Upregulation of NOX-2 and Nrf-2 Promotes 5-Fluorouracil Resistance of Human Colon Carcinoma (HCT-116) Cells
Bhargav N. Waghela1, Foram U. Vaidya1, and Chandramani Pathak1,2,a*
1School of Biological Sciences & Biotechnology, Indian Institute of Advanced Research, Koba Institutional Area, 382426 Gujarat, Gandhinagar, India2Amity Institute of Biotechnology, Amity University Haryana, 122413 Gurgaon, India
* To whom correspondence should be addressed.
Received July 14, 2020; Revised September 21, 2020; Accepted September 21, 2020
Altered expression of cellular redox genes and proteins contributes to invasion, metastasis, and drug resistance in cancer. NADPH oxidase (NOX) isoforms are the pro-oxidant enzymes that generate ROS as a primary product. Dysregulation of NOX activity and expression alters ROS generation, which either directly or indirectly modulates cell death and survival signaling during the progression of cancer. Nuclear factor erythroid 2-related factor 2 (Nrf-2) is an inducible transcription factor, which transcribes an array of antioxidant genes and protects cancer cells from the oxidative stress. Both NOXs and Nrf-2 participate in the regulation of cellular redox homeostasis; but their dysregulation promotes oxidative stress, which contributes to the progression of different types of cancer. Indeed, the role of NOX isoforms and Nrf-2 in developing the drug resistance in cancer is largely unknown. In the present study, we have explored the association of NOX isoforms and Nrf-2 signaling with the MDR1 gene expression in colon carcinoma cells (HCT-116/R). The MDR1 gene was overexpressed to develop resistant HCT-116/R cells and the NOX activation and ROS generation were monitored. We also assessed the role of NOX isoforms and Nrf-2 in the 5-fluorouracil (5-FU) mediated apoptotic cell death of HCT-116/R cells. The HCT-116/R cells demonstrated higher expression of HIF-1α, Nrf-2, and HO-1 and were highly resistant to 5-FU; they also displayed upregulated expression and activity of NOX-2, as well as elevated ROS levels. Interestingly, the treatment with HDC, a specific NOX-2 inhibitor, reduced the ROS levels in HCT-116/R cells. The treatment with HDC and ML-385 (specific inhibitor of Nrf-2) augmented the 5-FU-mediated apoptotic cell death of HCT-116/R cells, which suggests that NOX-2 and Nrf-2 are involved in the development of the chemoresistant phenotype of these cells. Taken together, NOX-2 and Nrf-2 are associated with developing drug resistance of colorectal cancer cells and might be potential targets to overcome drug resistance during cancer therapy.
KEY WORDS: 5-fluorouracil, drug resistance, HO-1, MDR1, NOX-2, Nrf-2, ROSDOI: 10.1134/S0006297921030044
Abbreviations: 5-FU, 5-fluorouracil; ABC, ATP-binding cassette; DPBS, Dulbecco’s phosphate buffered saline; HCT-116/R, MDR resistant colon cancer cells; NOX, NADPH oxidase; Nrf-2, nuclear factor erythroid 2-related factor 2; ROS, reactive oxygen species;
INTRODUCTION
Colon cancer is responsible for a higher mortality rate and one of the leading causes of death worldwide [1]. Chemotherapy and surgery are commonly offered against cancer, but cancer cells become chemoresistant over a short period of treatment with anticancer drugs. The acquisition of resistance to chemotherapy is a major hurdle in the successful application of cancer therapy [2]. The development of drug resistance cancer cells has complex molecular mechanisms [3]. Mechanistically, the development of drug resistance is associated with various cellular phenomena and processes, such as drug inactivation, drug target alteration, drug efflux, DNA damage repair, cell death inhibition, epithelial to mesenchymal transition (EMT), inherent cell heterogeneity, epigenetic effects, or any combination of these mechanisms [4, 5].
Compelling evidence suggests that the development of drug resistance in cancer cells occurs via upregulation of drug efflux proteins or reduction in the cellular uptake of anti-cancer drugs. One of the most studied mechanisms of drug resistance in cancer involves the overexpression of the ATP-binding cassette (ABC) transporter family proteins, which pump out the anticancer drugs and reduce their accumulation in the cells [6]. ABC transporters utilize energy from ATP hydrolysis to transport substrates across the plasma membrane against the concentration gradient [7]. Forty-nine members of the ABC transporter family are known that are divided into 7 subfamilies, from ABCA to ABCG. ABC transporters contain two nucleotide-binding domains and two transmembrane domains. They can be topologically classified based on the sequence of the nucleotide-binding domain (ABC domain), which contains the conserved ABC sequence. ABC sequence is involved in the efflux of endogenous molecules and xenobiotics out of the cells [8]. Among all, an elevated expression of the MDR1 (multi-drug resistance protein 1) gene has been found in various types of cancer, such as cancers of the colon, liver, kidney, pancreas, etc. [9]. Overexpression of the MDR1 gene confers resistance to a wide variety of neutral and cationic hydrophobic chemotherapeutics, including anthracycline, alkaloids, taxanes, antibiotics, etc. [10].
Redox signaling and oxidative stress regulatory proteins, NADPH oxidase (NOX) family proteins, and their primary product reactive oxygen species (ROS) play an important role in tumorigenesis [11]. Previous reports revealed that ROS play an important role in determining the fate of cells [12]. ROS at low concentrations coordinate various cellular signaling pathways, but at higher levels, they induce oxidative stress and cell death [13]. ROS are known to be generated by various cellular processes as the end metabolic products. NOX is a complex enzyme system that produces ROS as a primary product. NOX catalyzes the electron transfer from NADPH to molecular oxygen and generates ROS. The NOX enzyme family consists of seven members named NOX-1-5 and DUOX1-2 [14]. The existence of different NOX isoforms and their distribution in different types of tissues suggest a complex role of NOX in maintaining cellular redox homeostasis [15]. ROS generation resulting from the dysregulated activation and expression of NOX significantly contributes to the progression of tumorigenesis and metastasis [16]. The NOX isoform NOX-1 supports cell proliferation and is associated with the colon cancer progression [17]. It was suggested that the switch from NOX-1 to NOX-2 promotes invasive phenotypes of colon cancer cells [18]. Moreover, the nuclear factor erythroid 2-related factor 2 (Nrf-2), an inducible transcription factor, mainly transcribes anti-oxidant and cytoprotective genes. Nrf-2 protects cancer cells from the oxidative stress and helps to maintain the redox homeostasis [19]. However, the involvement of NOX isoforms and Nrf-2 in the deployment of drug-resistant phenotype of colon cancer cells is poorly understood. In the present study, we have investigated the role of redox proteins NOX-2 and Nrf-2 and their response after treatment with the anti-cancer drug 5-fluorouracil (5-FU) in the drug-resistant colon cancer cells (HCT-116/R). We also found that the inhibition of NOX-2 and Nrf-2 sensitizes HCT-116/R to 5-FU. These findings may help to establish direct involvement of NOX-2 and Nrf-2 in the redox regulation in drug resistance.
