The complete hippocampal genome is screened for CS responsive genes, which are potential targets for drugs promoting restorative capacity still present in the diseased brain [5].
Another approach may be to use the known association between depression and cognitive disturbances. Tene et al. [91] suggested that depression screening among stroke and TIA (transitory ischemic attacks) survivors is a tool to identify patients who are prone to have a worse cognitive and functional outcome. A selected panel of biomarkers may be useful for paraclinical underpinning of post stroke depression diagnosis, clarifying various aspects of its multifactorial pathogenesis, optimizing therapeutic interventions, and assessing treatment effectiveness [84]. Unfortunately, we still lack substantial, rigorous and comprehensive follow-up studies to better identify possible subtypes of depression that may represent a major predictor for later cognitive decline. This would enable specific interventions during critical episodes of these subtypes that should reduce this substantial risk [82].
Thus, the first message is that we are able to easily evaluate HPAA (mal)functioning and individual stress-reactivity of the patient using a challenge and this is a practical approach to estimate the risks of remote hippocampal damage and potential development of post-stroke or posttraumatic cognitive and depressive disturbances.
HPAA hyperactivity can be present in chronic diseases, affecting endocrine, cardiovascular and nervous system (dementia, depression), particularly during comorbid conditions. The spectrum of molecules interacting at different levels of the HPAA is exponentially increasing, ranging from supra-hypothalamic targets to post-receptor mechanisms and it includes agents acting on the suprachiasmatic nucleus, CRH-R1 receptor, adrenal steroidogenesis, GR and peripheral/central 11HSD type 1 enzyme. This area of research is rapidly advancing in order to develop therapeutic strategies to counteract HPAA hyperactivity and to reduce the burden of stress-related disorders [98]. The cumulative evidence makes a strong case implicating CS receptors dysfunction in the pathogenesis of affective disorders. Although definitive controlled trials remain to be conducted, there is evidence indicating that CS-lowering or CS receptors antagonist treatments may be of clinical benefit in selected individuals with depression. Recently, refined molecular technologies and the generation of genetically engineered mice have allowed to specifically target individual genes involved in CS receptor signaling and stress hormone regulation. The identification and detailed characterization of these molecular pathways will ultimately lead to the development of novel neuropharmacological intervention strategies [99].
Pharmacological tools for modulating MRs and GRs, as well as decreasing CS levels are available, mostly for experimental studies, and this is encouraging. No doubt, translation of the experimental results and performing respective clinical studies would bring us closer to the ultimate goal – prevention of dementia and depression induced by focal brain damage. Antiglucocorticoid agents appear to have therapeutic value in particular conditions, potentially in disorders prominently and invariably characterized by early hippocampal lesions and memory impairment, like AD or other types of dementia [97].
The first MR antagonist, spironolactone, was developed more than 60 years ago to treat primary aldosteronism and pathological edema. The knowledge of the scope of MR function was expanded along with clinical evidence of the therapeutic importance of MR antagonists to prevent inappropriate MR activation [100]. About 40 years elapsed between the first and MR-selective second generation of MR antagonists [101]. Twenty years later, a third-generation antagonist has yet to be marketed; progress has been slowed by the lack of appreciation of the large variety of cell types that express the MR and its diverse cell-type-specific actions, and also its unique complex interactions at the molecular level. Fourth generation antagonists and selective agonists based on structural determinants of tissue and ligand-specific MR activation should be considered [101].
Addition of MR antagonists to standard therapy for heart failure, kidney disease, metabolic syndrome and diabetes is increasing steadily in response to clinical trials demonstrating clear benefits. In addition to blocking deleterious activity of MR within the heart, vessels and kidneys, MR antagonists target MR in hemodynamic regulatory centers in the brain, thereby decreasing excessive sympathetic nervous system drive, vasopressin release, abnormal baroreceptor function, and circulating and tissue pro-inflammatory cytokines. However, brain MR are also involved in cognition, memory, and this should be borne in mind considering their potential use [101]. MR-mediated signaling in the brain has been suggested as a protective factor in the development of psychopathology, in particular mood disorders. Activation of the MR therefore may offer a target for alleviating depression and cognitive dysfunction. Accordingly, the MR agonist fludrocortisone was found to enhance the efficacy of antidepressants and to improve memory and executive functions in young depressed patients. CS coordinate via MR the networks underlying individual stress coping, and this action is complemented by the widely distributed lower affinity GR involved in the subsequent management of stress adaptation. In this MR/GR regulation, the MR is an important target for promoting resilience [97].
