Effect of His6-tag Position on the Expression and Properties of Phenylacetone Monooxygenase from Thermobifida fusca
P. D. Parshin1,2, A. A. Pometun1,2,3,a*, U. A. Martysuk4, S. Yu. Kleymenov3,5, D. L. Atroshenko1,2,3, E. V. Pometun6, S. S. Savin1,3, and V. I. Tishkov1,2,3,b*
1Lomonosov Moscow State University, Faculty of Chemistry, 119234 Moscow, Russia2Innovations and High Technologies MSU Ltd., 109451 Moscow, Russia
3Bach Institute of Biochemistry, Research Center of Biotechnology of the Russian Academy of Sciences, 119071 Moscow, Russia
4Mendeleev University of Chemical Technology of Russia, 125047 Moscow, Russia
5Koltzov Institute of Developmental Biology of Russian Academy of Sciences, 119334 Moscow, Russia
6Sechenov First Moscow State Medical University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received March 23, 2020; Revised March 27, 2020; Accepted March 27, 2020
Phenylacetone monooxygenase (EC 1.14.13.92, PAMО) catalyzes oxidation of ketones with molecular oxygen and NADPH with the formation of esters. PAMО is a promising enzyme for biotechnological processes. In this work, we generated genetic constructs coding for PAMO from Thermobifida fusca, containing N- or C-terminal His6-tags (PAMO N and PAMO C, respectively), as well as PAMO L with the His6-tag attached to the enzyme C-terminus via a 19-a.a. spacer. All PAMO variants were expressed as catalytically active proteins in Escherichia coli BL21(DE3) cells; however, the expression level of PAMO N was 3 to 5 times higher than for the other two enzymes. The catalytic constants (kcat) of PAMO C and PAMO L were similar to that published for PAMO L produced in a different expression system; the catalytic constant for PAMO N was slightly lower (by 15%). The values of Michaelis constants with NADPH for all PAMО variants were in agreement within the published data for PAMO L (within the experimental error); however, the KM for benzylacetone was several times higher. Thermal inactivation studies and differential scanning calorimetry demonstrated that the thermal stability of PAMO N was 3 to 4 times higher compared to that of the enzymes with the C-terminal His6-tag.
KEY WORDS: phenylacetone monooxygenase, His-tag, gene expression, mutagenesis, catalytic properties, temperature stabilityDOI: 10.1134/S0006297920050065
Abbreviations: BVMO, Baeyer–Villiger monooxygenase; DSC, differential scanning calorimetry; IPTG, isopropyl β-D-1-thiogalactopyranoside; FDH, formate dehydrogenase; PAMO, phenylacetone monooxygenase.
INTRODUCTION
Baeyer–Villiger monooxygenases (BVMOs) is a family of enzymes catalyzing a large variety of oxidative reactions that are impossible or very difficult to conduct in classic organic synthesis. Traditional methods for performing Baeyer–Villiger reactions require strong peroxide oxidants, such as 3-chloroperbenzoic acid or trifluoroperacetic acid, which conflicts with the principles of green chemistry. Although the catalyzed reaction is oxidation, BVMOs require an additional source of reducing equivalents in a form of NADPH. High regioselectivity and enantioselectivity make BVMOs extremely attractive for application in biotechnological processes [1].
A disadvantage of BVMOs that hinders their practical application is a low stability of these enzymes. BVMO from the thermophilic bacterium Thermobifida fusca, whose gene was cloned in 2004, lacks this drawback [2]. This enzyme exhibits the highest catalytic efficiency in the reaction of phenylacetone conversion into benzylacetate; hence the name phenylacetone monooxygenase (PAMO, EC 1.14.13.92). PAMO also demonstrates high catalytic constants in the oxidation of benzylacetone and other structurally similar aromatic ketones and can be used in the synthesis of flavoring substances, since the ester formed by benzylacetone oxidation has a pronounced fruity smell [2].
PAMО, like a majority of BVMOs, contains FAD and uses NADPH as an additional source of energy. PAMO is a monomeric enzyme with a molecular mass of 62 kDa. The 3D crystal structure of PAMO has been solved in [3]. The catalytic properties of this enzyme were described earlier [2], as well as its kinetic mechanism [4] and effects of organic solvents [5]. An attempt to change the coenzyme specificity from NADPH to NADH (because of lower cost of the latter) by site-directed mutagenesis has been undertaken in [6].
