REVIEW: Protein Aggregation and Cataract: Role of Age-Related Modifications and Mutations in α-Crystallins*
Prashanth Budnar1, Ramakrishna Tangirala1, Raman Bakthisaran1, and Ch. Mohan Rao1,a,b**
1Centre for Cellular and Molecular Biology (CCMB), Council of Scientific and Industrial Research (CSIR), Uppal Road, Hyderabad 500007, India* The article is published as a part of the Special Issue “Protein Misfolding and Aggregation in Cataract Disorders” (Vol. 87, No. 2).
** To whom correspondence should be addressed.
Received October 5, 2021; Revised February 2, 2022; Accepted February 2, 2022
Cataract is a major cause of blindness. Due to the lack of protein turnover, lens proteins accumulate age-related and environmental modifications that alter their native conformation, leading to the formation of aggregation-prone intermediates, as well as insoluble and light-scattering aggregates, thus compromising lens transparency. The lens protein, α-crystallin, is a molecular chaperone that prevents protein aggregation, thereby maintaining lens transparency. However, mutations or post-translational modifications, such as oxidation, deamidation, truncation and crosslinking, can render α-crystallins ineffective and lead to the disease exacerbation. Here, we describe such mutations and alterations, as well as their consequences. Age-related modifications in α-crystallins affect their structure, oligomerization, and chaperone function. Mutations in α-crystallins can lead to the aggregation/intracellular inclusions attributable to the perturbation of structure and oligomeric assembly and resulting in the rearrangement of aggregation-prone regions. Such rearrangements can lead to the exposure of hitherto buried aggregation-prone regions, thereby populating aggregation-prone state(s) and facilitating amorphous/amyloid aggregation and/or inappropriate interactions with cellular components. Investigations of the mutation-induced changes in the structure, oligomer assembly, aggregation mechanisms, and interactomes of α-crystallins will be useful in fighting protein aggregation-related diseases.
KEY WORDS: cataract, aggregation, α-crystallinDOI: 10.1134/S000629792203004X
Abbreviations: ACD, alpha crystallin domain; CTE, C-terminal extension; NTD, N-terminal domain; sHsp, small heat shock protein.
INTRODUCTION
Protein misfolding due to mutations, covalent modifications, or environmental factors, leads to the loss of function and protein aggregation, which are detrimental to cells. Cells have therefore evolved various quality control mechanisms to prevent aggregation (chaperones) or to remove protein aggregates (proteasome and autophagosome). However, the failure of quality control mechanisms due to aging or mutations results in progressive accumulation of protein aggregates and might cause protein misfolding diseases, such as neurodegenerative disorders (Alzheimer’s, Parkinson’s, Huntington’s diseases), amyotrophic lateral sclerosis, cystic fibrosis, Gaucher’s disease, and cataract.
CATARACT AND PROTEIN AGGREGATION
Cataract is one of the leading causes of blindness [1]. Cataract is a result of pathological opacity of crystalline lens, brought about by changes in its transparency and refractive index. Eye lens, together with cornea, focus the incident light on the retina for image formation. The lens needs to be transparent, with a high refractive index and appropriate refractive index gradient, for an aberration-free image. The lens has evolved a unique structure and composition to meet these requirements. It is avascular. Further, fiber cells present in the central region of the lens (nucleus) lose organelles such as nuclei, mitochondria, endoplasmic reticulum, and ribosomes. Moreover, tight organization of fiber cells reduces the intercellular space. These strategies remove or decrease the content of light-scattering structures and enable lens transparency [2].
Lens is made mostly of proteins. Lens fiber cells have a very high protein content (over 70% w/v). Crystallins are highly upregulated during fiber cell differentiation and constitute ∼90% of the total protein. Vertebrate lenses contain three groups of crystallins viz. – α-, β-, and γ [2]. Crystallins are commonly considered as structural proteins. However, α-crystallin is not only a structural protein but also has molecular chaperone activity. Lens transparency and high refraction index are achieved by dense and short-ranged systematic packing of crystallins at high concentrations with a gradient of protein concentration of about 250-400 mg/ml from the outer cortex to the core [2, 3]. The refractive index is high in the nuclear region and decreases at periphery, averaging to ∼1.4 [2]. Separation of the protein matrix into protein-rich and low protein phases can result in light scattering. This is avoided by the presence of α-crystallin in eye lenses, which forms short-range contacts with other crystallin proteins [2]. The probability of protein crystallization among crystallin components is also reduced due to the highly polydisperse nature of α-crystallins [2]. Since, there is no protein turnover in lenses, crystallins synthesized in utero remain until death. Therefore, there is a need for a chaperone system to retain lens proteins in their native conformation and to prevent phase separation/precipitation. α-Crystallins serve as molecular chaperones that bind to partially unfolded proteins and prevent their misfolding and aggregation.
Many theories have been proposed to explain cataract formation. From the physicochemical point of view, spatial variations in the protein density that cause lens opacification, can be due to effects of osmotic pressure at the cellular level. Benedek and co-workers proposed phase separation to explain the molecular mechanisms of cataract development [4]. At high concentrations, crystallins and other proteins undergo spontaneous phase separation into coexisting protein-rich and protein-poor phases. Phase separation also depends on changes in temperature and thus can be induced in intact lenses by decreasing the temperature, giving rise to cold cataracts [2]. Oxidative damage, which is caused by multiple factors including exposure to UV radiation, can induce formation of non-native disulfide bonds between protein subunits, leading to the formation of large, irreversible aggregates causing cataract [5]. Over the lifetime, crystallins endure various irreversible covalent modifications, such as those caused by UV irradiation, deamidation, oxidation, proteolysis, and Maillard reaction [6]. These covalent modifications destabilize the native structure of crystallins, thus decreasing the chaperone activity of α-crystallin and promoting partial unfolding of βγ-crystallins, leading to the development of age-related cataract [5, 6]. There is also evidence of amyloid type aggregation in cataractous lenses [7-9].
α-CRYSTALLIN: CHAPERONE FUNCTION AND STRUCTURAL
ASPECTS
Crystallins are known for more than 100 years. Mörener, in the year 1894, showed that lens contains four protein fractions – an insoluble form that he called albuminoid and three water-soluble fractions, α-, β-, and γ-crystallins [10]. α-Crystallin is isolated from the eye lens as a large polydisperse oligomer of two closely related proteins αA- and αB-crystallins. Roy and Spector observed that the low-molecular-weight fraction of α-crystallin disappears from the nuclear region of older human lenses [11], while it is present there in younger lenses. Subsequent studies found that such water-insoluble fractions consisted of α-crystallin complexes with other crystallins [12]. In retrospect, these observations could be taken as indications for the interactions of α-crystallins with other proteins and, perhaps, their role in age-related changes, including loss of lens transparency. In mammalian cells, α-crystallin exists as large oligomers composed of two gene products, αA- and αB-crystallins, at a 3 : 1 ratio in most vertebrate lenses. However, the ratio of αA- to αB-crystallin varies depending on a species and age [2, 12]. α-Crystallin is a member of the small heat shock protein (sHsp) family. It has the characteristic α-crystallin domain (ACD, ∼80-100 conserved amino acids) flanked by the variable N-terminal domain (NTD) and C-terminal extension (CTE) [13, 14]. αA- and αB-crystallins share ∼58% sequence homology with each other and ∼45% with the other human sHsps, such as HspB6 [13, 14].
α-Crystallins were first recognized as members of the sHsp family in early 1980s, when their sequence homology with sHsps from Drosophila was observed [15]. Surprisingly, no experimental verification of this observation had been obtained for a decade. The first experimental evidence that α-crystallin functions as a sHsp/molecular chaperone came in 1992 from in vitro studies by Horwitz, who showed that α-crystallin prevented thermal aggregation of βγ-crystallins and alcohol dehydrogenase [16]. The studies from our laboratory demonstrated that eye lens α-crystallin prevents photo-aggregation of γ-crystallin [17], as well as heat-induced aggregation of ζ-crystallin and carbonic anhydrase [18], DTT-induced aggregation of α-lactalbumin [19], and refolding-induced aggregation of β-crystallin [20]. Homooligomers of αA- and αB-crystallins were found to exhibit the chaperone activity against different target proteins, albeit to different extent [21, 22]. Our laboratory showed for the first time that the chaperone activity of α-crystallin is temperature-dependent, as it is very low below 30°C and steeply increases at temperatures above 30°C [17]. Such temperature-dependent activity appears to be a general phenomenon among Hsps, since these proteins need to function at non-permissible temperatures. The solubility of the hydrophobic dye pyrene increased in the presence of α-crystallin as a function of temperature, indicating that the temperature increase promoted exposure of hydrophobic surfaces in α-crystallin [17]. The temperature-induced increase in the chaperone-like activity of α-crystallin is attributable to various factors, including changes in the quaternary structure, increase in the content or reorganization of hydrophobic surfaces, and increase in the subunit exchange rate [18, 20]. The studies from our laboratory have also shown that α-crystallin binds to the molten globule state of target proteins [19] and helps the refolding of early intermediates [23].