Low affinity GR, whose expression is ubiquitous, is only activated when CS levels rise (during circadian peak and in response to stress). GR modulates neuronal functions and viability through both genomic and non-genomic actions, and, importantly, its transcriptional regulatory activity is tightly associated with CS secretory pattern [52]. Both genetic and environmental factors can contribute to impaired GR function [68]. Recent evidence shows that CS–GR can exert neuroprotective effects; for any potential therapeutic strategies in neurodegenerative diseases we need to understand the precise modifications both in HPAA and in GR activity and find ways to harness their protective actions [52]. In rodent experiments, mifepristone (RU 486), a GR antagonist, improved susceptibility to chronic stress [102]. Notably, GR signaling in interneurons is differentially important in females, which may have implications for GR-directed gender-specific therapies for stress-related affective disease states [103].
Now at least two antagonists of CS receptors are widely used in clinical practice in Russia. Both demonstrate rather broad specificity towards different steroid receptors and none of them is routinely used for treatment of cognitive or affective disorders. Spironolactone, a synthetic 17-spironolactone CS with potassium-sparing diuretic, antihypertensive, and antiandrogen activities, competitively inhibits MRs. Mifepristone is an antagonist of GR and progesterone receptors. In any case, these CS receptor antagonists are safe to be applied in treatment of different pathologies in clinical conditions. Thus, the second message is that there are many CS receptor ligands accessible, some of them with proven safety are used clinically (though not for treatment of cognitive or affective diseases). The available compounds can be studied in respective experiments and in clinical situations, and new tissue-specific ones should be designed with higher specificity towards definite CS receptors.
Understanding the role of the immune system and inflammation in patients with major depression or cognitive deficits is essential in order to develop efficacious treatments potentially targeting inflammation to reduce a patient’s symptomatology and comorbidities. Since most if not all antidepressants have specific anti-inflammatory effects, restoration of decreased neurogenesis, which may be induced by inflammatory processes, may be related to the therapeutic efficacy of antidepressant treatments [72].
An alternative approach to prevent remote stress-related hippocampal damage is to analyze known properties of drugs used in clinical conditions, find the compounds with desired properties and study their potential benefits. For instance, there is a wide variety of presumably adaptogenic drugs for the claimed stabilization of physiological processes and promotion of homeostasis. Though the adaptogen concept requires additional clinical and preclinical research, and is therefore not accepted into strict current terminology, some drugs allowed for clinical use have confirmed adaptogenic properties. In particular, drugs containing neuropeptides, e.g., Cerebrolysin, are pleiotropic and possess adaptogenic properties confirmed at molecular level [104]. It may be practical to perform a meta-analysis of adaptogenic properties of drugs used in treatment of cerebral lesions (in particular, stroke, trauma), find the most promising compounds and investigate experimentally the effects of these drugs on the HPAA disturbed by different stress factors. Therefore, the third message relates to the assumption that some of the numerous drugs possessing adaptogenic properties and used in the clinic may have a potential for normalizing the HPAA and other mechanisms of stress response; the most effective drugs can be found by experimental screening and verified in preclinical and clinical studies.
There are still many points within the field which should be clarified, the new understanding potentially providing insights into the pathophysiology and potential new treatments of stress-related cognitive and neuropsychiatric disorders. In particular, a better understanding of the CS actions could lead to a more rational treatment strategy of stress-related disorders [105]. The epigraph to this chapter is the words of William James, a leading thinker of the late nineteenth century, “father of American psychology”. Let’s follow the winner’s attitude: it may be difficult to prevent delayed hippocampus-mediated post-lesion disturbances, but it is possible based on known mechanisms and those which will be discovered in the near future. A key message is the statement that a search for potential means of preventing post-lesion remote hippocampal damage as well as the development of delayed cognitive and emotional disturbance should be performed, the perspectives looking quite optimistic.
CONCLUSION
The distant damage to the hippocampus associated with a primary lesion of another brain structure was described many years ago (see [4] for a review), however, so far, no rational concept has been proposed to explain selective vulnerability of the hippocampus to such kind of injury. Recently, we have proposed a new viewpoint which may contribute to the explanation why the hippocampus is so vulnerable to damage of this kind [4]. We hypothesized that this phenomenon may be mediated by relatively high vulnerability to neuroinflammation related to impairments of local CS metabolism and signaling. We suggested at least two major different lines of events following focal brain damage (stroke or traumatic brain injury): the first one, related to the direct primary damage to the brain tissue, and the second one associated with HPAA-mediated stress response events, in particular excessive CS release, activation of the HPAA and hippocampal MRs and GRs. In other words, we proposed that remote hippocampal damage and the following cognitive/affective disturbances after different brain injuries is not just a direct consequence of primary injury, but to a big extent a result of stress load and interaction of CSs with CS receptors of the hippocampus, hippocampal damage being associated with both post-injury dementia and depression. Obviously, individual stress-responsivity becomes a key factor defining the risk of these pathological events. In this article, we tried (i) to construct the rationale which would help to understand more deeply the system of multi-level intertwining and interdependent mechanisms of hippocampus-mediated stress response; (ii) to describe possible causes and nature of the deregulation of this comprehensive system; (iii) to show how malfunctioning of this system affects hippocampal operation and how it influences human cognitive and emotional status; (iv) to suggest which pathogenetically justified approaches can be considered to prevent or repair this malfunctioning. The “magic box” of stress response regulation (Fig. 5) contains many mechanisms integrating multiple systems (HPAA/CSs/CS receptors, neurotrophins and their receptors, inflammatory machinery, neurotransmitters and their receptors and many others) at multiple levels which, in case the system is imbalanced, makes preventive/therapeutic measures complicated since a primary key target may be not obvious. However, fine mutual regulation between systems and operational neuroplasticity mechanisms [106] give a hope that fixing one precise key link would help to repair the whole machine.