MATERIALS AND METHODS
Chemicals. All chemicals were of molecular biology grade. RPMI 1640 medium, Dulbecco’s phosphate buffered saline (DPBS), fetal bovine serum (FBS), PSN (penicillin, streptomycin, and neomycin) antibiotic solution, 5,6-CFDA dye, Annexin V Alexa Fluor 488 conjugate, propidium iodide (PI) stain, cDNA synthesis kit, SYBR green, anti-Nrf-2 antibody, and horseradish peroxidase (HRP)-conjugated secondary anti-rabbit and anti-goat antibodies were purchased from GIBCO, Life Technologies (USA). LDH Cytotoxicity Detection kit was purchased from Takara (Japan). Nitro Blue Tetrazolium (NBT), 2′,7′-dichlorofluorescin diacetate (H2DCFDA), ML171, N-[4-[2,3-dihydro-1-(2-methylbenzoyl)-1H-indol-5-yl]-5-methyl-2-thiazolyl]-1,3-benzodioxole-5 acetamide (ML385), histamine dihydrochloride (HDC), Thiazolyl Blue Tetrazolium Bromide (MTT), Trypan Blue dye, and anti-mouse HRP-conjugated antibody were purchased from Sigma-Aldrich (USA). Diphenyleneiodonium (DPI) was purchased from APExBIO (USA). Anti-NOX-1 (1 : 1000) and anti-NOX-2 (1 : 500) antibodies were purchased from Abd Serotec (BioRad, USA); anti-MDR1 (1 : 1000), anti-HIF1α (1 : 1000) and anti-β-actin (1 : 1000) antibodies were purchased from Cell Signaling Technology (USA).
Cell lines and culture. Human colorectal carcinoma (HCT-116) cell line was obtained from the National Center for Cell Science (NCCS), Pune, India. The cells were grown in RPMI 1640 medium containing L-glutamine (2 mM). The medium was supplemented with 10% fetal bovine serum and antibiotic cocktail containing penicillin (5 mg/ml), streptomycin (5 mg/ml), and neomycin (10 mg/ml) (GIBCO, Invitrogen, UK). The cells were kept in a humidified atmosphere of 95% air and 5% CO2 in a CO2 incubator at 37°C. Exponentially growing cultured cells were used for all experiments.
Transfection. The cells were transfected with the plasmids coding for MDR1 (pHaMDRwt Addgene plasmid #10957) and GFP-Nrf-2 (pcDNA3-EGFP-C4-Nrf-2 Addgene plasmid #21549) using Lipofectamine LTX as per manufacturer’s instructions. Briefly, HCT-116 cells were seeded in culture dishes and incubated for 16 h. Before the transfection, the culture medium was replaced by the fresh medium. The DNA and Lipofectamine mixture was prepared separately in tubes as per manufacturer’s protocol and incubated for 8 min. Next, the mixture was added to the cells, and the cells were incubated for 48 h at 37°C in a CO2 incubator. Hereafter, HCT-116 cells overexpressing MDR1 gene are denoted as HCT-116/R cells
Treatment with 5-FU. The stock solution of the anti-cancer agent 5-FU (50 mM) was prepared in the cell culture grade DMSO. HCT-116/R cells were treated with various doses of 5-FU (1, 10, 25, 50, 100, 500, 1000, and 2000 µM) for 24 h. For the inhibition studies, HCT-116/R cells were treated with 5-FU (100 µM) with or without inhibitors for 24 h.
Inhibition of NOX-2 and Nrf-2. The activity of NOX-2 and Nrf-2 was blocked using specific inhibitors. The stock solutions of the NOX inhibitor DPI (10 mM), selective NOX-1 inhibitor ML171 (0.5 mM), selective NOX-2 inhibitor HDC (5 mM), and selective Nrf-2 inhibitor ML385 (10 mM) were prepared in the cell culture grade DMSO. HCT-116/R cells were treated with DPI (10 µM) for 2 h or with ML171 (0.5 µM), HDC (10 µM), and ML385 (10 µM) for 4 h. For the cell death analysis, HCT-116/R cells were treated with 5-FU (100 µM) alone or in combination with ML171 (0.5 µM), HDC (10 µM), or ML385 (10 µM) for 24 h. HCT-116/R cells were pre-treated with DPI (10 µM) for 2 h followed by 5-FU (100 µM) treatment for 24 h.
Morphological analysis. The changes in the cellular morphology of HCT 116 cells after transfection were observed under the bright-field microscope using a DIC filter. Briefly, HCT-116 and HCT-116/R cells were washed with DPBS, and at least 150 cells from three different fields were observed using the DIC filter (Olympus IX81/DP-71). The images were acquired with the Image-Pro MC 6.1 software (USA) and analyzed with the Image J software (NIH, USA).
Rhodamine 123 accumulation assay. The staining with rhodamine 123 was performed as described earlier with minor modification [20]. In brief, HCT-116 and HCT-116/R cells were washed with DPBS and stained with 10 µM rhodamine 123 (in HBSS) for 1 h; the images were acquired with the Image-Pro MC 6.1 software (USA) under an inverted fluorescence microscope (Olympus IX81/DP-71). The images were analyzed with the Image J software (NIH, USA).
Doxorubicin intracellular distribution and accumulation assay. The assay was performed as reported earlier [21]. In brief, HCT-116 and HCT-116/R cells were washed with DPBS and treated with 5 µM doxorubicin for 2 h and observed under an inverted fluorescence microscope (Olympus IX81/DP-71).
Intracellular ROS assay. The intracellular generation of ROS was monitored using the H2DCFDA dye. In brief, HCT-116 and HCT-116/R cells were incubated with H2DCFDA (25 µM) for 20 min at 37°C in the dark. Next, the cells were washed with DPBS, and the fluorescence of DCF molecules was assessed at the excitation at 485 nm and emission at 525 nm using a Multimode microplate reader (Molecular Devices, USA). For qualitative analysis, the images were acquired with the Image-Pro MC 6.1 software (USA) using an inverted fluorescence microscope (Olympus IX81/DP-71).
Nitro Blue Tetrazolium (NBT) assay. NADPH oxidase (NOX) activity was determined by Nitro Blue Tetrazolium (NBT) assay as described earlier [22]. The NBT assay is based on the reduction of NBT to formazan by the superoxide produced in the NOX-catalyzed reaction. Briefly, HCT-116 and HCT-116/R cells were incubated with 100 µl NBT (1 mg/ml in RPMI culture medium) at 37°C for 1 h. Then, the cells were disrupted with 100 µl of DMSO, and the absorbance was recorded at 570 nm. The results were presented as the mean percentage of absorbance in different groups.
Determination of IC50 value and cell proliferation assay. The half-maximal inhibitory concentration (IC50) for 5-FU in HCT-116 and HCT-116/R was determined by the MTT assay as described earlier [23]. Briefly, HCT-116 and HCT-116/R cells were treated with various doses of 5-FU (1, 10, 25, 50, 100, 500, 1000, 2000 µM) for 24 h. The cells were then incubated with 0.5 mg/ml MTT solution for 4 h at 37°C. Next, 0.1 ml of SDS-HCl (10% SDS in 0.01 M HCl) was added to each well, mixed thoroughly, and incubated in the dark for 20 min at 37°C. The absorbance in the wells was recorded at 570 nm with a reference wavelength of 650 nm using a multimode microplate reader (SpectraMax M2e, Molecular Devices). The effect of 5-FU and inhibitors on the proliferation of HCT-116 and HCT-116/R cells was examined by the MTT assay. The results are presented as the percentage of cell proliferation in different treatment groups.
Cell death assay. Cell death was evaluated by Trypan Blue exclusion assay as described previously [24]. In brief, HCT-116/R cells were treated with 5-FU and combination with NOX, NOX-1, NOX-2, and Nrf-2 inhibitor DPI (10 µM), ML171 (0.5 µM), HDC (10 µM), or ML385 (10 µM) with 5-FU (100 µM) alone or 5-FU (100 µM) in combination with DPI (10 µM), ML171 (0.5 µM), HDC (10 µM), or ML385 (10 µM) for 24 h. After completion of incubation, the cells were harvested and washed once with DPBS. Equal volumes of cell suspension and Trypan Blue solution were mixed, and live and dead cells were counted under a bright-field microscope. The percentage of cell death was determined by the following formula: number of dead cells/total number of cells ×100%.