Homooligomers of αA- and αB-crystallins showed differences in the temperature-dependent activity, as the structure of αB-crystallin is more sensitive to temperature than the structures of αA-crystallin or α-crystallin heterooligomer isolated from lens [21]. The mechanism of α-crystallin chaperone activity against target proteins has been intensively investigated by our laboratory and other research groups. Our studies have shown that αA- and αB-crystallins interact with the heat-induced unfolding intermediates of citrate synthase in two modes: (i) weakly with early unfolding intermediates, which facilitates enzyme refolding to its active state, and (ii) strongly and irreversibly with late unfolding intermediates with the formation of soluble complexes, thus preventing protein aggregation [22]. Both αA- and αB-crystallins bind with low-affinity to the compact native-like states and with high affinity to more unfolded states. The studies from other laboratories using model aggregation proteins such as β-crystallin, T4 lysozyme and its mutants suggested that both αA- and αB-crystallins bind to the substrates with both low and high affinity [24, 25]. The transition to the high-affinity mode can be induced by temperature, pH, or phosphorylation [26]. α-Crystallin also prevents amyloid aggregation of apolipoprotein C-II and serpin by interacting with partially structured intermediates, suggesting that α-crystallin inhibits the nucleation step of amyloidogenesis [27, 28]. We have shown that αB-crystallin prevents Aβ40 and Aβ42 fibril formation by interacting with Aβ40 fibril seeds [29]. The studies using quartz crystal microbalance, analytical ultracentrifugation, and single molecule detection systems, such as confocal two-color coincidence detection and total internal reflection microscopy, revealed that αB-crystallin prevents Aβ fibrillation by binding to Aβ oligomeric and fibrillar nucleus [30, 31]. The studies from several laboratories including ours have demonstrated that αB-crystallin also prevents α-synuclein fibrillation [13].
The polydisperse nature of α-crystallins has made their crystallization and structural studies difficult. However, the α-crystallin domain (ACD) alone could be crystallized and have been investigated in the X-ray diffraction studies. It was found that the ACDs (∼80 amino acids) of both crystallins are characterized by the presence of β-strands. A total of seven β-strands (β3-β9) are arranged into the immunoglobulin-like β-sandwich fold, with three strands (β3-β9-β8) arranged in the anti-parallel manner with other four β-strands (β4-β5-β6+β7) connected by a short loop [13, 32]. Biochemical studies have shown expression of the αA- or αB-crystallin ACDs resulted in the formation of dimers and loss of higher-molecular-weight oligomers [13, 32]. NTD is ∼65 amino acids long in both αA and αB-crystallins; it is enriched with hydrophobic amino acids and disordered. Studies involving either truncation of the NTD or deletion of conserved motifs SRLFDQFFG and FLRAPSWF affected the oligomer assembly in αB-crystallin, giving rise to the dimers to hexamers or smaller oligomers [33, 34]. Solid-state NMR studies provided insights into the structure of the N-terminal region, which contains two short α-helices (residues 14-17 and 27-31) and a two-stranded antiparallel β-sheet (residues 48-50 and 61-63) connected by a long loop [35]. CTE in αA- and αB-crystallins is ∼20 amino acids long, flexible, polar, and has a highly conserved IXI motif which facilitates oligomer formation by acting as an “anchor point”, thereby contributing to the polydispersity of α-crystallin [13]. Several studies have shown that deletion of the CTE in αA- and αB-crystallins affects the quaternary structure and distribution of oligomers. Deletions or mutations in the CTE of αB-crystallin decrease the size of the oligomers and affect solubilization of the substrates [36, 37]. Since it has been shown that the loss of chaperone activity is associated with pathology, we have attempted to increase this activity, prompted by our observation that it can be upregulated by the temperature-dependent structural perturbations. We have produced genetically engineered chimeric proteins with a significantly higher chaperone activity, thus demonstrating that the structural organization and chaperone activity of sHsps depend on the sequence and length of the CTE, NTD, and conserved motifs [36, 38].
Based on the data of solid-state NMR, SAXS (small angle X-ray scattering), and EM (electron microscopy), a model was proposed for the full-length αB-crystallin as a symmetric multimer of 24 subunits associated through a hierarchy of interactions involving the ACD, CTE, and NTD: two ACDs bind to form a dimer. Three dimers associate with each other to form a hexamer through interactions of the conserved IXI motif of the C-terminus in one dimer with the hydrophobic pockets (β4 and β8 strands) in the other dimer [32, 39]. The hexamers associate into higher-order multimers through various interactions involving the NTD [35, 39]. This model is consistent with the structural data; however, the polydispersity often reported for αB-crystallin needs to be reconciled with the symmetric 24-mer.
In human lenses, αA-crystallin exists in both oxidized and reduced forms due to the presence of two cysteine residues. Kaiser et al. [40] studied the structure of αA-crystallin oligomers in both reduced and oxidized form using a combination of cryo-EM, crosslinking/mass-spectrometry, NMR spectroscopy, molecular modeling. They showed that αA-crystallin oligomers exist mainly in 12-, 16-, and 20-meric assemblies that were interconvertible via addition or subtraction of tetramers. The assembly proceeds through the tetramer formation, wherein two β7-interface dimers associate through their N-terminal fragments. The tetrameric units link through further N-terminal interactions into higher-order assemblies. An enhanced dynamics of the CTE leads to the cross-dimer domain swapping and is a determinant of oligomer heterogeneity. Although αA- and αB-crystallins demonstrate high sequence similarity with a similar sequence length, they form different oligomeric assemblies using similar type of interactions. The dimerization is mediated by the β7 interface, whereas oligomerization is mediated by the N-terminal interactions and IXI binding to a nearby protomer. However, the CTE of αB-crystallin is involved in the formation of hexameric species, whereas the CTE of αA-crystallin is involved in a higher-order oligomeric assembly in the 3D domain-swapped form. Hence, the N-terminal interactions are important for oligomer formation in αA-crystallin [40].
AGE-RELATED MODIFICATIONS IN α-CRYSTALLIN AND
CATARACT
Crystallins, including α-crystallin, are long-lived lens proteins and thus are subjects of age-related modifications. Epidemiological studies showed that environmental and occupational factors increase the risk of age-related cataracts [41]. A large number of studies have demonstrated that the water-insoluble fraction of lens increases in aged and cataract lenses [42, 43]. Many post-translational modifications, such as deamidation, oxidation, acetylation, methylation, isomerization, carbamylation, and phosphorylation, have been observed in both αA- and αB-crystallins from aged and cataract lenses (Table 1).
Table 1. Post-translational/age-related
modifications in α-crystallins
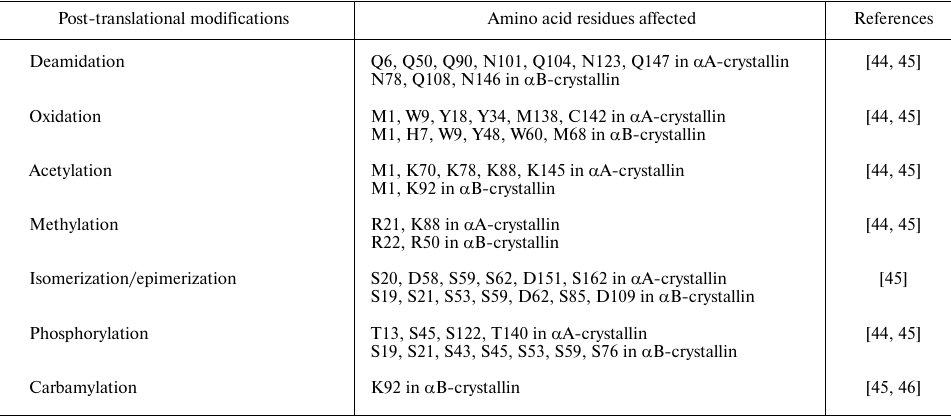
UV irradiation promotes the development of age-related cataracts [47]. Lens has tryptophan-derived UV filters, such as kynurenine, 3-hydroxykynurenine, and 3-hydroxykynurenine glucoside, which absorb UV radiation. However, UV-induced photoexcitation of these UV filters leads to the production of free radicals and reactive oxygen species, which oxidize methionine, tryptophan, and cysteine residues of α-crystallin and lead to the non-disulfide covalent crosslinking and oligomerization [48, 49]. Exposure to UV irradiation causes significant secondary and tertiary structural changes and impairs the chaperone-like activity of α-crystallin [50].
Oxidation is one of the most common causes of post-translational modification observed in the lens, where it commonly affects methionine, tryptophan, tyrosine, cysteine, and histidine residues in α-crystallin (Table 1). In the UV-exposed bovine lenses, oxidation occurred at M1 and M168 residues in αA-crystallin and at M1, W9, W60, and M68 residues in αB-crystallin [48]. In vitro studies also showed that oxidation of αA- and αB-crystallins resulted in significant changes in their secondary and tertiary structure and formation of low-molecular-weight species lacking chaperone activity [51].
Deamidation is another typical post-translational modification, which is higher in the water-insoluble fraction of both normal and cataract lenses. Deamidation converts amide in the side chain of asparagine and glutamine residues to the carboxylic acid residue and introduces a negative charge to the protein, thus destabilizing protein structure. Mass spectrometry studies revealed that deamidation occurs at Q6, Q50, Q90, N101, Q104, N123, and Q147 in αA-crystallin and at N78, Q108, N146 in αB-crystallin (Table 1). It was found that α-crystallin in cataract lenses is highly deamidated in comparison to the proteins in normal lenses. The effect of age-related deamidation on the structural and functional properties of α-crystallin was studied using different deamidation-mimicking mutants of αA-crystallin (N101D, N123D, and N101D/N123D) and αB-crystallin (N78D, N146D, and N78D/N146D) [52]. αA-crystallin mutants showed altered secondary structure and microenvironment; the homomers with deamidation at N123 exhibited a significant decrease in the chaperone activity [52, 53]. Deamidation at N146 affected both structural and functional properties of αB-crystallin [54].
