REVIEW: Latent Inflammation and Defect in Adipocyte Renewal as a Mechanism of Obesity-Associated Insulin Resistance
A. V. Vorotnikov1,a#*, I. S. Stafeev1,b#*, M. Yu. Menshikov1, M. V. Shestakova2, and Ye. V. Parfyonova1
1Institute of Experimental Cardiology, National Medical Research Center of Cardiology, Ministry of Healthcare of the Russian Federation, 121552 Moscow, Russia2Diabetes Institute, National Medical Research Center of Endocrinology, 117036 Moscow, Russia
# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received June 25, 2019; Revised August 9, 2019; Accepted August 10, 2019
Obesity is a major risk factor for type 2 diabetes and metabolic syndrome and an essential medical and social problem. In the first part of the review, we briefly highlight the biochemical basis of metabolic disbalance in obesity and evolution of our views on the mechanisms of insulin resistance development in insulin-sensitive tissues. Because obesity relates to the disturbance in the normal physiology of fat tissue, the second part of the review focuses on latent inflammation that develops in obesity and is supported by immune cells. Finally, the problem of adipocyte hypertrophy, reduced regenerative potential of fat progenitor cells, and impaired renewal of fat depots is discussed in the context of type 2 diabetes pathogenesis.
KEY WORDS: obesity, inflammation, adipose tissue, adipocyte renewal, insulin resistance, type 2 diabetesDOI: 10.1134/S0006297919110099
Abbreviations: AMPK, AMP-dependent protein kinase; BMI, body mass index; DAG, diacylglycerol; FFA, free fatty acid; GLUT4, glucose transporter type 4; IL, interleukin; ILC, innate lymphoid cell; IR, insulin resistance; IRS, insulin receptor substrate; MSC, mesenchymal stromal cell; PI3 kinase, phosphoinositide 3-kinase; T2DM, type 2 diabetes mellitus; TAG, triacylglycerol; TLR2/TLR4, Toll-like receptor type 2 or 4, respectively; TNFα, tumor necrosis factor alpha; TNFR, TNFα receptor.
Obesity and type 2 diabetes mellitus (T2DM) are the key problems in
modern endocrinology. According to the Russian Federal Diabetes
register, ~3% of the population have T2DM [1], yet
>30% suffer from obesity [2]. Obesity is the
major risk factor for insulin resistance (IR) early in T2DM and
critically contributes to the metabolic syndrome development. Obesity
and T2DM increase the risk of micro- and macrovascular disorders,
cancer and cardiovascular complications, chronic kidney disease, limb
amputation, and blindness, all leading to physical disabilities,
impaired quality of life, and decreased work efficiency. Understanding
how IR and T2DM develop and relate to obesity is a central goal in
molecular endocrinology, as well as in cardiology and oncology.
Tackling this goal requires true integration of fundamental and clinical science. Now it has become clear that obesity-related metabolic disorders (briefly described in the first section of this review) have biochemical basis. Over the last decades, the use of biochemical, physiological, and cell biology techniques has helped to identify the general mechanisms of IR development; these results are summarized in the second section. During the last years, there has been a strong trend towards the studies at the system level, including those in knockout and high-fat diet animal models, as well as analysis of tissue biopsies from healthy donors, obese individuals, and T2DM patients. This approach offers unique possibilities that go beyond the laboratory context but requires the results of laboratory experiments to be verified in clinical studies, which is discussed in the last sections. The major focus is placed on the adipose tissue because obesity is primarily linked to its dysfunction. Apparently, immune cells, inflammatory and regenerative processes critically contribute to the T2DM pathogenesis by gradually changing this tissue functions. We hypothesize that these changes include impaired renewal of fat depots that greatly contributes to the IR development in the adipose tissue and to the overall pathogenesis of obesity-associated T2DM.
OBESITY, IR, AND T2DM
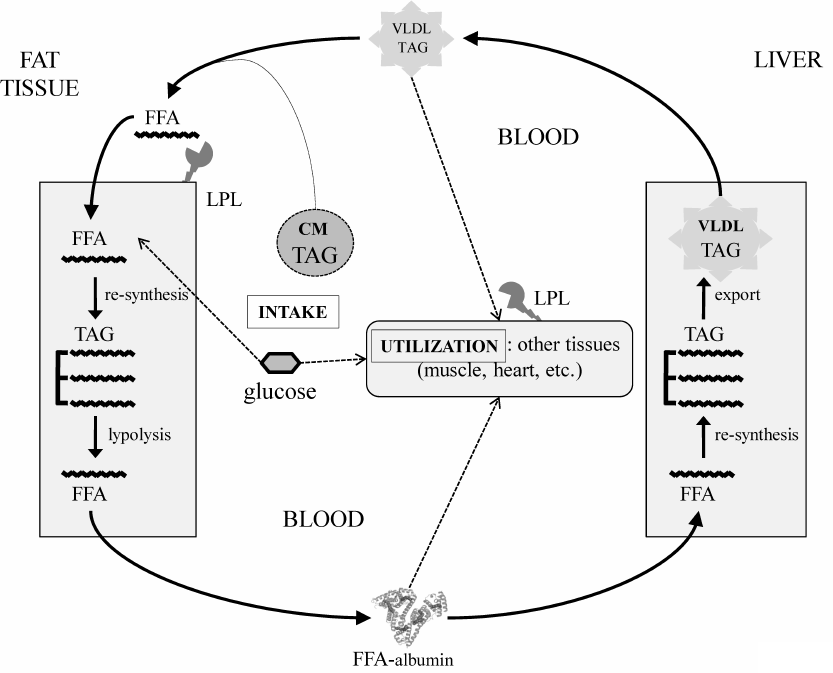
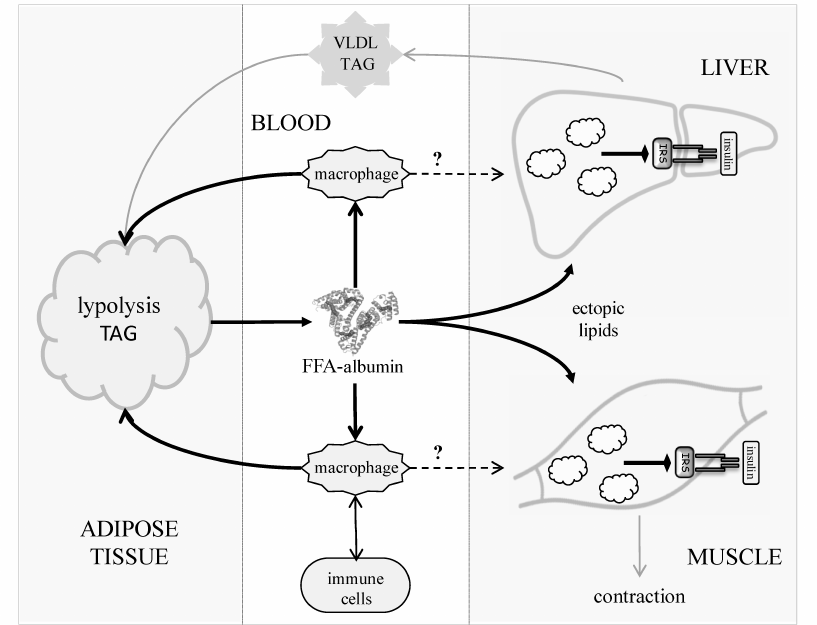
Primary obesity results from the excessive dietary input [3]. As an anabolic hormone, insulin facilitates energy deposition in two major forms – limited short-term glycogen in muscles and liver and the long-term triacylglycerols (TAGs) in the fat tissue adipocytes. Normally, energy reserves are utilized between the meals mainly during physical activity (Fig. 1). Adrenaline and glucagon stimulate lipolysis in the fat tissue, causing release of free fatty acids (FFAs) to the blood. FFAs circulate in a complex with albumin and provide energy for the cells of peripheral tissues. Together with glycerol produced by lipolysis in the fat tissue, FFAs are carried to the liver, where they are re-synthesized into TAGs and packed into very-low-density lipoproteins (VLDLs) to be transported to the peripheral tissues via the bloodstream. Lipoprotein lipases present on the surface of vascular endothelial cells hydrolyze TAGs in the lipoproteins. Thus, liberated from VLDLs, or those circulated in blood in complexes with albumin, FFAs are carried to the underlying tissues through the vascular endothelium (transcytosis) by specific FFA-binding proteins. Adipocytes also express lipoprotein lipase and uptake FFAs, thereby closing the lipid transport cycle (TAG-FFA cycle) between the liver and adipose tissue. This process is well known [4] and described in detail in classic biochemistry textbooks [5]. Importantly, it operates continuously regardless of the nutrient excess or deficit, which allows to balance the supply, usage, and storage of energy at the system level in accordance to the delivery of energy sources or their utilization during physical activity. Obesity develops as a result of chronic overload of the TAG-FFA flux with nutritional glucose and FFAs that greatly exceeds their utilization in tissues (Fig. 1, dashed arrows). Fat depots become filled up; muscles and liver accumulate glycogen. When maintained, this imbalance drives the hypertrophy of adipocytes and adipogenesis in the fat tissue, i.e., formation of new fat depots from the resident progenitors, adipose-derived mesenchymal stromal cells (MSCs) [6].
Fig. 1. TAG-FFA cycle between the adipose tissue and the liver. The TAG-FFA cycle governs the balance between the energy influx, consumption, and storage as lipids (TAGs) [5]. Lipids constantly circulate between the fat and the liver as FFAs either in a complex with albumin or components of VLDLs. Chylomicrons (CMs) ensure energy influx from the dietary sources; the brain and the muscle tissues consume most of the energy. Lipids are delivered from VLDLs and CMs into cells as FFAs, which are released from TAGs by lipoprotein lipase (LPL). The residual VLDLs (low density lipoproteins, LDL) and CM remnants are uptaken by the liver.
Obesity aggravates metabolic syndrome and provokes IR, thus representing the major risk factor for T2DM. IR is manifested at both system and cellular levels. The first sign of IR is the delayed glucose utilization from the blood in response to a bolus of insulin, which is regarded as glucose intolerance and prediabetes. As IR intensifies, the blood levels of glucose and insulin become elevated even after the overnight fast. Eventually, hyperglycemia causes dysfunction of pancreatic β-cells because of glucotoxicity and impairs insulin production. Together with low insulin sensitivity of the insulin-targeted cells, impaired insulin production results in sustained hyperglycemia shortly followed by diagnosis of T2DM.