In most studies, PAMO-encoding genetic constructs were cloned in pBAD expression vectors under control of the arabinose promoter [7]. To obtain highly purified enzyme preparations, the His6-tag was attached to the protein C-terminus. To minimize the effect of His6-tag on the PAMО properties, the tag was separated from the enzyme C-terminus with the 19-a.a. sequence KLGPEQKLISEEDLNSAVD (PAMО L cloned in pBAD/myc-HisA vector) [2].
Here, we used a robust expression system based on the pET28a vector and Escherichia coli BL21(DE3) cells to generate recombinant PAMО. Protein expression was induced with lactose or its analog isopropyl β-D-1-thiogalactopyranoside (IPTG). To simplify enzyme purification, the expression constructs contained the pamo gene fused with a sequence coding for the His6-tag on the N- or C-terminus of the enzyme (PAMO N and PAMO C, respectively). For the purpose of comparison of the novel enzyme variants, the PAMО L construct was also cloned and expressed in the pET28a vector.
MATERIALS AND METHODS
Genetic constructs were produced by polymerase chain reaction (PCR) using as a template the pamo gene synthesized according to the AAZ55526.1 nucleotide sequence (GenBank). The primers (Evrogen, Russia) used to introduce the His6-tag into the gene sequence are shown in Table 1.
Table 1. Primers used in the work

PCR was carried out in a T100 Thermal Cycler (Bio-Rad, USA); the reaction mixture (25 μl) contained 2.5 μl of 10× PCR buffer supplied by the manufacturer along with the enzyme, 1 μl of MgCl2 (25 mM), 2 μl of dNTP solution (2.5 mM each), 1 μl of genomic DNA (50 ng/μl), 0.5 μl of Phusion DNA polymerase (5 u/μl; Thermo Fisher Scientific, USA), 1 μl of primers (20 pmol each), and 17 μl of deionized water. PCR was carried out in the following regime: 30 cycles (10 s at 98°C, 10 s at 72°C, 1.5 min at 72°C), followed by elongation for 10 min at 72°C.
PCR products were purified by electrophoresis in 1% agarose gel and digested with NcoI and HindIII endonucleases (Thermo Fisher Scientific, USA) (PAMO N and PAMO C) or NcoI and XhoI endonucleases (Thermo Fisher Scientific) (PAMO L). DNA digestion products were purified by electrophoresis in 1% agarose gel and ligated into the pET28a plasmid digested with the corresponding restriction endonucleases. E. coli DH5α cells were transformed with the ligation mixture, plated on Petri dishes with agar medium (Difco, Germany) containing kanamycin (30 μg/ml), and incubated for 16 h at 37°C. Three clones were selected from each Petri dish to isolate the plasmids. The obtained constructs were verified by DNA sequencing at the Genome Collective Use Center (Engelhardt Institute of Molecular Biology, Russian Academy of Sciences).
Protein expression. E. coli BL21(DE3) cells were transformed with the plasmid DNA. The resulting clones were transferred to 4 ml of LB medium (Difco) containing 30 μg/ml kanamycin and incubated overnight at 37°C in a shaker (180 rpm). The cells from the overnight culture were seeded in 200 ml of fresh TB medium [24 g/liter yeast extract, 12 g/liter bactotriptone (Difco), 4 ml/liter glycerol, 0.017 M KH2PO4, 0.072 M K2HPO4, pH 7.0] with 30 μg/ml kanamycin and grown at 37°C at 120 rpm until the optical absorbance A600 of 0.8. Protein expression was induced by adding IPTG to a final concentration of 200 μM, and the cells were incubated for another 24 or 7 h at 120 rpm at various temperatures (30 or 37°C). The cells were then pelleted by centrifugation in a Beckman J21 centrifuge (Beckman, USA) for 20 min at 6000 rpm at 4°C. The pellet was resuspended in a cold buffer (50 mM Tris-HCl, 500 mM NaCl and 20 mM imidazole, pH 7.5).
Protein purification. Aqueous FAD solution was added to the cell suspension to a final concentration of 10 μM and the cells were disintegrated by sonication with a Branson Sonifier 250 ultrasonic disintegrator (Thermo Fisher Scientific, USA) in 4-5 cycles of 90 s on ice with interruptions for cooling. The lysate was incubated at 55°C for 10 min; the cell debris was removed by centrifugation in a Beckman J21 centrifuge (Beckman, USA) (40 min, 10,000 rpm, 4°C). The resulting cell-free extract was applied onto a 1-ml HisTrap HP column (GE Healthcare Life Science, USA) equilibrated with 50 mM Tris-HCl buffer (pH 7.5) containing 500 mM NaCl and 20 mM imidazole, and proteins were eluted with a 20-500 mM linear gradient of imidazole concentration. Fractions containing the target protein were transferred to 50 mM sodium phosphate buffer (pH 7.5) by gel filtration on a Sephadex G-25 column (Pharmacia Biotech, Sweden). The efficiency of purification was monitored by denaturing electrophoresis using a Mini Protean 2 device (BioRad, Austria) according to the manufacturer's protocol.