Funding. This work was supported by the Russian Foundation for Basic Research, grant no. 18-00-00125 (stress, depression), and the Russian Academy of Sciences Presidium Program “Fundamental bases of physiological adaptation technologies” (remote hippocampal damage).
Conflict of interest. The author declares no conflict of interest.
Compliance with ethical norms. This article does not contain any studies with human participants or animals performed by the author.
REFERENCES
1.McEwen, B. S. (1996) Gonadal and adrenal steroids
regulate neurochemical and structural plasticity of the hippocampus via
cellular mechanisms involving NMDA receptors, Cell. Mol.
Neurobiol., 16, 103-116.
2.De Kloet, E. R., Han, F., and Meijer, O. C. (2008)
From the stalk to down under about brain glucocorticoid receptors,
stress and development, Neurochem. Res., 33, 637-642.
3.Uchoa, E. T., Aguilera, G., Herman, J. P., Fiedler,
J. L., Deak, T., and de Sousa, M. B. (2014) Novel aspects of
glucocorticoid actions, J. Neuroendocrinol., 26,
557-572.
4.Gulyaeva, N. V. (2019) Functional neurochemistry of
the ventral and dorsal hippocampus: stress, depression, dementia and
remote hippocampal damage, Neurochem. Res., 44,
1306-1322.
5.De Kloet, E. R. (2000) Stress in the brain, Eur.
J. Pharmacol., 405, 187-198.
6.De Kloet, E. R., Vreugdenhil, E., Oitzl, M. S., and
Joels, M. (1998) Brain corticosteroid receptor balance in health and
disease, Endocr. Rev., 19, 269-301.
7.Meijer, O. C., Buurstede, J. C., and Schaaf, M. J.
M. (2019) Corticosteroid receptors in the brain: transcriptional
mechanisms for specificity and context-dependent effects, Cell. Mol.
Neurobiol., 39, 539-549.
8.Joels, M. (2018) Corticosteroids and the brain,
J. Endocrinol., 238, R121-R130.
9.Groeneweg, F. L., Karst, H., de Kloet, E. R., and
Joels, M. (2011) Rapid non-genomic effects of corticosteroids and their
role in the central stress response, J. Endocrinol., 209,
153-167.
10.Nishi, M., and Kawata, M. (2006) Brain
corticosteroid receptor dynamics and trafficking: implications from
live cell imaging, Neuroscientist, 12, 119-133.
11.De Kloet, E. R, Oitzl, M. S., and Schobitz, B.
(1994) Cytokines and the brain corticosteroid receptor balance:
relevance to pathophysiology of neuroendocrine-immune communication,
Psychoneuroendocrinology, 19, 121-134.
12.De Kloet, E. R., Meijer, O. C., de Nicola, A. F.,
de Rijk, R. H., and Joels, M. (2018) Importance of the brain
corticosteroid receptor balance in metaplasticity, cognitive
performance and neuro-inflammation, Front. Neuroendocrinol.,
49, 124-145.
13.Murakami, G., Hojo, Y., Kato, A., Komatsuzaki,
Y., Horie, S., Soma, M., Kim, J., and Kawato, S. (2018) Rapid
nongenomic modulation by neurosteroids of dendritic spines in the
hippocampus: androgen, oestrogen and corticosteroid, J.
Neuroendocrinol., 30, doi: 10.1111/jne.12561.
14.Giordano, R., Pellegrino, M., Picu, A., Bonelli,
L., Balbo, M., Berardelli, R., Lanfranco, F., Ghigo, E., and Arvat, E.
(2006) Neuroregulation of the hypothalamus-pituitary-adrenal (HPA) axis
in humans: effects of GABA-, mineralocorticoid-, and
GH-secretagogue-receptor modulation, Sci. World J., 6,
1-11.
15.Joels, M., and de Kloet, E. R. (2017) 30 years of
the mineralocorticoid receptor: the brain mineralocorticoid receptor: a
saga in three episodes, J. Endocrinol., 234, T49-T66.
16.Le Menuet, D., and Lombes, M. (2014) The neuronal
mineralocorticoid receptor: from cell survival to neurogenesis,
Steroids, 91, 11-19.
