Direct Effect of the Synthetic Analogue of Glucagon-Like Peptide Type 1, Liraglutide, on Mature Adipocytes Is Realized through Adenylate-Cyclase-Dependent Enhancing of Insulin Sensitivity
Elizaveta D. Mamontova1,2,3, Svetlana S. Michurina1,2, Iurii S. Stafeev2,a*, Ekaterina L. Sorkina3, Igor A. Sklyanik3, Ekaterina O. Koksharova3, Mikhail Y. Menshikov2, Marina V. Shestakova3, and Yelena V. Parfyonova2,4
1Faculty of Biology, Lomonosov Moscow State University, 119234 Moscow, Russia2Institute of Experimental Cardiology, National Medical Research Centre for Cardiology, 121552 Moscow, Russia
3Diabetes Institute, Endocrinology Research Centre, 117036 Moscow, Russia
4Faculty of Basic Medicine, Lomonosov Moscow State University, 119234 Moscow, Russia
* To whom correspondence should be addressed.
Received November 2, 2020; Revised December 7, 2020; Accepted December 7, 2020
Incretin hormones analogues, including glucagon-like peptide type 1 (GLP-1), exhibit complex glucose-lowering, anorexigenic, and cardioprotective properties. Mechanisms of action of GLP-1 and its analogues are well known for pancreatic β-cells, hepatocytes, and other tissues. Nevertheless, local effects of GLP-1 and its analogues in adipose tissue remain unclear. In the present work effects of the GLP-1 synthetic analogue, liraglutide, on adipogenesis and insulin sensitivity of the 3T3-L1 adipocytes were examined. Enhancement of insulin sensitivity of mature adipocytes by the GLP-1 synthetic analogue liraglutide mediated by adenylate cyclase was demonstrated. The obtained results imply existence of the positive direct insulin-sensitizing effect of liraglutide on mature adipocytes.
KEY WORDS: insulin resistance, adipocyte, GLP-1, incretinsDOI: 10.1134/S000629792103010X
Abbreviations: CREB, cAMP responsive element binding protein; GLP-1, glucagon-like peptide-1; cAMP, cyclic adenosine monophosphate; Erk, extracellular signal regulated kinase; GLUT4, glucose transporter type 4; IR, insulin resistance; IRS-1, insulin receptor substrate type 1; JNK, c-Jun NH2-terminal kinase; PKA, protein kinase A, cAMP-dependent protein kinase; PPARgamma, peroxisome proliferator-activated receptor type gamma; SQ22536, adenylate cyclase inhibitor; T2DM, type 2 diabetes mellitus; UCP-1, uncoupling protein type 1.
INTRODUCTION
Obesity is considered as one of the leading causes of diseases among humans worldwide and is a risk factor for developing insulin resistance (IR) and type 2 diabetes mellitus (T2DM) [1, 2] that primarily develops due to increased adipose tissue mass. Obesity and T2DM also increase risk of development and severity of cancer [3], cardiovascular [4] and many other diseases. Examining physiology of adipose tissue represents a crucial scientific and medical issue.
Glucagon-like peptide-1 (GLP-1) and its analogues comprise one of the latest and most promising agents used for correcting T2DM. Physiologically, GLP-1 is a peptide belonging to the incretin hormone family originating from the limited proglucagon proteolysis occurring in the small intestine L-cells [5]. Free GLP-1 is rapidly broken down by the dipeptidyl peptidase-4 in the blood stream, so that its half-life is only 1-2 min [6], hence, its synthetic analogues are used in T2DM therapy. Liraglutide is a synthetic analogue of GLP-1 containing several modifications not affecting its specificity to GLP-1 receptor. The extended half-life of liraglutide in blood is 13 h [7].
GLP-1 and other agents based on incretin hormones comprise a group of drugs exerting simultaneously metabolic and cardioprotective effects in T2DM therapy. It is believed that incretin hormone-based agents including GLP-1 analogues act more as cardioprotective drugs with antidiabetic effect rather than solely antidiabetic agents [8, 9]. GLP-1 and its analogues demonstrate potential to lower the rate of developing of cardiovascular and cerebrovascular complications such as ischemic stroke, myocardial infarction, and acute coronary syndrome [10]. Recently, it was also shown that GLP-1 was involved in enhancing endogenous antioxidant defense, inhibited cardiomyocyte apoptosis, alleviated endothelial inflammation and dysfunction [11].
The GTP-binding protein-dependent exocytosis of insulin-bearing vesicles occurring due to the stimulated adenylate cyclase and subsequent production of cyclic adenosine monophosphate (cAMP) that activates calcium signaling represents the most investigated mechanism of the effects of GLP-1 on pancreatic β-cells [12]. Despite the existence of substantial evidence regarding the systemic effects of GLP-1 on the pancreas, central nervous, and cardiovascular systems [13, 14], the data on the mode of action of GLP-1 and its analogues on adipose tissue are scarce. The fact that the therapy with liraglutide and GLP-1 analogues is often conducted by administration of such agents directly into the subcutaneous adipose tissue is another issue that generates interest to the effects of locally applied medicinal agents both on adipose cell turnover and differentiation of adipocyte progenitors. It is acknowledged that GLP-1 promotes glucose uptake and its storage in adipose tissue [15]. By now, two quite distinct mechanisms of liraglutide effects on adipose tissue have been proposed. On the one hand, GLP-1 promotes activation of both white and beige adipocyte differentiations [16, 17]. Other studies demonstrate potential for GLP-1 to stimulate activity of adenylate cyclase and related cAMP production resulting in the subsequently stimulated protein kinase A (PKA)-dependent lipolysis [18].
Here we examined the effects of the synthetic GLP-1 analogue liraglutide on the de novo adipocyte generation (by assessing activity of beige and white 3T3-L1 preadipocyte differentiation), on inflammatory state of mature adipocytes (by evaluating activity of the c-Jun N-terminal kinase, JNK), as well as on insulin sensitivity of mature adipocytes.
MATERIALS AND METHODS
Overall study design. 3T3-L1 preadipocyte cell culture was used as a model. Examining white and beige preadipocyte differentiation was performed by exposure to various induction mixtures with and without liraglutide. Differentiation efficacy was assessed by calculating mean number of mature adipocyte per surface unit, be measuring adsorbed OilRedO dye in the cell extracts, as well as by measuring the level of expression of white/beige adipogenic marker with immunoblotting.
