REVIEW: The Mechanisms of L-Arginine Metabolism Disorder in Endothelial Cells
Jennet T. Mammedova1, Alexey V. Sokolov1, Irina S. Freidlin1, and Eleonora A. Starikova1,a*
1Institute of Experimental Medicine, 197376 Saint-Petersburg, Russia* To whom correspondence should be addressed.
Received August 6, 2020; Revised September 14, 2020; Accepted September 14, 2020
L-arginine is a key metabolite for nitric oxide production by endothelial cells, as well as signaling molecule of the mTOR signaling pathway. mTOR supports endothelial cells homeostasis and regulates activity of L-arginine-metabolizing enzymes, endothelial nitric oxide synthase, and arginase II. Disruption of the L-arginine metabolism in endothelial cells leads to the development of endothelial dysfunction. Conflicting results of the use of L-arginine supplement to improve endothelial function reveals a controversial role of the amino acid in the endothelial cell biology. The review is aimed at analysis of the current data on the role of L-arginine metabolism in the development of endothelial dysfunction.
KEY WORDS: L-arginine, endothelium, nitric oxide, eNOS, arginase, mTORDOI: 10.1134/S0006297921020036
Abbreviations: ADMA, asymmetric dimethyl arginine; BH4, tetrahydrobiopterin; CaM, calmodulin; EC, endothelial cells; eNOS, endothelial nitric oxide synthase; Hsp90, heat shock protein 90; HUVEC, human umbilical vein endothelial cells; mTOR, mechanical target of rapamycin; NADP, nicotinamide adenine dinucleotide phosphate; NO, nitric oxide; ROS, reactive oxygen species.
INTRODUCTION
Endothelial nitric oxide synthase (eNOS) is an enzyme that catalyzes the reaction of nitric oxide (NO) synthesis from L-arginine. NO controls blood vessels tone and permeability by activating guanylyl cyclase and increasing the level of cyclic 3′,5′-guanosine monophosphate (cGMP) in smooth muscle cells [1]. NO inhibits proliferation of the vascular smooth muscle cells, peroxidation of cell membranes and blood lipoproteins, aggregation of platelet, leukocyte adhesion, and regulates the blood gas transport properties [2]. The key role of NO in maintaining vascular homeostasis was proved in the studies of eNOS knockout mice that developed hypertension [3], demonstrated increased proliferation of vascular smooth muscle cells in response to damage [4], leukocyte adhesion to the endothelium [5], hypercoagulopathy [6], atherosclerosis exacerbation [7], and impaired angiogenesis regulation [8]. It was found that the L-arginine supplementation increased production of NO by endothelial cells (EC) [9, 10], hence, the use of the amino acid as a dietary supplement correcting endothelial function was studied in various pathologies in humans and in animal models [11-14]. However, a number of studies have shown that L-arginine supplementation has no positive effect [15], and in some cases even exacerbates endothelial dysfunction [16]. This indicates an ambiguous role of L-arginine in the regulation of eNOS function.
Tonic NO production by ECs is provided by a variety of processes that directly depend on the metabolism of L-arginine. L-arginine is not only a common substrate for eNOS and arginase which regulate each other [17], but also serves as a signaling molecule of mechanical target of rapamycin (mTOR) signaling pathway [18] closely related to the metabolism of the amino acid [19, 20]. All this makes L-arginine one of the key factors regulating vascular homeostasis. The review analyzes the literature data on the role of L-arginine metabolism disorder in the development of endothelial dysfunction.
eNOS ACTIVITY REGULATION
eNOS is constitutively expressed in ECs. Activity of the enzyme changes in response to hydrodynamic stress, coagulation, vasoconstriction, and hypoxia and is regulated by phosphorylation of its amino acid residues causing either activation (S615, S633, and S1177) or inhibition (S114, T495, Y656) of the enzyme [21]. Ca2+/calmodulin (CaM) mediated eNOS activation is caused by bradykinin, estradiol, and vascular endothelial growth factor (VEGF) [21]. Serotonin induces the enzyme activation, when platelets are activated and coagulation processes are enhanced [22]. The increase in the concentration of intracellular calcium leads to the CaM activation, which interacts with the CaM-binding site of eNOS, and blocks T495 – the enzyme inhibiting phosphorylation site. Simultaneously, CaM-mediated phosphorylation of calcium/Calmodulin-dependent protein Kinase II (CaMKII) leads to phosphorylation of the eNOS activating phosphorylation site (S1177) [23]. Shear stress, production of reactive oxygen species (ROS), and growth factors induce Akt1-mediated phosphorylation of eNOS at two sites S615 and S1177 [24]. Increased concentrations of AMP and intracellular calcium, as well as nutrient deficiency activate AMPK, induce eNOS phosphorylation at S1177, and NO production [25]. In studies on animals and EC cultures, activation of cAMP-dependent protein kinase A has been shown to restore endothelial barrier function by phosphorylating eNOS at S1177 [21, 26]. According to the recent data, the metabolic regulator mTOR, which integrates signals from Akt, AMPK, and CAMKII, also activates eNOS by direct phosphorylation at S1177 [19]. Under shear stress, Pp60src and PYK2 kinases regulate eNOS activity by phosphorylating at Y81 and Y657, respectively [26-28]. When exposed to damaging factors, such as oxidative, heat, or shear stress, the expression of heat shock protein 90 (Hsp90) increases in cells. The main function of Hsp90 is to stabilize conformational folding of proteins. In ECs, Hsp90 stabilizes the structure and supports the function of eNOS by binding to the enzyme oxygenase domain. In the study by Förstermann et al., Hsp90 was shown to increase the affinity of eNOS for L-arginine, CaM, and NADPH, which in turn increased the enzyme activity [29].
Located in the Ca2+/CaM-binding site of eNOS, T495 is the main site that negatively regulates activity of the enzyme [26]. T495 phosphorylation mediated by PKC, Rho kinases [30] and AMPK [31] prevents formation of CaM complex with eNOS. Also, Rho kinase suppresses eNOS activity in an indirect way, through inhibition of CaM [32], Akt, and AMPK signaling molecules [30, 33]. Localization of eNOS in caveolae makes negative regulation of the enzyme activity by caveolin-1 [33] possible, which prevents CaM binding to the enzyme at low concentrations of intracellular calcium [21, 27].
THE ROLE OF L-ARGININE DEFICIENCY IN eNOS REGULATION
In its fully functional state, eNOS is a homodimer with two domains in each of monomers: the oxygenase domain that interacts with heme, L-arginine, and tetrahydrobiopterin (BH4) and the reductase domain that interacts with NADPH synthase and CaM (Fig. 1).
Fig. 1. Schematic illustration of eNOS functional activity in the presence or absence of the substrate. a) eNOS activity when sufficient amount of the substrate is present; b) eNOS activity when the substrate is deficient; c) eNOS activity when the enzyme dissociates.