LDH cytotoxicity detection assay. The cytotoxic effect of 5-FU and inhibitors was examined using the LDH assay kit (Takara) according to manufacturer’s instruction. Briefly, 1.5 × 104 HCT-116/R cells were treated as mentioned in the figure legend. The medium was collected and mixed with the LDH reaction mixture containing diaphorase (catalyst) and dye solution and incubated for 30 min at room temperature in the dark. The absorbance was recorded at 490 nm with a reference wavelength of 650 nm using a multimode microplate reader (SpectraMax M2e, Molecular Devices). The data were represented as the percentage of cytotoxicity (LDH release).
Annexin V/PI staining. Apoptotic cell death was evaluated by Annexin V/PI staining as described previously [24]. In brief, HCT-116/R cells were grown on poly-L-lysine-coated coverslips and treated as described earlier. Next, the cells were stained with Annexin V Alexa Fluor 488 for 20 min followed by propidium iodide (PI) staining for 5 min. At least 150 cells from three random fields were observed under the inverted fluorescence microscope (Olympus IX81/DP-71).
Analysis of cell viability. The cell viability was analyzed by CFDA staining. Briefly, HCT-116/R cells were treated as described in figure legends, stained with the CFDA dye (25 µM) for 20 min, washed twice with DPBS, and observed under an inverted fluorescence microscope (Olympus IX81/DP-71).
Nrf-2 intracellular translocation. The translocation of GFP-Nrf-2 between the nucleus and the cytoplasm was examined by fluorescence microscopy. In brief, 4 × 104 cells were transfected with the MDR1 gene; after 24 h, the cells were transfected with the plasmid coding for GFP-Nrf-2 using Lipofectamine LTX with Plus reagent and incubated for 24 h. Next, the cells were washed with DPBS and examined under an inverted fluorescence microscope (Olympus IX81/DP-71).
Western blotting. Expression of MDR1, HIF-1α, Nrf-2, NOX-1, NOX-2, and β-actin proteins was evaluated by Western blotting. Briefly, 1 × 106 cells were transfected with the MDR1 plasmid for 48 h. The cells were harvested at different time intervals, washed twice with ice-cold DPBS, and lysed in the lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM MgCl2, 150 mM CaCl2, 1 mM Na vanadate, 0.05% Nonidet P-40, protease inhibitor cocktail) and kept on ice for 30 min followed by disruption and centrifugation at 8000 rpm for 10 min at 4°C. The supernatant (whole cell lysate) was collected and the protein concentration was determined with the BCA protein concentration kit. Fifty micrograms of protein was resolved by electrophoresis in 12 and 10% SDS-PAG and transferred to a PVDF membrane by the wet electroplating method at 4°C. The membrane was blocked with 3% fat-free milk in TBST for 2 h at room temperature followed by overnight incubation with the primary antibodies at 4°C. The membrane was probed with the HRP-conjugated secondary antibodies (1 : 3000). The proteins were detected using the EZ-ECL kit according to the manufacturer’s instructions, and the signal was captured on Kodak X-Omat blue film in the dark.
RNA extraction and real-time PCR. Total RNA was extracted using Trizol reagent (Invitrogen, Life Technologies, USA) as per manufacturer’s instruction. Total RNA (1 µg) was reversely transcribed to cDNA using the cDNA synthesis kit (Invitrogen, Life Technologies). Real-time PCR was performed in MicroAmp fast optical 96-well PCR plates (Applied Biosystems, USA) using Step One plus real-time PCR detection system (Applied Biosystems). The reaction mixture contained cDNA (100 pmol), SYBR Green mastermix, 1 µM of each primer, and nuclease-free water up to 20 µl of the final reaction volume (20 µl). The expression of NOX-1, NOX-2, and HO-1 mRNAs was normalized to18S rRNA (endogenous control). The sequences of the primers used in the study are presented in the table.
List of primers used in the study
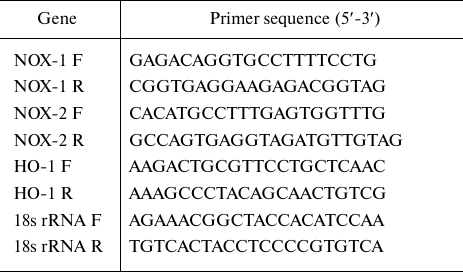
Note. F, forward primer; R, reverse prime.
Statistical analysis. The data were analyzed with the Student’s t-test and one-way analysis of variance (ANOVA) followed by the Student–Newman–Keulas (SNK) test for multiple comparisons using the Sigma plot 12.0 statistical analysis software. The difference between the groups at * p ≤ 0.05, ** p ≤0.01, and *** p ≤ 0.001 was considered statistically significant.
RESULTS
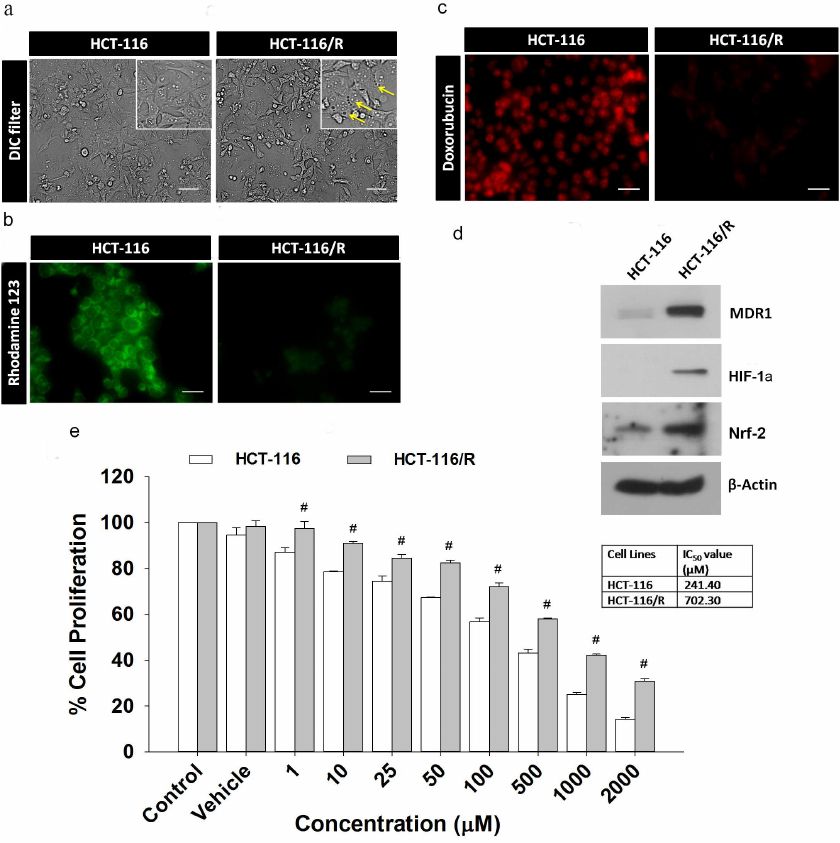
Overexpression of MDR1 induces drug resistance phenotype in HCT-116 cells. Here, we examined the effects of the MDR1 gene overexpression in HCT-116 cells. We transfected the MDR1 gene into HCT-116 cells and analyzed the resulting transfected cells (HCT-116/R) for the drug-resistant phenotype under a microscope. We found that the cells overexpressing MDR1 had more intracellular vacuoles compared to the wild-type cells (HCT-116) (Fig. 1a). Next, we analyzed the ability of HCT-116/R cells to reduce the drug uptake using rhodamine 123 staining. Most of HCT-116 cells showed intense rhodamine 123 fluorescence, whereas the fluorescence of HCT-116/R cells was weaker, which indicates that the uptake of rhodamine 123 by the MDR1-overexpressing cells was reduced (Fig. 1b). We also analyzed the intracellular distribution and accumulation of doxorubicin in both cell lines and found that most of HCT-116 cells displayed prominent doxorubicin fluorescence in the nucleus, while HCT-116/R cells showed no doxorubicin fluorescence in the nucleus and diminished fluorescence in the cytoplasm (Fig. 1c). The overexpression of MDR1 in HCT-116/R cells was also confirmed by immunoblotting (Fig. 1d). Moreover, expression of HIF-1α and Nrf-2 was also upregulated. These results suggest that the elevated expression of MDR1, HIF-1α, and Nrf-2 is associated with the development of drug resistance.