In the postabsorptive period, up to 50% of blood glucose is utilized by the brain, while other visceral organs use up to 25% glucose [7]. The rest 25% is utilized via the insulin-stimulated uptake (80-90% in muscles and ~5% in fat tissues). These figures illustrate why muscle tissues contribute most to the whole-body IR. However, the contribution of adipose tissue may significantly increase in obesity, reaching 30-50% of the prandial glucose [8]. Importantly, appearance of IR in the adipose compartment is associated with chronic (latent) inflammation, which maintains metabolic shifts and pathological changes both locally and at the system level. How latent inflammation induces and maintains IR is discussed below. Here, it should be noted that lipolysis is upregulated in the fat tissue in IR, shifting the balance in the TAG-FFA cycle towards the increased blood levels of FFAs. This shift is further promoted by low utilization of FFAs in the case of physical inactivity. Thus, overnutrition and sedentary lifestyle significantly increase the risk of imbalance in the TAG-FFA cycle, latent inflammation and IR development in the fat tissue of obese individuals. It is therefore likely that obesity, latent inflammation, and IR form a pathologic triumvirate in prediabetes and create conditions for development of T2DM. Although the cause-and-effect relationships between these events are not entirely understood and have yet to be fully established, there is no doubt they are versatile and likely realized at the molecular level of intracellular signaling in adipocytes, at the paracrine level in the adipose tissue, and at the whole-body level. The latter involves interactions of different organs (as illustrated by the example of the TAG-FFA cycle), which are controlled by the endocrine system and feedback loops formed by hormones produced by the peripheral tissues, such as adipokines and myokines.
At the cellular level, insulin acts in a tissue-specific manner, but always via specific receptors and several signaling pathways, including the PI3 kinase pathway and protein kinase B/Akt as the major of them [9-11]. All variations in cell responses to insulin arise at the postreceptor level and depend on the cell type. They mainly have a metabolic signature and are less coupled to transcriptional regulation. Generally, insulin receptor signaling targets glucose uptake, synthesis of glycogen, fatty acids, TAGs, and proteins. In parallel, insulin represses the reverse processes, such as glycogen breakdown, gluconeogenesis, and FFA delivery to the mitochondria for β-oxidation. A hallmark of insulin receptor signaling is involvement of a specific scaffold protein, the insulin receptor substrate (IRS). IRS binds and triggers upstream signaling mediators, which are described elsewhere in more details [11, 12]. IRS is also a target for the negative feedback and crosstalk regulation that switches off the downstream signaling [12-15]. These mechanisms vary in different insulin-dependent tissues and are discussed in more detail in the next section.
MOLECULAR MECHANISMS OF IR DEVELOPMENT
T2DM has been long known as a disease. Yet, only in the last few decades it has become increasingly common in the developed countries, likely because of abundance of unhealthy food and sedentary lifestyle. Similarly, relationships between T2DM, glucose tolerance, obesity, and increased blood levels of FFAs have been long noticed. Since then, many studies, from non-trivial recruit selection [16] to clinically well-characterized patients [17], have revealed that the blood levels of FFAs are normally less than 0.6 mM, but increase to 0.6-0.7 mM and more in the T2DM patients. Accordingly, many studies have employed animals or volunteers for intravenous infusion of 0.3-2 mM lipid mixtures (for example, Intralipid), similar in composition to those in the blood plasma. These studies have led to the conclusion that obesity and dyslipidemia (dysfunction of lipid metabolism) associated with the increased blood levels of FFAs, are critical to IR development and subsequent T2DM [18].
More than 50 years ago, examination of the relationship between FFAs and IR had led Philip Randle to the idea of glucose-fatty acid cycle and hypothesis that FFAs and glucose compete as the energy sources in animal cells [19]. Actually, Randle considered the left and the middle parts of the cycle shown in Fig. 1, with no lipid turnover in the liver. Based on the notion that increased (excessive) levels of FFAs in the blood suppress glucose utilization by muscles, Randle hypothesized that (i) cells reciprocally use lipid and carbohydrate metabolism for respiration; (ii) increased blood FFAs suppress catabolism of glucose in muscles; (iii) the effect of FFAs is realized via inhibition of pyruvate dehydrogenase (PDH) complex, phosphofructokinase 1 and hexokinase (i.e., glycolysis) due to accumulation of acetyl-CoA and NADH in the mitochondria, and citrate and glucose 6-phosphate in the cytoplasm; and (iv) preferential oxidation of fatty acids decreases the ability of insulin to stimulate glucose uptake by cells [20].
In fact, Randle primarily focused on the metabolic effects of FFAs towards the carbohydrate metabolism [21] (Fig. 2a). His key provision was that FFA oxidation in the mitochondria increases acetyl-CoA/CoA and NADH/NAD+ ratios, which diminishes the PDH complex activity, whereas the efflux of citrate to the cytoplasm inhibits phosphofructokinase 1. As a result, the glycolytic flux is inhibited and cells accumulate glucose 6-phosphate, which reduces hexokinase activity and glucose phosphorylation. Glucose accumulates in the cytoplasm and prevents its uptake from the outside. Although the effect of FFAs on the insulin-stimulated glucose uptake was not ruled out, its contribution was considered insignificant on the basis of some experimental results [20].
Fig. 2. The hypotheses for mechanism of lipid-induced IR in muscles: a) the metabolic hypothesis by P. Randle [20, 21]; b) the signaling hypothesis by G. Shulman [10, 25]. a) FFAs bypass glycolysis-derived pyruvate to enter the Krebs cycle (TCA) and increase the levels of acetyl-CoA (Ac-CoA), citrate, and NADH (not shown). Ac-CoA inhibits pyruvate dehydrogenase (PDH), whereas citrate enters the cytosol where it inhibits phosphofructokinase 1 (PFK1). As a result, the glycolytic flux is inhibited and the level of glucose 6-phosphate is increased, which reduces the hexokinase (HK) activity. This increases the intracellular glucose level and shifts the balance towards reduced glucose uptake. However, direct measurements of 31P- and 13C-glucose isotopes by NMR spectroscopy revealed a decrease, but not an increase, in the glucose and glucose 6-phosphate levels. Hence, an alternative mechanism has been proposed (b) suggesting that FFAs interfere with the insulin-induced glucose uptake by interrupting insulin cascade activation (receptor > IRS > PI3 kinase (PI3K) > Akt) and incorporation of glucose transporter GLUT4 into the cell membrane.
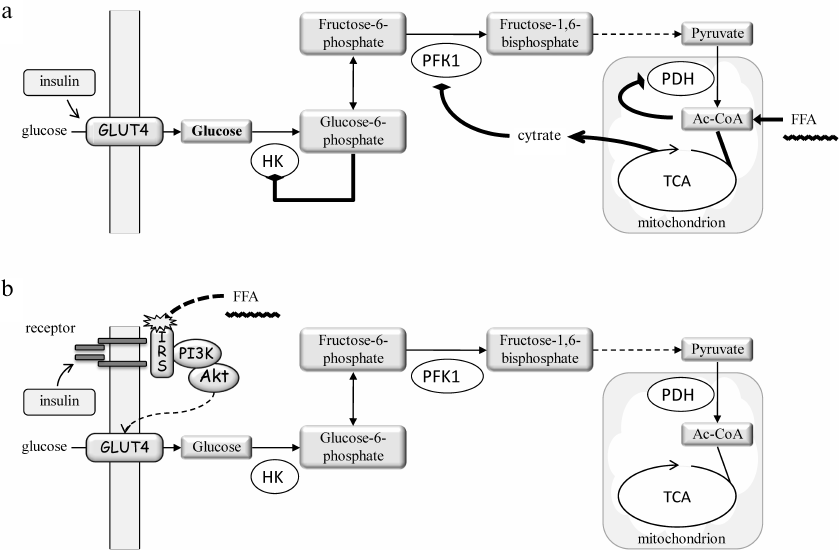
The first indications that FFAs inhibit insulin-stimulated glucose uptake were provided in 1990s by Boden et al. (reviewed in [22]). However, the key data were soon reported by Gerald Shulman and colleagues. Using 31P and 13C isotopes and nuclear magnetic resonance spectroscopy, they demonstrated that intravenous infusion of FFAs in fact reduced the levels of intracellular glucose and glucose 6-phosphate rather than elevated them, as it was predicted by the Randle hypothesis [23, 24]. The following studies also demonstrated reduced PI3 kinase pathway activity and led to the conclusion that FFAs induce IR largely via impairment of insulin signaling to glucose transport into the cells (reviewed in [25]) (Fig. 2b).
Currently, this view dominates, while the Randle hypothesis (Fig. 2a) subsided, but in reality, the truth may well lie somewhere between, and each mechanism may contribute [26]. Moreover, later studies have demonstrated that GLUT4, which has been earlier considered as solely the insulin-dependent glucose transporter, is also regulated by the AMP-dependent kinase (AMPK) in the insulin-independent manner. Taking into account that AMPK is a cellular energy sensor and reduction in its activity is critical for the TAG-FFA cycle imbalance and T2DM, the role of AMPK likely needs to be taken into account to complement the mechanism of IR development [27].
A significant step towards understanding the mechanisms of IR was the finding that IR is associated with the appearance of ectopic lipids, i.e., the lipids deposited in “wrong” locations in an organism. The phenomenon is linked to obesity and related to decreased mitochondrial function, which might occur naturally (e.g., in ageing), or as a result of genetic predisposition, or as an anomaly such as lipodystrophy [25]. At this background, overnutrition may interfere with mitochondrial oxidation of fatty acids and cause the TAG-FFA cycle overload (Fig. 1). This was experimentally demonstrated in transgenic mice overexpressing lipoprotein lipase in the liver [28] or skeletal muscles [29]. Excessive lipids accumulated in the form of droplets specifically in these organs. In humans, these changes may lead to the non-alcoholic fatty liver disease (NAFLD) and sarcopenia. Importantly, ectopic lipids are found inside (rather than outside) of the cells. Such a location creates conditions for aberrant signaling involving lipid-dependent and inflammatory kinases, the two groups of enzymes that most critically interfere with signal transduction from the insulin receptor to the glucose transport machinery in the cells.