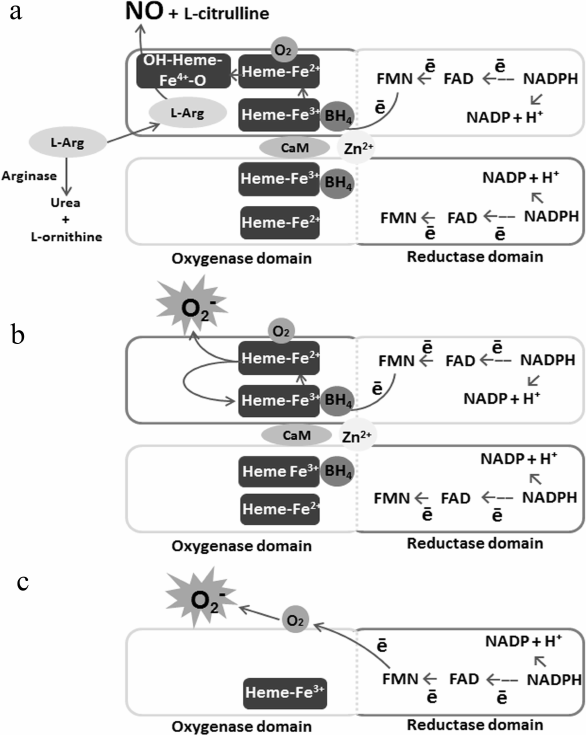
eNOS performs sequential electron transfer from the NADPH reductase domain of one monomer, via flavin adenine nucleotide (FAD) and flavin mononucleotide (FMN), to the heme oxygenase domain of the other monomer, where L-arginine is oxidized with BH4 and oxygen to form L-citrulline and NO [34, 35]. Under the effect of proinflammatory factors, oxidative stress, and L-arginine deficiency, the dimeric structure of eNOS is disrupted [36]. Due to the fact that electron transport is carried out in the trans-position from the reductase domain of one subunit to the oxygenase domain of the other eNOS subunit, the enzyme dissociation leads to separation of NADPH oxidation processes and a decrease in NO synthesis. Suppression of tonic production of NO leads to increased inflammation and thrombosis in blood vessels [37]. These processes are enhanced under the conditions of substrate deficiency, since oxygen becomes the terminal electron acceptor instead of L-arginine, hence, eNOS synthesizes the superoxide anion radical – O2•– in large amounts. The interaction of O2•– with NO leads to formation of peroxynitrite (ONOO–) oxidizing BH4 into BH2, which, in turn, causes eNOS uncoupling and amplification of the process. Hyperproduction of O2•– and ONOO– results in ECs activation, induces expression of adhesion molecules and development of inflammation [36]. High ROS concentration has cytotoxic and mutagenic effects, causes ECs senescence and apoptosis, increases the risk of atherosclerosis and hypertension development [38]. Thus, phosphorylation of dissociated eNOS at activating sites is seen as one of the factors bringing about oxidative stress, inflammation, coagulation, and vascular tone deregulation. Therefore, under conditions of the substrate deficiency, eNOS activity should to be inhibited. However, a decrease in NO production with activation of the enzyme is often observed in the pathologies associated with endothelial dysfunction [39].
ABSOLUTE L-ARGININE DEFICIENCY
A number of studies on cell cultures and laboratory animals have shown that the eNOS activity depends on the concentration of extracellular L-arginine [9, 10]. The synthesis of NO in human umbilical vein endothelial cells (HUVECs) depends on the concentration of L-arginine in the culture medium. It was shown that under conditions of L-arginine deficiency, NO production induced by bFGF was completely inhibited. L-arginine supplementation restored the production of the metabolite [40]. In experiments on the EA.hy926 EC line, it was shown that partial removal of the L-arginine transporter CAT-1 resulted in the decrease in NO production [41]. The condition of cardiovascular system of a person suffering a rare genetic L-arginine metabolism disorder has been described in the literature. Concentration of L-arginine in the plasma of this patient was reduced by 79% compared to the reference values due to mutation in the SLC7A7 gene encoding the amino acid transporter y + LAT-1. The consequences of L-arginine deficiency included development of myocardial ischemia, disruption of vasomotor vascular function, decrease in the absolute number of platelets, increase in the concentration of the thrombin-antithrombin III complex and the products of fibrin degradation in plasma [42]. The results obtained in this study raise many questions. In particular, absence of the amino acid transporter y + LAT-1 can lead to disruption of the transport of not only L-arginine, but a number of other amino acids. Therefore, the changes detected in this patient may be caused by various reasons. On the other hand, the changes expected for the blood from a patient with L-arginine deficiency were not detected indicating the existence of a compensatory mechanism. Despite the impossibility of unambiguous interpretation, this single observation provides important information about the role of L-arginine and NO in maintenance of vascular homeostasis.
Given that eukaryotic cells predominantly exist in a microenvironment with an excess of nutrients, it seems unlikely that the vascular endothelium should be deprived of one conditionally essential amino acid. However, a decrease in the blood L-arginine concentration as a result of arginine-metabolizing enzymes (arginases and iNOS) activity has been described in hemolysis [43], sepsis [44], surgical injuries [45], pulmonary arterial hypertension [46], as well as in the model of Alzheimer’s disease in mice [47], and is considered as one of the possible causes of endothelial dysfunction [48].
RELATIVE L-ARGININE DEFICIENCY
It has been observed that in spite of high concentration of the intracellular L-arginine sufficient to meet eNOS needs, L-arginine supplement increases NO production in ECs. This shows that eNOS has a limited access to the intracellular L-arginine and uses predominantly extracellular pool of the amino acid for NO synthesis. This phenomenon was termed as “arginine paradox” [41]. To explain the “arginine paradox”, two hypotheses were suggested for a mechanism that regulates the substrate’s bioavailability for eNOS [41].
Asymmetric dimethylarginine (ADMA) is an endogenous eNOS inhibitor that restricts access of the enzyme to L-arginine and NO production, even if L-arginine concentration in ECs is sufficient [49]. ADMA suppresses eNOS activity by competing with L-arginine for binding to the enzyme active site [50] and amino acid transporter CAT, decreasing L-arginine transport into endothelial cells [51]. Inhibition of NOS by ADMA can only be overcome by a relative excess of L-arginine [49]. An increase in ADMA concentration is observed in various pathologies associated with endothelial dysfunction (preeclampsia, diabetes mellitus, hypertension) [52, 53]. ADMA production is also enhanced in apoptotic and senescent ECs [54].
ADMA is an endogenous product of methylated proteins degradation. Methylation of L-arginine residues in proteins is catalyzed by a family of enzymes, protein-arginine methyltransferases (PRMTs), that use S-adenosyl-L-methionine as a source of methyl groups [55]. An increase in expression of PRMTs genes in ECs occurs under the effect of oxidized low-density lipoproteins (LDL) [56]. Hyperproduction of NO in inflammation leads to S-nitrosylation of cysteine in the active center of dimethyl arginine dimethyl amino hydrolase (DDAG), an enzyme that catalyzes ADMA degradation to L-citrulline and dimethylamine [57]. Inhibition of DDAG not only inhibits ADMA catabolism [58], but also reduces the production of L-citrulline, which can be used by arginine succinate synthases (ASS) and arginine succinate lyases (ASL) as a precursor for L-arginine resynthesis [59]. eNOS colocalization in caveola with ASS and ASL facilitates access of the enzyme to resynthesized L-arginine.