Next, phosphorylation kinetics of the pro-inflammatory JNK and pro-mitogenic Erk (extracellular signal regulated kinase) kinases was assessed after mature adipocytes were exposed to liraglutide at concentration of 100 nM for 30 min, 1, 6, 18, and 24 h as well as in combination with the adenylate cyclase inhibitor (SQ22536) at concentration of 500 µM followed by estimation of adipocyte insulin sensitivity.
Insulin sensitivity was assessed by treating 3T3-L1 mature adipocytes with liraglutide for 24 h followed by measuring phosphorylation and expression level of the major insulin signaling components [insulin receptor substrate-1 (IRS-1) and Akt kinase] using immunoblotting, as well as measuring insulin-stimulated [3H]-2-deoxyglucose uptake.
Materials. Murine 3T3-L1 preadipocyte cells (ATCC, USA) were cultured in the DMEM-F12 medium (glucose concentration – 4.5 g/liter) (Gibco, USA) supplemented with 10% fetal bovine serum (FBS, HyClone, USA) and 60 U/ml penicillin/streptomycin mixture (Gibco). 3T3-L1 preadipocytes underwent differentiation after adding insulin, isobutylmethylxanthine, dexamethasone, rosiglitazone, triiodothyronine and isoproterenol (Sigma-Aldrich, USA). Adipocytes were incubated with synthetic GLP-1 analogue liraglutide (Selleckchem, Germany) and SQ22536 adenylate cyclase inhibitor (Fluka, Germany). Immunoblotting analysis was performed by using primary monoclonal antibodies against k pJNK1/2-T183/Y185 (R&D, USA), tJNK1/2 (R&D, USA), pErk1/2-T202/Y204 (Cell Signaling, USA), tErk1/2 (Cell Signaling), uncoupling protein type 1 (UCP-1, Thermo Scientific, USA), peroxisome proliferator-activated receptor type gamma (PPARgamma, Cell Signaling), pIRS-Y612 (Thermo Scientific), tIRS (Cell Signaling), pAkt-S473 (Cell Signaling), tAkt (Abcam, USA), glucose transporter type 4 (GLUT4, Abcam), beta-actin (Abcam), and secondary rabbit anti-mouse mouse IgG antibodies conjugated with horseradish peroxidase (Abcam). Insulin-induced uptake of [3H]-2-deoxy-glucose was assessed by using 2-deoxy-glucose (Sigma-Aldrich) and [3H]-2-deoxy-glucose (Perkin Elmer, USA) solutions. A cell lysate sample was dissolved in an UltimaGold liquid scintillation cocktail (Perkin Elmer) for measuring decay rate with a LKB RackBeta counter (LKB, Sweden). Protein concentration was measured with a Pierce BCA Protein Assay Kit (Thermo Scientific).
3T3-L1 preadipocyte cultivation and differentiation. 3T3-L1 preadipocytes were cultured in a DMEM-F12 medium (4.5 g/liter glucose) supplemented with 10% FBS and 60 U/ml penicillin/streptomycin mixture.
White adipogenesis was induced according to the protocol proposed by Zebisch et al. [19] with modifications. Cells were cultured to ~100% confluency, maintained in tight confluency for 2 more days, followed by replacing the medium with a DMEM medium supplemented with 10% FBS, 0.5 mM dexamethasone, 0.25 µM isobutylmethylxanthine, 2 µM rosiglitazone, and 100 nM insulin. Next, the culture medium was replaced with the standard medium for 2-4 days, and the obtained adipocyte culture was used in further experiments. Lypophilic OilRedO dye (Merck Millipore, Germany) was used to assess the level of lipid accumulation followed by imaging of 4 random fields of view on an AxioVertA1 microscope (Zeiss, Germany) in a phase-contrast mode followed by counting the number of mature adipocytes per area unit. OilRedO dye was extracted from the stained adipocytes by using 100% isopropanol followed by assessing absorption of the extract on a Multiscan Microplate Reader (Labsystems, USA).
Beige adipogenesis was induced according to the protocol proposed by Miller et al. [20] with modifications. Cells were cultured to ~100% confluency and maintained in tight confluency for 2 more days, followed by replacing the medium for the next 4 days with DMEM supplemented with 10% FBS, 0.5 mM dexamethasone, 0.25 µM isobutylmethylxanthine, 2 µM rosiglitazone, and 100 nM insulin, 1 nM triiodothyronine, and 100 µM isoproterenol. Next, the medium was replaced with the standard medium for 2-4 days followed by using the obtained adipocyte culture in experiments.
Assessing phosphorylation level in the major components of insulin signaling pathway in the mature 3T3-L1 adipocytes. Mature adipocytes were deprived of fetal bovine serum for 4 h in the serum-free DMEM-F12 medium in order to assess phosphorylation levels of the major components of insulin signaling. Next, the cells were incubated with 100 nM insulin for 20 min followed by lysing them in a radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1% (w/v) Triton-X100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris-HCl at pH 8.0) supplemented with a protease/phosphatase inhibitor cocktail (Roche, Sweden). Cell lysates were homogenized using insulin syringe with a 29G gauge size needle.
Insulin-induced [3H]-2-deoxyglucose uptake. Mature 3T3-L1 adipocytes were stimulated with insulin at concentration of 100 nM for 20 min followed by adding 100 µM 2-deoxyglucose and 0.5 µCi [3H]-2-deoxyglucose for 5 min to assess insulin-induced glucose uptake. Next, the cells were washed three times with cold phosphate buffered saline and lysed with the RIPA buffer. Finally, cell lysate was dissolved in a liquid scintillation cocktail for measuring decay rate in a β-radiation counter.
Examining effects of liraglutide on activity of pro-mitogenic Erk and pro-inflammatory JNK kinases. A relation between phosphorylation and expression of pro-mitogenic Erk and pro-inflammatory JNK kinases was investigated depending on the time of incubation in the presence of 100 nM liraglutide. 3T3-L1 mature adipocytes were serum deprived in the serum-free DMEM-F12 medium for 4 h followed by exposure to liraglutide for 30 min, 1, 6, 18, and 24 h. After that, the cells were lysed in the RIPA buffer followed by immunoblotting. To investigate the effects of liraglutide on the kinase expression in the 3T3-L1 mature adipocytes, the serum-starved for 4 h adipocytes were incubated with SQ22536 adenylate cyclase inhibitor (500 µM) followed by addition of liraglutide.