Activation of insulin receptor and downstream signaling requires tyrosine phosphorylation of the receptor and IRS [9, 10]. Insulin binding triggers receptor autophosphorylation and IRS binding, followed by IRS phosphorylation by the receptor at several sites. This event allows for the binding of the adaptor proteins that recognize phosphotyrosine-containing sequences in IRS. In particular, Tyr612 binds the regulatory subunit of PI3 kinase, thus releasing the catalytic subunit, which phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to yield phosphatidylinositol 3,4,5-trisphosphate (PIP3). PIP3 recruits to the plasma membrane proteins with pleckstrin homology (PН) domains that bind inositol rings phosphorylated at 3′-position. PH domain is present in the phosphoinositide-dependent kinase PDK1 and protein kinase B (Akt), the major targets of the PI3 kinase pathway. PDK1 is the master regulator of the AGC family kinases [30], including cAMP-dependent kinases (A), cGMP-dependent (G) kinases, and protein kinase C (C), which gave the name to this kinase family. PDK1 activates these enzymes by phosphorylating a residue in their activation loop. In Akt, this residue is Thr308, whose phosphorylation is absolutely required for the Akt activation. Besides, additional phosphorylation of a residue within the so-called hydrophobic motif of the substrate kinases is needed for their effective recognition by PDK1 [30]. In Akt, this site is Ser473; it is phosphorylated by the mTOR kinase as a part of the mTORC2 protein complex formed by Rictor. Because mTORC2 is also activated by PDK1, full activation of Akt completely depends on the PI3 kinase pathway.
Akt has many cell substrates, and the PI3 kinase pathway is implicated in a wide variety of cell responses ranging from metabolic to transcriptional regulation [31]. With regard to metabolism, the key Akt function is regulation of glucose transport in the muscle and adipose cells and of gluconeogenesis in the liver. In all these tissues, impairments in the insulin-dependent activation of Akt and PI3 kinases are now considered as the major molecular mechanisms of IR and metabolic disturbances associated with T2DM [10, 25].
The mechanism of lipid-induced suppression of IRS is the simplest in muscle, where it has been studied in more detail [23, 32] (Fig. 3a). In 1980-90s, many studies have shown that accumulation of ectopic lipids in muscle leads to increased levels of diacylglycerol (DAG) and activation of protein kinase C (PKC) (see [10] for a review). The PKC family includes three groups of related enzymes that differ in the mode of activation. The classic PKCs (cPKC; isoforms α, β, γ) are Ca2+-, phospholipid- and DAG-dependent; the “novel” PKCs (nPKC; isoforms δ, ε, θ, η) need only DAG for activation, whereas atypical PKCs (aPKC; isoforms ζ, λ, ι) do not require Ca2+ or DAG. Because of a single amino acid substitution in the DAG-binding region, activation of nPKC is more effective and prolonged. It is nPKC that is responsible for IR in muscle and the liver.
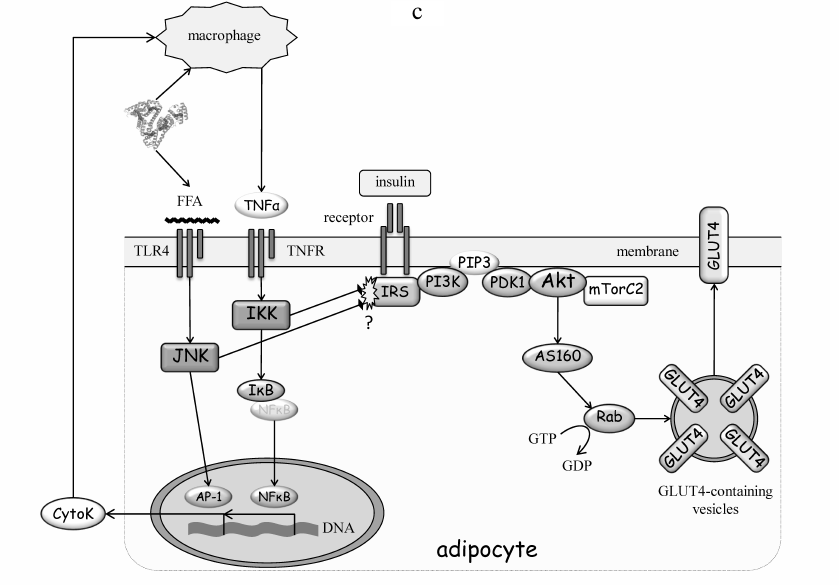
Fig. 3. Molecular mechanisms of IR development. a) IR development in muscles. Accumulation of TAG in ectopic lipids increases diacylglycerol (DAG) content and promotes nPKC activation in the cytoplasm. PKCθ phosphorylates Ser1101 in IRS and inhibits tyrosine phosphorylation of IRS in response to insulin receptor activation. As a result, insulin fails to activate the PI3 kinase pathway, Akt, and mTOR complex 2 (mTORC2). AS160 is the substrate of Akt and GDP-to-GTP exchange factor of small GTPase Rab that controls translocation of GLUT4-containing vesicles to the plasma membrane. Inactivation of insulin cascade prevents mobilization GLUT4 and glucose uptake stimulation by insulin. b) IR development in the liver. Accumulation of ectopic lipids leads to activation of PKCε that phosphorylates insulin receptor at Thr1160 and prevents activation of PI3 kinase. The PI3 kinase pathway targets phosphorylase kinase (PhK), glycogen synthase kinase 3 (GSK3), and glycogen synthase (GS), as well as the transcription factor FOXO1 that regulates expression of phosphoenolpyruvate carboxykinase (PEPCK), glucose 6-phosphatase (G6Pase) and glucokinase (GCK). Collectively, inactivation of insulin signaling in the liver increases glycogen breakdown and gluconeogenesis flux, resulting in the increased glucose production. c) IR development in the adipose tissue. FFAs activate the inflammatory pathways both in adipocytes and macrophages, thereby creating an autonomous cycle, the activity of which is maintained by pro-inflammatory cytokines (CytoK) and TNFα (typical representative of macrophage-derived cytokines). JNK and IKK inactivate insulin signaling by phosphorylating IRS. See the text for more details.
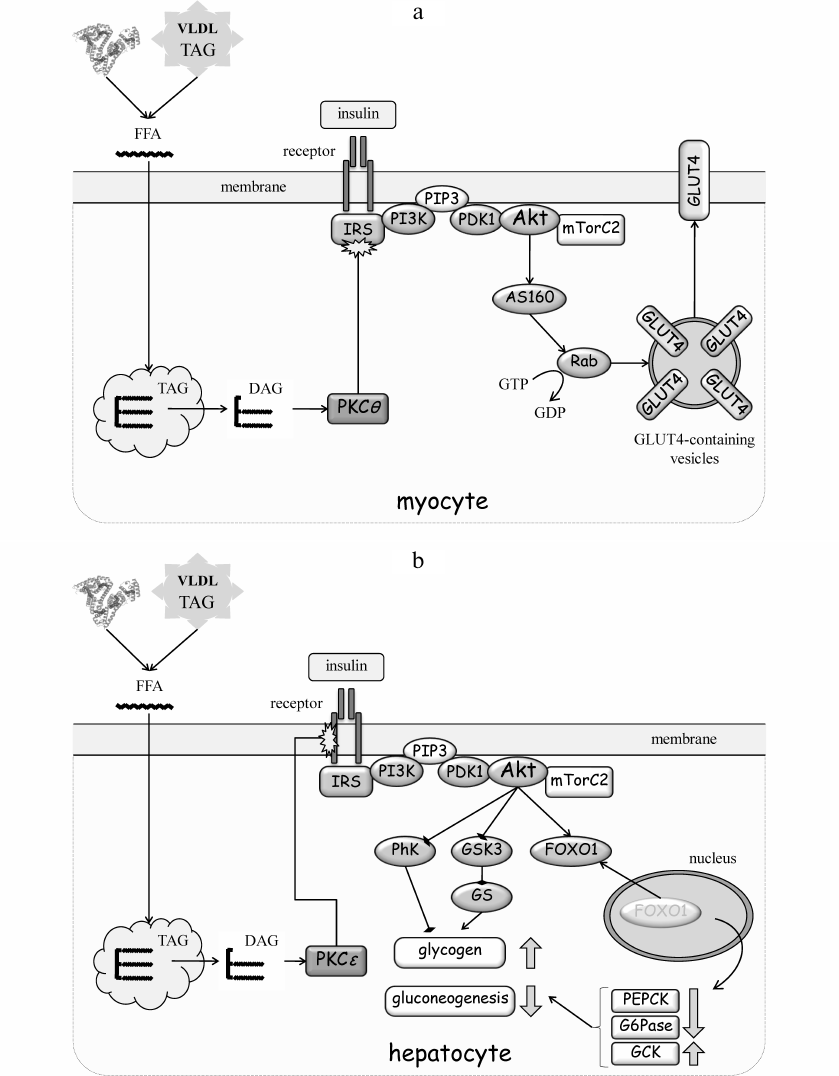
Many studies using cellular, animal, and knockout models have identified nPKCθ and, to some extent, nPKCδ as responsible for IR in muscles, and nPKCε – for IR in liver [10, 25]. Mechanistically, nPKCθ has been shown to phosphorylate Ser1101 in IRS1 and to interfere with tyrosine phosphorylation of IRS in skeletal muscles in response to insulin [33]. This phosphorylation was increased in human muscles after lipid infusion, which caused IR [34].
The mechanism of lipid-induced IR in the liver is more complex. This is likely because liver is a multifunctional organ and the major metabolic hub in the organism. It is targeted by insulin; however, insulin does not activate glucose uptake, but rather promotes glucose storage in the form of glycogen and inhibits de novo synthesis of glucose. Glycogen synthesis is stimulated by a combined Akt-dependent inactivation of phosphorylase kinase and glycogen synthase kinase 3 (Fig. 3b). This leads to suppression of glycogen breakdown and re-activation of glycogen synthase and glycogen synthesis. Suppression of gluconeogenesis by insulin is due to the Akt-mediated retention of the transcription factor FOXO1 in the cytosol; otherwise, FOXO1 translocates to the nucleus, where it activates expression of the key gluconeogenesis enzymes phosphoenolpyruvate carboxykinase and glucose 6-phosphatase and inhibits expression of glucokinase.