PAMO tryptic digestion in polyacrylamide gel. After Coomassie Brilliant Blue staining, a piece of the gel containing the enzyme (3-4 mm3) was cut out, washed twice with 100 μl of 40% acetonitrile in 0.1 M NH4HCO3 for 20 min at 37°C to remove the dye, and transferred to 100 μl of acetonitrile for dehydration. After dehydration, the gel piece was dried to remove acetonitrile and 3.5 μl of modified trypsin solution (Promega, USA) in 0.05 M NH4HCO3 was added. Hydrolysis was carried out for 20 h at 37°C; next, 5.25 μl of 0.5% trifluoroacetic acid (TFA) in 50% aqueous acetonitrile solution was added tom the reaction mixture and thoroughly mixed. The resulting supernatant was used for MALDI mass spectroscopy analysis.
MALDI mass spectrometry analysis was performed using equipment of the Industrial Biotechnologies Center for Collective Use of the Fundamentals of Biotechnology Federal Research Center, Russian Academy of Sciences.
The samples for mass spectrometry were prepared by mixing 1.5 μl of the tryptic hydrolysate with 0.5 μl of 2,5-dihydroxybenzoic acid (10 mg/ml in 20% aqueous acetonitrile, 0.5% TFA; Sigma, Germany). The resulting mixture was air-dried.
Mass spectra were recorded with an Ultraflextreme MALDI-TOF/TOF mass spectrometer (Bruker, Germany) equipped with a positive-ion UV laser (Nd) and a reflectron. The accuracy of measurement of monoisotopic masses after recalibration with the peaks for trypsin autolysate was 0.002-0.011% (20-110 ppm). The spectra were obtained within the mass range of 500-6500 m/z; laser power was selected to achieve the best resolution.
Proteins were identified using the Mascot software (www.matrixscience.com). Mass spectra were processed with the FlexAnalysis 3.3 software package (Bruker Daltonics, Germany). Using the Mascot program (“peptide fingerprint” option), we searched a local database with the above accuracy, taking into account the following possible modifications: acetylation (protein N-terminus), Gln pyroGlu (N-terminal Q), oxidation (M), propionamide (C). Candidate proteins with the confidence score >42 in the NCBI database were considered as reliably identified (p < 0.05).
Determination of catalytic parameters. The concentration of the enzyme active sites was determined spectrophotometrically with a Shimadzu 1800PC spectrophotometer (Shimadzu GmbH, Germany) from FAD absorbance at 441 nm (ε = 12.4 mM–1 cm–1). PAMO solution was incubated at 55°C for 10 min before the assay; the reaction was carried out at 30°C and monitored by NADPH consumption that was measured by the absorbance decrease at 340 nm. The total reaction volume was 1 ml; the reaction mixture contained 50 mM sodium phosphate buffer (pH 7.5), 50 μM NADPH, and 10 mM benzylacetone. To determine KM, the concentrations of NADPH and benzylacetone were varied in the 5-500 μM and 0.25-15 mM concentration ranges, respectively.
Kinetics of thermal inactivation. The thermal stability of the PAMO enzymes was studied in 0.05 M sodium phosphate buffer (pH 7.5) at varied temperatures. For each experiment, a series of 0.5 ml plastic tubes with 100 μl of enzyme solution (0.2 mg/ml) was prepared. The tubes were placed in a water bath (Waters, USA) preheated to the required temperature (thermostat accuracy, ± 0.1°C). At certain time points, the tubes were removed from the water bath and placed on ice for 5 min, followed by centrifugation for 3 min at 12,000 rpm in an Eppendorf 5415D centrifuge (Eppendorf, Germany). The supernatant was assayed for the PAMO residual activity as described above. The thermal inactivation rate constant (kin) was determined as a slope of the linear regression plot in the semi-logarithmic coordinates ln(A/A0) versus time using the Origin Pro 8.5 software (OriginLab Corporation, USA).