Immunoblotting assay. Proteins in cell lysates were separated using 10% polyacrylamide gel electrophoresis under denaturing conditions according to Laemmli procedure [21] and next electrotransferred onto wet polyvinylidene fluoride membranes (Amersham, USA) according to the manufacturer’s recommendations. Membranes were blocked for 6 h in the 50 mM Tris-HCl (pH 8.0) containing 5% skim milk (Applichem, Germany) supplemented with 0.1% (v/v) Tween 20 followed by incubation with primary antibodies, according to the manufacturer’s recommendations. Next the membranes were treated with secondary antibodies according to manufacturer’s instructions. Chemiluminescence was visualized using a Clarity ECL kit (Bio-Rad, USA) and recorded with a gel-documentation system FusionX (Vilber-Lourmat, France). Next, images were processed using ImageJ software (https://imagej.nih.gov/ij/) and GelAnalyzer (http://www.gelanalyzer.com/). Finally, phosphorylated protein/total protein ratio was calculated or total protein level was normalized to β-actin used as endogenous control.
Statistical analysis. The data are presented as mean ± standard deviation. Each experimental point was obtained by carrying out three independent repeats, each of which was analyzed in three replicates. Significance of differences was calculated by using Student’s t-test with statistical significance threshold set at p < 0.05.
RESULTS
Liraglutide exerts no profound effect on beige adipocyte differentiation, but contributes to upregulated UCP-1 expression during white adipocyte differentiation of 3T3-L1 preadipocytes. Initially, we assessed impact of the synthetic GLP-1 analogue liraglutide on events underlying white and beige differentiation of preadipocytes. This was important due to the common use of subcutaneous injection of liraglutide in clinical practice and its potential local effects on differentiation of preadipocytes inside the adipose tissue. Experiments were carried out by adding liraglutide at concentration of 100 nM to the induction mixture and the culture medium following induction (see “Materials and Methods” section).
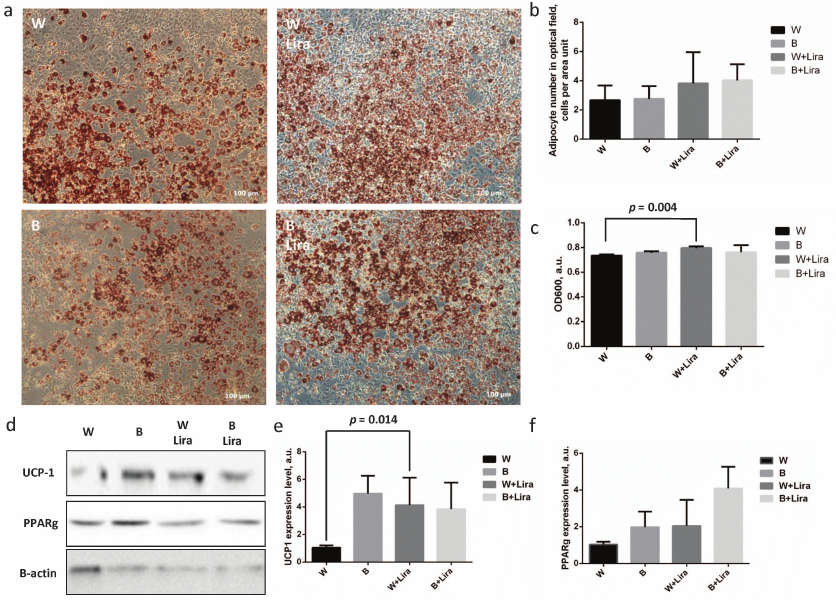
The level of 3T3-L1 preadipocyte differentiation with and without liraglutide was assessed by cell staining with lyophilic dye OilRedO (Fig. 1a). Enumeration of mature adipocytes per surface area unit demonstrated no significant differences between the groups, but exposure of white and beige adipocytes to liraglutide caused statistically insignificant increase of their numbers (Fig. 1b). Similar situation was observed during assessment of the level of OilRedO dye absorption in the cell extracts showing only slight increase of absorption in the extracts of the liraglutide-treated adipocytes. Such effect was significant in the case of white adipocyte differentiation (p = 0.004), but the rise in extract absorption after liraglutide exposure comprised less than 10% out of the total magnitude (Fig. 1c). The obtained data cast a doubt on the ability of liraglutide to regulate white adipocyte differentiation and lipid accumulation. Nonetheless, expression of the major components involved in the white (PPARgamma) and beige (UCP-1) adipogenesis was analyzed using immunoblotting. It was found that liraglutide significantly upregulated expression of the white adipogenesis marker UCP-1 (p = 0.032) (Fig. 1, d and e). In contrast, liraglutide exposure exerted no significant effect on the expression of both UCP-1 and PPARgamma related to beige adipogenesis (Fig. 1, d and f). Thus, it can be concluded that liraglutide has a potential to enhance expression of the markers of beige adipogenesis within white adipocytes, but it does not affect expression of the white adipogenesis markers. Overall, the obtained data imply that liraglutide does not produce any effect on white adipogenesis. However, it is worth noting that the liraglutide exposure resulted in the significantly upregulated level of UCP-1 in white adipocytes suggesting its potential for modulation of the adipocyte phenotype towards the thermogenic one. Further experiments were conducted on 3T3-L1 mature adipocytes following white adipogenesis.
Fig. 1. Liraglutide exposure does not produce any marked impact on beige adipogenesis, but contributes to upregulation of UCP-1 expression during white adipogenesis in 3T3-L1 preadipocytes. a) 3T3-L1 adipocyte microimaging panel (phase-contrast) after white/beige (W/B) adipogenesis with/without liraglutide (Lira), stained with OilRedO lipophilic dye; b) adipocyte number per microimage area unit shown in panel (a); c) absorption of OilRedO dye extract; d) representative immunoblots; e) statistical analysis assessing UCP-1 expression level; f) statistical analysis assessing PPARgamma expression level. The data from three independent experiments are presented; Student’s t-test was used to calculate significance of differences: significance threshold was set at p < 0.05.