We also determined the chemosensitivity of HCT-116 and HCT-116/R cells to 5-FU. The half-maximal inhibitory concentration (IC50) of 5-FU for HCT-116 and HCT-116/R was ~241.40 and ~702.30 µM, respectively (Fig. 1e), which indicates that the HCT-116/R cells are highly resistant to 5-FU. Therefore, the MDR1 overexpression confers drug resistance in human colorectal carcinoma (HCT-116) cells.
Fig. 1. Characterization of drug-resistant HCT-116/R cells obtained by transfection of HCT-116 cells with the MDR1 gene: a) morphological analysis; b) rhodamine 123 staining; c) intracellular doxorubicin distribution and accumulation; scale bar: 20 µm. d) Western blotting of MDR1, HIF-1α, and Nrf-2 (β-actin was used as a loading control). e) Effect of 5-FU on the proliferation of HCT-116 and HCT-116/R cells as determined by the MTT assay. Error bars represent ±SEM of three independent experiments; # p ≤ 0.001, significant difference between HCT-116 and HCT-116/R cells (one-way ANOVA followed by the SNK test).
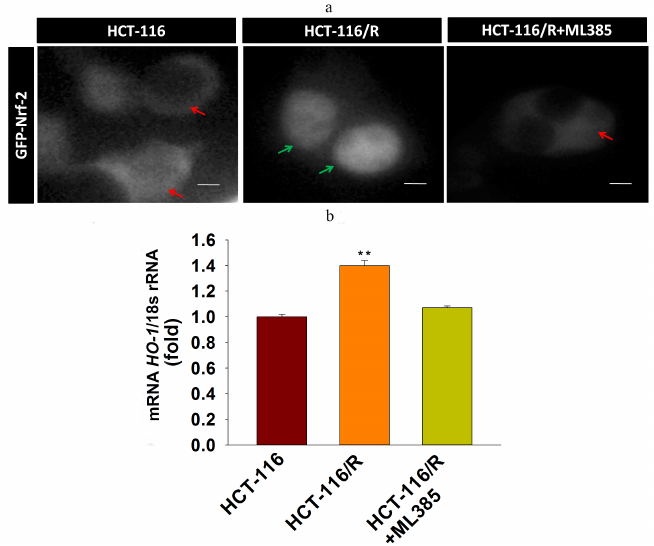
Upregulated expression of Nrf-2 and HO-1 mRNAs is involved in the drug resistance of HCT-116/R cells. Nrf-2 is mainly associated with the anti-oxidant response and cellular detoxification, as it regulates antioxidant response element (ARE)-dependent target genes [25]. The Nrf-2/HO-1 axis participates in the development of drug resistance in various malignancies [26]. In order to explore whether the Nrf-2/HO-1 signaling axis is functional in the HCT-116/R cells, we analyzed Nrf-2 expression and activation and found that Nrf-2 was significantly upregulated in HCT-116/R cells (Fig. 1d). We also investigated the activity of Nrf-2 by studying its intracellular localization. We found a prominent expression of GFP-Nrf-2 in the nuclei of HCT-116/R cells (Fig. 2a), while in HCT-116 cells, GFP-Nrf-2 was mostly accumulated in the cytoplasm. To evaluate the activity of Nrf-2 in the nucleus, we analyzed the expression of the Nrf-2 target gene HO-1 at the mRNA level. Our results showed a significantly higher expression of HO-1 mRNA in the HCT-116/R cells as compared to HCT-116 cells (Fig. 2b). The treatment with ML385 (specific Nrf-2 inhibitor) significantly decreased the activation of the Nrf-2 and HO-1 mRNA expression in HCT-116/R cells. (Fig. 2, a and b) Taken together, these results showed that the activation of Nrf-2 and the downstream HO-1 gene are associated with the development of drug resistance in HCT-116/R cells.
Fig. 2. Activation of Nrf-2 signaling and Nrf-2 intracellular localization. a) Nrf-2 distribution in HCT-116 cells and drug-resistant HCT-116/R cells before and after treatment with ML385. Green and red arrows indicate nuclear and cytoplasmic accumulation, respectively, of GFP-Nrf-2; scale bar: 5 µm. b) RT-qPCR analysis of HO-1 mRNA. Error bars represent ±SEM of three independent experiments; ** p ≤ 0.01, significant difference between HCT-116 and HCT-116/R cells (Student’s t-test).
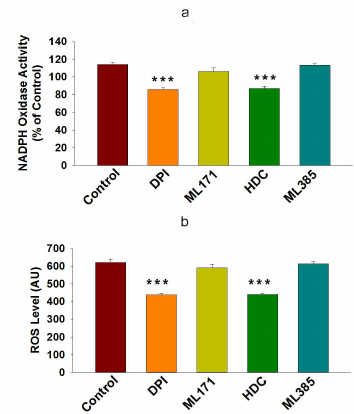
Higher NOX activity and NOX-2 expression activate ROS generation and promote drug resistance in cancer cells. ROS are known to contribute to oxidative stress and progression of cancer [27]. NOX enzymes are one of the major sources of cellular ROS [28]. Here, we were interested to examine the involvement of NOX-mediated ROS generation into the drug resistance in cancer. First, we examined the NOX activity in HCT-116 and HCT-116/R cells and found that it was significantly higher in the HCT-116/R cells (Fig. 3a). This result raised the question whether the elevated activity of NOX correlated with the expression of its isoforms, NOX1 and NOX2. We examined expression of NOX-1 and NOX-2 at the protein and mRNA levels and found that NOX-2 was drastically upregulated in HCT-116/R cells (Fig. 3, b-d). However, the expression of NOX-1 remained unchanged, which suggests that the isoform-specific function of NOX is involved in the formation of the drug-resistant phenotype of human colorectal cancer (Fig. 3, b and c). We also monitored the level of intracellular ROS in HCT-116/R cells using the fluorescent dye H2DCFDA and observed a significant increase in the intracellular ROS level (Fig. 3e), which was confirmed by the microscopic analysis of the cells (Fig. 3, f and g). Moreover, to verify the role of specific NOX isoforms, we treated the HCT-116/R cells with specific inhibitors, such as DPI (inhibitor of NOX), ML171 (NOX-1 specific inhibitor), HDC (NOX-2 specific inhibitor), and ML385 (inhibitor of Nrf-2). DPI and HDC inhibited NOX activity and ROS production as compared to the control HCT-116/R cells. However, ML171 and ML385 only slightly inhibited NOX activation and ROS generation (Fig. 4, a and b). Therefore, our results suggest that NOX activation and upregulated expression of NOX-2 facilitate ROS generation in the drug-resistant HCT-116/R cells.
Fig. 3. Activation of the NOX enzyme system and intracellular ROS generation in HCT-116/R cells. a) Analysis of NOX activity by the NBT assay. b) Western blot analysis of NOX-1 and NOX-2 (β-actin was used as a loading control). RT-qPCR analysis of (c) NOX-1 and (d) NOX-2. The intracellular ROS generation was evaluated using H2DCFDA. e) Quantitative spectrophotometric analysis; f and g) qualitative analysis by fluorescence microscopy; scale bar, 50 µm. Error bars represent ±SEM of three independent experiments; ** p ≤ 0.01, *** p ≤ 0.001, significant differences between HCT-116 and HCT-116/R cells (Student’s t-test).