Another post-translational modification that occurs in aged and cataractous lenses is phosphorylation of α-crystallin (Table 1). The studies of α-crystallin in human lenses and its modification as a function of age and cataract development using MALDI-MS revealed an increased amount of phosphorylated αB-crystallin in the cortical regions of older lenses [55]. Proteomic analysis of congenital cataractous lenses demonstrated that phosphorylation occurs at residues T13, S45, S122, T140 in αA-crystallin and at S19, S21, S43, S45, S53, S59, S76 in αB-crystallin [56]. Phosphoproteomics characterization of normal and cataractous lenses followed by the quantitative analysis indicated higher phosphorylation levels of α-crystallin (T153 in αA-crystallin and S19, S76 in αB-crystallin) in cataractous lenses [57]. Phosphorylation of αB-crystallin increases with aging and is promoted by stress and disease [58]. Several studies have been carried out to determine the levels of αB-crystallin phosphorylation. The extent of αB-crystallin phosphorylation in cultured rat lenses was approximately 0.25 mol phosphate per mol protein [59]. The level of αB-crystallin phosphorylation in bovine lenses increases up to 0.25 mol phosphate per mol protein during the first three years of life and remains unchanged thereafter [59, 60]. αA-crystallin, on the other hand, can get phosphorylated up to 0.5 mol phosphate per mol protein [59, 60]. Phosphorylation in αB-crystallin commonly occurs at S19, S45, and S59. Phosphorylation of αB-crystallin can be beneficial or deleterious, depending on its level. Phosphorylation at a single site, either at S45 or S59, increases the chaperone activity of this protein, whereas hyperphosphorylation at two residues (S45 and S59) increases its partition to the insoluble fraction [58]. The R120G mutant of αB-crystallin undergoes hyperphosphorylation and partitions into insoluble fractions [61, 62]. Earlier investigation from our laboratory using phosphomimicking mutants indicated that hyperphosphorylation of R120G αB-crystallin (S19D/S45D/S59D-R120G αB-crystallin or 3D-R120G αB-crystallin) increases its propensity to aggregation [58].
Another commonly observed post-translational modification in aged lenses is racemization/isomerization, when the L-forms of serine and aspartic acid change to their D-forms. Amino acid residues that undergo isomerization in αA- and αB-crystallins are listed in Table 1. αA-crystallin is more susceptible to isomerization than αB-crystallin due to a high content of aspartic acid residues [63]. The D58 and S59/62 residues in αA-crystallin undergo racemization/isomerization during aging and their D-isoforms are more common in cataractous lenses than in normal lenses [64, 65]. Isomerization alters the structure of α-crystallin. Thus, isomerization of D58, D84, and D151 residues of αA-crystallin observed in aged lenses results in the disruption of its higher-order heteropolymeric structure and decreased solubility, eventually leading to the cataract development [64]. In αB-crystallin, isomerization of D109 residue disrupts the crucial salt bridge with R120 at the dimer interface, leading to aberrant oligomerization. Residues S59 and D62 are involved in the interface generation by forming a salt bridge cluster with R163 and E165 of the adjacent monomer [66]. Isomerization at D62 affects phosphorylation of S59, thus perturbing formation of the salt bridge and disrupting the interface, which alters the oligomerization dynamics [66]. Epimerization of S162 significantly weakens the intersubunit binding [66]. In diabetes, hyperglycemia promotes non-enzymatic reactions, such as glycation and oxidation, that result in the formation of advanced glycation end-products (AGEs) and accelerate Maillard reactions in lens crystallins, leading to the age-related cataract [67, 68].
In vitro studies in bovine and rat lenses showed that glycation, which occurs at lysine side chains, increases the susceptibility of crystallins to sulfhydryl oxidation and leads to the formation of high-molecular-weight aggregates with disulfide crosslinks [69]. Although glycation does not noticeably alter the structure of α-crystallin, it significantly decreases its chaperone activity [70]. Truncation is one of the most common modifications occurring in lens, wherein removal of a part of protein sequence either from the N- or C-terminus results in several peptide fragments. Modifications, such as deamidation and isomerization, increase the probability of protein truncation. α-Crystallins undergo several truncations, mostly at the C-terminus: truncation sites include S172, S162, N101, D58, and L40 in αA-crystallin and K174, T170, T40, and E34 in αB crystallin (see [43, 55] and review [71]). It was found that in aged lenses, the N-terminal region of αA-crystallin is truncated by 3 residues, while the N-terminal region of αB-crystallin can be truncated by 1 or 6 residues [72]. The studies from our laboratory in collaboration with the Garland group revealed a truncated form of αB-crystallin with the loss of the C-terminal lysine in human cataract lenses [73]. This truncated form, designated as αBg, was more acidic and migrated slightly lower than the full-size protein in SDS-PAGE. The amount of αBg varied from 10-90% of total αB-crystallin. In vitro studies showed that the chaperone activity of αBg was comparable to that of the normal αB-crystallin [73]. Removal of the C-terminal residues in the recombinant truncated (151 residues) αA-crystallin affected its oligomerization and subunit exchange [44]. Mutants of αA- and αB-crystallins with truncation of 16 C-terminal amino acids exhibit decreased chaperone activity [74]. Truncation of α-crystallin also generates several low-molecular-weight peptides, which are observed in aging human lens and associated with lens opacity and cataract [75, 76]. The αA66-80 peptide from αA-crystallin was shown to bind to soluble α-crystallin, impair its chaperone activity, and induce its aggregation [77]. αA66-80 also formed amyloid fibrils, promoted generation of hydrogen peroxide, and induced apoptosis [78].
Other modifications in α-crystallin are acetylation and carbamylation of lysine residues (Table 1). In human lenses, acetylation occurs at K70 and K99 in αA-crystallin and K92 and K166 in αB-crystallin [44]. The assays of the chaperone activity of the acetylation-mimicking mutant K70Q of α-crystallin revealed both altered structure and varying chaperone activity of this protein that depended on the client protein [79]. Carbamylation of lens proteins, which is believed to be an age-dependent process, was found to be increased in the lens nucleus compared to the cortex region [71]. Carbamylation of K92 residue in αB-crystallin was observed in cataractous lenses [46].
Protein–protein crosslinking is associated with the aggregation process taking place during cataract development. Truncation and post-translational modifications, such as oxidation, deamidation, and isomerization, trigger protein–protein crosslinking of crystallins that can proceed through different mechanisms [via dehydroalanine (DHA), Asn succinimide intermediate and truncation, or C-terminal succinic anhydride intermediate] (see review [71]). Proteomic studies revealed that α-crystallin undergoes protein–protein crosslinking that involves reaction of lysine residues of the non-enzymatically truncated protein to the C-terminal aspartate residues [71, 80]. This biphasic mechanism includes two spontaneous events – protein cleavage and crosslinking. Protein cleavage can take place at pH 5-7.4 and involves peptide cleavage at the C-terminal side of aspartate residue, which then undergoes spontaneous cyclization to form C-terminal aspartate anhydride intermediate. The next step is crosslinking, when the C-terminal Asp anhydride intermediate either reacts with water to form aspartate or with a nucleophile such as lysine ε-amino group or N-terminal amine group to form a covalent protein–protein crosslink [80]. Proteomic analysis of cataract lenses identified different crosslinks in αA-crystallin, αB-crystallin, γC/D-crystallin, and γS-crystallin. The identified crosslinks were αA-crystallin K78–αA-crystallin D58, αA-crystallin D151–αB-crystallin K82, αA-crystallin D151–αB-crystallin K150, αB-crystallin D129–αB-crystallin K82, αB-crystallin D129–αB-crystallin K90, αB-crystallin D129–αB-crystallin K121, αA-crystallin D151–γC- or γD-crystallin G2 [71, 80]. Most of the crosslinks were amide bonds between the lysine side chain of one peptide and C-terminal aspartate residue of another peptide. Interestingly, the αA-crystallin D151–γC/γD-crystallin G2 crosslink involved an amide bond formed by the N-terminal group of γC/D-crystallin with the C-terminal aspartate (D151) of αA-crystallin [71, 80].
Therefore, during aging, lens proteins, including α-crystallin, undergo multiple covalent modifications, truncations, and crosslinks. These modifications can occur simultaneously or independently and affect the structure, oligomerization, and chaperone function of α-crystallin, leading to the decreased solubility and increased protein aggregation and making the lens prone to the development of age-related cataract.
MUTATIONS IN α-CRYSTALLIN AND CATARACT
Mutations in αA- and αB-crystallins can lead to the development of autosomal congenital cataracts of different phenotypes (Tables 2 and 3). Cataract-associated mutations in α-crystallin can be missense, frameshift, or nonsense mutations. Most of these mutations are autosomal dominant and cause congenital cataracts, whereas a few mutations are recessive (Tables 2 and 3). The first reported cataract-causing mutation in α-crystallin was a single missense mutation, R116C, in αA-crystallin, which induced autosomal dominant cataract [81]. Vicart et al. reported the second case, in which cataract and desmin-related myopathy were caused by the missense R120G mutation in αB-crystallin.