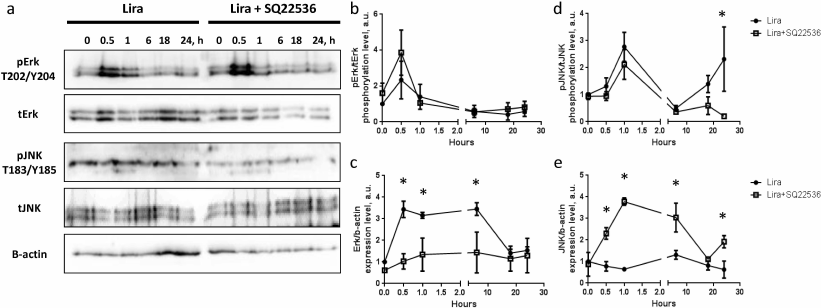
Liraglutide-activated adenylate cyclase results in upregulated expression of pro-mitogenic Erk and down-regulated expression of pro-inflammatory JNK. Impact of time of exposure to liraglutide on levels of activation and expression of pro-mitogenic (Erk) and pro-inflammatory (JNK) kinase in 3T3-L1 mature white adipocytes was investigated.
Analysis of kinetics of Erk and JNK (Fig. 2, a, b, and d) phosphorylation demonstrated similar transitory activation (Erk phosphorylation: p = 0.04 within 0-0.5 h; p = 0.117 within 0.5-1 h; and p = 0.04 within 1-6 h; same pattern of changes in significance levels was observed in the presence of adenylate cyclase inhibitor SQ22536). Analysis of Erk expression revealed robust upregulation in response to liraglutide exposure. The effect was observed 0.5 h after the start of exposure, which was a rather short time for realization of the expression-related response. Nonetheless, the level of Erk expression was significantly elevated during 6-h incubation with liraglutide and returned back to the baseline after 18-24 h exposure (Fig. 2, a and c). Expression of JNK kinase did not display any significant changes in time in the presence of liraglutide compared to the baseline level (Fig. 2, a and e). Hence, liraglutide is capable of transitory activation of Erk and JNK kinases, which is accompanied by the rather stable increase (up to 6 h) of Erk expression. In connection with this, it was suggested that liraglutide exerts divergent effects on gene expression in 3T3-L1 mature adipocytes.
Fig. 2. Liraglutide exposure does not produce significant impact on activation of Erk and JNK signaling, but affects their expression via adenylate cyclase activity. a) Representative immunoblots; b) time-dependent liraglutide-mediated effects on pErk/tErk phosphorylation level; c) time-dependent liraglutide-mediated effects on Erk1/β-actin expression level; d) time-dependent liraglutide-mediated effects on pJNK1/2-T183/Y185 kinase phosphorylation level; e) time-dependent liraglutide-mediated effects on pJNK1/2 kinase expression level. All experiments were performed by using 100 nM liraglutide ± 500 µM SQ22536. The data from three independent experiments are presented; Student’s t-test was used to calculate significance of differences: * p < 0.05, significance level was calculated for each paired value at each time point (±500 µM SQ22536).
CREB (cAMP response element-binding protein) is the major transcription factor involved in liraglutide-mediated effects, which is activated at elevated cAMP levels [22-24]. Adenylate cyclase is the main enzyme producing intracellular cAMP and it also able to activate CREB transcription factor. Therefore, the liraglutide mode of action was investigated further by using adenylate cyclase (SQ22536) inhibitor at concentration of 500 µM [25-27].
It was shown that the kinetics of Erk and JNK phosphorylation after exposure to liraglutide in the presence of SQ22536 demonstrated similar transient pattern. In the case of JNK it was found that its phosphorylation level was elevated after 24 h of incubation with liraglutide vs. phosphorylation level in the presence of SQ22536 (Fig. 2, a, b, and d). Furthermore, SQ22536 fully suppressed upregulated Erk expression after 0.5-6 h incubation with liraglutide. While liraglutide per se did not affect expression of JNK, statistically significant activation of the JNK expression was observed in the presence of SQ22536 (Fig. 2, a, c, and e). This result implies that liraglutide exerts divergent cAMP-dependent effects on Erk and JNK expression. Hence, it could be presumed that liraglutide might be potentially involved in controlling proliferation and inflammation in the adipose tissue.
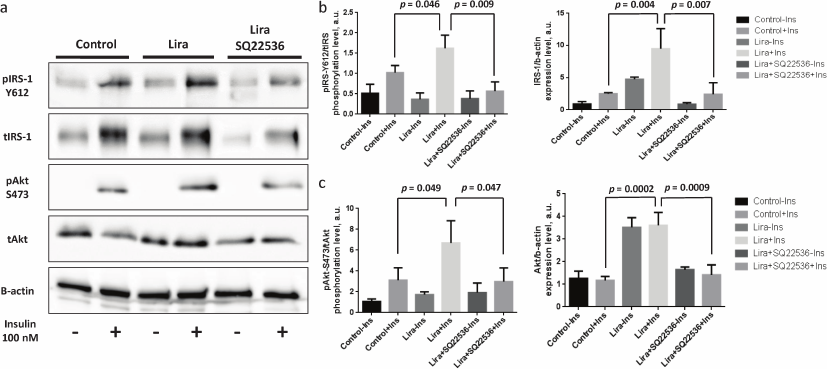
Liraglutide enhances phosphorylation and expression level of IRS-1 and Akt by activating adenylate cyclase. It was suggested that along with regulating proliferation and inflammation in the adipose tissue, liraglutide might exert systemic sugar-lowering effect due to elevated adipocyte insulin resistance upon subcutaneous injection.
It was found that liraglutide was able to elevate both phosphorylation level and expression of the scaffold protein IRS-1 promoting signaling from the insulin receptor to the kinases (Fig. 3, a and b) as well as kinase Akt (Fig. 3, a and c) when the mature adipocytes were affected directly. It must be mentioned that the baseline level of Akt protein kinase phosphorylation was virtually unchanged after liraglutide exposure, which together with the upregulated Akt expression triggered by liraglutide may indicate the decreased level of Akt phosphorylation observed after adding liraglutide without stimulation with insulin (Fig. 3, a and c). SQ22536 facilitated full elimination of liraglutide effects and return of phosphorylation and expression levels of IRS-1 and Akt to the baseline (Fig. 3, a and b). The obtained data demonstrate that liraglutide exerts stimulatory effects on insulin-dependent signaling pathway, which allows expecting associated changes in the insulin-dependent glucose uptake.
Fig. 3. Liraglutide enhances IRS-1 and Akt phosphorylation and expression via adenylate cyclase axis. All experiments were performed by using 100 nM liraglutide ± 500 µM SQ22536. a) Representative immunoblots; b) statistical analysis of IRS-1 phosphorylation and expression; c) statistical analysis of Akt phosphorylation and expression. The data from three independent experiments are presented; Student’s t-test was used to calculate significance of differences: significance level was set at p < 0.05.