Arginase catalyzes hydrolysis of L-arginine producing urea and L-ornithine. eNOS affinity to the substrate is 1000 times higher than that of arginases (Km = 2 µM vs. Km = 2 mM). However, the maximum reaction rate (Vmax) of arginases is 1000 times higher than that of eNOS [60]. Therefore, when intracellular L-arginine is deficient, due to the higher reaction rate (Vmax) carried out by arginase II, the equilibrium shifts towards polyamine production, and NO production decreases [17, 41]. Inhibition of eNOS is further enhanced by spermin, an arginase metabolism intermediate product, which inhibits calcium ions release from mitochondria and CAMKII-mediated eNOS activation [61].
The second hypothesis of the “different intracellular pools of arginine” proposed to explain the arginine paradox is based on the following facts. It is assumed that the extracellular pool of L-arginine is mainly used by eNOS for NO production, while the intracellular pool is equally available for both eNOS and arginase. Therefore, increases in arginase activity results in a relative substrate deficiency for eNOS and a decrease in NO production. The hypothesis of “different intracellular pools of arginine” explains both the arginine paradox and the competitive suppression of eNOS activity by arginase [17, 60].
Indeed, the activity of arginase and eNOS are regulated reciprocally [61]. As shown on human senescent ECs and aortic endothelium of elderly mice, the increased gene expression, protein synthesis, and activity of arginase II lead to dissociation of eNOS, promote ECs senesce with increased adhesive molecules expression. In contrast, shRNA-mediated silencing of arginase II or destruction of the arginase II gene restores eNOS function and reduces expression of the key markers of ECs senescence. Induction of the arginase II gene expression in the culture of “young” ECs triggers their senescence and emergence of the phenotype, characteristic of this state [62].
Human ECs express two isoforms of arginases with different localization. Arginase I is a cytosolic form, and arginase II is localized in mitochondria. Latter is the predominant isoform in ECs [60, 63]. Arginase activation is induced by damaging and proinflammatory factors – oxidized low-density lipoproteins, peroxynitrite [64], lipopolysaccharide, cytokines (TNF-α, IFN-γ), 8-bromo-cGMP, thrombin [65], and hypoxia [66]. Often, the development of endothelial dysfunction is accompanied by a simultaneous increase in arginase activity and ADMA production [26, 67]. ADMA increases arginase activity by releasing the intracellular pool L-arginine and reducing the production of N-hydroxy-L-arginine, an intermediate eNOS metabolite that suppresses arginase activity [61].
THE ROLE OF INTRACELLULAR L-ARGININE SENSORS IN REGULATION OF
eNOS ACTIVITY
Amino acids are structural monomers of proteins, so their bioavailability affects anabolic processes in cells. Currently, an active search for intracellular amino acid sensors and study of signaling pathways that integrate and convert signals from these sensors are underway. Of great interest are the sensors within the serine/threonine protein kinase mTOR pathway, which regulates the processes of anabolism, cell growth, proliferation [68], and arginine metabolizing enzymes activity [19, 20]. mTOR is in the composition of two separate multi-protein complexes mTORC1 and mTORC2, which perform different but partially overlapping functions [68]. A large amount of data has been accumulated in the literature proving the dependence of mTORC1 activation on L-arginine bioavailability [69]. Amino acid signaling sensors SLC38A9 and CASTOR1 were detected within mTORC1 [18, 69]. It is noteworthy that while CASTOR1 is a signaling molecule, SLC38A9 is a transceptor, i.e., it is both an L-arginine receptor and a lysosomal transporter [70]. Recent studies show that mTORC1 activity critically depends on the L-arginine bioavailability, at least in undifferentiated stem cells of human embryos (hESC) and immortal HeLa, MEF, HEK293T, U2OS, MRC5 cell lines [18]. Targeted studies of the effect of the amino acid deficiency on the mTOR regulation in ECs have not been conducted yet. In the literature, there are only indirect data indicating a possible role of L-arginine in the regulation of mTORC1 and EC function. It was shown that L-arginine deficiency reduced EC proliferation, adhesion [71, 72], migration [73, 74], suppressed tube formation on Matrigel in vitro [75], and angiogenesis in vivo [40, 75]. The possibility of positive mTOR regulation by L-arginine indicates the important role of this amino acid in maintaining endothelial cell homeostasis.
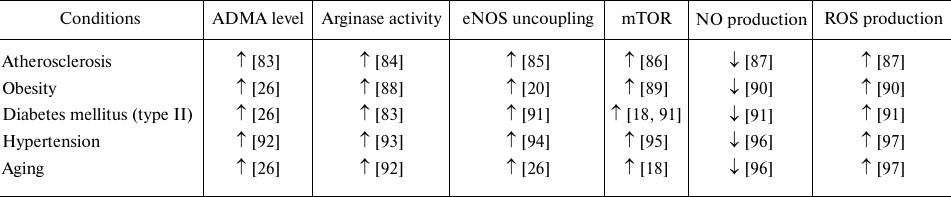
However, numerous literature data show that mTOR pathway activation accompanies many pathologies associated with endothelial dysfunction (table) [76]. As shown for the HUVECs in vitro and in the culture of aging rats aorta ex vivo, mTOR activation contributes to ECs senescence and eNOS uncoupling with increased ROS, and decreased NO production [77]. Inhibition of this signaling pathway, on the contrary, produces a vasoprotective effect observed in patients with kidney transplantation. The use of rapamycin, specific inhibitor of mTOR, for the purpose of immunosuppression, reduced cases of hypertension in this pathology [78, 79]. Inhibition of mTORC1 in vivo by rapamycin administration enhanced NO production and vasodilation [80, 81]. It was shown that rapamycin has cardioprotective properties [76], reduces expression of mRNA of the endothelial activation markers, VCAM-1, and E-selectin in HUVECs [82]. The use of rapamycin and resveratrol in the senescent EC culture and aorta of old rats restored eNOS function [62]. The aortic EC senescent phenotype in C56BL/6J mice with induced obesity was due to Akt/mTOR activation [77]. Rapamycin-induced Akt/mTOR inhibition restored EC proliferation, sprouting, eNOS activity, and vasodilation. In vivo, this correlated with enhanced angiogenesis, restored blood flow and increased capillary density after lower limb ischemia [77]. Inhibition of mTORC1 by rapamycin under shear stress increased the basal level of carotid artery eNOS expression in mice in dose-dependent manner [77].