Table 2. Cataract-causing mutations in
αA-crystallin (CRYAA)
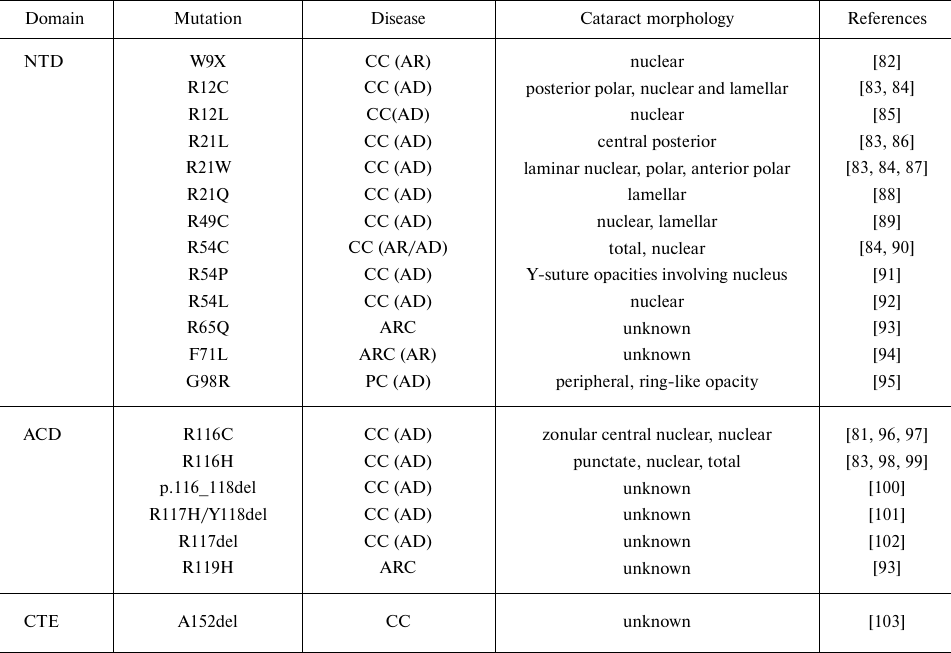
Notes. Designations: NTD, N-terminal domain; ACD, α-crystallin
domain; CTE, C-terminal extension; CC, congenital cataract; ARC,
age-related cataract; PC, presenile cataract (AD, autosomal dominant;
AR, autosomal recessive); n/a, not applicable.
Table 3. Disease-causing mutations in
αB-crystallin (CRYAB)
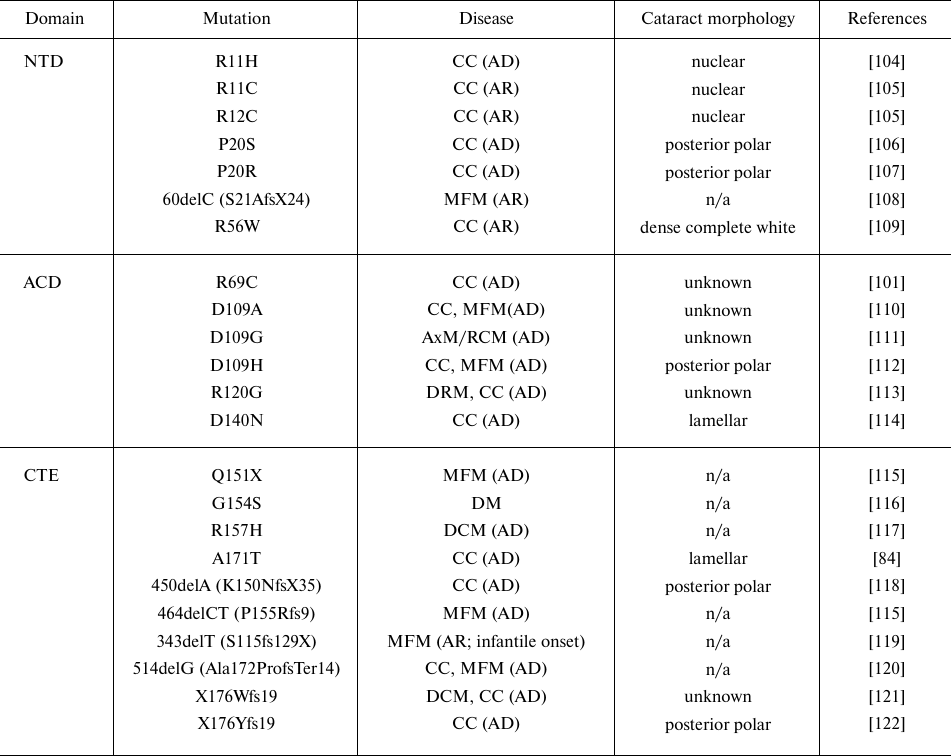
Notes. Designations: NTD, N-terminal domain; ACD, α-crystallin
domain; CTE, C-terminal extension; CC, congenital cataract; DCM,
dilated cardiomyopathy; DRM, desmin-related myopathy; AxM/DM/MFM,
axial/distal/myofibrillar myopathy; RCM, restrictive cardiomyopathy
(AD, autosomal dominant; AR, autosomal recessive); n/a, not
applicable.
Most cataract-causing mutations in the αA-crystallin NTD are dominant missense mutations of conserved arginine residues. One nonsense mutation (W9X) results in a truncated form of αA-crystallin, giving rise to a recessive congenital cataract (Table 2). Similarly, cataract-causing mutations in the NTD of αB-crystallin are also missense mutations of conserved arginine residues (R11H, R12C, R56W) and proline residue (P20R), resulting in either dominant or recessive congenital cataract. Biophysical studies of the N-terminal cataract-causing mutants (R11H, P20S, R56W) showed that the R11H mutant formed smaller oligomers, whereas P20S and R56W mutants formed larger oligomers than those generated by the wild-type αB-crystallin. The P20S mutant showed lower thermal stability than the wild-type protein. Both P20S and R56W mutants exhibited lower chaperone activity than the wild-type αB-crystallin when tested against UV-irradiated βL-crystallin as a substrate [123]. In both αA- and αB-crystallins, the ACD is involved in the dimer formation and is composed of conserved residues that make up the β-sandwich domain. Cataract-causing mutations in the ACD of αA-crystallin occur at the conserved glycine and arginine residues (G98R and R116C, respectively) (Table 2). The recombinant αA-crystallin with the R116C mutation shows structural alterations leading to increase in polydispersity and decrease in the protein subunit exchange and chaperone activity [124, 125]. The G98R mutation in αA-crystallin causes pre-senile cataract with the onset at the age of ∼18 years [95]. The studies from our laboratory demonstrated that the G98R mutant undergoes impaired folding and forms inclusion bodies [126]. When solubilized and refolded, the mutant protein exhibited changes in the secondary, tertiary, and quaternary structure, and formed larger oligomers lacking the chaperone activity [126]. In αB-crystallin, cataract- and/or myopathy-causing mutations occur in the ACD at the arginine and aspartate residues (R69C, D109H, R120G, and D140N) (Table 3). Biophysical and biochemical studies of the R120GαB mutant showed changes in the secondary and tertiary structure of this protein along with the reduced chaperone activity [125, 127]. The R120G mutant of αB-crystallin interacted abnormally with cellular components (e.g., desmin) resulting in the generation of aggregates leading to the cell toxicity [128]. The studies of the R69C and D109H mutants revealed their altered structure (secondary, tertiary, and quaternary), increased amyloidogenic propensity, and decreased chaperone-like activity [129]. The D140N mutation affected the tertiary/quaternary structure of αB-crystallin, resulting in larger aggregates and decreased thermal stability [114]. The D140N mutant lacked the chaperone-like activity and inhibited the chaperone function of the wild-type αB-crystallin, thus behaving as a dominant negative mutant [114]. Residues D109, R120, and D140 are highly conserved and form a crucial part of the β-sandwich domain: the dimer interface forms the base of the shared groove with the R120 residues present at either end interacting with D109 to form a salt bridge that helps to generate a shared interface groove in αB-crystallin [130]. Dominant mutations, such as R120G or D109G, disturb this salt bridge, thereby disturbing the groove and conformation of the extended β-sandwich domain [112, 130]. Similarly, D140 residue situated on the hairpin loop connecting β8-9 forms a salt bridge with H83 situated on the β3/β4 arch in the β-sandwich domain [114, 130]. Therefore, all three mutations could be expected to affect the ion-pair interactions occurring in the loop and the arch connected to the shape of the extended dimer bottom sheet.
The C-terminal region in α-crystallin is important for the protein flexibility and solubility. Dominant mutations G154S and R157H in this region cause mild cardiomyopathy without cataract development, whereas the A171T mutation causes congenital lamellar cataract (Table 3). The studies of the C-terminal mutants (G154S, R157H, and A171T) showed that the R157H mutant formed smaller oligomers than the wild-type αB-crystallin. The mutants showed lower thermostability and formed aggregates at lower temperature than the wild-type protein. They also differed in the formation of heterooligomeric complexes with HspB4 and HspB6. The chaperone activity of G154S and A171T mutants was lower than that of the wild-type αB-crystallin [131]. However, the nonsense mutation A152del in αA-crystallin [103] and the frameshift mutations Ala172ProfsTer14, X176Wfs19, and X176Yfs19 in αB-crystallin [121, 122] are associated with the autosomal dominant congenital cataracts. Recently a novel dominant frameshift mutation c.514delG (p.Ala172ProfsTer14) was identified, which causes a multisystem disorder characterized by congenital cataracts, hypotonia, myopathy, respiratory failure, and dysphagia [120]. This novel mutation results in the frameshift at Ala172 leading to the addition of 12 new amino acids and protein elongation to 184 residues. Bioinformatic analysis indicated an increased hydrophobicity and decreased flexibility of the C-terminal domain in the mutant protein, which eventually affects its solubility and chaperone activity [120]. The p.X176Y mutation leads to the loss of the stop codon and results in the addition of 19 amino acids to the wild-type αB-crystallin [122]. Bioinformatic analysis predicted that addition of 19 amino acids causes formation of novel secondary structures (α-helix and random coil) and generates an extended strand in the tertiary structure, making the mutant protein pathogenic due to the increased hydrophobicity and reduced protein stability [122].