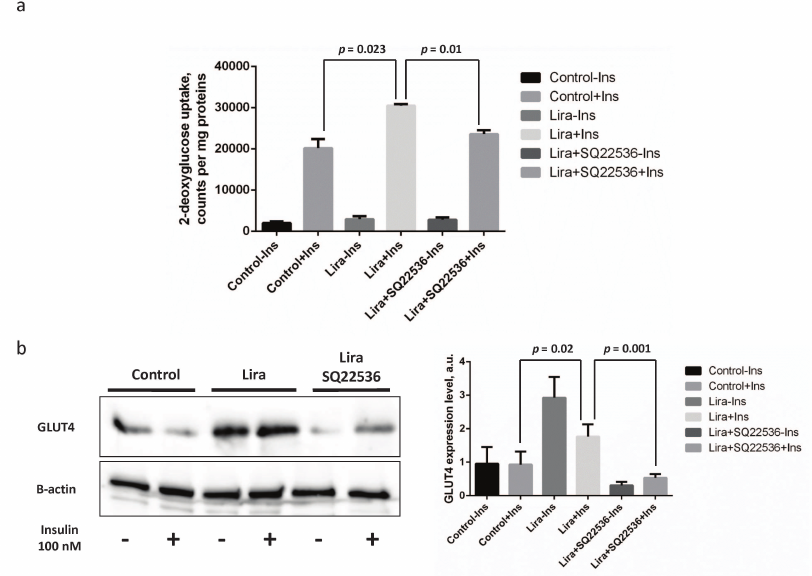
Liraglutide enhances insulin-stimulated adenylate cyclase-dependent glucose uptake in 3T3-L1 mature adipocytes. After assessing activity of insulin signaling cascade, we investigated physiological response to insulin in the 3T3-L1 mature adipocytes. Previously, it was observed that the rate of glucose uptake represents a real adipocyte response to insulin exposure.
It was found that the glucose uptake in the insulin-treated vs. control 3T3-L1 mature adipocytes was stimulated by approximately 9.5-fold, whereas this process was enhanced further (up to 11-fold) after liraglutide addition. It is worth mentioning that the absolute glucose uptake rate after adding insulin together with liraglutide increased by 30% vs. insulin alone (p = 0.023, Fig. 4a). Addition of SQ22536 fully suppressed the liraglutide-associated effects (p = 0.01, Fig. 4a), whereas no significant differences were observed in the absolute glucose uptake rate in the insulin- vs. liraglutide/SQ22536-treated cells (Fig. 4a). In addition, we also investigated potential mechanisms underlying enhanced insulin sensitivity of adipocytes triggered by liraglutide by assessing expression of the glucose transporter type 4 (GLUT4). It was shown that exposure to liraglutide resulted in substantially increased GLUT4 expression that was fully abolished by adding SQ22536 (Fig. 4b). It is worth noting that combining liraglutide with insulin lowers GLUT4 expression compared to the insulin-unstimulated cells but not to the control or inhibitor-treated cells. These results could be associated with the extremely pleotropic effects of liraglutide on adipocytes. To summarize, we could suggest that liraglutide contributes to the improved adipocyte insulin sensitivity via the adenylate cyclase-dependent pathway.
Fig. 4. Liraglutide enhances insulin-dependent glucose uptake via adenylate cyclase-mediated upregulation of expression of the insulin-dependent glucose transporter GLUT4. All experiments were performed by using 100 nM liraglutide ± 500 µM SQ22536. a) Histogram depicting insulin-stimulated 2-deoxyglucose uptake; b) representative immunoblots and relevant densitometry data. The data from three independent experiments are presented; Student’s t-test was used to calculate significance of differences: significance level was set at p < 0.05.
DISCUSSION
In this work we demonstrated direct effects of the synthetic GLP-1 analogue liraglutide on preadipocytes and mature 3T3-L1 adipocytes. In particular, liraglutide at concentration of 100 nM did not profoundly affect preadipocyte potential to adipogenesis, but exposure to liraglutide caused upregulation of UCP-1 expression in white adipocytes thereby directing them towards beige differentiation. Liraglutide was able to divergently regulate adenylate cyclase-dependent expression of pro-mitogenic Erk and pro-inflammatory JNK kinases in mature adipocytes. Moreover, liraglutide displayed a potential to enhance expression of a number of components involved in insulin signaling as well as insulin-dependent glucose transporter GLUT4 via adenylate cyclase-driven mechanism. In addition, liraglutide stimulated the insulin-dependent glucose uptake that also proceeded with participation of adenylate cyclase (Fig. 5).
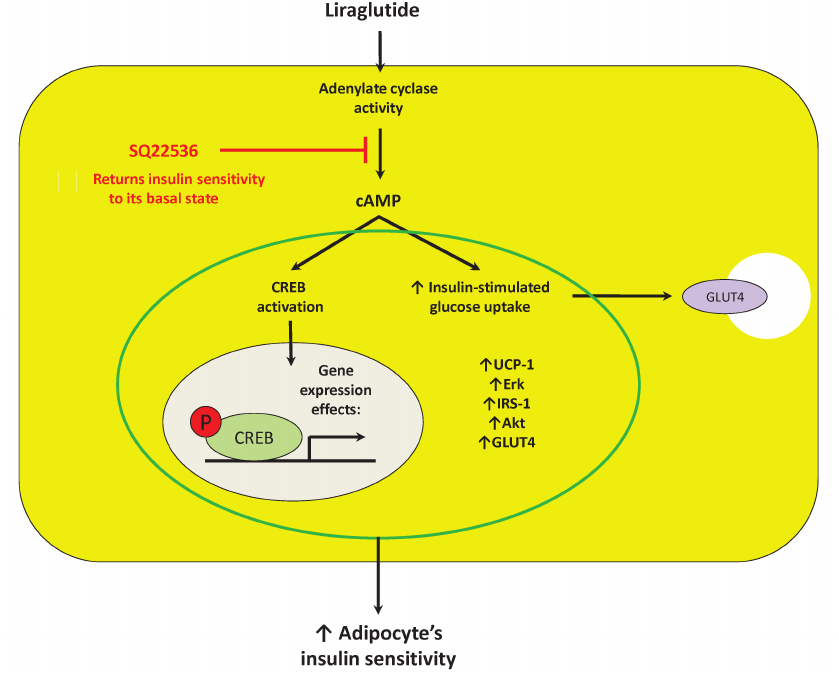
Fig. 5. Suggested mechanism of direct effects of liraglutide on mature adipocytes is associated with control of adenylate cyclase activity, which, in turn, is able to affect expression of crucial components in thermogenesis (UCP-1), inflammation (JNK), proliferation and survival (Erk), as well as insulin sensitivity (IRS-1, Akt, and GLUT4) by activating transcription factor CREB. Designations: Akt, protein kinase B; cAMP, cyclic adenosine monophosphate; CREB, cAMP response element-binding protein; Erk, extracellular signal-regulated kinase; GLUT4, glucose transporter type 4; IRS, insulin receptor substrate; SQ22536, adenylate cyclase inhibitor; UCP-1, uncoupling protein 1.