Changes in activity of the arginine-dependent enzymes and production of
their metabolites in the course of different pathologies and aging
It is remarkable that mTORC1 activates both eNOS and arginase, despite the fact that these enzymes reciprocally regulate each other (Fig. 2). Activation of mTOR leads to eNOS phosphorylation at S1177 [19]. This enhances interaction of eNOS with Ca2+-binding proteins and makes activity of the enzyme less dependent on Ca2+ concentration [98]. It is also assumed that the Ca2+/calmodulin complex can serve as a structure that stabilizes the interaction of eNOS with mTORC1 [98]. It is important to emphasize that eNOS phosphorylation at the activating sites does not necessarily increases NO production and restores endothelial function. On the contrary, phosphorylation of uncoupling eNOS at S1177 can increase ROS production, oxidative stress, and induce inflammation associated with cell aging [40].
Fig. 2. Arginine metabolism and endothelial dysfunction. a) Physiological conditions. mTOR maintains the balance between arginase II and eNOS activities. b) Endothelial dysfunction. Under the effects of proinflammatory factors and oxidative stress, arginase II activity increases, and a loop of mutual activation between arginase and mTOR is formed. mTOR-mediated eNOS phosphorylation at S1117 amplifies ROS production. This, combined with increased ADMA production, leads to a relative substrate deficiency for eNOS, the enzyme uncoupling and production of ROS instead of NO.
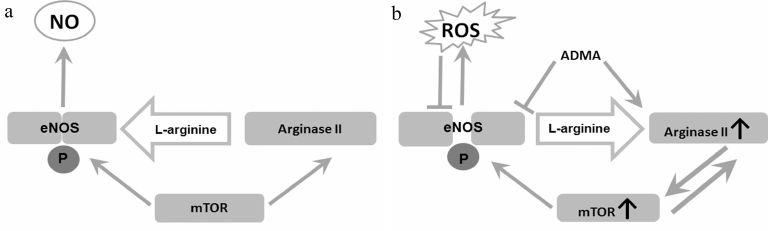
An additional factor contributing to eNOS dysfunction could be formation of a positive feedback loop between arginase II and mTORC1 [99]. mTOR/S6K1 hyperactivation leads to an increase in arginase II mRNA synthesis and stabilization [99]. It was found in the recent studies on the mouse AML12 hepatoma cells that arginase II can also increase mTORC1 activity, and this does not depend on L-arginine hydrolysis [20]. Induction of arginase II expression in hepatoma cells led to mTORC1 activation due to its association with lysosomes mediated by non-conventional class I myosin (Myo1b) [20]. It was established that interaction of Arg-II-Myo1b-mTORC1-S6K1 triggered human vascular smooth muscle cells (HUVSMC) senescence and apoptosis [20]. Arginase II activated mTOR/S6K1 signaling pathway in isolated mesenteric arteries in mice [77]. It is assumed that the synergy of arginase and mTOR activation is necessary for synchronization of glycolysis and activation of the urea cycle enzymes [100]. These data cast doubts on the dependence of mTOR on L-arginine bioavailability in endothelial cells. Otherwise, arginase activation should lead to mTOR inhibition by creating deficiency of intracellular L-arginine.
CONCLUSION
Numerous experimental data show that disruption of the arginine-metabolizing enzymes balance with predominant activation of arginases causes eNOS dissociation, which results in an increase in production of superoxide anion radical instead of vasoprotective NO. This mechanism underlies the development of vascular dysfunction in various pathologies and aging. Experiments aimed at inhibiting mTOR in endothelial dysfunction demonstrate that this intracellular signaling pathway is the central hub responsible for disrupting the balance of L-arginine metabolism in ECs. In the case of mTORC1 hyperactivation, a loop of mutual positive regulation is formed between mTORC1 and arginase, which finally leads to the eNOS uncoupling. The functional state of eNOS in ECs is determined by a variety of factors, including signaling molecules, substrate bioavailability, and enzyme conformation. The data currently accumulated in this area are to a large degree contradictory. It remains unclear how arginase can cause substrate deficiency for eNOS if the latter primarily uses extracellular arginine. Paradoxically, the mTOR pathway can synergistically activate eNOS and arginase, although these enzymes reciprocally regulate activity of each other. Another contradiction is that L-arginine deficiency caused by arginase suppresses eNOS activity, but increases activation of the L-arginine-dependent mTOR pathway. These issues require clarification to provide a more comprehensive understanding of the endothelial dysfunction mechanisms.
Ethics declarations. The authors declare no conflict of interests in financial or any other sphere. This article does not contain any studies with human participants or animals performed by any of the authors.
REFERENCES
1.Freed, J. K., and Gutterman, D. D. (2017)
Communication is key: mechanisms of intercellular signaling in
vasodilation, J. Cardiovasc. Pharmacol., 69, 264-272,
doi: 10.1097/FJC.0000000000000463.
2.Tousoulis, D., Kampoli, A. M., Tentolouris, C.,
Papageorgiou, N., and Stefanadis, C. (2012) The role of nitric oxide on
endothelial function, Curr. Vasc. Pharmacol., 10,
4-18.
3.Huang, P. L., Huang, Z., Mashimo, H., Bloch, K. D.,
Moskowitz, M. A., et al. (1995) Hypertension in mice lacking the gene
for endothelial nitric oxide synthase, Nature, 377,
239-242, doi: 10.1038/377239a0.
4.Moroi, M., Zhang, L., Yasuda, T., Virmani, R.,
Gold, H. K., Fishman, M. C., and Huang, P. L. (1998) Interaction of
genetic deficiency of endothelial nitric oxide, gender, and pregnancy
in vascular response to injury in mice, J. Clin. Invest.,
101, 1225-1232, doi: 10.1172/JCI1293.
5.Lefer, D. J., Jones, S. P., Girod, W. G., Baines,
A., Grisham, M. B., et al. (1999) Leukocyte-endothelial cell
interactions in nitric oxide synthase-deficient mice, Am. J.
Physiol., 276, H1943-H1950, doi:
10.1152/ajpheart.1999.276.6.H1943.
6.Freedman, J. E., Sauter, R., Battinelli, E. M.,
Ault, K., Knowles, C., Huang, P. L., and Loscalzo, J. (1999) Deficient
platelet-derived nitric oxide and enhanced hemostasis in mice lacking
the NOSIII gene, Circ. Res., 84, 1416-1421, doi:
10.1161/01.res.84.12.1416.
7.Kuhlencordt, P. J., Gyurko, R., Han, F.,
Scherrer-Crosbie, M., Aretz, T. H., et al. (2001) Accelerated
atherosclerosis, aortic aneurysm formation, and ischemic heart disease
in apolipoprotein E/endothelial nitric oxide synthase double-knockout
mice, Circulation, 104, 448-454, doi:
10.1161/hc2901.091399.
8.Ha, J. M., Jin, S. Y., Lee, H. S., Shin, H. K.,
Lee, D. H., et al. (2016) Regulation of retinal angiogenesis by
endothelial nitric oxide synthase signaling pathway, Kor. J.
Physiol. Pharmacol. Official J. Kor. Physiol. Soc. Kor. Pharmacol.,
20, 533-538, doi: 10.4196/kjpp.2016.20.5.533.