Similar to the muscle tissue, accumulation of ectopic lipids in the liver results in the DAG-dependent activation of nPKCε. However, nPKCε does not phosphorylate IRS (as in muscles), but targets Thr1160 of the insulin receptor [35] (Fig. 3b). This is a highly conserved residue; its phospho-mimicking mutation confers complete inactivity to the receptor, whereas its substitution with a residue uncapable of phosphorylation prevents hepatic IR in animals fed with high-fat diet [35]. The loss of receptor activity inactivates insulin signaling and relieves Akt-mediated inhibition of glycogen phosphorylase and FOXO1. When released, FOXO1 translocates to the nucleus and upregulates expression of phosphoenolpyruvate carboxykinase and glucose 6-phosphatase, while downregulating expression of glucokinase. The net result is the reduced uptake of postprandial glucose from the blood and increased rates of glycogenolysis and gluconeogenesis with a subsequent release of glucose into the blood. Contribution of hepatic glucose production becomes principal for sustained hyperglycemia under IR conditions in T2DM, especially in the fasting states.
Homeostasis of the adipose tissue allows for a long-term storage of considerable lipid amounts under continuous operation of the TAG-FFA cycle and constant lipid exchange (Fig. 1). Adipocytes are well protected from the lipid-induced damage. The concept of ectopic lipids does not apply to adipocytes; nPKC is likely to play a minor, if any, role in adipose IR induction. Rather, the paracrine regulation that involves other cells, mainly macrophages, contributes primarily to changes in adipocyte metabolism and its dysfunction, such as IR. While this regulation is described in detail in the next sections, it follows the same logic of turning off the insulin signaling to Akt and glucose uptake via PI3 kinase pathway.
Resident macrophages are targeted by multiple cytokines and adipokines produced by fat cells (Fig. 3c). Various environmental factors, including FFA complexes with albumin, induce cytokine secretion. By acting through the inflammatory receptors, such as tissue necrosis factor receptor (TNFR) and Toll-like receptors type 2 and 4 (TLR2/TLR4), they activate signaling that involves IKK, JNK, and transcription factors NF-κB and AP-1. The FFA levels in the blood plasma are elevated in obesity, thus promoting TLR4 activation. In addition, adipose tissue hypertrophy and hypoxia caused by obesity enhance production of pro-inflammatory cytokines that act as chemoattractants and recruit blood monocytes into the fat tissue. Subsequent differentiation of monocytes into pro-inflammatory macrophages (M1) boosts the paracrine effects of macrophages on adipocytes. This completes the vicious cycle thus increasing the inflammatory background and activity of the inflammatory IKK and JNK kinases in adipocytes. IKK and JNK induce IR by suppressing insulin signaling (Fig. 3c). The role of these kinases has been clearly demonstrated in animal models of IKKβ [36], IKKε [37], JNK1 [38], or JNK2 [39] knockout. Yet surprisingly, the site phosphorylated in IRS has not been precisely identified. Mutation of the most expected Ser302 and Ser307 [40] did not produce anticipated results in mice fed with a high-fat diet [41, 42]. Thus, despite the substantial evidence that inflammation and inflammatory kinases IKK and JNK mediate adipose IR development, their substrates in adipocytes, and mechanistic involvement of serine phosphorylation of IRS, remain uncertain.
Summing up the mechanisms of IR development in obesity, a principal conclusion can be drawn that all of them are tightly associated with disturbed lipid metabolism and increased blood and interstitial levels of FFAs, which is typical of dyslipidemia. However, in contrast to the widely accepted view on serine phosphorylation of IRS as a key event in IR induction via inactivation of insulin signaling, final evidence is still missing for all the insulin-targeted tissues. Bearing in mind potential contribution of other mechanisms, such as the metabolic hypothesis by Randle, AMPK and decreased muscle activity, the prospects for further studies to develop a unifying theory remain wide open.
IMMUNE CELLS AND LATENT INFLAMMATION IN ADIPOSE TISSUE
While many protein kinases are able to phosphorylate IRS at Ser residues in vitro and in cells [12, 14, 15], the role of IRS phosphorylation in the adipose tissue in vivo has been reliably demonstrated only for JNK and IKK. These kinases are activated by various stress stimuli that directly or indirectly induce the inflammatory signaling in cells. Latent inflammation in adipose tissue, which leads to IR, results from a combination of different factors associated with excessive lipid accumulation in the cells. There are four classic components that link obesity, latent inflammation, and IR in the adipose tissue: hyperlipidemia, hypoxia, oxidative stress, and endoplasmic reticulum (ER) stress. All they develop via different mechanisms, but activate the same transcription factors NF-κB and AP-1 that upregulate expression of pro-inflammatory genes and secretion of pro-inflammatory cytokines, which act in an auto- and paracrine manner to lock the pathologic loop shown in Fig. 3c, resulting in a self-sustained autonomous cycle of latent inflammation in adipose tissue.
Typically, inflammation develops as a response of immune cells to foreign agents that appear in the blood during infection, or to the products of disrupted and dead cells, or as an aberrant autoimmune response. These stimuli are correspondingly classified into pathogen-associated (PAMP), damage-associated (DAMP), or stress-associated molecular patterns (SAMP) [43]. FFAs act non-specifically through the TLR4 receptors to activate inflammation (Fig. 3c). Inside the cells, all receptor-dependent inflammatory pathways converge at the key kinase level, including stress-activated JNK1/2 and three IKK isoforms (α, β and γ) with different functions [11]. JNK activates the AP-1 transcription factor, whereas IKK targets NF-κB to trigger expression of pro-inflammatory genes. TLR4 receptors are also expressed by the macrophages, endothelial and other cells that reside in the adipose tissue. TLR4 activation in macrophages upregulates production of pro-inflammatory cytokines, including prototypical tumor necrosis factor (TNFα). Macrophage-derived cytokines act as paracrine activators of TNFR and other cytokine receptors on adipocytes, maintaining the latent inflammation cycle [44].
Immune cells function as additional modulators of adipose tissue homeostasis and contribute to the development and maintenance of latent inflammation (Fig. 4). All of them contain Toll-like and cytokine receptors and become inevitably involved in the inflammatory response. The innate lymphoid cells (ILCs) are on the top of the hierarchy as the primary immune cells. There are three major ILC phenotypes that have different surface markers, gene expression profiles, and secreted factors. Their classification is based on the dominant action of a certain transcription factor that specifies cell development and function: group 1 expresses transcription factor T-bet; group 2 – GATA3; and group 3 – RORγT. ILCs from groups 1 and 3 are involved in development of obesity-associated latent inflammation, whereas group 2 ILCs maintain adipose tissue homeostasis by secreting the anti-inflammatory cytokines, such as IL-4 and IL-13. The group 1 ILCs additionally contribute to maintenance of latent inflammation and IR in the adipose tissue by controlling monocyte differentiation and polarization of macrophages into the pro-inflammatory M1 phenotype [45-47].
Fig. 4. Role of immune cells in the regulation of immune status and adipocyte state in the fat tissue. This regulation is hierarchal: ILCs play the major role by regulating secretion profiles of other immune cells, including T cells, B cells, and natural killer (NK) cells, which together modulate the phenotype (immune status) of macrophages. Macrophages are the most effector cells; they regulate the adipocyte state and insulin sensitivity in the adipose tissue.
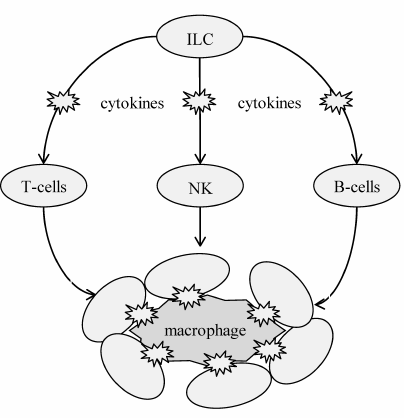
T lymphocytes demonstrate close relationship to ILCs (Fig. 4). They also can polarize with a change in their secretion profile. The subtypes of T helper (Th) cells differ in the expression of transcription factors and effector genes. Thus, Th1 cells express transcription factors T-bet and Blimp1; Th2 cells express GATA3 and Blimp1; and Th17 cells express RORγT and IRF4. Th2 and Treg lymphocytes maintain the adipose tissue homeostasis. They secrete anti-inflammatory growth factors and cytokines (IL-4, IL-5, IL-13 by Th2 and IL-10 and transforming growth factor TGFβ by Treg), thus limiting the population of inflammatory Th1 cells. Obesity is associated with considerable infiltration of adipose tissue by Th1 and Th17 lymphocytes that secrete pro-inflammatory factors (interferon-γ by Th1; IL-17, IL-21 and IL-22 by Th17), thus maintaining constitutive inflammation [48-50].
B cells are also involved in the adipose tissue homeostasis (Fig. 4). They are likely recruited and activated by molecules produced by cell damage [51]. In obesity, adipose tissue is infiltrated by B lymphocytes that actively participate in the pro-inflammatory polarization of macrophages, thus maintaining latent inflammation. In addition to lymphocytes, NK cells contribute to the adipose tissue IR, mainly by driving macrophage polarization to M1 phenotype [52, 53].
Dendritic cells remain the least explored group of immune cells in the pathogenesis of T2DM, mostly because they are difficult to distinguish from M1 macrophages that express similar surface markers. Dendritic cells contribute to the development of latent inflammation by regulating infiltration of adipose tissue by T lymphocytes and differentiation of these cells [54, 55].
All the above groups of immune cells regulate activity, metabolism, and polarization of macrophages that have the highest effector potential. There are two major macrophage phenotypes, pro- (M1) and anti-inflammatory (M2), which are the result of polarization, i.e., the process of further differentiation marked by changes in the profiles of cytokine expression and secretion. The type of polarization critically impacts obesity and IR development. It is controlled by immune cells, cytokines, and other microenvironmental cues that create a network of signaling cascades, transcription factors, and epigenetic patterns inside the cells. While the canonical IRF/STAT signaling cascade activated by interferons and Toll-like receptors shifts the macrophage phenotype toward M1, the cytokines IL-4 and IL-13 drive polarization towards the M2 phenotype in a STAT6-dependent manner [56]. In obesity, adipose tissue actively secretes cytokines that stimulate macrophage recruitment and polarization into pro-inflammatory M1 phenotype, as well as promote the M2-to-M1 transition, thereby maintaining latent inflammation in this tissue [57].
LATENT INFLAMMATION AND SYSTEMIC IR
The above data clearly demonstrate that, at least in the adipose tissue, inflammation is tightly associated with obesity and lipid-induced IR. However, the causative relationship between inflammation and IR in the adipose tissue and, most importantly, between inflammation and systemic IR, is all but apparent. The observations that chronic (latent) inflammation typical for the adipose tissue hypertrophied in obesity often results in IR [3, 44] and that the inflammatory cascade activity is increased under in vitro conditions that lead to IR [58] suggest that latent inflammation precedes IR. However, latent inflammation does not always lead to IR and T2DM (as in case of autoimmune diseases), and obesity-unrelated T2DM may not involve the inflammatory component. This means that the link between latent inflammation and IR is not mandatory a priori but may be self-sufficient for the development of lipid-induced IR in obesity.