Differential scanning calorimetry (DSC). The dependence of PAMO heat capacity on temperature was studied using a MicroCal VP DSC differential adiabatic scanning microcalorimeter (Malvern Panalytical Ltd, Great Britain). The working volume of tantalum capillary cells was 140 μl. To prevent bubble formation and solution boiling with a temperature increase, excessive pressure (3 atm) was maintained in the calorimeter cells; calibration was carried out by applying a fixed power (25 μW) per cell. The instrumental baseline was determined prior to the experiment. The working cell was filled with the PAMO solution and the control cell was filled with a buffer. The enzyme concentration was 1 mg/ml; the heating rate was 1°C/min.
RESULTS
PAMO expression and purification. Metal-affinity chromatography is one of the most efficient and commonly used protein purification methods. This purification procedure requires the presence of 6 to 12 histidine residues (His-tag) at the N- or C-terminus of a protein [8]. Although His-tag makes purification much easier, its introduction may also cause significant changes (and sometimes worsening) of protein properties, as it has been demonstrated for various enzymes [9-12]. Therefore, the effect of His-tag on the enzyme catalytic properties and stability must be studied empirically. The goal of this work was to generate PAMO constructs with the His-tag located at the N- and C-termini of the enzyme (PAMO N and PAMO C, respectively) and to compare the properties of the modified enzymes. For more comprehensive comparison, we also reproduced previously described PAMO L construct with the His-tag distanced from the enzyme C-terminus with an additional 19-a.a. sequence [2]. Expression vectors for the three PAMO variants were generated by inserting nucleotides encoding the His6-tag into the pamo gene sequence by PCR. Sequencing of the resulted plasmids confirmed the absence of undesired nucleotide replacements in the generated sequences.
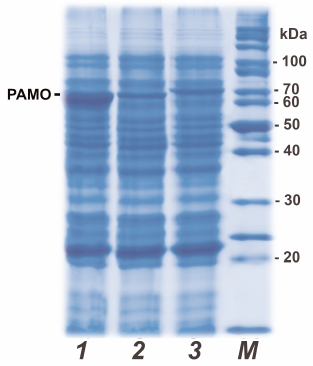
In this work, we avoided the use of expression systems with the arabinose promoter because of high costs of L-arabinose (expression inducer). Instead, we used a robust system based on the pET28a expression vector and E. coli BL21(DE3) cells to produce recombinant PAMO variants. In this case, expression of the target gene is induced with IPTG or lactose, which is significantly cheaper. To optimize the expression conditions for PAMO, we varied the growth medium composition, IPTG concentration, temperature, and duration of cultivation. The highest yield was achieved for 24-h expression after induction with 0.2 mM IPTG in TB medium at 30°C. Early cell lysis was observed at 37°C, while shortening of the protein synthesis time to 8 h or switching to 2YT medium resulted in lower protein yields. It should be noted that even if changes in the growth conditions equally affected the expression levels of all three PAMO variants, the level of PAMО N expression was always 3 to 5 times higher (Fig. 1, line 1) than the expression levels of PAMO C and PAMО L under the same conditions (Fig. 1, lines 2 and 3, respectively). As seen from Fig.1, the presence of additional 19 a.a. residues in PAMO L (lane 3) slightly increased its molecular mass in comparison to PAMО N and PAMО C (lanes 1 and 2).
Fig. 1. Analytical denaturing electrophoresis of E. coli cells after induction of PAMO expression: (1-3) cells expressing PAMO N, PAMO C and PAMO L, respectively; M, molecular weight markers.
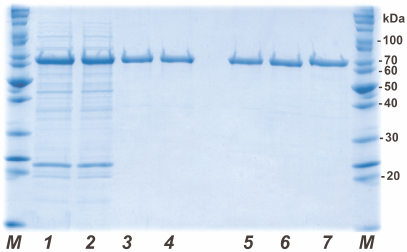
His6-tagged PAMО proteins were purified by metal-affinity chromatography. Figure 2 shows the results of analytical denaturing electrophoresis of PAMО N preparations at different purification stages (lanes 1-4) and purified preparations of PAMО N, PAMО C, and PAMО L (lanes 5-7, respectively). All three variants of recombinant His6-tagged PAMО were purified to near-homogeneity in one step. To confirm the authenticity of the purified proteins, the obtained preparations were purified from the gel and analyzed by MALDI-TOF/TOF. Mass spectrometry analysis revealed no post-translational modification in the PAMO proteins expressed in E. coli cells and confirmed correct positioning of the His6-tag.
Fig. 2. Analytical denaturing electrophoresis of enzyme preparations at different stages of purification. Stages of PAMO N purification: (1) cell lysate, (2) thermal treatment of cell lysate, (3) metal-affinity chromatography, and (4) desalting. Purified PAMO preparations: (5) PAMO N, (6) PAMO C, and (7) PAMO L; M, molecular weight markers.