9.Zani, B. G., and Bohlen, H. G. (2005) Transport of
extracellular l-arginine via cationic amino acid transporter is
required during in vivo endothelial nitric oxide production,
Am. J. Physiol. Heart. Circ. Physiol., 289,
H1381-H1390.
10.MacKenzie, A., and Wadsworth, R. M. (2003)
Extracellular L-arginine is required for optimal NO synthesis by eNOS
and iNOS in the rat mesenteric artery wall, Br. J. Pharmacol.,
139, 1487-1497, doi: 10.1038/sj.bjp.0705380.
11.Oludare, G. O., Jinadu, H. D., and Aro, O. O.
(2018) L-arginine attenuates blood pressure and reverses the
suppression of angiogenic risk factors in a rat model of preeclampsia,
Pathophysiology, 25, 389-395, doi:
10.1016/j.pathophys.2018.08.001.
12.Mariotti, F. (2020) Arginine supplementation and
cardiometabolic risk, Curr. Opin. Clin. Nutr. Metab. Care,
23, 29-34, doi: 10.1097/MCO.0000000000000612.
13.Moretto, J., Guglielmetti, A. S.,
Tournier-Nappey, M., Martin, H., Prigent-Tessier, A., Marie, C., and
Demougeot, C. (2017) Effects of a chronic l-arginine supplementation on
the arginase pathway in aged rats, Exp. Gerontol., 90,
52-60, doi: 10.1016/j.exger.2017.01.023.
14.Kabat, A., and Dhein, S. (2006) L-arginine
supplementation prevents the development of endothelial dysfunction in
hyperglycaemia, Pharmacology, 76, 185-191.
15.Rodrigues-Krause, J., Krause, M., Rocha, I.,
Umpierre, D., and Fayh, A. (2018) Association of L-arginine
supplementation with markers of endothelial function in patients with
cardiovascular or metabolic disorders: a systematic review and
meta-analysis, Nutrients, 11, 15, doi:
10.3390/nu11010015.
16.Xiong, Y., Fru, M. F., Yu, Y., Montani, J. P.,
Ming, X. F., and Yang, Z. (2014) Long term exposure to L-arginine
accelerates endothelial cell senescence through arginase-II and S6K1
signaling, Aging (Albany NY), 6, 369-379, doi:
10.18632/aging.100663.
17.Morris, S. M., Jr. (2016) Arginine metabolism
revisited, J. Nutr., 146, 2579S-2586S, doi:
10.3945/jn.115.226621.
18.Saxton, R. A., and Sabatini, D. M. (2017) mTOR
signaling in growth, metabolism, and disease, Cell, 168,
960-976, doi: 10.1016/j.cell.2017.02.004.
19.Decker, B., and Pumiglia, K. (2018) mTORc1
activity is necessary and sufficient for phosphorylation of
eNOSS1177, Physiol. Rep., 6, e13733, doi:
10.14814/phy2.13733.
20.Yu, Y., Xiong, Y., Montani, J. P., Yang, Z., and
Ming, X. F. (2018) Arginase-II activates mTORC1 through myosin-1b in
vascular cell senescence and apoptosis, Cell Death Dis.,
9, 313, doi: 10.1038/s41419-018-0356-9.
21.Rafikov, R., Fonseca, F. V., Kumar, S., Pardo,
D., Darragh, C., Elms, S., and Black, S. M. (2011) eNOS activation and
NO function: structural motifs responsible for the posttranslational
control of endothelial nitric oxide synthase activity, J.
Endocrinol., 210, 271-284, doi: 10.1530/JOE-11-0083.
22.Govers, R., and Rabelink, T. J. (2001) Cellular
regulation of endothelial nitric oxide synthase, Am. J. Physiol.
Renal. Physiol., 280, F193-F206.
23.Murthy, S., Koval, O. M., Ramiro Diaz, J. M.,
Kumar, S., Nuno, D., et al. (2017) Endothelial CaMKII as a regulator of
eNOS activity and NO-mediated vasoreactivity, PLoS One,
12, e0186311, doi: 10.1371/journal.pone.0186311.
24.Lee, M. Y., Gamez-Mendez, A., Zhang, J., Zhuang,
Z., Vinyard, D. J., et al. (2018) Endothelial cell autonomous role of
Akt1: regulation of vascular Tone and ischemia-induced arteriogenesis,
Arterioscler. Thromb. Vasc. Biol., 38, 870-879, doi:
10.1161/ATVBAHA.118.310748.
25.Cacicedo, J. M., Gauthier, M. S., Lebrasseur, N.
K., Jasuja, R., Ruderman, N. B., and Ido, Y. (2011) Acute exercise
activates AMPK and eNOS in the mouse aorta, Am. J. Physiol. Heart
Circ. Physiol., 301, H1255-H1265, doi:
10.1152/ajpheart.01279.2010.
26.Vanhoutte, P. M., Zhao, Y., Xu, A., and Leung, S.
W. S. (2016) Thirty years of saying NO: sources, fate, actions, and
misfortunes of the endothelium-derived vasodilator mediator, Circ.
Res. Am. Heart Assoc., 119, 375-396, doi:
10.1161/CIRCRESAHA.116.306531.
27.Ghimire, K., Altmann, H. M., Straub, A. C., and
Isenberg, J. S. (2017) Nitric oxide: what’s new to NO? Am. J.
Physiol., 312, C254-C262, doi:
10.1152/ajpcell.00315.2016.
28.Bibli, S. I., Zhou, Z., Zukunft, S., Fisslthaler,
B., Andreadou, I., et al. (2017) Tyrosine phosphorylation of eNOS
regulates myocardial survival after an ischaemic insult: role of PYK2,
Cardiovasc. Res., 113, 926-937, doi:
10.1093/cvr/cvx058.
29.Förstermann, U., and Sessa, W. C. (2012)
Nitric oxide synthases: regulation and function, Eur. Heart J.,
33, 829-837, doi: 10.1093/eurheartj/ehr304.
30.Sugimoto, M., Nakayama, M., Goto, T. M., Amano,
M., Komori, K., and Kaibuchi, K. (2007) Rho-kinase phosphorylates eNOS
at threonine 495 in endothelial cells, Biochem. Biophys. Res.
Commun., 361, 462-467, doi: 10.1016/j.bbrc.2007.07.030.
31.Chen, Z. P., Mitchelhill, K. I., Michell, B. J.,
Stapleton, D., Rodriguez-Crespo, I., et al. (1999) AMP-activated
protein kinase phosphorylation of endothelial NO synthase, FEBS
Lett., 443, 285-289.
32.Greif, D. M., Sacks, D. B., and Michel, T. (2004)
Calmodulin phosphorylation and modulation of endothelial nitric oxide
synthase catalysis, Proc. Natl. Acad. Sci. USA, 101,
1165-1170, doi: 10.1073/pnas.0306377101.