17.Odermatt, A., and Kratschmar, D. V. (2012)
Tissue-specific modulation of mineralocorticoid receptor function by
11β-hydroxysteroid dehydrogenases: an overview, Mol. Cell.
Endocrinol., 350, 168-186.
18.Kellendonk, C., Gass, P., Kretz, O., Schutz, G.,
and Tronche, F. (2002) Corticosteroid receptors in the brain: gene
targeting studies, Brain Res. Bull., 57, 73-83.
19.Saaltink, D. J., and Vreugdenhil, E. (2014)
Stress, glucocorticoid receptors, and adult neurogenesis: a balance
between excitation and inhibition? Cell. Mol. Life Sci.,
71, 2499-2515.
20.Vitellius, G., Trabado, S., Bouligand, J.,
Delemer, B., and Lombes, M. (2018) Pathophysiology of glucocorticoid
signaling, Ann. Endocrinol. (Paris), 79, 98-106.
21.Van Weert, L. T. C. M., Buurstede, J. C., Sips,
H. C. M., Mol, I. M., Puri, T., Damsteegt, R., Roozendaal, B.,
Sarabdjitsingh, R. A., and Meijer, O. C. (2019) Mechanistic insights in
NeuroD potentiation of mineralocorticoid receptor signaling, Int. J.
Mol. Sci., 20, doi: 10.3390/ijms20071575.
22.Joels, M., and Karst, H. (2012) Corticosteroid
effects on calcium signaling in limbic neurons, Cell. Calcium,
51, 277-283.
23.Lapp, H. E., Bartlett, A. A., and Hunter, R. G.
(2019) Stress and glucocorticoid receptor regulation of mitochondrial
gene expression, J. Mol. Endocrinol., 62, R121-R128.
24.Reul, J. M. (2014) Making memories of stressful
events: a journey along epigenetic, gene transcription, and signaling
pathways, Front. Psychiatry, 5, 5.
25.Mifsud, K. R., Gutierrez-Mecinas, M., Trollope,
A. F., Collins, A., Saunderson, E. A., and Reul, J. M. (2011)
Epigenetic mechanisms in stress and adaptation, Brain Behav.
Immun., 25, 1305-1315.
26.Bennett, M. R., and Lagopoulos, J. (2014) Stress
and trauma: BDNF control of dendritic-spine formation and regression,
Prog. Neurobiol., 112, 80-99.
27.Gulyaeva, N. V. (2017) Interplay between brain
BDNF and glutamatergic systems: a brief state of the evidence and
association with the pathogenesis of depression, Biochemistry
(Moscow), 82, 301-307.
28.Leal, G., Bramham, C. R., and Duarte, C. B.
(2017) BDNF and hippocampal synaptic plasticity, Vitam. Horm.,
104, 153-195.
29.Suri, D., and Vaidya, V. A. (2013) Glucocorticoid
regulation of brain-derived neurotrophic factor: relevance to
hippocampal structural and functional plasticity, Neuroscience,
239, 196-213.
30.Linz, R., Puhlmann, L. M. C., Apostolakou, F.,
Mantzou, E., Papassotiriou, I., Chrousos, G. P., Engert, V., and
Singer, T. (2019) Acute psychosocial stress increases serum BDNF
levels: an antagonistic relation to cortisol but no group differences
after mental training, Neuropsychopharmacology, 44,
1797-1804, doi: 10.1038/s41386-019-0391-y.
31.Daskalakis, N. P., De Kloet, E. R., Yehuda, R.,
Malaspina, D., and Kranz, T. M. (2015) Early life stress effects on
glucocorticoid–BDNF interplay in the hippocampus, Front. Mol.
Neurosci., 8, 68.
32.Finsterwald, C., and Alberini, C. M. (2014)
Stress and glucocorticoid receptor-dependent mechanisms in long-term
memory: from adaptive responses to psychopathologies, Neurobiol.
Learn. Mem., 112, 17-29.
33.Sapolsky, R. M. (1990) Glucocorticoids,
hippocampal damage and the glutamatergic synapse, Prog. Brain
Res., 86, 13-23.
34.Popoli, M., Yan, Z., McEwen, B. S., and Sanacora,
G. (2011) The stressed synapse: the impact of stress and
glucocorticoids on glutamate transmission, Nat. Rev. Neurosci.,
13, 22-37.
35.Chaouloff, F., and Groc, L. (2011) Temporal
modulation of hippocampal excitatory transmission by corticosteroids
and stress, Front. Neuroendocrinol., 32, 25-42.
36.Goujon, E., LayE, S., Parnet, P., and Dantzer, R.
(1997) Regulation of cytokine gene expression in the central nervous
system by glucocorticoids: mechanisms and functional consequences,
Psychoneuroendocrinology, 22 (Suppl. 1), S75-S80.