Considering the central role of adipose tissue in energy metabolism (Fig. 1) and the importance of FFAs in IR in insulin-dependent tissues (Fig. 3), it is plausible that the pathological process may begin in the fat tissue, actively develop there due to the auto- and paracrine interactions between adipocytes, macrophages, and other leukocytes (Figs. 3c and 4), and then expand to other tissues. Although this process should not be considered as an infection in the strict sense, these two phenomena have much in common. FFAs could be regarded as physical inducers of inflammatory response similar to DAMPs or SAMPs both in size and the mechanism of action via the inflammatory receptors and signaling pathways in the recipient cells. FFAs are not antigens; they act directly on the receptors, bypassing the binding to antibodies. FFA complexes with albumin constantly circulate in the blood, normally amounting up to 0.6 mM (with respect to FFAs) with no significant inflammatory response. Remarkably, a relatively minor increase in the blood FFA level (about 30%, exceeding 0.7 mM in T2DM patients [17]) leads to dramatic events with a major functional outcome. It is possible that this effect is determined not only by increased FFA levels, but also by its duration, or limited capacity of albumin as an FFA carrier, or other factors (see the next section). Although FFA action is clearly different from the classic immune response, this does not exclude the possibility that FFAs may affect B cells and induce or alter their responses, similarly to the antigen-presenting cells. These possibilities, as well as the question whether these cells can participate in the systemic expansion of IR, clearly require separate studies.
Latent inflammation is widely accepted as the major risk factor for IR in adipose tissue [3, 44, 57, 59]. In the last section, we discuss feasibility of the anti-inflammatory approaches in treating IR both experimentally and clinically. Earlier, we demonstrated a potential of the anti-inflammatory IL-4 that demonstrated positive results in the in vitro IR model using 3T3-L1 adipocytes [60]. In addition, different strategies to induce IR in 3T3-L1 adipocytes were successful only in the case of sustained activation of inflammatory signaling [58]. Collectively, all these data argue in favor of inflammation as the major inducer of IR.
At the same time, the recent study used the cell lines, animal models, and human adipose biopsies to demonstrate that inflammation in the adipose tissue may be a result of IR [61]. AdRiKO mice (adipocyte-specific knockout of Rictor, the scaffold organizer of mTORC2) fed with a high-fat diet developed systemic IR, macrophage infiltration of white adipose tissue, and inflammation, but not the other way around. Obesity induced IR, leading to increased expression of the monocyte chemoattractant peptide MCP-1 and migration into the fat tissue of mainly M1 macrophages that caused inflammation. The authors concluded that mTORC2 activation counteracts IR independently of obesity. The effect of the high-fat diet was more pronounced in AdRiKO mice, while activation of mTORC2 in the wild-type mice was protective. An obvious question as to whether obesity affects the mTORC2 activity and predisposes to IR development in human tissues has been also addressed. mTORC2 was less active in obese vs. healthy individuals; the phosphorylation level of Ser473 in Akt (the mTORC2 substrate) was lower and negatively correlated with the body mass index (BMI) in obese individuals [61]. Although expression of MCP-1 and CD68 was higher and correlated with BMI in patients with morbid obesity, the cause-and-effect relationship could not be established, as the experiments on human subjects were not possible. Whether the mTORC2 activity is different between healthy individuals and T2DM patients also remained unexplored.
Therefore, the mechanism for maintaining IR that had once developed as a result of dyslipidemia and signaling response in different tissues (Figs. 2 and 3) seems to play the principal role in T2DM pathogenesis. This mechanism is likely to involve cyclic interactions between dyslipidemia, chronic inflammation, and IR that maintain its activity for indefinite time (Fig. 5). In obesity, FFA blood levels increase and stimulate IR development in the muscles and liver by interrupting insulin signaling inside the cells. At the same time, FFAs trigger resident macrophages and their inflammatory responses in the adipose tissue. In addition, FFAs impact circulating immune cells and cause their infiltration into the adipose tissue. This results in the increased inflammatory background, which is further maintained by sustained pro-inflammatory reactivity of macrophages and other immune cells. This chronic latent inflammatory process promotes lipolysis and FFA release by adipocytes, closing up this self-sustaining inflammatory cycle.
Fig. 5. Proposed mechanism of systemic IR development. Continuous excessive energy supply characteristic for obesity causes imbalance in the TAG-FFA cycle and increases the circulating FFAs levels, leading to activation of macrophages and formation of autonomous cycle of latent inflammation. Excessive FFAs are uptaken by other tissues, where they accumulate as ectopic lipids and activate lipid-dependent nPKC, which inhibits insulin signaling at the receptor or IRS levels. Systemic IR is maintained by latent inflammation mediated by macrophages and other immune cells in the adipose tissue.
ADIPOCYTE HYPERTROPHY AND MSC DYSFUNCTION
Adipocytes are responsible for the long-term storage of energy surplus. Under the excessive energy supply conditions, such as in obesity, and low physical activity, fat deposition may occur either due to adipocyte hypertrophy or hyperplasia and formation of new fat depots [6]. An increase in the number of cells is provided by proliferation, whereas formation of new fat depots results from the adipogenic differentiation of MSCs (adipocyte precursors). These activities of MSCs are likely not compromised in obesity, and adipocyte renewal is not impaired [62]. At the same time, the total number of adipocytes in the body is set in an early age and does not much change thereafter, with a halftime of adipocyte renewal approximately 8 years independently of the fat volume [62]. This indirectly indicates that adipocyte renewal occurs in a one-to-one fashion, and obesity is largely due to the adipocyte hypertrophy, i.e., an increase in the cell volume and amount of deposited lipids.
In obese T2DM patients, adipocytes are hypertrophied even more than in obese, but otherwise healthy subjects [63]. However, proliferative and adipogenic activities of MSCs isolated from these donors are very different. Diabetic MSCs proliferate much slower and their differentiation potential is considerably lower [63]. Figure 6 summarizes the results of comparative studies on healthy patients with different BMI [6] and obese T2DM patients vs. obese, but metabolically healthy donors [63]. Collectively, these data suggest that T2DM may be specifically linked to latent inflammation in the adipose tissue and dysfunctional renewal of fat depots regardless of BMI.
Fig. 6. Proposed role of latent inflammation and impaired renewal of fat depots in IR and T2DM pathogenesis. The capacity of fat depots may be increased by increased number of adipocytes (hyperplasia) or their size (hypertrophy) [6]. The number of adipocytes is likely set in early childhood and does not much change thereafter, with the half-period of adipocyte renewal about 8 years, independently of the fat tissue size [62]. Therefore, contribution of hyperplasia is manifested at the stage of MSC (adipocyte precursor) proliferation, thus determining the potential capacity of fat depots. The ability to store excessive lipids is determined by the renewal of fat depots, including recruitment of new adipocytes and disposal of spent ones. A combination of impaired proliferation and adipogenic differentiation with the increased inflammatory background in adipose tissue of obese and T2DM patients [63] suggest the involvement of latent inflammation and impaired regenerative potential of MSCs in the development of IR and T2DM.
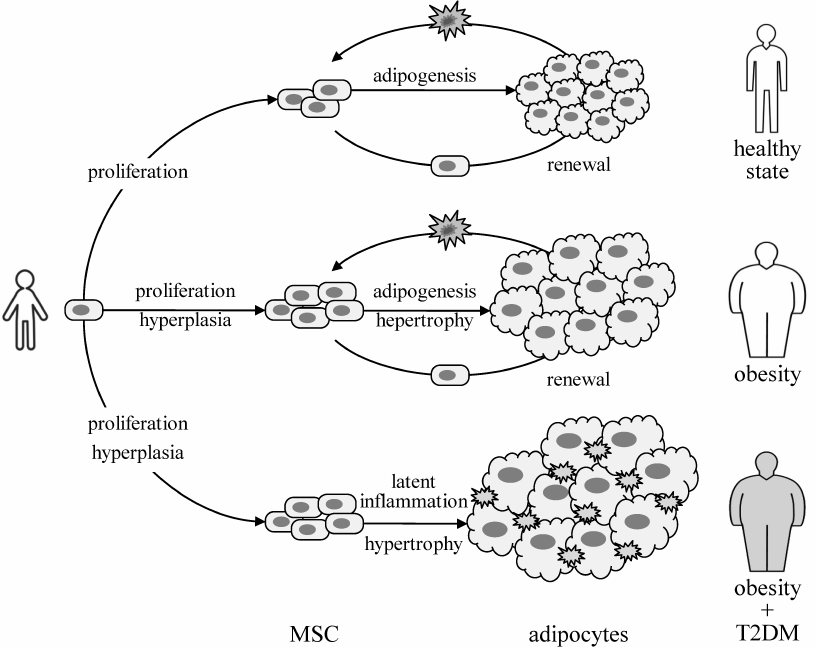
Interestingly, BMI and other overweight-related parameters positively correlate with the plasma levels of FFAs; however, plasma levels of FFA only slightly differ (by ~6%) in obese individuals with or without IR (НOMA-index) and T2DM [17]. These data obtained in a large cohort of test subjects together with the retrospective analysis of earlier studies have shed a doubt on the credibility of association between the blood levels of FFAs and T2DM but supported association of FFA levels with obesity [17, 64]. Counterintuitive as these observations may seem, they perfectly fit the above hypothesis (Fig. 6). Latent inflammation and dysfunctional renewal of fat depots can be more critical for IR and T2DM development than the blood levels of FFAs.
Adipose tissue is a complex multicomponent system composed of different cell types that mutually regulate each other’s activities; its homeostasis needs regulation for proper tissue functioning. Both immune cells and MSCs can regulate the state of adult adipocytes. MSCs have an immuno-privileged status and exert immunomodulatory activity. Systemic introduction of MSCs isolated from healthy animals into the high-fat diet animal models increases insulin sensitivity, which is associated with changes in the phenotype of resident immune cells. The mechanisms of the donor MSC action may involve secretion of exosomes that contain STAT3 and activate the M2 phenotype of macrophages [65] and/or suppress ubiquitin-dependent degradation of IRS1 in muscles [66].