33.Oliveira-Paula, G. H., Lacchini, R., and
Tanus-Santos, J. E. (2016) Endothelial nitric oxide synthase: From
biochemistry and gene structure to clinical implications of NOS3
polymorphisms, Gene, 575 (2 Pt 3), 584-599, doi:
10.1016/j.gene.2015.09.061.
34.Gebhart, V., Reiß, K., Kollau, A., Mayer,
B., and Gorren, A. (2019) Site and mechanism of uncoupling of
nitric-oxide synthase: Uncoupling by monomerization and other
misconceptions, Nitric Oxide, 89, 14-21, doi:
10.1016/j.niox.2019.04.007.
35.Luo, S., Lei, H., Qin, H., and Xia, Y. (2014)
Molecular mechanisms of endothelial NO synthase uncoupling, Curr.
Pharm. Des., 20, 3548-3553, doi:
10.2174/13816128113196660746.
36.Li, H., and Förstermann, U. (2017) Nitric
Oxide (Third Edition), Biology and Pathobiology, 117-124, doi:
10.1016/B978-0-12-804273-1.00009-0.
37.Sharma, J. N., Al-Omran, A., and Parvathy, S. S.
(2007) Role of nitric oxide in inflammatory diseases,
Inflammopharmacology, 15, 252-259, doi:
10.1007/s10787-007-0013-x.
38.Liang, P., Jiang, B., Li, Y., Liu, Z., Zhang, P.,
et al. (2018) Autophagy promotes angiogenesis via AMPK/Akt/mTOR
signaling during the recovery of heat-denatured endothelial cells,
Cell Death Dis., 9, 1152, doi:
10.1038/s41419-018-1194-5.
39.Vanhoutte, P. M. (2018) Nitric oxide: from good
to bad, Ann. Vasc. Dis., 11, 41-51, doi:
10.3400/avd.ra.17-00134.
40.Park, I. S., Kang, S. W., Shin, Y. J., Chae, K.
Y., Park, M. O., et al. (2003) Arginine deiminase: a potential
inhibitor of angiogenesis and tumour growth, Br. J. Cancer,
89, 907-914, doi: 10.1038/sj.bjc.6601181.
41.Chen, F., Lucas, R., and Fulton, D. (2013) The
subcellular compartmentalization of arginine metabolizing enzymes and
their role in endothelial dysfunction, Front. Immunol.,
4, 184, doi: 10.3389/fimmu.2013.00184.
42.Loscalzo, J. (2001) An experiment of nature:
genetic L-arginine deficiency and NO insufficiency, J. Clin.
Invest., 108, 663-664, doi: 10.1172/JCI13848.
43.Morris, C. R., Kato, G. J., Poljakovic, M., Wang,
X., Blackwelder, W. C., et al. (2005) Dysregulated arginine metabolism,
hemolysis-associated pulmonary hypertension, and mortality in sickle
cell disease, JAMA, 294, 81-90, doi:
10.1001/jama.294.1.81.
44.Luiking, Y. C., and Deutz, N. E. (2007) Exogenous
arginine in sepsis, Crit. Care. Med., 35, 557-563, doi:
10.1097/01.CCM.0000279191.44730.A2.
45.Pribis, J. P., Zhu, X., Vodovotz, Y., and Ochoa,
J. B. (2012) Systemic arginine depletion after a murine model of
surgery or trauma, JPEN J. Parenter. Enteral. Nutr., 36,
53-59, doi: 10.1177/0148607111414579.
46.Kao, C., Wedes, S., Hsu, J., Bohren, K., Comhair,
S., Jahoor, F., and Erzurum, S. (2015) Arginine metabolic endotypes in
pulmonary arterial hypertension, Pulmonary Circ., 5,
124-134, doi: 10.1086/679720.
47.Kan, M. J., Lee, J. E., Wilson, J. G., Everhart,
A. L., Brown, C. M., et al. (2015) Arginine deprivation and immune
suppression in a mouse model of Alzheimer’s disease, J.
Neurosci., 35, 5969-5982, doi:
10.1523/JNEUROSCI.4668-14.2015.
48.Morris, S. M., Jr. (2012) Arginases and arginine
deficiency syndromes, Curr. Opin. Clin. Nutr. Metab. Care,
15, 64-70, doi: 10.1097/MCO.0b013e32834d1a08.
49.Di Franco, M., Lucchino, B., Conti, F., Valesini,
G., and Spinelli, F. R. (2018) Asymmetric dimethyl arginine as a
biomarker of atherosclerosis in rheumatoid arthritis, Mediat.
Inflamm., 2018, 3897295, doi: 10.1155/2018/3897295.
50.Jung, C. H., Lee, W. J., Hwang, J. Y., Lee, M.
J., Seol, S. M., et al. (2012) Vaspin increases nitric oxide
bioavailability through the reduction of asymmetric dimethylarginine in
vascular endothelial cells, PLoS One, 7, e52346, doi:
10.1371/journal.pone.0052346.
51.Watson, C. P., Pazarentzos, E., Fidanboylu, M.,
Padilla, B., Brown, R., and Thomas, S. A. (2016) The transporter and
permeability interactions of asymmetric dimethylarginine (ADMA) and
L-arginine with the human blood-brain barrier in vitro, Brain
Res., 1648 (Pt A), 232-242, doi:
10.1016/j.brainres.2016.07.026.
52.Yokoro, M., Minami, M., Okada, S., Yano, M.,
Otaki, N., Ikeda, H., and Fukuo, K. (2018) Urinary sodium-to-potassium
ratio and serum asymmetric dimethylarginine levels in patients with
type 2 diabetes, Hypertens. Res., 41, 913-922, doi:
10.1038/s41440-018-0098-1.
53.Gać, P., Poręba, M., Jurdziak, M.,
Trzmielewska, E., Gocławska, K., et al. (2020) Cardiovascular
risk factors and the concentration of asymmetric dimethylarginine,
Adv. Clin. Exp. Med., 29, 63-70, doi:
10.17219/acem/111808.
54.Hou, L., Guo, J., Xu, F., Weng, X., Yue, W., and
Ge, J. (2018) Cardiomyocyte dimethylarginine dimethylaminohydrolase1
attenuates left-ventricular remodeling after acute myocardial
infarction: involvement in oxidative stress and apoptosis, Basic
Res. Cardiol., 113, 28, doi: 10.1007/s00395-018-0685-y.
55.Fulton, M. D., Brown, T., and Zheng, Y. G. (2019)
The biological axis of protein arginine methylation and asymmetric
dimethylarginine, Int. J. Mol. Sci., 20, 3322, doi:
10.3390/ijms20133322.
56.Wielkoszyński, T., Zalejska-Fiolka, J.,
Strzelczyk, J. K., Owczarek, A. J., Cholewka, A., Furmański, M.,
and Stanek, A (2018) Oxysterols increase inflammation, lipid marker
levels and reflect accelerated endothelial dysfunction in experimental
animals, Mediators Inflamm., 2018, 2784701, doi:
10.1155/2018/2784701.