37.Duque Ede, A., and Munhoz, C. D. (2016) The
pro-inflammatory effects of glucocorticoids in the brain, Front.
Endocrinol. (Lausanne), 7, 78.
38.Pariante, C. M. (2017) Why are depressed patients
inflamed? A reflection on 20 years of research on depression,
glucocorticoid resistance and inflammation, Eur.
Neuropsychopharmacol., 27, 554-559.
39.Walker, F. R., Nilsson, M., and Jones, K. (2013)
Acute and chronic stress-induced disturbances of microglial plasticity,
phenotype and function, Curr. Drug Targets, 14,
1262-1276.
40.Gadek-Michalska, A., Tadeusz, J., Rachwalska, P.,
and Bugajski, J. (2013) Cytokines, prostaglandins and nitric oxide in
the regulation of stress-response systems, Pharmacol. Rep.,
65, 1655-1662.
41.Johnson, J. D., Barnard, D. F., Kulp, A. C., and
Mehta, D. M. (2019) Neuroendocrine regulation of brain cytokines after
psychological stress, J. Endocr. Soc., 3,
1302-1320.
42.Deak, T., Quinn, M., Cidlowski, J. A., Victoria,
N. C., Murphy, A. Z., and Sheridan, J. F. (2015). Neuroimmune
mechanisms of stress: sex differences, developmental plasticity, and
implications for pharmacotherapy of stress-related disease,
Stress, 18, 367-380.
43.Bekhbat, M., Rowson, S. A., and Neigh, G. N.
(2017) Checks and balances: the glucocorticoid receptor and NFκB
in good times and bad, Front. Neuroendocrinol., 46,
15-31.
44.De Kloet, E. R., and Joels, M. (2017) Brain
mineralocorticoid receptor function in control of salt balance and
stress-adaptation, Physiol. Behav., 178, 13-20.
45.Brocca, M. E., Pietranera, L., de Kloet, E. R.,
and De Nicola, A. F. (2019) Mineralocorticoid receptors,
neuroinflammation and hypertensive encephalopathy, Cell. Mol.
Neurobiol., 39, 483-492.
46.Frank, M. G., Weber, M. D., Watkins, L. R., and
Maier, S. F. (2015) Stress sounds the alarmin: the role of the
danger-associated molecular pattern HMGB1 in stress-induced
neuroinflammatory priming, Brain Behav. Immun., 48,
1-7.
47.Pearson-Leary, J., Osborne, D. M., and McNay, E.
C. (2016) Role of glia in stress-induced enhancement and impairment of
memory, Front. Integr. Neurosci., 9, 63.
48.Van Olst, L., Bielefeld, P., Fitzsimons, C. P.,
de Vries, H. E., and Schouten, M. (2018) Glucocorticoid-mediated
modulation of morphological changes associated with aging in microglia,
Aging Cell, 17, e12790.
49.McEwen, B. S. (1997) Possible mechanisms for
atrophy of the human hippocampus, Mol. Psychiatry, 2,
255-262.
50.Vyas, S., Rodrigues, A. J., Silva, J. M.,
Tronche, F., Almeida, O. F., Sousa, N., and Sotiropoulos, I. (2016)
Chronic stress and glucocorticoids: from neuronal plasticity to
neurodegeneration, Neural Plast., 2016, 6391686.
51.Libro, R., Bramanti, P., and Mazzon, E. (2017)
Endogenous glucocorticoids: role in the etiopathogenesis of
Alzheimer’s disease, Neuro Endocrinol. Lett., 38,
1-12.
52.Vyas, S., and Maatouk, L. (2013) Contribution of
glucocorticoids and glucocorticoid receptors to the regulation of
neurodegenerative processes, CNS Neurol. Disord. Drug Targets,
12, 1175-1193.
53.Ouanes, S., and Popp, J. (2019) High cortisol and
the risk of dementia and Alzheimer’s disease: a review of the
literature, Front. Aging Neurosci., 11, 43.
54.Paul, S., Jeon, W. K., Bizon, J. L., and Han, J.
S. (2015) Interaction of basal forebrain cholinergic neurons with the
glucocorticoid system in stress regulation and cognitive impairment,
Front. Aging Neurosci., 7, 43.
55.Kootar, S., Frandemiche, M. L., Dhib, G., Mouska,
X., Lorivel, T., Poupon-Silvestre, G., Hunt, H., Tronche, F., Bethus,
I., Barik, J., and Marie, H. (2018) Identification of an acute
functional cross-talk between amyloid-β and glucocorticoid
receptors at hippocampal excitatory synapses, Neurobiol. Dis.,
118, 117-128.
56.Bisht, K., Sharma, K., and Tremblay, M. E. (2018)
Chronic stress as a risk factor for Alzheimer’s disease: roles of
microglia-mediated synaptic remodeling, inflammation, and oxidative
stress, Neurobiol. Stress, 9, 9-21.