MUTATIONS IN α-CRYSTALLIN AND THEIR EFFECT ON PROTEIN
AGGREGATION
Most mutations in α-crystallins mentioned in Table 2 and 3 are autosomal dominant negative mutations. Mutations in α-crystallins might induce protein unfolding and formation of homo- or hetero-aggregates that can be toxic to cells. Different αA- and αB-crystallin mutants form aggregates/inclusion bodies when expressed in bacterial/cells (Table 4).
Table 4. Aggregates formed by different
αA- and αB-crystallin mutants

As shown in Table 4, various cataract-causing mutants of αA-crystallin form inclusion bodies/aggregates. Some mutants of αA-crystallins (e.g., R49C and R54C) form nuclear aggregates. Studies in the knock-in mouse model revealed that the R49C mutation decreases solubility and increases protein hydrophobicity; the mutant protein aggregates with the formation of cataract likely due to the increased hydrophobicity and binding to β- and γ-crystallins [141].
Various studies, including our recent results, indicate that the R54C mutant exhibits only slight structural changes and has a chaperone activity comparable to that of the wild-type protein [133, 142]. We found that unlike the diffused localization of the wild-type αA-crystallin in the cytoplasm of SRA01/04 lens epithelial cells, the R54C mutant of αA-crystallin formed speckles in both the cytoplasm and the nucleus. It also associated more strongly with nucleosomal histones than the wild-type protein and elicited stress-like response and apoptosis [133]. The study on the congenital cataract-causing mutations at R54 (R54C, R54L, R54P) showed that these mutations resulted in the structural changes, decreased thermal stability, lower chaperone-like activity, and increased propensity to amyloidogenesis [143].
We showed earlier that G98R αA-crystallin undergoes aggregation and forms inclusion bodies when produced in bacteria [126]. It also shows altered chaperone-like activity, which is modulated by Cu2+ ion [126, 144, 145].
Similarly, different disease-causing mutations in αB-crystallin result in the formation of inclusion bodies/aggregates (Table 4). The study by Raju and Abraham [135] of different congenital cataract- and cardiomyopathy-causing mutants of αB-crystallin in mammalian cells showed that the D109H, R120G, D140N, and R157H mutants formed more aggregates than other mutants [135]. However, it should be noted that the authors used yellow fluorescent protein (YFP) to tag the mutants. Therefore, there is a possibility that the fused fluorescent protein could affect aggregation of the mutant proteins. Various studies performed in vitro, ex vivo, or in vivo (in transgenic mice) showed aggregation and formation of inclusion bodies by R120G αB-crystallin interacting with desmin [128, 137, 146].
There are two types of protein aggregations – amorphous and well-ordered amyloid fibrillar aggregation. As mentioned earlier, amyloid aggregates have been found in the cataract eye lens. In vitro studies have shown that crystallins, including α-crystallin, form amyloid aggregates under denaturing conditions [147]. This raises the question whether amyloids are formed in cataract in addition to amorphous aggregation. The competition between amorphous and ordered amyloid aggregation is observed in protein aggregation [148]. The study of cataracts in mouse showed that the recombinant murine γBnop mutant (truncated protein formed due to 11-base pair deletion in the third exon at Ser138 followed by 4-base pair insertion) formed amyloid fibrils in vitro (unlike the wild-type protein) [149]. Analysis of lenses by the 2D infrared spectroscopy (2DIR) identified amyloid β-sheet structures in the UV-irradiated porcine lenses [7]. Similar studies in the non-cataract and cataractous human lenses revealed the presence of amyloid β-sheet secondary structure in mature and cataract lenses, whereas juvenile lenses lacked any amyloid structure [8]. Mutant R120G αB-crystallin tends to form higher amyloid fibrils in vitro compared to the wild-type protein [150]. Recent study using 2DIR spectroscopy showed the presence of amyloid-like structure in the Cryab-R120G knock-in mice lenses indicating that mutant α-crystallin can indeed form amyloid aggregates in vivo [9]. Anti-amyloid oligomer antibodies have been shown to recognize the aggregates of R120G αB-crystallin in transgenic mice model [137]. These studies indicate that both amyloid formation and amorphous aggregation can take place during cataract development. Therefore, it would be interesting to understand the aggregation mechanisms of α-crystallins per se and the effect of disease-causing mutations on the propensity to and mechanisms of aggregation of α-crystallins.
Further studies are required to dissect the mechanisms of α-crystallin aggregation induced by the disease-causing mutations. It is possible that perturbation of the native structure by mutations leads to the exposure of already existing aggregation-prone regions which are otherwise buried or masked in the folded structure, as these regions are important contributing factors not only to aggregation but also to inappropriate interactions of α-crystallin with cellular components. Therefore, studies involving experimental crosslinking to identify protein segments involved in intermolecular interactions, their alterations caused by mutations, native oligomeric interface sequences/regions, and protein fragments mediating aggregation process, will contribute to our understanding of the mechanism of α-crystallin aggregation in cataract formation.
CONCLUSIONS
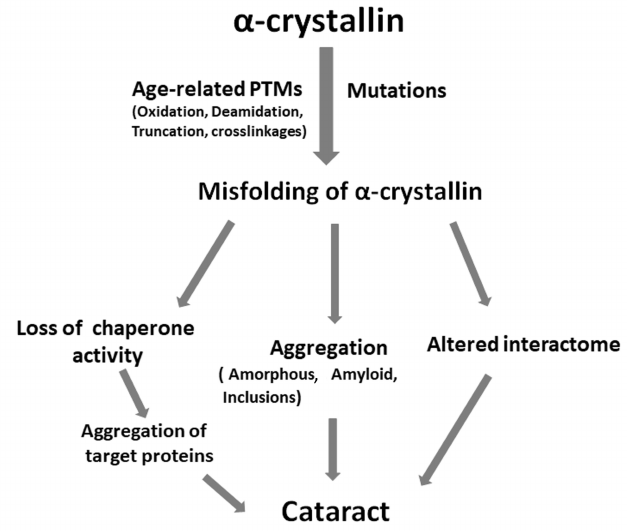
Studies conducted in recent years have shown that α-crystallin maintains the crystalline nature and transparency of eye lenses by participating in the packing of βγ-crystallins and preventing their aggregation. The figure depicts how post-translational modifications and mutations can lead to cataract. Different age-related modifications and mutations in α-crystallins either perturb the structure of these proteins or decrease their chaperone function, which results in the aggregation of target proteins, formation of aggregates (amyloid, amorphous, inclusions), and altered interactome. These processes can occur simultaneously or independently, leading to the cell toxicity and cataract development (figure). Mutations in αA-crystallin often result in the development of congenital cataracts. Since expression of αB-crystallin is ubiquitous, mutations in αB-crystallin can lead not only to cataract but to other diseases, such as myopathies. Therefore, development of strategies to prevent aggregation or formation of inclusions due to the age-related modifications or mutations is important for treatment and prevention of cataract and protein aggregation diseases. Some sterols have shown promising effects in reversing cataract in experimental animals [151, 152]. Interestingly, peptides derived from the chaperone sites of αA- and αB-crystallins (the so-called mini-chaperones) exhibit therapeutic potential against cataract and other protein aggregation diseases [153]. Identification of segments that participate in the intermolecular interactions (both native oligomer interfaces and regions mediating aggregation) and elucidation of effects of mutations on those interactions will be useful in developing approaches to prevent the onset and/or progression of cataract.
Role of age-related modifications and mutations in α-crystallins in the development of cataract
Ethics declarations. The authors declare no conflicts of interest. This article does not contain description of studies with the involvement of humans or animal subjects.
REFERENCES
1.Chou, C. F., Cotch, M. F., Vitale, S., Zhang, X.,
Klein, R., et al. (2013) Age-related eye diseases and visual impairment
among U.S. adults, Am. J. Prev. Med., 45, 29-35, doi:
10.1016/j.amepre.2013.02.018.
2.Bloemendal, H., de Jong, W., Jaenicke, R., Lubsen,
N. H., Slingsby, C., et al. (2004) Ageing and vision: structure,
stability and function of lens crystallins, Prog. Biophys. Mol.
Biol., 86, 407-485, doi:
10.1016/j.pbiomolbio.2003.11.012.
3.Delaye, M., and Tardieu, A. (1983) Short-range
order of crystallin proteins accounts for eye lens transparency,
Nature, 302, 415-417, doi: 10.1038/302415a0.
4.Benedek, G. B. (1971) Theory of transparency of the
eye, Appl. Opt., 10, 459-473, doi:
10.1364/AO.10.000459.