57.Knipp, M., Braun, O., Gehrig, P. M., Sack, R.,
and Vasák, M. (2003) Zn(II)-free dimethylargininase-1 (DDAH-1)
is inhibited upon specific Cys-S-nitrosylation, J. Biol. Chem.,
278, 3410-3416.
58.Bollenbach, A., and Tsikas, D. (2019)
Pharmacological activation of dimethylarginine dimethylaminohydrolase
(DDAH) activity by inorganic nitrate and DDAH inhibition by
NG-hydroxy-L-arginine, Nω,Nω-dimethyl-L-citrulline and
Nω,Nω-dimethyl-Nδ-hydroxy-L-citrulline: results and
overview, Amino Acids, 51, 483-494, doi:
10.1007/s00726-018-2684-6.
59.Morita, M., Hayashi, T., Ochiai, M., Maeda, M.,
Yamaguchi, T., Ina, K., and Kuzuya, M. (2014) Oral supplementation with
a combination of L-citrulline and L-arginine rapidly increases plasma
L-arginine concentration and enhances NO bioavailability, Biochem.
Biophys. Res. Commun., 454, 53-57, doi:
10.1016/j.bbrc.2014.10.029.
60.Elms, S., Chen, F., Wang, Y., Qian, J., Askari,
B., et al. (2013) Insights into the arginine paradox: evidence against
the importance of subcellular location of arginase and eNOS, Am. J.
Physiol. Heart Circ. Physiol., 305, H651-666, doi:
10.1152/ajpheart.00755.2012.
61.Koo, B. H., Won, M. H., Kim, Y. M., and Ryoo, S.
(2020) p32-Dependent p38 MAPK activation by arginase II downregulation
contributes to endothelial nitric oxide synthase activation in HUVECs,
Cells, 9, 392, doi: 10.3390/cells9020392.
62.Rajapakse, A. G., Yepuri, G., Carvas, J. M.,
Stein, S., Matter, C. M., et al. (2011) Hyperactive S6K1 mediates
oxidative stress and endothelial dysfunction in aging: inhibition by
resveratrol, PLoS One, 6, e19237, doi:
10.1371/journal.pone.0019237.
63.Morris, S. M., Jr. (2009) Recent advances in
arginine metabolism: roles and regulation of the arginases, Br. J.
Pharmacol., 157, 922-930, doi:
10.1111/j.1476-5381.2009.00278.x.
64.Chandra, S., Romero, M. J., Shatanawi, A.,
Alkilany, A. M., Caldwell, R. B., and Caldwell, R. W. (2012) Oxidative
species increase arginase activity in endothelial cells through the
RhoA/Rho kinase pathway, Br. J. Pharmacol., 165, 506-519,
doi: 10.1111/j.1476-5381.2011.01584.x.
65.Ming, X. F., Barandier, C., Viswambharan, H.,
Kwak, B. R., Mach, F., et al. (2004) Thrombin stimulates human
endothelial arginase enzymatic activity via RhoA/ROCK pathway:
implications for atherosclerotic endothelial dysfunction,
Circulation, 110, 3708-3714, doi:
10.1161/01.CIR.0000142867.26182.32.
66.Morris, S. M., Jr., Kepka-Lenhart, D., and Chen,
L. C. (1998) Differential regulation of arginases and inducible nitric
oxide synthase in murine macrophage cells, Am. J. Physiol.,
275, E740-747, doi: 10.1152/ajpendo.1998.275.5.E740.
67.Chandrasekharan, U. M., Wang, Z., Wu, Y., Tang,
W., Hazen, S. L., Wang, S., and Husni, M. E. (2018) Elevated levels of
plasma symmetric dimethylarginine and increased arginase activity as
potential indicators of cardiovascular comorbidity in rheumatoid
arthritis, Arthritis Res. Ther., 20, 123, doi:
10.1186/s13075-018-1616-x.
68.Liu, G. Y., and Sabatini, D. M. (2020) mTOR at
the nexus of nutrition, growth, ageing and disease, Nat. Rev. Mol.
Cell Biol., 21, 183-203, doi: 10.1038/s41580-019-0199-y.
69.Jung, J. W., Kim, J. E., Kim, E., and Lee, J. W.
(2020) Amino acid transporters as tetraspanin TM4SF5 binding partners,
Exp. Mol. Med., 52, 7-14, doi:
10.1038/s12276-019-0363-7.
70.Scalise, M., Galluccio, M., Pochini, L., Cosco,
J., Trotta, M., et al. (2019) Insights into the transport side of the
human SLC38A9 transceptor, Biochim. Biophys. Acta Biomembr.,
1861, 1558-1567, doi: 10.1016/j.bbamem.2019.07.006.
71.Starikova, E. A., Lebedeva, A. M., Burova, L. A.,
and Freidlin, I. S. (2012) Regulation of endothelial cells functions by
ultrasonic supernatant of Streptococcus pyogenes,
Tsitologiya, 1, 49-46.
72.Starikova, E. A., Sokolov, A. V., Vlasenko, A.
Y., Burova, L. A., Freidlin, I. S., and Vasilyev, V. B. (2016)
Biochemical and biological activity of arginine deiminase from
Streptococcus pyogenes M22, Biochem. Cell Biol., 94,
129-137, doi: 10.1139/bcb-2015-0069.
73.Starikova, E. A., Mammedova, J. T., Burova, L.
A., Sokolov, A. V., Vasilyev, V. B., and Freidlin, I. S. (2017) Effect
of arginine deiminase from Streptococcus pyogenes on
cytoskeleton structure and migration activity of human endothelial
cells, Meditsinskaya Immunologiya, 19, 521-528, doi:
10.15789/1563-0625-2017-5-521-528.
74.Mammedova, J. T., Starikova, E. A., Burova, L.
A., Malashicheva, A. B., Semenova, D. S., and Freidlin, I. S. (2017)
Effect of arginine deiminase from Streptococcus pyogenes on
proliferation and migration of human umbilical vein endothelial cells,
Tsitokiny Vospaleniye, 3, 48-51.
75.Beloussow, K., Wang, L., Wu, J., Ann, D., and
Shen, W. C. (2002) Recombinant arginine deiminase as a potential
antiangiogenic agent, Cancer Lett., 183, 155-162, doi:
10.1016/S0304-3835(01)00793-5.
76.Donato, A. J., Machin, D. R., and Lesniewski, L.
A. (2018) Mechanisms of dysfunction in the aging vasculature and role
in age-related disease, Circ. Res., 123, 825-848, doi:
10.1161/CIRCRESAHA.118.312563.
77.Galvan, V., and Hart, M. J. (2016) Vascular
mTOR-dependent mechanisms linking the control of aging to
Alzheimer’s disease, Biochim. Biophys. Acta, 1862,
992-1007, doi: 10.1016/j.bbadis.2015.11.010.
78.Johnson, R. W., Kreis, H., Oberbauer, R.,
Brattstrom, C., Claesson, K., and Eris, J. (2001) Sirolimus allows
early cyclosporine withdrawal in renal transplantation resulting in
improved renal function and lower blood pressure,
Transplantation, 72, 777-786.