57.Pomara, N., Greenberg, W. M., Branford, M. D.,
and Doraiswamy, P. M. (2003) Therapeutic implications of HPA axis
abnormalities in Alzheimer’s disease: review and update,
Psychopharmacol. Bull., 37, 120-134.
58.Numakawa, T., Odaka, H., and Adachi, N. (2017)
Actions of brain-derived neurotrophic factor and glucocorticoid stress
in neurogenesis, Int. J. Mol. Sci., 18, E2312,
doi: 10.3390/ijms18112312.
59.Fitzsimons, C. P., Herbert, J., Schouten, M.,
Meijer, O. C., Lucassen, P. J., and Lightman, S. (2016) Circadian and
ultradian glucocorticoid rhythmicity: implications for the effects of
glucocorticoids on neural stem cells and adult hippocampal
neurogenesis, Front. Neuroendocrinol., 41, 44-58.
60.Lucassen, P. J., Oomen, C. A., Naninck, E. F.,
Fitzsimons, C. P., van Dam, A. M., Czeh, B., and Korosi, A. (2015)
Regulation of adult neurogenesis and plasticity by (early) stress,
glucocorticoids, and inflammation, Cold Spring Harb. Perspect.
Biol., 7, a021303.
61.Schoenfeld, T. J., and Gould, E. (2012). Stress,
stress hormones, and adult neurogenesis, Exp. Neurol.,
233, 12-21.
62.Lupien, S. J., and Lepage, M. (2001) Stress,
memory, and the hippocampus: can’t live with it, can’t live
without it, Behav. Brain Res., 127, 137-158.
63.Herman, J. P., Ostrander, M. M., Mueller, N. K.,
and Figueiredo, H. (2005) Limbic system mechanisms of stress
regulation: hypothalamo-pituitary-adrenocortical axis, Prog.
Neuropsychopharmacol. Biol. Psychiatry, 29, 1201-1213.
64.Joels, M., Pasricha, N., and Karst, H. (2013) The
interplay between rapid and slow corticosteroid actions in brain,
Eur. J. Pharmacol., 719, 44-52.
65.Zunszain, P. A., Anacker, C., Cattaneo, A.,
Carvalho, L. A., and Pariante, C. M. (2011) Glucocorticoids, cytokines
and brain abnormalities in depression, Prog. Neuropsychopharmacol.
Biol. Psychiatry, 35, 722-729.
66.Leonard, B. (2000) Stress, depression and the
activation of the immune system, World J. Biol. Psychiatry,
1, 17-25.
67.Oglodek, E., Szota, A., Just, M., Mos, D., and
Araszkiewicz, A. (2014) The role of the neuroendocrine and immune
systems in the pathogenesis of depression, Pharmacol. Rep.,
66, 776-781.
68.Silverman, M. N., and Sternberg, E. M. (2012)
Glucocorticoid regulation of inflammation and its functional
correlates: from HPA axis to glucocorticoid receptor dysfunction,
Ann. N. Y. Acad. Sci., 1261, 55-63.
69.Leonard, B. E. (2018) Inflammation and
depression: a causal or coincidental link to the pathophysiology?
Acta Neuropsychiatr., 30, 1-16.
70.Makhija, K., and Karunakaran, S. (2013) The role
of inflammatory cytokines on the etiopathogenesis of depression,
Aust. N. Z. J. Psychiatry, 47, 828-839.
71.Bauer, M. E., and Teixeira, A. L. (2019)
Inflammation in psychiatric disorders: what comes first? Ann. N. Y.
Acad. Sci., 437, 57-67.
72.Maes, M., Yirmyia, R., Noraberg, J., Brene, S.,
Hibbeln, J., Perini, G., Kubera, M., Bob, P., Lerer, B., and Maj, M.
(2009) The inflammatory and neurodegenerative (I&ND) hypothesis of
depression: leads for future research and new drug developments in
depression, Metab. Brain Dis., 24, 27-53.
73.Kim, Y. K., Na, K. S., Myint, A. M., and Leonard,
B. E. (2016) The role of pro-inflammatory cytokines in
neuroinflammation, neurogenesis and the neuroendocrine system in major
depression, Prog. Neuropsychopharmacol. Biol. Psychiatry,
64, 277-284.
74.Barnard, D. F., Gabella, K. M., Kulp, A. C.,
Parker, A. D., Dugan, P. B., and Johnson, J. D. (2019) Sex differences
in the regulation of brain IL-1β in response to chronic stress,
Psychoneuroendocrinology, 103, 203-211.
75.Cattaneo, A., and Riva, M. A. (2016)
Stress-induced mechanisms in mental illness: a role for glucocorticoid
signaling, J. Steroid Biochem. Mol. Biol., 160,
169-174.