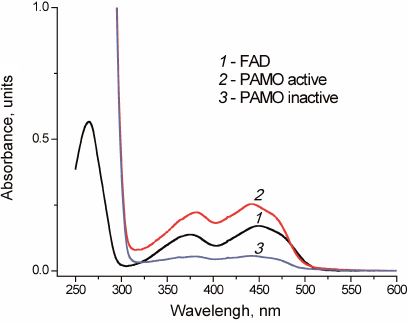
All three variants of recombinant PAMO lost some FAD during purification, however, their activity was restored by incubation with free FAD. Successful replenishment with FAD was confirmed by the enzyme spectrum after desalting used to remove unbound FAD (Fig. 3).
Fig. 3. Absorbance spectra of free FAD (1) and PAMO before (3) and after (2) FAD addition in 0.05 M Na-phosphate buffer, pH 7.5.
Kinetic properties. Table 2 shows kinetic parameters of PAMO proteins with different position of the His6-tag, which proves that position of the His6-tag has no significant effect on the enzyme catalytic parameters. The catalytic constant for PAMO N was slightly lower, but the difference was within the limits of experimental error. The Michaelis constants of the obtained enzymes with NADPH were similar to that for PAMO L produced in a different expression system [2] (within the experimental error), However, for all the three enzymes, the KM value with benzylacetone was several times higher than the one published in [2]. The activities of all three enzymes linearly depended on the enzyme concentration.
Table 2. Kinetic parameters for three PAMO
enzymes with different position of the His6-tag (50 mM
Tris-HCl, pH 7.5, 30°C)*
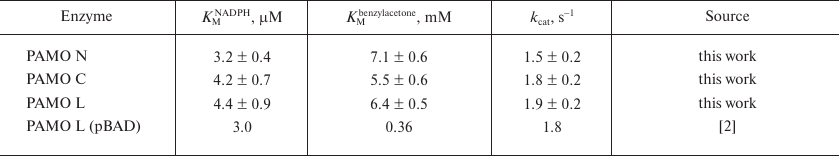
* The data are mean values from triple independent experiments.
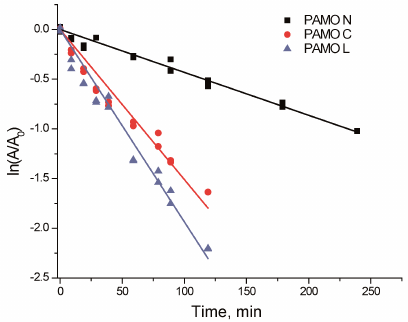
Thermal stability. Figure 4 shows the time course of PAMO inactivation in semi-logarithmic coordinates upon incubation at 59°C. The linear character of the observed dependencies indicates the first-order kinetics of the enzyme thermal inactivation. The apparent rate constants of enzyme inactivation (kin) were calculated from the linear dependence slope. The apparent inactivation rate constant of PAMO N was 3 to 4 times lower compared to those for the enzymes with the C-terminal His6-tag.
Fig. 4. Residual activity of three PAMO variants upon incubation at 59°C (0.05 M Na-phosphate, pH 7.5).
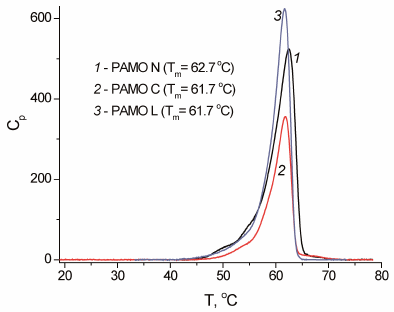
The results on the enzyme inactivation kinetics were in agreement with the data generated by DSC (Fig. 5). The melting temperature of the PAMO N protein globule was 1°C higher compared to the other two enzymes (Fig. 5). Considering that the rate constant for the protein thermal inactivation increases 10-fold on average with the temperature rise by 3°C, the observed 1°C difference in the melting temperature of PAMО variants agrees with the observed difference in the apparent rate constants of thermal inactivation for these enzymes.
Fig. 5. Melting curves for three PAMO enzymes with different position of the His6-tag (0.05 M Na-phosphate buffer, pH 7.5; enzyme concentration, 0.7-1.0 mg/ml; scan rate, 1°C/min).