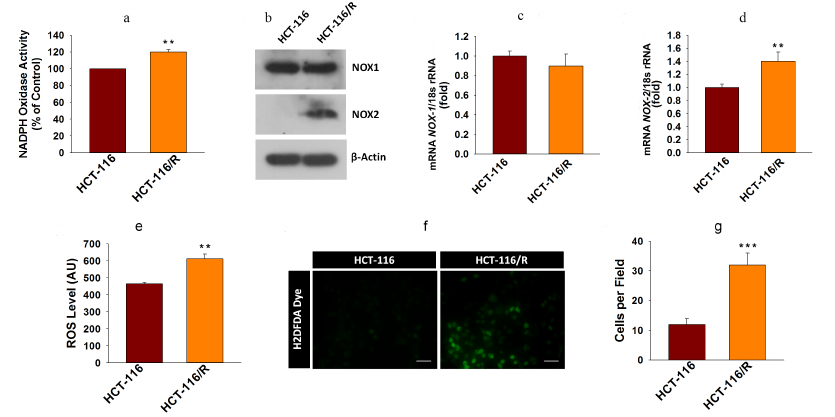
Fig. 4. NOX-2-mediated ROS generation in HCT-116/R cells. HCT-116/R cells were treated with DPI (10 µM), ML171 (0.5 µM), HDC (10 µM), and ML385 (10 µM) and assayed for the NOX activation. a) Analysis of NOX activity by the NBT assay. b) Spectrophotometric analysis of ROS generation. Error bars represent ±SEM of three independent experiments; *** p ≤ 0.001, significant difference between untreated and treated cells (one-way ANOVA followed by the SNK test).
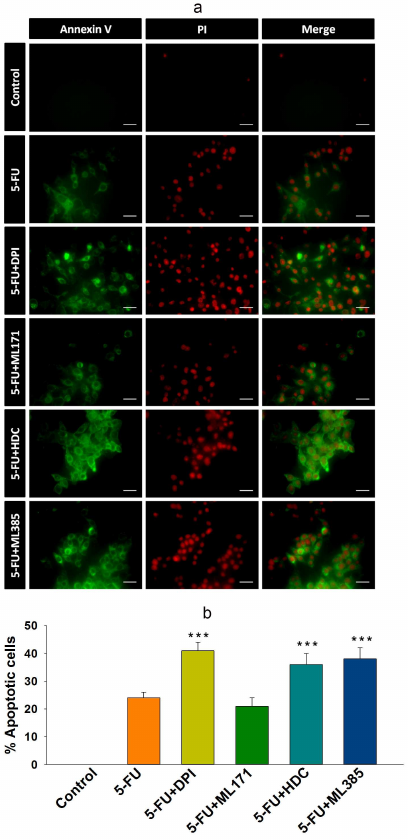
Inhibition of NOX-2 and Nrf-2 sensitized HCT-116/R cells to 5-FU and promoted their death. 5-FU is an antimetabolite that interferes with the DNA and RNA synthesis in cancer cells [29]. It is widely used to treat various malignancies, including breast, head, neck, and colon cancers [30, 31]. Currently, 5-FU in combination with other chemotherapeutic agents is mainly used in the treatment of colon cancer, which has significantly improved patient survival rate [32]. However, the acquisition of drug resistance limits the efficacy of 5-FU. Based on the observed elevated NOX-2 and Nrf-2 expression levels and activation in HCT-116/R cells, we investigated the role of NOX-2 and Nrf-2 activation in the 5-FU resistance in these cells. HCT-116/R cells were treated with 5-FU alone or in combination with the NOX inhibitor DPI and Nrf-2 inhibitor ML385. The treatment with 5-FU in combination with DPI and ML385 significantly inhibited cell proliferation and induced cell death as compared to 5-FU alone, which suggests that NOX and Nrf-2 are involved in the emergence of the resistance to 5-FU (Fig. 5, a and b). To identify the role of specific NOX isoforms in this process, we treated HCT-116/R cells with 5-FU and ML171 (specific NOX-1 inhibitor) and with 5-FU and HDC (specific NOX-2 inhibitor). Interestingly, the 5-FU + HDC treatment inhibited cell proliferation and induced cell death as compared to 5-FU alone (Fig. 5, a and b). However, the treatment with 5-FU + ML171 did not produce any significant effect on the cell proliferation and cell death as compared to 5-FU alone (Fig. 5, a and b). These results suggest that the inhibition of NOX-2 and Nrf-2 activities augments the 5-FU-mediated death of HCT-116/R cells. We also monitored the LDH release to analyze the necrotic cell death in all treatment groups, but observed no significant LDH release upon the treatment, which suggests that neither 5-FU alone nor the 5-FU in combination with the tested inhibitors caused necrotic cell death (Fig. 5c). Next, we studied the morphological changes and cell viability upon various treatments and found that the treatment with 5-FU in combination with DPI, HDC, or ML385 significantly altered the morphology and reduced the viability of HCT-116/R cells (Fig. 5, a-c). The treatment with 5-FU in combination with ML171 did not produce any significant effect on the cell morphology and viability as compared to the treatment with 5-FU alone, which suggests that the inhibition of the NOX-2 and Nrf-2 activity reduces the viability of HCT-116/R cells (Fig. 6, a-c). The mode of cell death induced by the treatment was determined by the Annexin V/PI staining. Our results revealed that the treatment with 5-FU in combination with DPI, HDC, or ML385 showed more Annexin V/PI-positive cells as compared to the treatment with 5-FU or with 5-FU in combination with ML171 (Fig. 7, a and b). This result indicates that the inhibition of NOX-2 and Nrf-2 activity promotes 5-FU-mediated apoptosis in HCT-116/R cells. Therefore, NOX-2 and Nrf-2 activation facilitates development of the 5-FU resistance in HCT-116/R cells.
Fig. 5. Inhibition of NOX-2 activity promotes cell death in HCT-116/R cells. HCT-116/R cells were treated with either 5-FU (100 µM) alone or 5-FU (100 µM) in combination with DPI (10 µM), ML171 (0.5 µM), HDC (10 µM), or ML385 (10 µM). a) Analysis of cell proliferation by the MTT assay. b) Cell death analysis by the Trypan Blue assay. c) LDH release assay with the LDH cytotoxicity detection kit. Error bars represent ±SEM of three independent experiments; *** p ≤ 0.001, significant difference between the cells treated with 5-FU alone and with 5-FU in combination with inhibitors (one-way ANOVA followed by the SNK test).
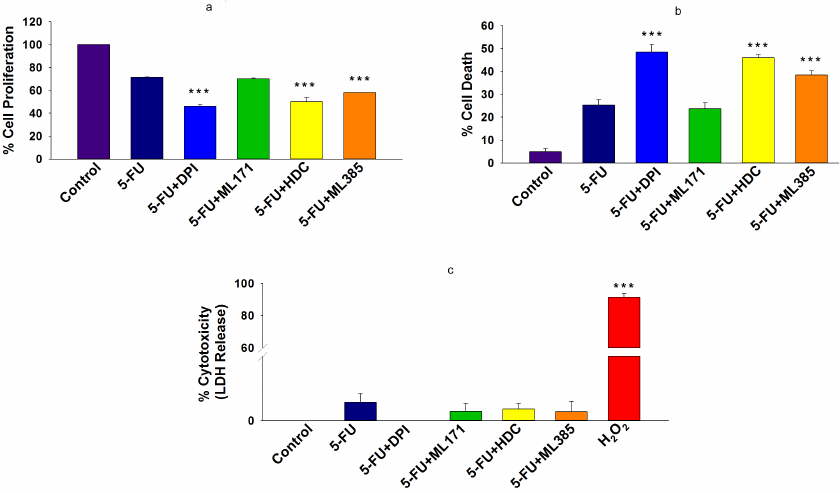
Fig. 6. Inhibition of NOX-2 activation promotes apoptotic cell death in HCT-116/R cells. HCT-116/R cells were treated with 5-FU (100 µM) alone or with 5-FU (100 µM) in combination with DPI (10 µM), ML171 (0.5 µM), HDC (10 µM), or ML385 (10 µM). a) Analysis of cell morphology. b and c) Cell viability analysis by CFDA staining; scale bar: 50 µm. Error bars represent ±SEM of three independent experiments; *** p≤ 0.001, significant difference between the cells treated with 5-FU alone and with 5-FU in combination with inhibitor (one-way ANOVA followed by the SNK test).