5.Hanson, S. R., Hasan, A., Smith, D. L., and Smith,
J. B. (2000) The major in vivo modifications of the human
water-insoluble lens crystallins are disulfide bonds, deamidation,
methionine oxidation and backbone cleavage, Exp. Eye Res.,
71, 195-207, doi: 10.1006/exer.2000.0868.
6.Harding, J. J. (2002) Viewing molecular mechanisms
of ageing through a lens, Ageing Res. Rev., 1, 465-479,
doi: 10.1016/s1568-1637(02)00012-0.
7.Zhang, T. O., Alperstein, A. M., and Zanni, M. T.
(2017) Amyloid beta-sheet secondary structure identified in UV-induced
cataracts of porcine lenses using 2D IR spectroscopy, J. Mol.
Biol., 429, 1705-1721, doi: 10.1016/j.jmb.2017.04.014.
8.Alperstein, A. M., Ostrander, J. S., Zhang, T. O.,
and Zanni, M. T. (2019) Amyloid found in human cataracts with
two-dimensional infrared spectroscopy, Proc. Natl. Acad. Sci.
USA, 116, 6602-6607, doi: 10.1073/pnas.1821534116.
9.Alperstein, A. M., Molnar, K. S., Dicke, S. S.,
Farrell, K. M., Makley, L. N., et al. (2021) Analysis of amyloid-like
secondary structure in the Cryab-R120G knock-in mouse model of
hereditary cataracts by two-dimensional infrared spectroscopy, PLoS
One, 16, e0257098, doi: 10.1371/journal.pone.0257098.
10.Mörner, C. T. (1894) Untersuchung der
Proteїnsubstanzen in den leichtbrechenden Medien des Auges I [in
German], bchm, 18, 61-106, doi:
10.1515/bchm1.1894.18.1.61.
11.Roy, D., and Spector, A. (1976) Absence of
low-molecular-weight alpha crystallin in nuclear region of old human
lenses, Proc. Natl. Acad. Sci. USA, 73, 3484-3487, doi:
10.1073/pnas.73.10.3484.
12.Horwitz, J., Bova, M. P., Ding, L. L., Haley, D.
A., and Stewart, P. L. (1999) Lens alpha-crystallin: Function and
structure, Eye (Lond), 13 (Pt. 3b), 403-408, doi:
10.1038/eye.1999.114.
13.Bakthisaran, R., Tangirala, R., and Rao, Ch. M.
(2015) Small heat shock proteins: Role in cellular functions and
pathology, Biochim. Biophys. Acta, 1854, 291-319, doi:
10.1016/j.bbapap.2014.12.019.
14.Kappe, G., Franck, E., Verschuure, P., Boelens,
W. C., Leunissen, J. A., et al. (2003) The human genome encodes 10
alpha-crystallin-related small heat shock proteins: HspB1-10, Cell
Stress Chaperones, 8, 53-61, doi:
10.1379/1466-1268(2003)8<53:thgecs>2.0.co;2.
15.Ingolia, T. D., and Craig, E. A. (1982) Four
small Drosophila heat shock proteins are related to each other and to
mammalian alpha-crystallin, Proc. Natl. Acad. Sci. USA,
79, 2360-2364, doi: 10.1073/pnas.79.7.2360.
16.Horwitz, J. (1992) Alpha-crystallin can function
as a molecular chaperone, Proc. Natl. Acad. Sci. USA, 89,
10449-10453, doi: 10.1073/pnas.89.21.10449.
17.Raman, B., and Rao, C. M. (1994) Chaperone-like
activity and quaternary structure of alpha-crystallin, J. Biol.
Chem., 269, 27264-27268.
18.Raman, B., Ramakrishna, T., and Rao, C. M. (1995)
Temperature dependent chaperone-like activity of alpha-crystallin,
FEBS Lett., 365, 133-136, doi:
10.1016/0014-5793(95)00440-k.
19.Rajaraman, K., Raman, B., Ramakrishna, T., and
Rao, C. M. (1998) The chaperone-like alpha-crystallin forms a complex
only with the aggregation-prone molten globule state of
alpha-lactalbumin, Biochem. Biophys. Res. Commun., 249,
917-921, doi: 10.1006/bbrc.1998.9242.
20.Raman, B., and Rao, C. M. (1997) Chaperone-like
activity and temperature-induced structural changes of
alpha-crystallin, J. Biol. Chem., 272, 23559-23564, doi:
10.1074/jbc.272.38.23559.
21.Datta, S. A., and Rao, C. M. (1999) Differential
temperature-dependent chaperone-like activity of alphaA- and
alphaB-crystallin homoaggregates, J. Biol. Chem., 274,
34773-34778, doi: 10.1074/jbc.274.49.34773.
22.Rajaraman, K., Raman, B., Ramakrishna, T., and
Rao, C. M. (2001) Interaction of human recombinant alphaA- and
alphaB-crystallins with early and late unfolding intermediates of
citrate synthase on its thermal denaturation, FEBS Lett.,
497, 118-123, doi: 10.1016/s0014-5793(01)02451-6.
23.Goenka, S., Raman, B., Ramakrishna, T., and Rao,
C. M. (2001) Unfolding and refolding of a quinone oxidoreductase:
alpha-crystallin, a molecular chaperone, assists its reactivation,
Biochem. J., 359, 547-556, doi:
10.1042/0264-6021:3590547.
24.Sathish, H. A., Koteiche, H. A., and McHaourab,
H. S. (2004) Binding of destabilized betaB2-crystallin mutants to
alpha-crystallin: The role of a folding intermediate, J. Biol.
Chem., 279, 16425-16432, doi: 10.1074/jbc.M313402200.
25.McHaourab, H. S., Dodson, E. K., and Koteiche, H.
A. (2002) Mechanism of chaperone function in small heat shock proteins.
Two-mode binding of the excited states of T4 lysozyme mutants by
alphaA-crystallin, J. Biol. Chem., 277, 40557-40566, doi:
10.1074/jbc.M206250200.
26.Koteiche, H. A., and McHaourab, H. S. (2003)
Mechanism of chaperone function in small heat-shock proteins.
Phosphorylation-induced activation of two-mode binding in
alphaB-crystallin, J. Biol. Chem., 278, 10361-10367, doi:
10.1074/jbc.M211851200.
27.Hatters, D. M., Lindner, R. A., Carver, J. A.,
and Howlett, G. J. (2001) The molecular chaperone, alpha-crystallin,
inhibits amyloid formation by apolipoprotein C-II, J. Biol.
Chem., 276, 33755-33761, doi: 10.1074/jbc.M105285200.
28.Devlin, G. L., Carver, J. A., and Bottomley, S.
P. (2003) The selective inhibition of serpin aggregation by the
molecular chaperone, alpha-crystallin, indicates a nucleation-dependent
specificity, J. Biol. Chem., 278, 48644-48650, doi:
10.1074/jbc.M308376200.
29.Raman, B., Ban, T., Sakai, M., Pasta, S. Y.,
Ramakrishna, T., et al. (2005) AlphaB-crystallin, a small heat-shock
protein, prevents the amyloid fibril growth of an amyloid beta-peptide
and beta2-microglobulin, Biochem. J., 392, 573-581, doi:
10.1042/BJ20050339.
30.Shammas, S. L., Waudby, C. A., Wang, S., Buell,
A. K., Knowles, T. P., et al. (2011) Binding of the molecular chaperone
alphaB-crystallin to Abeta amyloid fibrils inhibits fibril elongation,
Biophys. J., 101, 1681-1689, doi:
10.1016/j.bpj.2011.07.056.
31.Narayan, P., Meehan, S., Carver, J. A., Wilson,
M. R., Dobson, C. M., et al. (2012) Amyloid-beta oligomers are
sequestered by both intracellular and extracellular chaperones,
Biochemistry, 51, 9270-9276, doi: 10.1021/bi301277k.
32.Haslbeck, M., Peschek, J., Buchner, J., and
Weinkauf, S. (2016) Structure and function of alpha-crystallins:
Traversing from in vitro to in vivo, Biochim. Biophys.
Acta, 1860, 149-166, doi: 10.1016/j. bbagen.2015.06.008.
33.Santhoshkumar, P., Murugesan, R., and Sharma, K.
K. (2009) Deletion of (54)FLRAPSWF(61) residues decreases the
oligomeric size and enhances the chaperone function of
alphaB-crystallin, Biochemistry, 48, 5066-5073, doi:
10.1021/bi900085v.
34.Pasta, S. Y., Raman, B., Ramakrishna, T., and
Rao, Ch. M. (2003) Role of the conserved SRLFDQFFG region of
alpha-crystallin, a small heat shock protein. Effect on oligomeric
size, subunit exchange, and chaperone-like activity, J. Biol.
Chem., 278, 51159-51166, doi: 10.1074/jbc.M307523200.
35.Jehle, S., Vollmar, B. S., Bardiaux, B., Dove, K.
K., Rajagopal, P., et al. (2011) N-terminal domain of alphaB-crystallin
provides a conformational switch for multimerization and structural
heterogeneity, Proc. Natl. Acad. Sci. USA, 108,
6409-6414, doi: 10.1073/pnas.1014656108.
36.Pasta, S. Y., Raman, B., Ramakrishna, T., and
Rao, Ch. M. (2002) Role of the C-terminal extensions of
alpha-crystallins. Swapping the C-terminal extension of
alpha-crystallin to alphaB-crystallin results in enhanced chaperone
activity, J. Biol. Chem., 277, 45821-45828, doi:
10.1074/jbc.M206499200.