The main function of GLP-1 is mediation of insulinotropic effects after food intake (elevated insulin secretion) as well as suppression of glucagon secretion by activation of the specific GLP-1 receptor. Insulinotropic effect combined with other fast-developing GLP-1 effects contributes to the decrease of the blood glucose level [28-30]. Thus, it could be concluded that GLP-1 acts as an enhancer that additionally stimulates postprandial insulin secretion causing maximal activation of the cell glucose uptake. Nonetheless, the reduced GLP-1 level and altered total profile of incretin hormone secretion is observed in patients with IR and T2DM [31-33]. That is why therapeutic approaches based on the use of GLP-1 and other incretin hormone analogues for correcting IR and T2DM attracted so much attention in the beginning of 21th century [34, 35]. Currently, the synthetic GLP-1 analogues such as liraglutide-based drugs are the most commonly used therapeutic agents. The majority of GLP-1 analogue-based agents is administered subcutaneously into the adipose tissue, regulated generation of which is a crucial problem associated with obesity and T2DM.
Interaction between GLP-1 and the adipose tissue has been thoroughly investigated in terms of unveiling major regulatory mechanisms. GLP-1 can modulate activity of hypothalamic-pituitary system and affect metabolism in the adipose tissue by enhancing beige adipogenesis [36-38]. Nonetheless, the recent study exploring mechanisms of efficient bariatric surgery revealed importance of the local GLP-1 receptor expressed exclusively in the subcutaneous adipose tissue for improving metabolic parameters in patients with obesity and T2DM [39]. Thus, it may be expected that local activity of GLP-1 and its analogues could improve metabolic profile and insulin sensitivity. Our study attempted to elucidate the mechanisms underlying GLP-1-related effects (modelled by using the synthetic GLP-1 analogue liraglutide) on white and beige adipogenesis.
Our first finding was lack of the effects of liraglutide on white adipogenesis in combination with the upregulated UCP-1 expression in white adipocytes after exposure to liraglutide. No convincing data on the local effects of GLP-1 on activated beige adipogenesis were observed before. It is known that the response of the hypothalamic-pituitary system involved in regulation of thermogenesis is crucial for control of beige adipogenesis upon systemic exposure to liraglutide [36-38]. However, activated white adipogenesis in the 3T3-L1 preadipocytes after exposure to liraglutide and GLP-1 was demonstrated in some studies [16, 17]. It must be mentioned, however, that these studies used different models of adipogenesis without using rosiglitazone. The latter is known to induce strong adipogenic differentiation that may annul potential effects of liraglutide on white adipogenesis. To explain lack of the effect of liraglutide on UCP-1 level it can be presumed that the induction mixture itself triggers maximum possible upregulation of UCP-1 expression under conditions of beige adipogenesis.
While discussing local effects of liraglutide on Erk and JNK (Fig. 5) kinase expression possible local pro-mitogenic and protective (pro-survival) (Erk) as well as pro-inflammatory (JNK) activities should be mentioned. Indeed, some studies noted pro-mitogenic effects of liraglutide and GLP-1 mediated by activated Erk kinase in various cell types [40, 41]. However, there is also evidence describing anti-inflammatory activity of liraglutide and GLP-1 observed, however, in animal models. In particular, native GLP-1 overexpressed in the adipose tissue was able to lower obesity level and macrophage infiltration in adipose tissue as well as promote anti-inflammatory macrophage polarization [42, 43]. However, it should be clarified that these studies described systemic rather than local liraglutide effects, whereas we presented the data on local anti-inflammatory effects of liraglutide.
Despite existing of multiple data pointing at central role of the GLP-1 and its analogues in the major mechanism regulating homeostasis in adipose tissue, in our study we demonstrated highly prominent direct effects of GLP-1 on adipocytes. Liraglutide is able to markedly upregulate expression of the components involved in insulin signaling pathway and promote glucose uptake in the subcutaneous adipose tissue. These results are in agreement with the data published by Gao et al. [44] demonstrating that the insulin signaling in adipocyte was regulated by GLP-1 affecting expression of insulin receptor, IRS-1 as well as insulin-dependent glucose transporter GLUT4. In our study we managed to demonstrate for the first time that such effects were driven by the adenylate cyclase activity and resulted in the elevated insulin-dependent glucose uptake and GLUT4 expression.
Moreover, it was clearly demonstrated that the elevated insulin-dependent glucose uptake occurred after direct exposure of 3T3-L1 mature adipocytes to liraglutide, and raised a question about the fate of such glucose molecules. Our data suggest that glucose was not directly transformed into the energy storage (lipids), because neither the adipocyte count nor the total adipocyte lipid content were changed (Fig. 1, a-c). Potentially, it might be explained by the elevated expression of the mitochondrial uncoupling protein 1 (UCP-1), which is the major component of thermogenesis capable of utilizing the cellular energy reserves. It could be suggested that the elevated insulin-dependent glucose uptake by the mature 3T3-L1 adipocytes initiated by liraglutide is associated with the enhanced thermogenesis and nutrient consumption.
Previously, the mechanisms of liraglutide-related expression effects in the adipose tissue were explored, but no consensus has been reached. There is no doubt that the transcription factor CREB plays a crucial role in regulating gene expression in the pancreas [22-24]. Previous studies revealed a 3-fold increase in the adipocyte cAMP level after GLP-1 exposure indirectly confirming that GLP-1 acted in a cAMP-dependent manner [18]. It was also demonstrated that addition of the cAMP-dependent protein kinase (PKA) blocked GLP-1-associated changes in the fatty acid synthase expression as well as activation of the transcription factor CREB in the mature 3T3-L1 adipocytes [17]. However, it should be noted that the transcription factor CREB may act not only as a homodimer, but also as an active heterodimer with the ATF transcription factors. Hence, it should be taken into consideration that other CREB/ATF family transcription factors could be also involved in mediating physiological effects of liraglutide [45]. The obtained data on adenylate cyclase-dependent expression responses to liraglutide exposure confirm participation of the adenylate cyclase-dependent PKA-CREB signaling pathway in realization of the local liraglutide-associated expression effects in the subcutaneous adipose tissue.