DISCUSSION
NAD(P)+-dependent oxidoreductases are highly efficient enzymes that can be used for a high-yield synthesis of >99% optically pure products that cannot be obtained using the methods of traditional organic chemistry. However, to make enzymatic catalysis cost-effective, one has to reduce the cost of the coenzyme NAD(P)H. This can be achieved by coupling the main reaction with the NAD(P)H regeneration system that would convert its oxidized form, NAD(P)+, back into NADPH. One of the enzymes widely used for the coenzyme regeneration is formate dehydrogenase (FDH). The first biocatalytic process for the production of ε-caprolactones in the Baeyer–Villiger reaction with the use of cyclohexanone monooxygenase and enzymatic regeneration of the reduced coenzyme was described in 1997 [13, 14]. To reduce NADP+ to NADPH, mutant FDH from Pseudomonas sp.101 (NP-PseFDH) was used, after its coenzyme specificity had been altered from NAD+ to NADP+ by site-directed mutagenesis [15-17].
The production capacity of a system involving two separate soluble biocatalysts is determined by the catalytic properties of individual enzymes. Creation of fusion proteins that combine enzymes catalyzing coupled reactions into a single polypeptide chain has become a popular approach for enhancing the efficiency of biocatalysis. Bringing two active sites in a close proximity in the same protein globule often results in a multiple increase in the catalytic efficiency compared to a mixture of two individual enzymes. This study was conducted within a framework of the project on constructing chimeric biocatalysts, in particular, a fusion enzyme combining mutant NADP+-specific FDH from Pseudomonas sp. 101 (PseFDH) and PAMО. We have demonstrated that the optimal arrangement for PseFDH included an additional polypeptide attached to the enzyme N-terminus [18]. Therefore, placing PAMO before PseFDH in the fusion enzyme seemed as a logical development. To purify this hybrid enzyme, His-tag has to be attached to the PAMО N-terminus. Since no information had been available on PAMO N, we produced and characterized this enzyme. It was also interesting to elucidate the need for a spacer peptide between the PAMО C-terminus and His6-tag, introduced earlier by others researchers in [2].
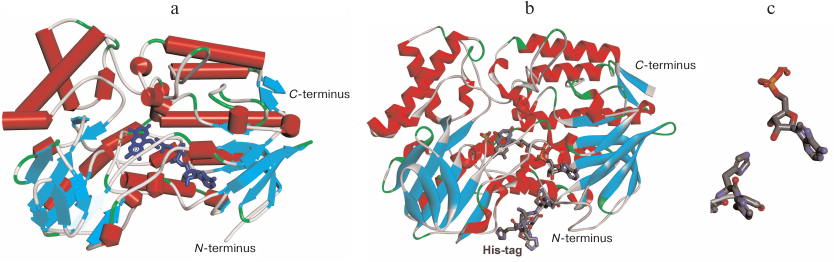
The results of our experiments prove one more time that there the effects of His-tag introduced at the N- or C-termini of an enzyme require a separate study. In the case of PAMО, addition of the His6-tag for affinity purification had almost no effect on the enzyme catalytic properties (the values of catalytic constants were similar within the experimental error). The absence of His6-tag positional effect on PAMО properties is in agreement with the data of X-ray structural analysis [3]. Figure 6a shows the crystal structure of the enzyme (PDB 1W4X) with indicated N- and C-termini. It is clearly seen that PAMO N- and C-termini are located equally far from the substrate-binding site and binding site for NADPH and FAD isoalloxazine moiety. The reason for the difference in the KM values with benzylacetone for the enzyme variants produced in this study and PAMO L expressed in a different system is not clear. It is possible that expression system can affect protein folding and conformation; however, worsening of one catalytic parameter only and not the others is difficult to explain. Additional research on the PAMO reaction with benzylacetone using gas chromatography may be necessary.
Fig. 6. a) 3D structure of PAMO (PDB 1W4X) indicating location of the N- and C-termini. b) PAMO N model with the addition of missing 10 a.a. residues and N-terminal His6-tag. c) Space orientation of FAD adenine group and the fifth residue of the His6-tag.
Unexpectedly, introduction of the His6-tag at the N-terminus of PAMO increased 3 to 5 times both the enzyme expression level and thermal stability. One of the reasons for a higher expression level in E. coli cells could be the optimal codon usage for the His6-tag histidine residues compared to the codons used for amino acid residues at the N-terminal sequence of the enzyme itself. A 3- to 5-fold increase in the enzyme expression level is an important factor for the practical applications of PAMO.