37.Treweek, T. M., Ecroyd, H., Williams, D. M.,
Meehan, S., Carver, J. A., and Walker, M. J. (2007) Site-directed
mutations in the C-terminal extension of human alphaB-crystallin affect
chaperone function and block amyloid fibril formation, PLoS One,
2, e1046, doi: 10.1371/journal.pone.0001046.
38.Kumar, L. V., and Rao, C. M. (2000) Domain
swapping in human alpha A and alpha B crystallins affects
oligomerization and enhances chaperone-like activity, J. Biol.
Chem., 275, 22009-22013, doi: 10.1074/jbc.M003307200.
39.Peschek, J., Braun, N., Franzmann, T. M.,
Georgalis, Y., Haslbeck, M., et al. (2009) The eye lens chaperone
alpha-crystallin forms defined globular assemblies, Proc. Natl.
Acad. Sci. USA, 106, 13272-13277, doi:
10.1073/pnas.0902651106.
40.Kaiser, C. J. O., Peters, C., Schmid, P. W. N.,
Stavropoulou, M., Zou, J., et al. (2019) The structure and oxidation of
the eye lens chaperone alphaA-crystallin, Nat. Struct. Mol.
Biol., 26, 1141-1150, doi: 10.1038/s41594-019-0332-9.
41.McCarty, C. A., and Taylor, H. R. (2002) A review
of the epidemiologic evidence linking ultraviolet radiation and
cataracts, Dev. Ophthalmol., 35, 21-31, doi:
10.1159/000060807.
42.Kamei, A. (1990) Characterization of
water-insoluble proteins in normal and cataractous human lens, Jpn.
J. Ophthalmol., 34, 216-224.
43.Harrington, V., McCall, S., Huynh, S.,
Srivastava, K., and Srivastava, O. P. (2004) Crystallins in water
soluble-high molecular weight protein fractions and water insoluble
protein fractions in aging and cataractous human lenses, Mol.
Vis., 10, 476-489.
44.Sharma, K. K., and Santhoshkumar, P. (2009) Lens
aging: Effects of crystallins, Biochim. Biophys. Acta,
1790, 1095-1108, doi: 10.1016/j.bbagen.2009.05.008.
45.Wilmarth, P. A., Tanner, S., Dasari, S., Nagalla,
S. R., Riviere, M. A., et al. (2006) Age-related changes in human
crystallins determined from comparative analysis of post-translational
modifications in young and aged lens: Does deamidation contribute to
crystallin insolubility? J. Proteome Res., 5, 2554-2566,
doi: 10.1021/pr050473a.
46.Lapko, V. N., Smith, D. L., and Smith, J. B.
(2001) In vivo carbamylation and acetylation of water-soluble
human lens alphaB-crystallin lysine 92, Protein Sci., 10,
1130-1136, doi: 10.1110/ps.40901.
47.Taylor, H. R., West, S. K., Rosenthal, F. S.,
Munoz, B., Newland, H. S., et al. (1988) Effect of ultraviolet
radiation on cataract formation, N. Engl. J. Med., 319,
1429-1433, doi: 10.1056/NEJM198812013192201.
48.Korlimbinis, A., Hains, P. G., Truscott, R. J.,
and Aquilina, J. A. (2006) 3-Hydroxykynurenine oxidizes
alpha-crystallin: Potential role in cataractogenesis,
Biochemistry, 45, 1852-1860, doi: 10.1021/bi051737+.
49.Anbaraki, A., Ghahramani, M., Muranov, K. O.,
Kurganov, B. I., and Yousefi, R. (2018) Structural and functional
alteration of human alphaA-crystallin after exposure to full spectrum
solar radiation and preventive role of lens antioxidants, Int. J.
Biol. Macromol., 118, 1120-1130, doi:
10.1016/j.ijbiomac.2018.06.136.
50.Lin, S. Y., Ho, C. J., and Li, M. J. (1999)
UV-B-induced secondary conformational changes in lens alpha-crystallin,
J. Photochem. Photobiol. B, 49, 29-34, doi:
10.1016/S1011-1344(99)00010-X.
51.Rajan, S., Horn, C., and Abraham, E. C. (2006)
Effect of oxidation of alphaA- and alphaB-crystallins on their
structure, oligomerization and chaperone function, Mol. Cell.
Biochem., 288, 125-134, doi: 10.1007/s11010-006-9128-4.
52.Chaves, J. M., Srivastava, K., Gupta, R., and
Srivastava, O. P. (2008) Structural and functional roles of deamidation
and/or truncation of N- or C-termini in human alpha A-crystallin,
Biochemistry, 47, 10069-10083, doi:
10.1021/bi8001902.
53.Gupta, R., and Srivastava, O. P. (2004)
Deamidation affects structural and functional properties of human
alphaA-crystallin and its oligomerization with alphaB-crystallin, J.
Biol. Chem., 279, 44258-44269, doi:
10.1074/jbc.M405648200.
54.Gupta, R., and Srivastava, O. P. (2004) Effect of
deamidation of asparagine 146 on functional and structural properties
of human lens alphaB-crystallin, Invest. Ophthalmol. Vis. Sci.,
45, 206-214, doi: 10.1167/iovs.03-0720.
55.Grey, A. C., and Schey, K. L. (2009) Age-related
changes in the spatial distribution of human lens alpha-crystallin
products by MALDI imaging mass spectrometry, Invest. Ophthalmol.
Vis. Sci., 50, 4319-4329, doi: 10.1167/iovs.09-3522.
56.MacCoss, M. J., McDonald, W. H., Saraf, A.,
Sadygov, R., Clark, J. M., et al. (2002) Shotgun identification of
protein modifications from protein complexes and lens tissue, Proc.
Natl. Acad. Sci. USA, 99, 7900-7905, doi:
10.1073/pnas.122231399.
57.Huang, C. H., Wang, Y. T., Tsai, C. F., Chen, Y.
J., Lee, J. S., et al. (2011) Phosphoproteomics characterization of
novel phosphorylated sites of lens proteins from normal and cataractous
human eye lenses, Mol. Vis., 17, 186-198.
58.Bakthisaran, R., Akula, K. K., Tangirala, R., and
Rao, Ch. M. (2016) Phosphorylation of alphaB-crystallin: Role in
stress, aging and patho-physiological conditions, Biochim. Biophys.
Acta, 1860, 167-182, doi: 10.1016/j.bbagen.2015.09.017.
59.Muranova, L. K., Sudnitsyna, M. V., and Gusev, N.
B. (2018) alphaB-crystallin phosphorylation: advances and problems,
Biochemistry (Moscow), 83, 1196-1206, doi:
10.1134/S000629791810005X.
60.Carver, J. A., Nicholls, K. A., Aquilina, J. A.,
and Truscott, R. J. (1996) Age-related changes in bovine
alpha-crystallin and high-molecular-weight protein, Exp. Eye
Res., 63, 639-647, doi: 10.1006/exer.1996.0158.
61.Den Engelsman, J., Gerrits, D., de Jong, W. W.,
Robbins, J., Kato, K., et al. (2005) Nuclear import of
{alpha}B-crystallin is phosphorylation-dependent and hampered by
hyperphosphorylation of the myopathy-related mutant R120G, J. Biol.
Chem., 280, 37139-37148, doi: 10.1074/jbc.M504106200.
62.Simon, S., Fontaine, J. M., Martin, J. L., Sun,
X., Hoppe, A. D., et al. (2007) Myopathy-associated alphaB-crystallin
mutants: Abnormal phosphorylation, intracellular location, and
interactions with other small heat shock proteins, J. Biol.
Chem., 282, 34276-34287, doi: 10.1074/jbc.M703267200.
63.Lyon, Y. A., Sabbah, G. M., and Julian, R. R.
(2018) Differences in alpha-Crystallin isomerization reveal the
activity of protein isoaspartyl methyltransferase (PIMT) in the nucleus
and cortex of human lenses, Exp. Eye Res., 171, 131-141,
doi: 10.1016/j.exer.2018.03.018.
64.Takata, T., and Fujii, N. (2016) Isomerization of
Asp residues plays an important role in alphaA-crystallin dissociation,
FEBS J., 283, 850-859, doi: 10.1111/febs.13635.
65.Hooi, M. Y., Raftery, M. J., and Truscott, R. J.
(2013) Age-dependent racemization of serine residues in a human
chaperone protein, Protein Sci., 22, 93-100, doi:
10.1002/pro.2191.
66.Lyon, Y. A., Collier, M. P., Riggs, D. L.,
Degiacomi, M. T., Benesch, J. L. P., et al. (2019) Structural and
functional consequences of age-related isomerization in
alpha-crystallins, J. Biol. Chem., 294, 7546-7555, doi:
10.1074/jbc.RA118.007052.
67.Lyons, T. J., Silvestri, G., Dunn, J. A., Dyer,
D. G., and Baynes, J. W. (1991) Role of glycation in modification of
lens crystallins in diabetic and nondiabetic senile cataracts,
Diabetes, 40, 1010-1015, doi: 10.2337/diab.40.8.1010.
68.Thorpe, S. R., and Baynes, J. W. (1996) Role of
the Maillard reaction in diabetes mellitus and diseases of aging,
Drugs Aging, 9, 69-77, doi:
10.2165/00002512-199609020-00001.