76.Zimmermann, C. A., Arloth, J., Santarelli, S.,
Loschner, A., Webe, P., Schmidt, M. V., Spengler, D., and Binder, E. B.
(2019) Stress dynamically regulates co-expression networks of
glucocorticoid receptor-dependent MDD and SCZ risk genes, Transl.
Psychiatry, 9, 41, doi: 10.1038/s41398-019-0373-1.
77.Gray, J. D., Kogan, J. F., Marrocco, J., and
McEwen, B. S. (2017) Genomic and epigenomic mechanisms of
glucocorticoids in the brain, Nat. Rev. Endocrinol., 13,
661-673.
78.Sousa, N., and Almeida, O. F. (2002)
Corticosteroids: sculptors of the hippocampal formation, Rev.
Neurosci., 13, 59-84.
79.Chen, J., Wang, Z. Z., Zhang, S., Chu, S. F.,
Mou, Z., and Chen, N. H. (2019) The effects of glucocorticoids on
depressive and anxiety-like behaviors, mineralocorticoid
receptor-dependent cell proliferation regulates anxiety-like behaviors,
Behav. Brain Res., 362, 288-298.
80.Li, Y., Qin, J., Yan, J., Zhang, N., Xu, Y., Zhu,
Y., Sheng, L., Zhu, X., and Ju, S. (2018) Differences of physical vs.
psychological stress: evidences from glucocorticoid receptor
expression, hippocampal subfields injury, and behavioral abnormalities,
Brain Imaging Behav., doi: 10.1007/s11682-018-9956-3 [Epub ahead
of print].
81.Leonard, B. E. (2007) Inflammation, depression
and dementia: are they connected? Neurochem. Res., 32,
1749-1756.
82.Herbert, J., and Lucassen, P. J. (2016)
Depression as a risk factor for Alzheimer’s disease: genes,
steroids, cytokines and neurogenesis – what do we need to
know? Front. Neuroendocrinol., 41, 153-171.
83.Kino, T. (2015) Stress, glucocorticoid hormones,
and hippocampal neural progenitor cells: implications to mood
disorders, Front. Physiol., 6, 230.
84.Levada, O. A., and Troyan, A. S. (2018)
Poststroke depression biomarkers: a narrative review, Front.
Neurol., 16, 577.
85.Ben Assayag, E., Korczyn, A. D., Giladi, N.,
Goldbourt, U., Berliner, A. S, Shenhar-Tsarfaty, S., Kliper, E.,
Hallevi, H., Shopin, L., Hendler, T., Baashat, D. B., Aizenstein, O.,
Soreq, H., Katz, N., Solomon, Z., Mike, A., Usher, S., Hausdorff, J.
M., Auriel, E., Shapira, I., and Bornstein, N. M. (2012) Predictors for
poststroke outcomes: the Tel Aviv Brain Acute Stroke Cohort (TABASCO)
study protocol, Int. J. Stroke, 7, 341-347.
86.Molad, J., Hallevi, H., Korczyn, A. D., Kliper,
E., Auriel, E., Bornstein, N. M., and Ben Assayag, E. (2019) Vascular
and neurodegenerative markers for the prediction of post-stroke
cognitive impairment: results from the TABASCO study, J.
Alzheimer’s Dis., 70, 889-898, doi:
10.3233/JAD-190339.
87.Molad, J., Ben-Assayag, E., Korczyn, A. D.,
Kliper, E., Bornstein, N. M., Hallevi, H., and Auriel, E. (2018)
Clinical and radiological determinants of transient symptoms associated
with infarction (TSI), J. Neurol. Sci., 390, 195-199.
88.Kliper, E., Ben Assayag, E., Tarrasch, R., Artzi,
M., Korczyn, A. D., Shenhar-Tsarfaty, S., Aizenstein, O., Hallevi, H.,
Mike, A., Shopin, L., Bornstein, N. M., and Ben Bashat, D. (2014)
Cognitive state following stroke: the predominant role of preexisting
white matter lesions, PLoS One, 9, e105461.
89.Ben Assayag, E., Tene, O., Korczyn, A. D.,
Shopin, L., Auriel, E., Molad, J., Hallevi, H., Kirschbaum, C.,
Bornstein, N. M., Shenhar-Tsarfaty, S., Kliper, E., and Stalder, T.
(2017) High hair cortisol concentrations predict worse cognitive
outcome after stroke: results from the TABASCO prospective cohort
study, Psychoneuroendocrinology, 82, 133-139.
90.Tene, O., Hallevi, H., Korczyn, A. D., Shopin,
L., Molad, J., Kirschbaum, C., Bornstein, N. M., Shenhar-Tsarfaty, S.,
Kliper, E., Auriel, E., Usher, S., Stalder, T., and Ben Assayag, E.
(2018) The price of stress: high bedtime salivary cortisol levels are
associated with brain atrophy and cognitive decline in stroke
survivors. Results from the TABASCO prospective cohort study, J.