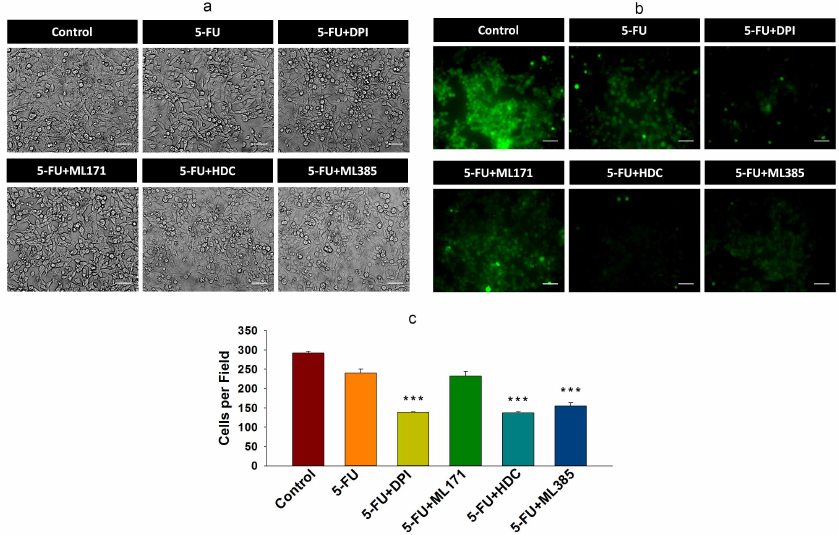
Fig. 7. Inhibition of NOX-2 and Nrf-2 activation promotes apoptotic cell death in HCT-116/R cells. HCT-116/R cells were treated with 5-FU (100 µM) alone or with 5-FU (100 µM) in combination with DPI (10 µM), ML171 (0.5 µM), HDC (10 µM), or ML385 (10 µM). a) and b) Analysis of apoptotic cell death by Annexin V/PI staining; scale bar: 20 µm. Error bars represent ±SEM of three independent experiments; *** p ≤ 0.001, significant difference between the cells treated with 5-FU alone and with 5-FU in combination with inhibitor (one-way ANOVA followed by the SNK test).
DISCUSSION
Drug resistance is a phenomenon when a pathological condition becomes tolerant to the therapeutics. The acquisition of drug resistance by cancer cells is the major hurdle in the use of anticancer agents. Although many types of cancers are initially susceptible to chemotherapeutic drugs, but, after sometime, cancer cells start developing drug resistance because of multiple intrinsic and extrinsic factors [5]. One of the most studied mechanisms of cancer drug resistance involves overexpression of the ABC transporter family proteins. Three transporters, such as the multidrug resistance protein 1 (MDR1), multidrug resistance-associated protein 1 (MRP1), and breast cancer resistance protein (BCRP), are involved in the drug resistance emergence in various malignancies [33] and protect cancer cells from a variety of chemotherapeutic agents. MDR1 (P-gp) was the first identified and extensively studied transporter protein in cancer models. In the recent past, compelling evidence has revealed that the overexpression of P-gp is associated with the development of multidrug resistance (MDR) in cancer cells. P-gp displays broad substrate specificity; therefore, P-gp-overexpressing cells display cross-resistance against multiple cytotoxic drugs and promote the MDR in cancer cells. The emergence of drug resistance in colon cancer cells is a major factor responsible for the tumor recurrence and decrease in the patient survival rate [34]. Hence, identification of new regulators of colon cancer resistance is greatly needed.
It was found that dysregulation of redox signaling proteins and genes promote drug resistance in cancer cells [35]. ROS are known to target redox-sensitive proteins and to regulate immune response, cell proliferation, cell differentiation, and cell death signaling. Intracellular ROS are mainly generated by NOXs and participate in the redox homeostasis of the cells. It is evident that the NOX-produced ROS are involved in cancer development and progression of various malignancies [36]. It was suggested that the switch from NOX-1 to NOX-2 expression is associated with malignancy [18]. Indeed, various oncogenes including RAS regulate NOX-1 expression and activation [37]. Apart from NOX-1, another isoform, NOX-2, was reported to participate in the development of invasive phenotypes of the colon cancer cells [18]. NOX-2 is the first identified isoform among all NOX isoforms which is mainly expressed by immune cells, such as macrophages, monocytes, and neutrophils [38]. The switch from NOX-1 to NOX-2 was found to promote invasion of colon cancer cells [18]. Collectively, these reports suggest that both NOX-1 and NOX-2 are involved in the colon cancer progression.
Nrf-2 is a major transcription factor that promotes transcription of multiple anti-oxidant genes. Under normal conditions, Nrf-2 is constantly degraded by Keap-1. During oxidative stress, Nrf-2 dissociates from Keap-1, which leads to its activation and translocation to the nucleus, where Nrf-2 initiates transcription of ARE-dependent genes [39]. It was suggested that mutations in Nrf-2 and Keap1 promote constitutive Nrf-2 activation and expression of cytoprotective (pro-survival) genes in various human cancers and eventually contribute to the drug resistance of cancer cells [40].
It was proposed that the activation of NOX enzymes and Nrf-2 is associated with the maintenance of cell homeostasis [41]. Mutations in the RAS gene promote NOX expression and activation, as well as ROS generation in the colon cancer cells [42, 43]. Besides, mutant RAS also promotes Nrf-2 activation [44]. Nrf-2 is known to regulate ROS generation by the NOX enzymes [45]. It was also suggested that the NOX activation is intricately associated with the antioxidants that regulate tumor microenvironment [46]. Moreover, the Nrf-2/HO-1 axis is often activated during progression of various malignancies. It is also associated with the chemoresistance and poor disease prognosis [26]. The contribution of NOX isoforms to the tumor invasion and growth has been reported, but the role of these proteins in the development of resistance in colon cancer remains elusive.
In this study, we explored the role of Nrf-2 in drug resistance. Thus, we found that Nrf-2 is upregulated in HCT-116/R cells. The activation of Nrf-2 signaling was confirmed by changes in the Nrf-2 intracellular location and expression of the target HO-1 gene, as we found a prominent accumulation of GFP-Nrf-2 in the nucleus and upregulation of HO-1 in the HCT-116/R cells. These results suggest that Nrf-2 is not only stabilized due to the elevated ROS generation in HCT-116/R cells, but is also activated and transcribes its target genes.
The accumulation of ROS resulted oxidative stress is associated with progression of cancer [27]. A possible link between cellular ROS and chemoresistance was suggested [47]. Since NOX enzymes are major producers of ROS, we investigated whether the activation of NOX enzymes is involved in the progression of resistance phenotypes and found elevated NOX activation and ROS generation in HCT-116/R cells. We also monitored the expression of NOX-1 and NOX-2 isoforms and observed that expression of NOX-2 but not of NOX-1 was upregulated in the drug-resistant HCT-116/R cells, which suggests the isoform-specific function of the NOX enzymes. Next, to assess the role of NOX-2, we used specific inhibitors of NOX-1 (ML171) and NOX-2 (HDC) and found that the NOX activity was inhibited by DPI and HDC, but not by ML171. These results suggest that the drug-resistance of HCT-116/R cells involves accelerating NOX-2 expression and activation of this protein.