The opposite case of MSC response to IR has been also demonstrated. MSCs from animals with IR demonstrated markedly altered expression profiles [67, 68]. As discussed above, obesity is the primary risk factor of IR development and T2DM, associated with increased inflammation in adipose tissue. Moreover, obesity is also associated with increased inflammatory state of MSCs themselves [63, 69]. Therefore, latent inflammation is a likely cause of MSC dysfunction and disturbed adipocyte renewal in T2DM. Other links between MSCs and T2DM remain yet unknown.
The current cell-centered paradigm of latent inflammation in adipose tissue and T2DM development is based on the cell–cell interactions between adipocytes and immune cells. A potential role of MSCs as critical regulators of the fat tissue homeostasis is often neglected. Little is known on the mechanisms that mediate interactions between MSCs and fat tissue cells other than adipocytes. Recently, several reports demonstrated altered properties of adipose-derived MSCs in T2DM [63, 69, 70]. Different mechanisms have been suggested to explain how MSCs affect the IR development, including immune regulation of the balance between macrophages and T cells by MSCs [69], compromised proliferation and adipogenesis of MSCs [63], and increased oxidative stress and autophagy in MSCs [70].
The possible causes of MSC dysfunction in T2DM are disputable. We found that MSCs have a “memory”, which allows them to retain some features altered by ageing or disease, even after the isolated cells had been cultured in vitro [63, 71-73]. Because these changes have remained for a relatively long period during culturing, they are unlikely solely due to signaling or metabolic effects. More likely, epigenetic mechanisms are involved in the formation of rather profound and lengthy pathological MSC phenotype. More efforts are clearly needed to explore this issue. Here, it is important that the above studies cast a new light on the problem of IR development in adipose tissue and, perhaps, T2DM in general. We hypothesize that T2DM pathogenesis may be associated with the dysfunction of adipose tissue progenitor cells caused by latent inflammation.
PROSPECTS OF ANTI-INFLAMMATORY T2DM THERAPY
Anti-inflammatory therapeutic approaches to IR and T2DM are actively studied. The first clinical trials were launched in 1996, when a TNFα-blocking antibody was used against T2DM [74]. The outcome was not exciting, as the antibody produced no significant effect on the clinical indicators in the test subjects. However, the results should be taken with some skepticism because only 10 patients were enrolled in the study. In the following study, TNFα inhibition was achieved using etanercept, a protein construct consisting of a soluble part of TNFα receptor and immunoglobulin Fc fragment. When used for 4-26 weeks, etanercept significantly reduced the fasting blood sugar and increased the level of high-molecular-weight adiponectin. This was the first, although modest, success of the anti-inflammatory therapy of T2DM [75-77].
Meanwhile, basic research has provided an information on the mechanism that links inflammation to IR and demonstrated the role of IKKβ in IR induction. This laid down the basis for using the next substance – salsalate (salicylate dimer), a non-steroid anti-inflammatory drug and non-specific IKKβ inhibitor. The trial duration was 12-48 weeks; regular intake of salsalate effectively reduced the fasting blood levels of sugar, triglycerides, and glycated hemoglobin, while increased levels of high-molecular-weight adiponectin [78-80].
Recent advances in the anti-inflammatory therapy of T2DM are based on the use of antibodies against IL-1β and its receptor. Earlier studies have demonstrated that the anti-IL-1β antibody reduces the levels of the C-reactive protein and glycated hemoglobin and activates secretion of insulin. The cardiovascular effects of anti-IL-1β antibody (canakinumab) are tested in the still ongoing CANTOS (Canakinumab ANti-inflammatory Thrombosis Outcomes Study) trial, that also monitors the antibody effects on T2DM. Canakinumab has been found to lower the incidence of cardiovascular complications in T2DM patients. However, despite a significant decrease in the C-reactive protein level in the treated subjects, no reduction in new T2DM incidents has been observed [81, 82].
Summing up the results of clinical studies, the anti-inflammatory therapy produces some positive effects on the carbohydrate metabolic parameters in T2DM patients; however, its current efficacy is still far from that of the classic sugar-lowering drugs such as metformin, dipeptidyl peptidase 4 inhibitors, glucagon-like peptide-1 (GLP-1) receptor agonists, etc. Nonetheless, recent experimental studies have exhibited a new trend in the anti-inflammatory approach based on the use of anti-inflammatory cytokines or gene-targeting techniques that may increase insulin sensitivity of adipose tissue and stimulate beige fat formation [60, 83-86]. So far, MSCs have not been considered as a potential target for the anti-inflammatory therapy; however, we believe that the above arguments prove the potential of this approach.
Funding. The authors’ research and this work are supported by the Russian Science Foundation (project 17-15-01435) and Russian Foundation for Basic Research (projects 17-04-02225a and 18-015-00398a).
Acknowledgements. The authors acknowledge a key contribution of the Academicians I. I. Dedov and V. A. Tkachuk in the initiation of national fundamental research in this area and are grateful to Prof. N. B. Gusev for critical contribution to the hypothesis of fat cell renewal in IR, to Prof. V. P. Shirinsky for continuous support and discussions, to S. S. Michurina, N. V. Podkuychenko, I. A. Sklyanik, and E. A. Shestakova for fruitful collaboration and discussion of the manuscript. We also deeply apologize to many authors of experimental studies that have not been cited due to the space limitations.
Conflict of interest. The authors declare no direct or potential conflict of interest related to the publication of this article.
Compliance with ethical standards. This paper does not involve experiments with human or animal subjects performed by any of the authors.
REFERENCES
1.Dedov, I. I., Shestakova, M. V., Vikulova, O. K.,
Zheleznyakova, A. V., and Isakov, M. A. (2018) Diabetes mellitus in
Russian Federation: prevalence, morbidity, mortality, parameters of
glycemic control and structure of glucose lowering therapy according to
the Federal Diabetes Register, status 2017, Diab. Mellit.,
21, 144-159.
2.Balanova, Yu. A., Shalnova, S. A., Deev, A. D.,
Imaeva, A. E., Kontsevaya, A. V., Muromtseva, G. A., Kapustina, A. V.,
Evstifeeva, S. E., and Drapkina, O. M. (2018) Obesity in Russian
population – prevalence and association with the
non-communicable diseases risk factors, Russ. J. Cardiol.,
2018, 123-130.
3.Saltiel, A. R. (2016) New therapeutic approaches
for the treatment of obesity, Sci. Transl. Med., 8,
323rv322.
4.Reshef, L., Olswang, Y., Cassuto, H., Blum, B.,
Croniger, C. M., Kalhan, S. C., Tilghman, S. M., and Hanson, R. W.
(2003) Glyceroneogenesis and the triglyceride/fatty acid cycle, J.
Biol. Chem., 278, 30413-30416.
5.Nelson, D. L., and Cox, M. C. (2004) Lehninger:
Principles of Biochemistry, 4th Edn., W. H. Freeman & Co, New
York.
6.Ghaben, A. L., and Scherer, P. E. (2019)
Adipogenesis and metabolic health, Nat. Rev. Mol. Cell Biol.,
20, 242-258.
7.Kowalski, G. M., and Bruce, C. R. (2014) The
regulation of glucose metabolism: implications and considerations for
the assessment of glucose homeostasis in rodents, Am. J. Physiol.
Endocrinol. Metab., 307, E859-E871.
8.Marin, P., Rebuffe-Scrive, M., Smith, U., and
Bjorntorp, P. (1987) Glucose uptake in human adipose tissue,
Metabolism, 36, 1154-1160.
9.Leto, D., and Saltiel, A. R. (2012) Regulation of
glucose transport by insulin: traffic control of GLUT4, Nat. Rev.
Mol. Cell Biol., 13, 383-396.
10.Petersen, M. C., and Shulman, G. I. (2018)
Mechanisms of insulin action and insulin resistance, Physiol.
Rev., 98, 2133-2223.
11.Stafeev, I. S., Vorotnikov, A. V., Ratner, E. I.,
Menshikov, M. Y., and Parfyonova, Y. V. (2017) Latent inflammation and
insulin resistance in adipose tissue, Int. J. Endocrinol.,
2017, 5076732.
12.Copps, K. D., and White, M. F. (2012) Regulation
of insulin sensitivity by serine/threonine phosphorylation of insulin
receptor substrate proteins IRS1 and IRS2, Diabetologia,
55, 2565-2582.
13.Boura-Halfon, S., and Zick, Y. (2009)
Phosphorylation of IRS proteins, insulin action, and insulin
resistance, Am. J. Physiol. Endocrinol. Metab., 296,
E581-E591.
14.Morino, K., Petersen, K. F., and Shulman, G. I.
(2006) Molecular mechanisms of insulin resistance in humans and their
potential links with mitochondrial dysfunction, Diabetes,
55, Suppl. 2, S9-S15.
15.Zick, Y. (2004) Uncoupling insulin signaling by
serine/threonine phosphorylation: a molecular basis for insulin
resistance, Biochem. Soc. Trans., 32, 812-816.
16.Hales, C. N., Walker, J. B., Garland, P. B., and
Randle, P. J. (1965) Fasting plasma concentrations of insulin,
non-esterified fatty acids, glycerol, and glucose in the early
detection of diabetes mellitus, Lancet, 1, 65-67.
17.Arner, P., and Ryden, M. (2015) Fatty acids,
obesity and insulin resistance, Obes. Facts, 8,
147-155.
18.Boden, G. (2008) Obesity and free fatty acids,
Endocrinol. Metab. Clin. North Am., 37, 635-646.
19.Randle, P. J., Garland, P. B., Hales, C. N., and
Newsholme, E. A. (1963) The glucose fatty-acid cycle. Its role in
insulin sensitivity and the metabolic disturbances of diabetes
mellitus, Lancet, 1, 785-789.
20.Randle, P. J. (1998) Regulatory interactions
between lipids and carbohydrates: the glucose fatty acid cycle after 35
years, Diab. Metab. Rev., 14, 263-283.
21.Randle, P. J., Kerbey, A. L., and Espinal, J.
(1988) Mechanisms decreasing glucose oxidation in diabetes and
starvation: role of lipid fuels and hormones, Diab. Metab. Rev.,
4, 623-638.
22.Boden, G. (1997) Role of fatty acids in the
pathogenesis of insulin resistance and NIDDM, Diabetes,
46, 3-10.