Alzheimer’s Dis., 65, 1365-1375.
91.Tene, O., Shenhar-Tsarfaty, S., Korczyn, A. D.,
Kliper, E., Hallevi, H., Shopin, L., Auriel, E., Mike, A., Bornstein,
N. M., and Assayag, E. B. (2016) Depressive symptoms following stroke
and transient ischemic attack: is it time for a more intensive
treatment approach? Results from the TABASCO cohort study, J. Clin.
Psychiatry, 77, 673-680.
92.Kliper, E., Ben Assayag, E., Korczyn, A. D.,
Auriel, E., Shopin, L., Hallevi, H., Shenhar-Tsarfaty, S., Mike, A.,
Artzi, M., Klovatch, I., Bornstein, N. M., and Ben Bashat, D. (2016)
Cognitive state following mild stroke: a matter of hippocampal mean
diffusivity, Hippocampus, 26, 161-169.
93.Herman, J. P., McKlveen, J. M., Ghosal, S., Kopp,
B., Wulsin, A., Makinson, R., Scheimann, J., and Myers, B. (2016)
Regulation of the hypothalamic-pituitary-adrenocortical stress
response, Compr. Physiol., 6, 603-621.
94.Pochigaeva, K., Druzhkova, T., Yakovlev, A.,
Onufriev, M., Grishkina, M., Chepelev, A., Guekht, A., and Gulyaeva, N.
(2017) Hair cortisol as a marker of hypothalamic-pituitary-adrenal axis
activity in female patients with major depressive disorder, Metab.
Brain Dis., 32, 577-583.
95.Druzhkova, T., Pochigaeva, K., Yakovlev, A.,
Kazimirova, E., Grishkina, M., Chepelev, A., Guekht, A., and Gulyaeva,
N. (2019) Acute stress {{anchor|GoBack}} response to a cognitive task
in patients with major depressive disorder: potential metabolic and
pro-inflammatory biomarkers, Metab. Brain Dis., 34,
621-629.
96.Joels, M., Karst, H., and Sarabdjitsingh, R. A.
(2018) The stressed brain of humans and rodents, Acta Physiol.
(Oxf.), 223, e13066.
97.De Kloet, E. R., Otte, C., Kumsta, R., Kok, L.,
Hillegers, M. H., Hasselmann, H., Kliegel, D., and Joels, M. (2016)
Stress and depression: a crucial role of the mineralocorticoid
receptor, J. Neuroendocrinol., 28, doi:
10.1111/jne.12379.
98.Martocchia, A., Curto, M., Toussan, L.,
Stefanelli, M., and Falaschi, P. (2011) Pharmacological strategies
against glucocorticoid-mediated brain damage during chronic disorders,
Recent Pat. CNS Drug Discov., 6, 196-204.
99.Muller, M., Holsboer, F., and Keck, M. E. (2002)
Genetic modification of corticosteroid receptor signaling: novel
insights into pathophysiology and treatment strategies of human
affective disorders, Neuropeptides, 36, 117-131.
100.Sutanto, W., and de Kloet, E. R. (1991)
Mineralocorticoid receptor ligands: biochemical, pharmacological, and
clinical aspects, Med. Res. Rev., 11, 617-639.
101.Gomez-Sanchez, E. P. (2016) Third-generation
mineralocorticoid receptor antagonists: why do we need a fourth? J.
Cardiovasc. Pharmacol., 67, 26-38.
102.Wang, W., Liu, L., Yang, X., Gao, H., Tang, Q.
K., Yin, L. Y., Yin, X. Y., Hao, J. R., Geng, D. Q., and Gao, C. (2019)
Ketamine improved depressive-like behaviors via hippocampal
glucocorticoid receptor in chronic stress induced-susceptible mice,
Behav. Brain Res., 364, 75-84.
103.Scheimann, J. R., Mahbod, P., Morano, R.,
Frantz, L., Packard, B., Campbell, K., and Herman, J. P. (2018)
Deletion of glucocorticoid receptors in forebrain GABAergic neurons
alters acute stress responding and passive avoidance behavior in female
mice, Front. Behav. Neurosci., 12, 325.
104.Stepanichev, M., Onufriev, M., Aniol, V.,
Freiman, S., Brandstaetter, H., Winter, S., Lazareva, N., Guekht, A.,
and Gulyaeva, N. (2017) Effects of Cerebrolysin on nerve growth factor
system in the aging rat brain, Restor. Neurol. Neurosci.,
35, 571-581.
105.Joels, M. (2008) Functional actions of
corticosteroids in the hippocampus, Eur. J. Pharmacol.,
583, 312-321.
106.Gulyaeva, N. V. (2017) Molecular mechanisms of
neuroplasticity: an expanding universe, Biochemistry (Moscow),
82, 237-242.