5-FU is a commonly used chemotherapeutic agent for the treatment of colorectal cancer [32, 48]. However, the preventive activity of 5-FU is reduced in the drug-resistant colon cancer cells [49]. Our result showed that the MDR1-overexpressing HCT-116/R cells were highly resistant to the 5-FU treatment. The half-maximal inhibitory concentration of 5-FU was approximately 2.9-fold higher in HCT-116/R cells as compared to that in HCT-116 cells, which may be related to the elevated P-gp (MDR1) expression. Recent studies suggest that the NOX-mediated changes in the cellular redox status promote chemoresistance [50]. Moreover, Nrf-2 is epigenetically modulated in the 5-FU-resistant colon cancer cells [51]. In support of previous findings, our results suggest that both NOX and Nrf-2 activation take place in HCT-116/R cells. Therefore, targeting NOX-2 and Nrf-2 may sensitize the HCT-116/R cells towards 5-FU. We showed that the combination of 5-FU with DPI (NOX enzyme inhibitor) and HDC (specific NOX-2 inhibitor) induced apoptotic death of HCT-116/R cells as compared to 5-FU alone. A combined treatment with 5-FU and ML385 (specific Nrf-2 inhibitor) produced the similar effects. However, no significant changes were observed upon the treatment with 5-FU and ML171 (specific NOX-1 inhibitor), suggesting the major role of NOX-2 and Nrf-2 in the developing 5-FU resistance in HCT-116/R cells.
CONCLUSION
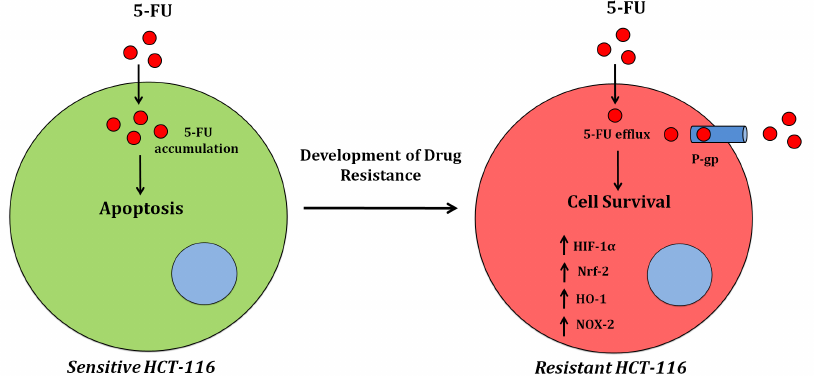
In conclusion, our results suggest that MDR1 plays a major role in the development of drug-resistant phenotypes of cancer cells. We also found a significant increase in the NOX activity and elevated ROS production in the drug-resistant HCT-116/R cells. Interestingly, a significant increase was also observed in the expression and activity of the redox proteins NOX-2 and Nrf-2 (Fig. 8), which promoted the 5-FU resistance in HCT-116/R cells. Therefore, drug resistance genes reprogram the cellular redox homeostasis by activating NOX-2 and Nrf-2 signaling, which can be used as a therapeutic target to reduce the drug resistance of colon cancer cells in the future.
Fig. 8. Alterations in the cellular redox signaling during the development of the 5-FU resistance in colon cancer (HCT-116) cells.
Funding. This study was supported by the SERB, Department of Science & Technology, New Delhi, Government of India (research grant EMR/2016/002574 to C. P.); ICMR, Gov of India (Senior Research fellowship to F. U. V.) and DST-INSPIRE (fellowship to B. N. W.).
Acknowledgments. The authors are grateful to the Indian Institute of Advanced Research, for providing the infrastructure and the research facility.
Ethics declarations. The authors declare no conflict of interest. This article does not contain description of studies with the involvement of humans or animal subjects.
REFERENCES
1.Rawla, P., Sunkara, T., and Barsouk, A. (2019)
Epidemiology of colorectal cancer: Incidence, mortality, survival, and
risk factors, Prz. Gastroenterol., 14, 89.
2.Beheshti, M. (2018) Different strategies to
overcome multidrug resistance in cancer. Research review, Int.
Pharm. Acta, 1, 88-89.
3.Longley, D., and Johnston, P. (2005) Molecular
mechanisms of drug resistance, J. Pathol., 205,
275-292.
4.Tanguturi, P., Kim, K.-S., and Ramakrishna, S.
(2020) The role of deubiquitinating enzymes in cancer drug resistance,
Cancer Chemother. Pharmacol., 85, 627-639.
5.Mansoori, B., Mohammadi, A., Davudian, S.,
Shirjang, S., and Baradaran, B. (2017) The different mechanisms of
cancer drug resistance: a brief review, Adv. Pharm. Bull.,
7, 339.
6.Choi, Y. H., and Yu, A. M. (2014) ABC transporters
in multidrug resistance and pharmacokinetics, and strategies for drug
development, Curr. Pharm. Des., 20, 793-807.
7.Locher, K. P., and Borths, E. (2004) ABC
transporter architecture and mechanism: implications from the crystal
structures of BtuCD and BtuF, FEBS Lett., 564,
264-268.
8.Holland, I. B. (2011) ABC transporters, mechanisms
and biology: an overview, Essays Biochem., 50, 1-17.
9.Gottesman, M. M., and Pastan, I. H. (2015) The role
of multidrug resistance efflux pumps in cancer: revisiting a JNCI
publication exploring expression of the MDR1 (P-glycoprotein)
gene, J. Natl. Cancer Inst.,107.
10.Gottesman, M. M., Fojo, T., and Bates, S. E.
(2002) Multidrug resistance in cancer: role of ATP-dependent
transporters, Nat. Rev. Cancer, 2, 48-58.
11.Snezhkina, A. V., Kudryavtseva, A. V., Kardymon,
O. L., Savvateeva, M. V., Melnikova, N. V., et al. (2019) ROS
generation and antioxidant defense systems in normal and malignant
cells, Oxid. Med. Cell. Longev., 2019, 6175804, doi:
10.1155/2019/6175804.
12.Maryanovich, M., and Gross, A. (2013) A ROS
rheostat for cell fate regulation, Trends Cell Biol., 23,
129-134.
13.Schieber, M., and Chandel, N. S. (2014) ROS
function in redox signaling and oxidative stress, Curr. Biol.,
24, R453-R462.
14.Crane, F., and Low, H. (2008) Reactive oxygen
species generation at the plasma membrane for antibody control,
Autoimmun. Rev., 7, 518-522.
15.Krause, K.-H. (2004) Tissue distribution and
putative physiological function of NOX family NADPH oxidases, Jpn.
J. Infect. Dis., 57, S28-S29.
16.Weyemi, U., Redon, C. E., Parekh, P. R., Dupuy,
C., and Bonner, W. M. (2013) NADPH oxidases NOXs and DUOXs as putative
targets for cancer therapy, Anticancer Agents Med. Chem.,
13, 502-514.
17.Juhasz, A., Markel, S., Gaur, S., Liu, H., Lu,
J., et al. (2017) NADPH oxidase 1 supports proliferation of colon
cancer cells by modulating reactive oxygen species-dependent signal
transduction, J. Biol. Chem., 292, 7866-7887.
18.Banskota, S., Regmi, S. C., and Kim, J.-A. (2015)
NOX1 to NOX2 switch deactivates AMPK and induces invasive phenotype in
colon cancer cells through overexpression of MMP-7, Mol. Cancer,
14, 123.
19.Jaramillo, M. C., and Zhang, D. D. (2013) The
emerging role of the Nrf2–Keap1 signaling pathway in cancer,
Genes Dev., 27, 2179-2191.
20.Kessel, D., Beck, W. T., Kukuruga, D., and
Schulz, V. (1991) Characterization of multidrug resistance by
fluorescent dyes, Cancer Res., 51, 4665-4670.