CONCLUSION
To sum up the results of our study it should be noted that the synthetic GLP-1 analogue liraglutide exerts positive direct effects on mature adipocytes by stimulating insulin sensitivity as well as displaying pro-mitogenic and anti-inflammatory activities. Such effects were mediated by adenylate cyclase likely via the PKA-CREB signaling pathway. The synthetic GLP-1 analogue liraglutide demonstrated no direct effect on both thermogenic and white adipocyte development. The data obtained indicate positive antidiabetic local effects of exposure to liraglutide, but the mechanisms controlling thermogenesis and storage of lipids seem to rely mostly on the systemic activity of liraglutide.
Funding. The study was financially supported by the Russian Science Foundation (project no. 19-75-00068) and by the collaboration framework between the Russian Science Foundation and Ministry of Science and Technology of Taiwan (project no. 20-45-08003).
Ethics declarations. Authors declare no conflict of interest in financial or any other sphere. This article does not contain any studies with human participants or animals performed by any of the authors.
REFERENCES
1.Olefsky, J. M., and Glass, C. K. (2010)
Macrophages, inflammation, and insulin resistance, Annu. Rev.
Physiol., 72, 219-246.
2.Vorotnikov, A. V., Stafeev, I. S., Menshikov, M.
Yu., Shestakova, M. V., and Parfyonova, Ye. V. (2019) Latent
inflammation and defect in adipocyte renewal as a mechanism of
obesity-associated insulin resistance, Biochemistry (Moscow),
84, 1329-1345.
3.Gallagher, E. J., and LeRoith, D. (2015) Obesity
and diabetes: the increased risk of cancer and cancer-related
mortality, Physiol. Rev., 95, 727-748.
4.Booth, G. L., Kapral, M. K., Fung, K., and Tu, J.
V. (2006) Recent trends in cardiovascular complications among men and
women with and without diabetes, Diabetes Care, 29,
32-37.
5.Mojsov, S., Weir, G. C., and Habener, J. F. (1987)
Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the
glucagon gene is a potent stimulator of insulin release in the perfused
rat pancreas, J. Clin. Invest., 79, 616-619.
6.Deacon, C. F., Nauck, M. A., Toft-Nielsen, M.,
Pridal, L., Willms, B., and Holst, J. J. (1995) Both subcutaneously and
intravenously administered glucagon-like peptide 1 are rapidly degraded
from the NH2-terminus in type II diabetic patients and in healthy
subjects, Diabetes, 44, 1126-1131.
7.Jackson, S. H., Martin, T. S., Jones, J. D., Seal,
D., and Emanuel, F. (2010) Liraglutide (victoza): the first once-daily
incretin mimetic injection for type 2 diabetes, P T, 35,
498-529.
8.Chou, C. Y., Chang, Y. T., Yang, J. L., Wang, J.
Y., Lee, T. E., et al. (2017) Effect of long-term incretin-based
therapies on ischemic heart diseases in patients with type 2 diabetes
mellitus: a network meta-analysis, Sci. Rep., 7,
15795.
9.Nathanson, D., Ullman, B., Löfström, U.,
Hedman, A., Frick, M., et al. (2012) Effects of intravenous exenatide
in type 2 diabetic patients with congestive heart failure: a
double-blind, randomised controlled clinical trial of efficacy and
safety, Diabetologia, 55, 926-935.
10.White, W. B., and Baker, W. L. (2016)
Cardiovascular effects of incretin-based therapies, Annu. Rev.
Med., 67, 245-260.
11.Erdogdu, O., Nathanson, D., Sjöholm, A.,
Nyström, T., and Zhang, Q. (2010) Exendin-4 stimulates
proliferation of human coronary artery endothelial cells through eNOS-,
PKA- and PI3K/Akt-dependent pathways and requires GLP-1 receptor,
Mol. Cell. Endocrinol., 325, 26-35.
12.Ding, W. G., and Gromada, J. (1997) Protein
kinase A-dependent stimulation of exocytosis in mouse pancreatic
beta-cells by glucose-dependent insulinotropic polypeptide,
Diabetes, 46, 615-621.
13.Knop, F. K., Visboll, T., and Holst, J. J. (2009)
Incretin-based therapy of type 2 diabetes mellitus, Curr. Protein.
Pept. Sci., 10, 46-55.
14.Campbell, J. E., and Drucker, D. J. (2013)
Pharmacology, physiology and mechanisms of incretin hormone action,
Cell. Metab., 17, 819-837.
15.Young, A. A., Gedulin, B. R., Bhavsar, S.,
Bodkin, N., Jodka, C., et al. (1999) Glucose-lowering and
insulin-sensitizing actions of exendin-4: studies in obese diabetic
(ob/ob, db/db) mice, diabetic fatty Zucker rats, and diabetic rhesus
monkeys (Macaca mulatta), Diabetes, 48,
1026-1034.
16.Challa, T. D., Beaton, N., Arnold, M., Rudofsky,
G., Langhans, W., and Wolfrum, C. (2012) Regulation of adipocyte
formation by GLP-1/GLP-1R signaling, J. Biol. Chem., 287,
6421-6430.
17.Chen, J., Zhao, H., Ma, X., Zhang, Y., Lu, S., et
al. (2017) GLP-1/GLP-1R signaling in regulation of adipocyte
differentiation and lipogenesis, Cell. Physiol. Biochem.,
42, 1165-1176.
18.Vendrell, J., Bekay, R. E., Peral, B.,
García-Fuentes, E., Megia, A., et al. (2011) Study of the
potential association of adipose tissue GLP-1 receptor with obesity and
insulin resistance, Endocrinology, 152, 4072-4079.
19.Zebisch, K., Voight, V., Wabitsch, M., and
Brandsch, M. (2012) Protocol for effective differentiation of 3T3-L1
cells to adipocytes, Anal. Biochem., 425, 8890.
20.Miller, C. N., Yang, J. Y., England, E., Yin, A.,
Baile, C. A., and Rayalam, S. (2015) Isoproterenol increases
uncoupling, glycolysis, and markers of beiging in mature 3T3-L1
adipocytes, PLoS One, 10, e0138344.