The higher thermal stability of PAMO N in comparison with the enzymes with the C-terminal His6-tag may result from the conformational changes induced by the N-terminal His6-tag. X-ray analysis indicated a very high flexibility of the N-terminus in the wild-type PAMO, since the resolved crystal structure lacked the first ten residues of the polypeptide chain [3], whereas the C-terminus was well structured (Fig. 6а). Our results and published data [2] suggest the presence of a comparatively loosely bound FAD in the active site. Hence, the most likely mechanism of PAMО thermal inactivation is FAD dissociation from the active site. Such mechanism is highly plausible because adenine moiety of FAD is exposed to the solvent (Fig. 6). We created a structural model of PAMO N by adding ten amino acid residues lacking in the crystal structure at the enzyme N-terminus and the His6-tag itself (Fig. 6b). Modeling and structural optimization were performed as described in detail in our study of His6-tagged PseFDH [18]. Analysis of the PAMO N model (Fig. 6b) demonstrated that the N-terminus of PAMO N containing positively charged His6-tag can come in close proximity to the adenine moiety of FAD. Modeling data support a possibility of hydrogen bond formation between the fifth residue of His6-tag and 3′-OH group and, to some extent, 2′-OH group of adenosine ribose (Fig. 6c), whereas electrostatic interaction between imidazoles in the His6-tag and negatively charged pyrophosphate of FAD are spatially restricted. The hydrogen bond suggested by the model may explain an increase in the enzyme thermal stability by the N-terminus structuring, which strengthens the binding of FAD ribose and ensures additional cofactor screening from the solvent. The stabilization effect corresponds to the energy of a newly formed hydrogen bond. On the contrary, the presence of a long spacer between the PAMO C-terminus and His6-tag in PAMО L compromises C-terminus structuring and results in a lower thermal stability of PAMO L vs. PAMO C (Fig. 4).
Based on the discovered positive effects of the introduced N-terminal His6-tag in PAMO N, we have started experiments on the construction and characterization of a fusion protein composed of PAMO and NP-PseFDH. The results of this ongoing work will be published in the nearest future.
Funding. The work was supported in part by a joint grant from the Russian Foundation for Basic Research and Moscow Government (project No. 19-34-70036). Some experiments were carried out using equipment from the Genome Shared-Access Equipment Center, Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, and Industrial Biotechnology Shared-Access Equipment Center, Fundamentals of Biotechnology Federal Research Center, Russian Academy of Sciences.
Acknowledgement. The authors express their gratitude to Prof. Irina Gazaryan for valuable comments on the manuscript.
Conflict of interests. The authors declare no conflict of interests.
Compliance with ethical standards. This article does not contain descriptions of studies involving humans or animals performed by any of the authors.
REFERENCES
1.Kamerbeek, N. M., Janssen, D. B., van Berkel, W. J.
H., and Fraaije, M. W. (2003) Baeyer–Villiger monooxygenases, an
emerging family of flavin-dependent biocatalysts, Adv. Synth.
Catal., 345, 667-678, doi: 10.1002/adsc.200303014.
2.Fraaije, M. W., Wu, J., Heuts, D. P. H. M., Van
Hellemond, E. W., Spelberg, J. H. L., and Janssen, D. B. (2005).
Discovery of a thermostable Baeyer–Villiger monooxygenase by
genome mining, Appl. Microbiol. Biotechnol., 66, 393-400,
doi: 10.1007/s00253-004-1749-5.
3.Malito, E., Alfieri, A., Fraaije, M. W., and
Mattevi, A. (2004) Crystal structure of a Baeyer–Villiger
monooxygenase, Proc. Natl. Acad. Sci., 101, 13157-13162,
doi: 10.1073/pnas.0404538101.
4.Torres Pazmiño, D. E., Baas, B. J., Janssen,
D. B., and Fraaije, M. W. (2008). Kinetic mechanism of phenylacetone
monooxygenase from Thermobifida fusca, Biochemistry,
47, 4082-4093, doi: 10.1021/bi702296k.
5.Secundo, F., Fialà, S., Fraaije, M. W., De
Gonzalo, G., Meli, M., Zambianchi, F., and Ottolina, G. (2011) Effects
of water miscible organic solvents on the activity and conformation of
the Baeyer–Villiger monooxygenases from Thermobifida fusca
and Acinetobacter calcoaceticus: a comparative study,
Biotechnol. Bioeng., 108, 491-499,
doi: 10.1002/bit.22963.
6.Jensen, C. N., Ali, S. T., Allen, M. J., and
Grogan, G. (2013) Mutations of an NAD(P)H-dependent flavoprotein
monooxygenase that influence cofactor promiscuity and
enantioselectivity, FEBS Open Bio, 3, 473-478,
doi: 10.1016/j.fob.2013.09.008.