79.Lebranchu, Y., Thierry, A., Toupance, O.,
Westeel, P. F., Etienne, I., et al. (2009) Efficacy on renal function
of early conversion from cyclosporine to sirolimus 3 months after renal
transplantation: concept study, Am. J. Transplant., 9,
1115-1123.
80.Carrizzo, A., Puca, A., Damato, A., Marino, M.,
Franco, E., et al. (2013) Resveratrol improves vascular function in
patients with hypertension and dyslipidemia by modulating NO
metabolism, Hypertension, 62, 359-366, doi:
10.1161/HYPERTENSIONAHA.111.01009.
81.Gordish, K. L, and Beierwaltes, W. H. (2014)
Resveratrol induces acute endothelium-dependent renal vasodilation
mediated through nitric oxide and reactive oxygen species scavenging,
Am. J. Physiol. Renal. Physiol., 306, F542-550.
82.Gholizadeh, S., Visweswaran, G. R. R., Storm, G.,
Hennink, W. E., Kamps, J. A. A. M., and Kok, R. J. (2018) E-selectin
targeted immunoliposomes for rapamycin delivery to activated
endothelial cells, Int. J. Pharm., 548, 759-770, doi:
10.1016/j.ijpharm.2017.10.027.
83.Mangiacapra, F., Conte, M., Demartini, C.,
Muller, O., Delrue, L., et al. (2016) Relationship of asymmetric
dimethylarginine (ADMA) with extent and functional severity of coronary
atherosclerosis, Int. J. Cardiol., 220, 629-633, doi:
10.1016/j.ijcard.2016.06.254.
84.Mahdi, A., Kövamees, O., and Pernow, J.
(2020) Improvement in endothelial function in cardiovascular disease
– is arginase the target? Int. J. Cardiol., 301,
207-214, doi: 10.1016/j.ijcard.2019.11.004.
85.Montezano, A. C., and Touyz, R. M. (2012)
Reactive oxygen species and endothelial function – role of nitric
oxide synthase uncoupling and Nox family nicotinamide adenine
dinucleotide phosphate oxidases, Basic Clin. Pharmacol.
Toxicol., 110, 87-94, doi:
10.1111/j.1742-7843.2011.00785.x.
86.Kurdi, A., De Meyer, G. R., and Martinet, W.
(2016) Potential therapeutic effects of mTOR inhibition in
atherosclerosis, Br. J. Clin. Pharmacol., 82, 1267-1279,
doi: 10.1111/bcp.12820.
87.Marchio, P., Guerra-Ojeda, S., Vila, J. M.,
Aldasoro, M., Victor, V. M., and Mauricio, M. D. (2019) Targeting early
atherosclerosis: a focus on oxidative stress and inflammation, Oxid.
Med. Cell. Longev., 2019, 8563845, doi:
10.1155/2019/8563845.
88.Arlouskaya, Y., Sawicka, A., Głowala, M.,
Giebułtowicz, J., Korytowska, N., et al. (2019) Asymmetric
Dimethylarginine (ADMA) and Symmetric Dimethylarginine (SDMA)
concentrations in patients with obesity and the risk of Obstructive
Sleep Apnea (OSA), J. Clin. Med., 8, 897, doi:
10.3390/jcm8060897.
89.Jia, G., Aroor, A. R., Martinez-Lemus, L. A., and
Sowers, J. R. (2014) Overnutrition, mTOR signaling, and cardiovascular
diseases, Am. J. Physiol. Regul. Integr. Compar. Physiol.,
307, R1198-R1206, doi: 10.1152/ajpregu.00262.2014.
90.Oguntibeju, O. O. (2019) Type 2 diabetes
mellitus, oxidative stress and inflammation: examining the links,
Int. J. Physiol. Pathophysiol. Pharmacol., 11, 45-63.
91.Daiber, A., Xia, N., Steven, S., Oelze, M., Hanf,
A., et al. (2019) New therapeutic implications of endothelial Nitric
Oxide Synthase (eNOS) function/dysfunction in cardiovascular disease,
Int. J. Mol. Sci., 20, 187, doi:
10.3390/ijms20010187.
92.Sandqvist, A., Schneede, J., Kylhammar, D.,
Henrohn, D., Lundgren, J., et al. (2018) Plasma L-arginine levels
distinguish pulmonary arterial hypertension from left ventricular
systolic dysfunction, Heart Vessels, 33, 255-263, doi:
10.1007/s00380-017-1055-7.
93.Rabelo, L. A., Ferreira, F. O., Nunes-Souza, V.,
da Fonseca, L. J., and Goulart, M. O. (2015) Arginase as a critical
prooxidant mediator in the binomial endothelial
dysfunction-atherosclerosis, Oxid. Med. Cell. Longev.,
2015, 924860, doi: 10.1155/2015/924860.
94.D’Uscio, L. V. (2011) eNOS uncoupling in
pulmonary hypertension, Cardiovasc. Res., 92, 359-360,
doi: 10.1093/cvr/cvr270.
95.Kumar, V., Evans, L. C., Kurth, T., Yang, C.,
Wollner, C., et al. (2019) Therapeutic suppression of mTOR (Mammalian
Target of Rapamycin) signaling prevents and reverses salt-induced
hypertension and kidney injury in dahl salt-sensitive rats,
Hypertension, 73, 630-639, doi:
10.1161/HYPERTENSIONAHA.118.12378.
96.Bruno, R. M., Masi, S., Taddei, M., Taddei, S.,
and Virdis, A. (2018) Essential hypertension and functional
microvascular ageing, High Blood Press Cardiovasc. Prev.,
25, 35-40, doi: 10.1007/s40292-017-0245-9.
97.Incalza, M. A., D’Oria, R., Natalicchio,
A., Perrini, S., Laviola, L., and Giorgino, F. (2018) Oxidative stress
and reactive oxygen species in endothelial dysfunction associated with
cardiovascular and metabolic diseases, Vascul. Pharmacol.,
100, 1-19, doi: 10.1016/j.vph.2017.05.005.
98.McCabe, T. J., Fulton, D., Roman, L. J., and
Sessa, W. C. (2000) Enhanced electron flux and reduced calmodulin
dissociation may explain “calcium‐independent” eNOS
activation by phosphorylation, J. Biol. Chem., 275,
6123-6128.
99.Yepuri, G., Velagapudi, S., Xiong, Y., Rajapakse,
A. G., Montani, J. P., et al. (2012) Positive crosstalk between
arginase-II and S6K1 in vascular endothelial inflammation and aging,
Aging Cell, 11, 1005-1016, doi: 10.1111/acel.12001.
100.Grandvuillemin, I., Buffat, C., Boubred, F.,
Lamy, E., Fromonot, J., et al. (2018) Arginase upregulation and eNOS
uncoupling contribute to impaired endothelium-dependent vasodilation in
a rat model of intrauterine growth restriction, Am. J. Physiol.
Regul. Integr. Comp. Physiol., 315, R509-R520, doi:
10.1152/ajpregu.00354.2017.