21.Laemmli, U. K. (1970) Cleavage of structural
proteins during the assembly of the head of bacteriophage T4,
Nature, 15, 680-685.
22.Bao, Y., Jiang, L., Chen, H., Zou, J., Liu, Z.,
and Shi, Y. (2015) The neuroprotective effect of liraglutide is
mediated by glucagon-like peptide 1 receptor-mediated activation of
cAMP/PKA/CREB pathway, Cell. Physiol. Biochem., 36,
2366-2378.
23.Kimura, T., Kaneto, H., Shimoda, M., Hirukawa,
H., Okauchi, S., et al. (2015) Protective effects of pioglitazone
and/or liraglutide on pancreatic β-cells in db/db mice: comparison
of their effects between in an early and advanced stage of diabetes,
Mol. Cell. Endocrinol., 400, 78-89.
24.Que, Q., Guo, X., Zhan, L., Chen, S., Zhang, Z.,
et al. (2019) The GLP-1 agonist, liraglutide, ameliorates inflammation
through the activation of the PKA/CREB pathway in a rat model of knee
osteoarthritis, J. Inflamm. (Lond), 16, 13.
25.Haslam, R. J., Davidson, M. M., and Desjardins,
J. V. (1978) Inhibition of adenylate cyclase by adenosine analogues in
preparations of broken and intact human platelets. Evidence for the
unidirectional control of platelet function by cyclic AMP, Biochem.
J., 176, 83-95.
26.Juan, C., Chang, C., Lai, Y., and Ho, L. (2005)
Endothelin-1 induces lipolysis in 3T3-L1 adipocytes, Am. J. Physiol.
Endocrinol. Metab., 288, E1146-E1152.
27.Li, F., Wang, D., Zhou, Y., Zhou, B., Yang, Y.,
et al. (2008) Protein kinase A suppresses the differentiation of 3T3-L1
preadipocytes, Cell. Res., 18, 311-323.
28.Wang, Q., and Brubacker, P. (2002) Glucagon-like
peptide 1 treatment delays the onset of diabetes in 8 week-old db/db
mice, Diabetologia, 45, 1263-1273.
29.Willard, F. S., and Sloop, K. W. (2012)
Physiology and emerging biochemistry of the glucagon-like peptide-1
receptor, Exp. Diab. Res., 2012, 470851.
30.Cho, Y. M., Fujita, Y., and Kieffer, T. J. (2014)
Glucagon-like peptide-1: glucose homeostasis and beyond, Annu. Rev.
Physiol., 76, 533-536.
31.Nauck, M. A., Vardarli, I., Deacon, C. F., Holst,
J. J., and Meier, J. J. (2011) Secretion of glucagon-like peptide 1
(GLP-1) in type 2 diabetes: what is up, what is down?
Diabetologia, 54, 10-18.
32.Woerle, H. J., Carneiro, L., Derani, A., Goke,
B., and Schirra, J. (2012) The role of endogenous incretin secretion as
amplifier of glucose-stimulated insulin secretion in healthy subjects
and patients with type 2 diabetes, Diabetes, 61,
2349-2358.
33.Ahren, B. (2013) Incretin dysfunction in type 2
diabetes: clinical impact and future perspectives, Diabetes.
Metab., 39, 195-201.
34.Peters, A. (2010) Incretin-based therapies:
review of current clinical trial data, Am. J. Med., 123,
S28-S37.
35.Drucker, D. J., Sherman, S. I., Gorelick, F. S.,
Bergenstal, R. M., Sherwin, R. S., and Buse, J. B. (2010)
Incretin-based therapies for the treatment of type 2 diabetes:
evaluation of the risks and benefit, Diabetes Care, 33,
428-433.
36.Lockie, S. H., Heppner, K. M., Chaudhary, N.,
Chabenne, J. R., Morgan, D. A., et al. (2012) Direct control of brown
adipose tissue thermogenesis by central nervous system glucagon-like
peptide-1 receptor signaling, Diabetes, 61,
2753-2762.
37.Beiroa, D., Imbernon, M., Gallego, R., Senra, A.,
Herranz, D., et al. (2014) GLP-1 agonism stimulates brown adipose
tissue thermogenesis and browning through hypothalamic AMPK,
Diabetes, 63, 3346-3358.
38.Kooijman, S., Wang, Y., Parlevliet, E. T., Boon,
M. R., Edelschaap, D., et al. (2015) Central GLP-1 receptor signalling
accelerates plasma clearance of triacylglycerol and glucose by
activating brown adipose tissue in mice, Diabetologia,
58, 2637-2646.
39.Ejarque, M., Guerrero-Pérez, F., de la
Morena, N., Casajoana, A., Virgili, N., et al. (2019) Role of adipose
tissue GLP-1R expression in metabolic improvement after bariatric
surgery in patients with type 2 diabetes, Sci. Rep., 9,
6274.
40.Quoyer, J., Longuet, C., Broca, C., Linck, N.,
Costes, S., et al. (2010) GLP-1 mediates antiapoptotic effect by
phosphorylating Bad through a β-arrestin 1-mediated ERK1/2
activation in pancreatic β-cells, J. Biol. Chem.,
285, 1989-2002.
41.Li, Y., Tweedie, D., Mattson, M. P., Holloway, H.
W., and Greig, N. H. (2010) Enhancing the GLP-1 receptor signaling
pathway leads to proliferation and neuroprotection in human
neuroblastoma cells, J. Neurochem., 113, 1621-1631.
42.Shiraishi, D., Fujiwara, Y., Komohara, Y.,
Mizuta, H., and Takeya, M. (2012) Glucagon-like peptide-1 (GLP-1)
induces M2 polarization of human macrophages via STAT3 activation,
Biochem. Biophys. Res. Commun., 425, 304-308.
43.Lee, Y. S., Park, M. S., Choung, J. S., Kim, S.
S., et al. (2012) Glucagon-like peptide-1 inhibits adipose tissue
macrophage infiltration and inflammation in an obese mouse model of
diabetes, Diabetologia, 55, 2456-2468.
44.Gao, H., Wang, X., Zhang, Z., Yang, Y., Yang, J.,
et al. (2007) GLP-1 amplifies insulin signaling by up-regulation of
IRbeta, IRS-1 and Glut4 in 3T3-L1 adipocytes, Endocrine,
32, 90-95.
45.Hai, T., and Curran, T. (1991) Cross-family
dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA
binding specificity, Proc. Natl. Acad. Sci. USA, 88,
3720-3724.