23.Roden, M., Price, T. B., Perseghin, G., Petersen,
K. F., Rothman, D. L., Cline, G. W., and Shulman, G. I. (1996)
Mechanism of free fatty acid-induced insulin resistance in humans,
J. Clin. Invest., 97, 2859-2865.
24.Petersen, K. F., Laurent, D., Rothman, D. L.,
Cline, G. W., and Shulman, G. I. (1998) Mechanism by which glucose and
insulin inhibit net hepatic glycogenolysis in humans, J. Clin.
Invest., 101, 1203-1209.
25.Shulman, G. I. (2014) Ectopic fat in insulin
resistance, dyslipidemia, and cardiometabolic disease, N. Engl. J
Med., 371, 1131-1141.
26.Hue, L., and Taegtmeyer, H. (2009) The Randle
cycle revisited: a new head for an old hat, Am. J. Physiol.
Endocrinol. Metab., 297, E578-E591.
27.Richter, E. A., and Hargreaves, M. (2013)
Exercise, GLUT4, and skeletal muscle glucose uptake, Physiol.
Rev., 93, 993-1017.
28.Kim, J. K., Fillmore, J. J., Chen, Y., Yu, C.,
Moore, I. K., Pypaert, M., Lutz, E. P., Kako, Y., Velez-Carrasco, W.,
Goldberg, I. J., Breslow, J. L., and Shulman, G. I. (2001)
Tissue-specific overexpression of lipoprotein lipase causes
tissue-specific insulin resistance, Proc. Natl. Acad. Sci. USA,
98, 7522-7527.
29.Ferreira, L. D., Pulawa, L. K., Jensen, D. R.,
and Eckel, R. H. (2001) Overexpressing human lipoprotein lipase in
mouse skeletal muscle is associated with insulin resistance,
Diabetes, 50, 1064-1068.
30.Pearce, L. R., Komander, D., and Alessi, D. R.
(2010) The nuts and bolts of AGC protein kinases, Nat. Rev. Mol.
Cell Biol., 11, 9-22.
31.Manning, B. D., and Cantley, L. C. (2007) AKT/PKB
signaling: navigating downstream, Cell, 129,
1261-1274.
32.Yu, C., Chen, Y., Cline, G. W., Zhang, D., Zong,
H., Wang, Y., Bergeron, R., Kim, J. K., Cushman, S. W., Cooney, G. J.,
Atcheson, B., White, M. F., Kraegen, E. W., and Shulman, G. I. (2002)
Mechanism by which fatty acids inhibit insulin activation of insulin
receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase
activity in muscle, J. Biol. Chem., 277, 50230-50236.
33.Li, Y., Soos, T. J., Li, X., Wu, J., Degennaro,
M., Sun, X., Littman, D. R., Birnbaum, M. J., and Polakiewicz, R. D.
(2004) Protein kinase C theta inhibits insulin signaling by
phosphorylating IRS1 at Ser1101, J. Biol. Chem., 279,
45304-45307.
34.Szendroedi, J., Yoshimura, T., Phielix, E.,
Koliaki, C., Marcucci, M., Zhang, D., Jelenik, T., Muller, J., Herder,
C., Nowotny, P., Shulman, G. I., and Roden, M. (2014) Role of
diacylglycerol activation of PKCtheta in lipid-induced muscle insulin
resistance in humans, Proc. Natl. Acad. Sci. USA, 111,
9597-9602.
35.Petersen, M. C., Madiraju, A. K., Gassaway, B.
M., Marcel, M., Nasiri, A. R., Butrico, G., Marcucci, M. J., Zhang, D.,
Abulizi, A., Zhang, X. M., Philbrick, W., Hubbard, S. R., Jurczak, M.
J., Samuel, V. T., Rinehart, J., and Shulman, G. I. (2016) Insulin
receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin
resistance, J. Clin. Invest., 126, 4361-4371.
36.Arkan, M. C., Hevener, A. L., Greten, F. R.,
Maeda, S., Li, Z. W., Long, J. M., Wynshaw-Boris, A., Poli, G.,
Olefsky, J., and Karin, M. (2005) IKK-beta links inflammation to
obesity-induced insulin resistance, Nat. Med., 11,
191-198.
37.Chiang, S. H., Bazuine, M., Lumeng, C. N.,
Geletka, L. M., Mowers, J., White, N. M., Ma, J. T., Zhou, J., Qi, N.,
Westcott, D., Delproposto, J. B., Blackwell, T. S., Yull, F. E., and
Saltiel, A. R. (2009) The protein kinase IKKepsilon regulates energy
balance in obese mice, Cell, 138, 961-975.
38.Hirosumi, J., Tuncman, G., Chang, L., Gorgun, C.
Z., Uysal, K. T., Maeda, K., Karin, M., and Hotamisligil, G. S. (2002)
A central role for JNK in obesity and insulin resistance,
Nature, 420, 333-336.
39.Tuncman, G., Hirosumi, J., Solinas, G., Chang,
L., Karin, M., and Hotamisligil, G. S. (2006) Functional in vivo
interactions between JNK1 and JNK2 isoforms in obesity and insulin
resistance, Proc. Natl. Acad. Sci. USA, 103,
10741-10746.
40.Lee, Y. H., Giraud, J., Davis, R. J., and White,
M. F. (2003) c-Jun N-terminal kinase (JNK) mediates feedback inhibition
of the insulin signaling cascade, J. Biol. Chem., 278,
2896-2902.
41.Copps, K. D., Hancer, N. J., Opare-Ado, L., Qiu,
W., Walsh, C., and White, M. F. (2010) Irs1 serine 307 promotes insulin
sensitivity in mice, Cell Metab., 11, 84-92.
42.Copps, K. D., Hancer, N. J., Qiu, W., and White,
M. F. (2016) Serine 302 phosphorylation of mouse insulin receptor
substrate 1 (IRS1) is dispensable for normal insulin signaling and
feedback regulation by hepatic S6 kinase, J. Biol. Chem.,
291, 8602-8617.
43.Bianchi, M. E. (2007) DAMPs, PAMPs and alarmins:
all we need to know about danger, J. Leukoc. Biol., 81,
1-5.
44.Lackey, D. E., and Olefsky, J. M. (2016)
Regulation of metabolism by the innate immune system, Nat. Rev.
Endocrinol., 12, 15-28.
45.Chalubinski, M., Luczak, E., Wojdan, K.,
Gorzelak-Pabis, P., and Broncel, M. (2016) Innate lymphoid cells type
2 – emerging immune regulators of obesity and
atherosclerosis, Immunol. Lett., 179, 43-46.
46.Boulenouar, S., Michelet, X., Duquette, D.,
Alvarez, D., Hogan, A. E., Dold, C., O’Connor, D., Stutte, S.,
Tavakkoli, A., Winters, D., Exley, M. A., O’Shea, D., Brenner, M.
B., von Andrian, U., and Lynch, L. (2017) Adipose type one innate
lymphoid cells regulate macrophage homeostasis through targeted
cytotoxicity, Immunity, 46, 273-286.
47.O'Sullivan, T. E., Rapp, M., Fan, X., Weizman, O.
E., Bhardwaj, P., Adams, N. M., Walzer, T., Dannenberg, A. J., and Sun,
J. C. (2016) Adipose-resident group 1 innate lymphoid cells promote
obesity-associated insulin resistance, Immunity, 45,
428-441.
48.Priceman, S. J., Kujawski, M., Shen, S.,
Cherryholmes, G. A., Lee, H., Zhang, C., Kruper, L., Mortimer, J.,
Jove, R., Riggs, A. D., and Yu, H. (2013) Regulation of adipose tissue
T cell subsets by Stat3 is crucial for diet-induced obesity and insulin
resistance, Proc. Natl. Acad. Sci. USA, 110,
13079-13084.
49.Bertola, A., Ciucci, T., Rousseau, D., Bourlier,
V., Duffaut, C., Bonnafous, S., Blin-Wakkach, C., Anty, R., Iannelli,
A., Gugenheim, J., Tran, A., Bouloumie, A., Gual, P., and Wakkach, A.
(2012) Identification of adipose tissue dendritic cells correlated with
obesity-associated insulin-resistance and inducing Th17 responses in
mice and patients, Diabetes, 61, 2238-2247.
50.Jung, C., Lichtenauer, M., Strodthoff, D.,
Winkels, H., Wernly, B., Burger, C., Kamchybekov, U., Lutgens, E.,
Figulla, H. R., and Gerdes, N. (2017) Alterations in systemic levels of
Th1, Th2, and Th17 cytokines in overweight adolescents and obese mice,
Pediatr. Diab., 18, 714-721.
51.Jounai, N., Kobiyama, K., Takeshita, F., and
Ishii, K. J. (2012) Recognition of damage-associated molecular patterns
related to nucleic acids during inflammation and vaccination, Front.
Cell Infect. Microbiol., 2, 168.
52.Lee, B. C., Kim, M. S., Pae, M., Yamamoto, Y.,
Eberle, D., Shimada, T., Kamei, N., Park, H. S., Sasorith, S., Woo, J.
R., You, J., Mosher, W., Brady, H. J., Shoelson, S. E., and Lee, J.
(2016) Adipose natural killer cells regulate adipose tissue macrophages
to promote insulin resistance in obesity, Cell Metab.,
23, 685-698.
53.Satoh, M., and Iwabuchi, K. (2018) Role of
natural killer T cells in the development of obesity and insulin
resistance: insights from recent progress, Front. Immunol.,
9, 1314.
54.Dominguez, P. M., and Ardavin, C. (2010)
Differentiation and function of mouse monocyte-derived dendritic cells
in steady state and inflammation, Immunol. Rev., 234,
90-104.
55.Stefanovic-Racic, M., Yang, X., Turner, M. S.,
Mantell, B. S., Stolz, D. B., Sumpter, T. L., Sipula, I. J., Dedousis,
N., Scott, D. K., Morel, P. A., Thomson, A. W., and O’Doherty, R.
M. (2012) Dendritic cells promote macrophage infiltration and comprise
a substantial proportion of obesity-associated increases in
CD11c+ cells in adipose tissue and liver, Diabetes,
61, 2330-2339.
56.Sica, A., and Mantovani, A. (2012) Macrophage
plasticity and polarization: in vivo veritas, J. Clin.
Invest., 122, 787-795.
57.Stafeev, I. S., Menshikov, M. Y., Tsokolaeva, Z.
I., Shestakova, M. V., and Parfyonova, Y. V. (2015) Molecular
mechanisms of latent inflammation in metabolic syndrome. possible role
of sirtuins and peroxisome proliferator-activated receptor type gamma,
Biochemistry (Moscow), 80, 1217-1226.