7.Van Bloois, E., Dudek, H. M., Duetz, W. A., and
Fraaije, M. W. (2012) A stepwise approach for the reproducible
optimization of PAMO expression in Escherichia coli for
whole-cell biocatalysis, BMC Biotechnol., 12, 12-31,
doi: 10.1186/1472-6750-12-31.
8.Arnau, J., Lauritzen, C., Petersen, G. E., and
Pedersen, J. (2006) Current strategies for the use of affinity tags and
tag removal for the purification of recombinant proteins, Protein
Expr. Purif., 48, 1-13,
doi: 10.1016/j.pep.2005.12.002.
9.Zhao, D., and Huang, Z. (2016) Effect of His-tag on
expression, purification, and structure of zinc finger protein,
ZNF191(243-368), Bioinorg. Chem. Appl., 2016, 8206854,
doi: 10.1155/2016/8206854.
10.Tishkov, V. I., Pometun, A. A., Stepashkina, A.
V., Fedorchuk, V. V., Zarubina, S. A., Kargov, I. S., Atroshenko, D.
L., Parshin, P. D., Kovalevski, R. P., Boiko, K. M., Eldarov, M. A.,
D'Oronzo, E., Facheris, S., Secundo, F., and Savin, S. S. (2018)
Rational design of practically important enzymes, Moscow Univ. Chem.
Bull., 73, 1-6, doi: 10.3103/S0027131418020153.
11.Booth, W. T., Schlachter, C. R., Pote, S., Ussin,
N., Mank, N. J., Klapper, V., Offermann, L. R., Tang, C., Hurlburt, B.
K., and Chruszcz, M. (2018) Impact of an N-terminal
polyhistidine tag on protein thermal stability, ACS Omega,
3, 760-768, doi: 10.1021/acsomega.7b01598.
12.Esen, H., Alpdağtaş, S., Mervan
Çakar, M., and Binay, B. (2019) Tailoring of recombinant FDH:
effect of histidine tag location on solubility and catalytic properties
of Chaetomium thermophilum formate dehydrogenase (CtFDH),
Prep. Biochem. Biotechnol., 49, 529-534,
doi: 10.1080/10826068.2019.1599394.
13.Rissom, S., Schwarz-Linek, U., Vogel, M.,
Tishkov, V.I., and Kragl, U. (1997) Synthesis of chiral
ε-lactones in a two-enzyme system of cyclohexanone
monooxygenase and formate dehydrogenase with integrated bubble-free
aeration, Tetrahedron: Assymetry, 8, 2523-2526,
doi: 10.1016/S0957-4166(97)00311-X.
14.Schwarz-Linek, U., Krödel, A., Ludwig,
F.-A., Schulze, A., Rissom, S., Kragl, U., Tishkov, V. I., and Vogel,
M. (2001) Synthesis of natural product precursors by
Baeyer–Villiger oxidation with cyclohexanone monooxygenase from
Acinetobacter, Synthesis, 33, 947-951,
doi: 10.1055/s-2001-13394.
15.Tishkov, V. I. (1993) Structure, catalytic
mechanism and protein engineering of NAD+-dependent formate
dehydrogenase, Doctor of Sciences Dissertation, Moscow State
University, Moscow.
16.Tishkov, V. I., Galkin, A. G., Fedorchuk, V. V.,
Savitsky, P. A., Rojkova, A. M., Gieren, H., and Kula, M.-R. (1999)
Pilot scale production and isolation of recombinant NAD+-
and NADP+-specific formate dehydrogenase, Biotechnol.
Bioeng., 64, 187-193,
doi: 10.1002/(SICI)1097-0290(19990720)64:2<187::AID-BIT7>3.0.CO;2-0.
17.Serov, A. E., Popova, A. S., Fedorchuk, V. V.,
and Tishkov, V. I. (2002) Engineering of coenzyme specificity of
formate dehydrogenase from Saccharomyces cerevisiae, Biochem.
J., 367, 841-847, doi: 10.1042/BJ20020379.
18.Pometun, A. A., Parshin, P. D., Galanicheva, N.
P., Uporov, I. V., Atroshenko, D. L., Savin, S. S., and Tishkov, V. I.
(2020) Influence of His6-sequence on properties of formate
dehydrogenase from bacterium Pseudomonas sp. 101, Moscow
Univ. Chem. Bull., 75, 4 [in English].