21.Shen, F., Chu, S., Bence, A. K., Bailey, B., Xue,
X., et al. (2008) Quantitation of doxorubicin uptake, efflux, and
modulation of multidrug resistance (MDR) in MDR human cancer cells,
J. Pharmacol. Exp. Ther., 324, 95-102.
22.Serrander, L., Cartier, L., Bedard, K., Banfi,
B., Lardy, B., et al. (2007) NOX4 activity is determined by mRNA levels
and reveals a unique pattern of ROS generation, Biochem. J.,
406, 105-114.
23.Waghela, B. N., Sharma, A., Dhumale, S., Pandey,
S. M., and Pathak, C. (2015) Curcumin conjugated with PLGA potentiates
sustainability, anti-proliferative activity and apoptosis in human
colon carcinoma cells, PLoS One, 10, e0117526.
24.Vaidya, F. U., Sharma, R., Shaikh, S., Ray, D.,
Aswal, V. K., and Pathak, C. (2019) Pluronic micelles encapsulated
curcumin manifests apoptotic cell death and inhibits pro-inflammatory
cytokines in human breast adenocarcinoma cells, Cancer Rep.,
2, e1133.
25.Tonelli, C., Chio, I. I. C., and Tuveson, D. A.
(2018) Transcriptional regulation by Nrf2, Antioxid. Redox
Signal., 29, 1727-1745.
26.Furfaro, A., Traverso, N., Domenicotti, C.,
Piras, S., Moretta, L., et al. (2016) The Nrf2/HO-1 axis in cancer cell
growth and chemoresistance, Oxid. Med. Cell. Longev.,
2016, 1958174, doi: 10.1155/2016/1958174.
27.Aggarwal, V., Tuli, H. S., Varol, A., Thakral,
F., Yerer, M. B., et al. (2019) Role of reactive oxygen species in
cancer progression: Molecular mechanisms and recent advancements,
Biomolecules, 9, 735.
28.Wang, X., Son, Y.-O., Chang, Q., Sun, L., Hitron,
J. A., et al. (2011) NADPH oxidase activation is required in reactive
oxygen species generation and cell transformation induced by hexavalent
chromium, Toxicol. Sci., 123, 399-410.
29.Das, D., Preet, R., Mohapatra, P., Satapathy, S.
R., Siddharth, S., et al. (2014) 5-Fluorouracil mediated anti-cancer
activity in colon cancer cells is through the induction of
Adenomatous Polyposis Coli: implication of the
long-patch base excision repair pathway, DNA Rep., 24,
15-25.
30.Grem, J. L. (2000) 5-Fluorouracil: forty-plus and
still ticking. A review of its preclinical and clinical development,
Invest. New Drugs, 18, 299-313.
31.Zhang, N., Yin, Y., Xu, S.-J., and Chen, W.-S.
(2008) 5-Fluorouracil: mechanisms of resistance and reversal
strategies, Molecules, 13, 1551-1569.
32.Pardini, B., Kumar, R., Naccarati, A., Novotny,
J., Prasad, R. B., Forsti, A., et al. (2011) 5-Fluorouracil-based
chemotherapy for colorectal cancer and MTHFR/MTRR genotypes, Br. J.
Clin. Pharmacol., 72, 162.
33.Kovalev, A. A., Tsvetaeva, D. A., and
Grudinskaja, T. V. (2013) Role of ABC-cassette transporters (MDR1,
MRP1, BCRP) in the development of primary and acquired multiple drug
resistance in patients with early and metastatic breast cancer, Exp.
Oncol., 35, 287-290.
34.Van der Jeught, K., Xu, H.-C., Li, Y.-J., Lu,
X.-B., and Ji, G. (2018) Drug resistance and new therapies in
colorectal cancer, World J. Gastroenterol., 24, 3834.
35.Liu, Y., Li, Q., Zhou, L., Xie, N., Nice, E. C.,
et al. (2016) Cancer drug resistance: redox resetting renders a way,
Oncotarget, 7, 42740.
36.Kamata, T. (2009) Roles of Nox1 and other Nox
isoforms in cancer development, Cancer Sci., 100,
1382-1388.
37.Adachi, Y., Shibai, Y., Mitsushita, J., Shang,
W., Hirose, K., and Kamata, T. (2008) Oncogenic Ras upregulates
NADPH oxidase 1 gene expression through MEK-ERK-dependent
phosphorylation of GATA-6, Oncogene, 27, 4921-4932.
38.Martner, A., Aydin, E., and Hellstrand, K. (2019)
NOX2 in autoimmunity, tumor growth and metastasis, J. Pathol.,
247, 151-154.
39.Taguchi, K., Motohashi, H., and Yamamoto, M.
(2011) Molecular mechanisms of the Keap1–Nrf2 pathway in stress
response and cancer evolution, Genes Cells, 16,
123-140.
40.Shibata, T., Kokubu, A., Gotoh, M., Ojima, H.,
Ohta, T., et al. (2008) Genetic alteration of Keap1 confers
constitutive Nrf2 activation and resistance to chemotherapy in
gallbladder cancer, Gastroenterology, 135,
1358-1368.e1354.
41.Sekhar, K. R., Crooks, P. A., Sonar, V. N.,
Friedman, D. B., Chan, J. Y., et al. (2003) NADPH oxidase activity is
essential for Keap1/Nrf2-mediated induction of GCLC in response to
2-indol-3-yl-methylenequinuclidin-3-ols, Cancer Res., 63,
5636-5645.
42.Wu, R. F., and Terada, L. S. (2009) Ras and Nox:
linked signaling networks? Free Radic. Biol. Med., 47,
1276-1281.
43.Laurent, E., McCoy, J. W. 3rd, Macina, R. A.,
Liu, W., Cheng, G., et al. (2008) Nox1 is over‐expressed in
human colon cancers and correlates with activating mutations in K-Ras,
Int. J. Cancer, 123, 100-107.
44.Tao, S., Wang, S., Moghaddam, S. J., Ooi, A.,
Chapman, E., et al. (2014) Oncogenic KRAS confers chemoresistance by
upregulating NRF2, Cancer Res.,74, 7430-7441.
45.Kovac, S., Angelova, P. R., Holmström, K.
M., Zhang, Y., Dinkova-Kostova, A. T., and Abramov, A. Y. (2015) Nrf2
regulates ROS production by mitochondria and NADPH oxidase, Biochim.
Biophys. Acta, 1850, 794-801.
46.Han, M., Zhang, T., Yang, L., Wang, Z., Ruan, J.,
and Chang, X. (2016) Association between NADPH oxidase (NOX) and lung
cancer: a systematic review and meta-analysis, J. Thorac. Dis.,
8, 1704.
47.Parekh, A., Das, S., Parida, S., Das, C. K.,
Dutta, D., et al. (2018) Multi-nucleated cells use ROS to induce breast
cancer chemo-resistance in vitro and in vivo,
Oncogene, 37, 4546-4561.
48.Longley, D. B., Harkin, D. P., and Johnston, P.
G. (2003) 5-Fluorouracil: mechanisms of action and clinical strategies,
Nat. Rev. Cancer, 3, 330-338.
49.He, L., Zhu, H., Zhou, S., Wu, T., Wu, H., et al.
(2018) Wnt pathway is involved in 5-FU drug resistance of colorectal
cancer cells, Exp. Mol. Med., 50, 1-12.
50.Kim, E.-K., Jang, M., Song, M.-J., Kim, D., Kim,
Y., and Jang, H. H. (2019) Redox-mediated mechanism of chemoresistance
in cancer cells, Antioxidants, 8, 471.
51.Kang, K., Piao, M., Kim, K., Kang, H., Chang, W.,
et al. (2014) Epigenetic modification of Nrf2 in
5-fluorouracil-resistant colon cancer cells: involvement of
TET-dependent DNA demethylation, Cell Death Dis., 5,
e1183-e1183.