58.Stafeev, I. S., Michurina, S. S., Podkuychenko,
N. V., Menshikov, M. Y., Parfyonova, Y. V., and Vorotnikov, A. V.
(2019) Chemical inducers of obesity-associated metabolic stress
activate inflammatory pathways and reduce insulin sensitivity in 3T3-L1
adipocytes, Biochemistry (Moscow), 84, 553-561.
59.Hotamisligil, G. S. (2006) Inflammation and
metabolic disorders, Nature, 444, 860-867.
60.Stafeev, I. S., Michurina, S. S., Podkuychenko,
N. V., Vorotnikov, A. V., Menshikov, M. Y., and Parfyonova, Y. V.
(2018) Interleukin-4 restores insulin sensitivity in lipid-induced
insulin-resistant adipocytes, Biochemistry (Moscow), 83,
498-506.
61.Shimobayashi, M., Albert, V., Woelnerhanssen, B.,
Frei, I. C., Weissenberger, D., Meyer-Gerspach, A. C., Clement, N.,
Moes, S., Colombi, M., Meier, J. A., Swierczynska, M. M., Jeno, P.,
Beglinger, C., Peterli, R., and Hall, M. N. (2018) Insulin resistance
causes inflammation in adipose tissue, J. Clin. Invest.,
128, 1538-1550.
62.Spalding, K. L., Arner, E., Westermark, P. O.,
Bernard, S., Buchholz, B. A., Bergmann, O., Blomqvist, L., Hoffstedt,
J., Naslund, E., Britton, T., Concha, H., Hassan, M., Ryden, M.,
Frisen, J., and Arner, P. (2008) Dynamics of fat cell turnover in
humans, Nature, 453, 783-787.
63.Stafeev, I., Podkuychenko, N., Michurina, S.,
Sklyanik, I., Panevina, A., Shestakova, E., Yah’yaev, K.,
Fedenko, V., Ratner, E., Vorotnikov, A., Menshikov, M., Yashkov, Y.,
Parfyonova, Y., and Shestakova, M. (2019) Low proliferative potential
of adipose-derived stromal cells associates with hypertrophy and
inflammation in subcutaneous and omental adipose tissue of patients
with type 2 diabetes mellitus, J. Diab. Compl., 33,
148-159.
64.Karpe, F., Dickmann, J. R., and Frayn, K. N.
(2011) Fatty acids, obesity, and insulin resistance: time for a
reevaluation, Diabetes, 60, 2441-2449.
65.Zhao, S., and Scherer, P. E. (2018) TLR4-induced
local adipose inflammation critically regulates glucose homeostasis,
Diabetes, 67 (Suppl. 1), 2032-P; doi:
10.2337/db18-2032-P.
66.Deng, Z., Xu, H., Zhang, J., Yang, C., Jin, L.,
Liu, J., Song, H., Chen, G., Han, W., and Si, Y. (2018) Infusion of
adipose-derived mesenchymal stem cells inhibits skeletal muscle
Mitsugumin 53 elevation and thereby alleviates insulin resistance in
type 2 diabetic rats, Mol. Med. Rep., 17, 8466-8474.
67.Pincu, Y., Huntsman, H. D., Zou, K., De Lisio,
M., Mahmassani, Z. S., Munroe, M. R., Garg, K., Jensen, T., and
Boppart, M. D. (2016) Diet-induced obesity regulates adipose-resident
stromal cell quantity and extracellular matrix gene expression, Stem
Cell Res., 17, 181-190.
68.Conley, S. M., Zhu, X. Y., Eirin, A., Tang, H.,
Lerman, A., van Wijnen, A. J., and Lerman, L. O. (2018) Metabolic
syndrome alters expression of insulin signaling-related genes in swine
mesenchymal stem cells, Gene, 644, 101-106.
69.Eljaafari, A., Robert, M., Chehimi, M., Chanon,
S., Durand, C., Vial, G., Bendridi, N., Madec, A. M., Disse, E.,
Laville, M., Rieusset, J., Lefai, E., Vidal, H., and Pirola, L. (2015)
Adipose tissue-derived stem cells from obese subjects contribute to
inflammation and reduced insulin response in adipocytes through
differential regulation of the Th1/Th17 balance and monocyte
activation, Diabetes, 64, 2477-2488.
70.Kornicka, K., Houston, J., and Marycz, K. (2018)
Dysfunction of mesenchymal stem cells isolated from metabolic syndrome
and type 2 diabetic patients as result of oxidative stress and
autophagy may limit their potential therapeutic use, Stem Cell
Rev., 14, 337-345.
71.Dzhoyashvili, N. A., Efimenko, A. Y., Kochegura,
T. N., Kalinina, N. I., Koptelova, N. V., Sukhareva, O. Y., Shestakova,
M. V., Akchurin, R. S., Tkachuk, V. A., and Parfyonova, Y. V. (2014)
Disturbed angiogenic activity of adipose-derived stromal cells obtained
from patients with coronary artery disease and diabetes mellitus type
2, J. Transl. Med., 12, 337.
72.Efimenko, A., Dzhoyashvili, N., Kalinina, N.,
Kochegura, T., Akchurin, R., Tkachuk, V., and Parfyonova, Y. (2014)
Adipose-derived mesenchymal stromal cells from aged patients with
coronary artery disease keep mesenchymal stromal cell properties but
exhibit characteristics of aging and have impaired angiogenic
potential, Stem Cells Transl. Med., 3, 32-41.
73.Efimenko, A. Y., Kochegura, T. N., Akopyan, Z.
A., and Parfyonova, Y. V. (2015) Autologous stem cell therapy: how
aging and chronic diseases affect stem and progenitor cells, Biores.
Open Access, 4, 26-38.
74.Ofei, F., Hurel, S., Newkirk, J., Sopwith, M.,
and Taylor, R. (1996) Effects of an engineered human anti-TNF-alpha
antibody (CDP571) on insulin sensitivity and glycemic control in
patients with NIDDM, Diabetes, 45, 881-885.
75.Dominguez, H., Storgaard, H., Rask-Madsen, C.,
Steffen Hermann, T., Ihlemann, N., Baunbjerg Nielsen, D., Spohr, C.,
Kober, L., Vaag, A., and Torp-Pedersen, C. (2005) Metabolic and
vascular effects of tumor necrosis factor-alpha blockade with
etanercept in obese patients with type 2 diabetes, J. Vasc.
Res., 42, 517-525.
76.Bernstein, L. E., Berry, J., Kim, S., Canavan,
B., and Grinspoon, S. K. (2006) Effects of etanercept in patients with
the metabolic syndrome, Arch. Intern. Med., 166,
902-908.
77.Stanley, T. L., Zanni, M. V., Johnsen, S.,
Rasheed, S., Makimura, H., Lee, H., Khor, V. K., Ahima, R. S., and
Grinspoon, S. K. (2011) TNF-alpha antagonism with etanercept decreases
glucose and increases the proportion of high molecular weight
adiponectin in obese subjects with features of the metabolic syndrome,
J. Clin. Endocrinol. Metab., 96, E146-E150.
78.Faghihimani, E., Aminorroaya, A., Rezvanian, H.,
Adibi, P., Ismail-Beigi, F., and Amini, M. (2013) Salsalate improves
glycemic control in patients with newly diagnosed type 2 diabetes,
Acta Diabetol., 50, 537-543.
79.Goldfine, A. B., Conlin, P. R., Halperin, F.,
Koska, J., Permana, P., Schwenke, D., Shoelson, S. E., and Reaven, P.
D. (2013) A randomized trial of salsalate for insulin resistance and
cardiovascular risk factors in persons with abnormal glucose tolerance,
Diabetologia, 56, 714-723.
80.Goldfine, A. B., Fonseca, V., Jablonski, K. A.,
Chen, Y. D., Tipton, L., Staten, M. A., and Shoelson, S. E.; Targeting
Inflammation Using Salsalate in Type 2 Diabetes Study Team (2013)
Salicylate (salsalate) in patients with type 2 diabetes: a randomized
trial, Ann. Intern. Med., 159, 1-12.
81.Ridker, P. M., Everett, B. M., Thuren, T.,
MacFadyen, J. G., Chang, W. H., Ballantyne, C., Fonseca,
F., Nicolau, J., Koenig, W., Anker, S.
D., Kastelein, J. J. P., Cornel, J. H., Pais,
P., Pella, D., Genest, J., Cifkova, R., Lorenzatti,
A., Forster, T., Kobalava, Z., Vida-Simiti,
L., Flather, M., Shimokawa, H., Ogawa,
H., Dellborg, M., Rossi, P. R. F., Troquay, R. P.
T., Libby, P., Glynn, R. J., and CANTOS Trial Group
(2017) Anti-inflammatory therapy with canakinumab for atherosclerotic
disease, N. Engl. J. Med., 377, 1119-1131.
82.Everett, B. M., Donath, M. Y., Pradhan, A. D.,
Thuren, T., Pais, P., Nicolau, J. C., Glynn, R. J., Libby, P., and
Ridker, P. M. (2018) Anti-inflammatory therapy with canakinumab for the
prevention and management of diabetes, J. Am. Coll. Cardiol.,
71, 2392-2401.
83.Odegaard, J. I., and Chawla, A. (2015) Type 2
responses at the interface between immunity and fat metabolism,
Curr. Opin. Immunol., 36, 67-72.
84.Odegaard, J. I., Lee, M. W., Sogawa, Y.,
Bertholet, A. M., Locksley, R. M., Weinberg, D. E., Kirichok, Y., Deo,
R. C., and Chawla, A. (2016) Perinatal licensing of thermogenesis by
IL-33 and ST2, Cell, 166, 841-854.
85.Lee, M. W., Odegaard, J. I., Mukundan, L., Qiu,
Y., Molofsky, A. B., Nussbaum, J. C., Yun, K., Locksley, R. M., and
Chawla, A. (2015) Activated type 2 innate lymphoid cells regulate beige
fat biogenesis, Cell, 160, 74-87.
86.Lee, S. E., Kang, S. G., Choi, M. J., Jung, S.
B., Ryu, M. J., Chung, H. K., Chang, J. Y., Kim, Y. K., Lee, J. H.,
Kim, K. S., Kim, H. J., Lee, H. K., Yi, H. S., and Shong, M. (2017)
Growth differentiation factor 15 mediates systemic glucose regulatory
action of T-helper type 2 cytokines, Diabetes, 66,
2774-2788.