69.Stevens, V. J., Rouzer, C. A., Monnier, V. M.,
and Cerami, A. (1978) Diabetic cataract formation: Potential role of
glycosylation of lens crystallins, Proc. Natl. Acad. Sci. USA,
75, 2918-2922, doi: 10.1073/pnas.75.6.2918.
70.Kumar, P. A., Kumar, M. S., and Reddy, G. B.
(2007) Effect of glycation on alpha-crystallin structure and
chaperone-like function, Biochem. J., 408, 251-258, doi:
10.1042/BJ20070989.
71.Schey, K. L., Wang, Z., Friedrich, M. G.,
Garland, D. L., and Truscott, R. J. W. (2020) Spatiotemporal changes in
the human lens proteome: Critical insights into long-lived proteins,
Prog. Retin. Eye Res., 76, 100802, doi:
10.1016/j.preteyeres.2019.100802.
72.Kamei, A., Iwase, H., and Masuda, K. (1997)
Cleavage of amino acid residue(s) from the N-terminal region of alpha
A- and alpha B-crystallins in human crystalline lens during aging,
Biochem. Biophys. Res. Commun., 231, 373-378, doi:
10.1006/bbrc.1997.6105.
73.Jimenez-Asensio, J., Colvis, C. M., Kowalak, J.
A., Duglas-Tabor, Y., Datiles, M. B., et al. (1999) An atypical form of
alphaB-crystallin is present in high concentration in some human
cataractous lenses. Identification and characterization of aberrant N-
and C-terminal processing, J. Biol. Chem., 274,
32287-32294, doi: 10.1074/jbc.274.45.32287.
74.Takemoto, L., Emmons, T., and Horwitz, J. (1993)
The C-terminal region of alpha-crystallin: involvement in protection
against heat-induced denaturation, Biochem. J., 294 (Pt.
2), 435-438, doi: 10.1042/bj2940435.
75.Santhoshkumar, P., Udupa, P., Murugesan, R., and
Sharma, K. K. (2008) Significance of interactions of low molecular
weight crystallin fragments in lens aging and cataract formation, J.
Biol. Chem., 283, 8477-8485, doi:
10.1074/jbc.M705876200.
76.Su, S. P., McArthur, J. D., and Andrew Aquilina,
J. (2010) Localization of low molecular weight crystallin peptides in
the aging human lens using a MALDI mass spectrometry imaging approach,
Exp. Eye Res., 91, 97-103, doi:
10.1016/j.exer.2010.04.010.
77.Kannan, R., Santhoshkumar, P., Mooney, B. P., and
Sharma, K. K. (2013) The alphaA66-80 peptide interacts with soluble
alpha-crystallin and induces its aggregation and precipitation: A
contribution to age-related cataract formation, Biochemistry,
52, 3638-3650, doi: 10.1021/bi301662w.
78.Raju, M., Santhoshkumar, P., and Sharma, K. K.
(2017) Lens endogenous peptide alphaA66-80 generates hydrogen peroxide
and induces cell apoptosis, Aging Dis., 8, 57-70, doi:
10.14336/AD.2016.0805.
79.Nagaraj, R. H., Nahomi, R. B., Shanthakumar, S.,
Linetsky, M., Padmanabha, S., et al. (2012) Acetylation of
alphaA-crystallin in the human lens: effects on structure and chaperone
function, Biochim. Biophys. Acta, 1822, 120-129, doi:
10.1016/j.bbadis.2011.11.011.
80.Wang, Z., Friedrich, M. G., Truscott, R. J. W.,
and Schey, K. L. (2019) Cleavage C-terminal to Asp leads to covalent
crosslinking of long-lived human proteins, Biochim. Biophys. Acta
Proteins Proteom., 1867, 831-839, doi:
10.1016/j.bbapap.2019.06.009.
81.Litt, M., Kramer, P., LaMorticella, D. M.,
Murphey, W., Lovrien, E. W., et al. (1998) Autosomal dominant
congenital cataract associated with a missense mutation in the human
alpha crystallin gene CRYAA, Hum. Mol. Genet., 7,
471-474, doi: 10.1093/hmg/7.3.471.
82.Pras, E., Frydman, M., Levy-Nissenbaum, E.,
Bakhan, T., Raz, J., et al. (2000) A nonsense mutation (W9X) in CRYAA
causes autosomal recessive cataract in an inbred Jewish Persian family,
Invest. Ophthalmol. Vis. Sci., 41, 3511-3515.
83.Hansen, L., Yao, W., Eiberg, H., Kjaer, K. W.,
Baggesen, K., et al. (2007) Genetic heterogeneity in
microcornea-cataract: Five novel mutations in CRYAA, CRYGD, and GJA8,
Invest. Ophthalmol. Vis. Sci., 48, 3937-3944, doi:
10.1167/iovs.07-0013.
84.Devi, R. R., Yao, W., Vijayalakshmi, P., Sergeev,
Y. V., Sundaresan, P., et al. (2008) Crystallin gene mutations in
Indian families with inherited pediatric cataract, Mol. Vis.,
14, 1157-1170.
85.Song, Z., Si, N., and Xiao, W. (2018) A novel
mutation in the CRYAA gene associated with congenital cataract and
microphthalmia in a Chinese family, BMC Med. Genet., 19,
190, doi: 10.1186/s12881-018-0695-5.
86.Graw, J., Klopp, N., Illig, T., Preising, M. N.,
and Lorenz, B. (2006) Congenital cataract and macular hypoplasia in
humans associated with a de novo mutation in CRYAA and compound
heterozygous mutations in P, Graefes Arch. Clin. Exp.
Ophthalmol., 244, 912-919, doi:
10.1007/s00417-005-0234-x.
87.Hansen, L., Mikkelsen, A., Nurnberg, P.,
Nurnberg, G., Anjum, I., et al. (2009) Comprehensive mutational
screening in a cohort of Danish families with hereditary congenital
cataract, Invest. Ophthalmol. Vis. Sci., 50, 3291-3303,
doi: 10.1167/iovs.08-3149.
88.Laurie, K. J., Dave, A., Straga, T., Souzeau, E.,
Chataway, T., et al. (2013) Identification of a novel oligomerization
disrupting mutation in CRYAlphaA associated with congenital cataract in
a South Australian family, Hum. Mutat., 34, 435-438, doi:
10.1002/humu.22260.
89.Mackay, D. S., Andley, U. P., and Shiels, A.
(2003) Cell death triggered by a novel mutation in the
alphaA-crystallin gene underlies autosomal dominant cataract linked to
chromosome 21q, Eur. J. Hum. Genet., 11, 784-793, doi:
10.1038/sj.ejhg.5201046.
90.Khan, A. O., Aldahmesh, M. A., and Meyer, B.
(2007) Recessive congenital total cataract with microcornea and
heterozygote carrier signs caused by a novel missense CRYAA mutation
(R54C), Am. J. Ophthalmol., 144, 949-952, doi:
10.1016/j.ajo.2007.08.005.
91.Su, D., Guo, Y., Li, Q., Guan, L., Zhu, S., et
al. (2012) A novel mutation in CRYAA is associated with autosomal
dominant suture cataracts in a Chinese family, Mol. Vis.,
18, 3057-3063.
92.Yang, Z., Su, D., Li, Q., Ma, Z., Yang, F., et
al. (2013) A R54L mutation of CRYAA associated with autosomal dominant
nuclear cataracts in a Chinese family, Curr. Eye Res.,
38, 1221-1228, doi: 10.3109/02713683.2013.811260.
93.Patel, R., Zenith, R. K., Chandra, A., and Ali,
A. (2017) Novel mutations in the crystallin gene in age-related
cataract patients from a North Indian population, Mol.
Syndromol., 8, 179-186, doi: 10.1159/000471992.
94.Bhagyalaxmi, S. G., Srinivas, P., Barton, K. A.,
Kumar, K. R., Vidyavathi, M., et al. (2009) A novel mutation (F71L) in
alphaA-crystallin with defective chaperone-like function associated
with age-related cataract, Biochim. Biophys. Acta, 1792,
974-981, doi: 10.1016/j.bbadis.2009.06.011.
95.Santhiya, S. T., Soker, T., Klopp, N., Illig, T.,
Prakash, M. V., et al. (2006) Identification of a novel, putative
cataract-causing allele in CRYAA (G98R) in an Indian family, Mol.
Vis., 12, 768-773.
96.Vanita, V., Singh, J. R., Hejtmancik, J. F.,
Nuernberg, P., Hennies, H. C., et al. (2006) A novel fan-shaped
cataract-microcornea syndrome caused by a mutation of CRYAA in an
Indian family, Mol. Vis., 12, 518-522.
97.Li, F. F., Yang, M., Ma, X., Zhang, Q., Zhang,
M., et al. (2010) Autosomal dominant congenital nuclear cataracts
caused by a CRYAA gene mutation, Curr. Eye Res., 35,
492-498, doi: 10.3109/02713681003624901.
98.Gu, F., Luo, W., Li, X., Wang, Z., Lu, S., et al.
(2008) A novel mutation in AlphaA-crystallin (CRYAA) caused autosomal
dominant congenital cataract in a large Chinese family, Hum.
Mutat., 29, 769, doi: 10.1002/humu.20724.
99.Richter, L., Flodman, P., Barria
von-Bischhoffshausen, F., Burch, D., Brown, S., et al. (2008) Clinical
variability of autosomal dominant cataract, microcornea and corneal
opacity and novel mutation in the alpha A crystallin gene (CRYAA),
Am. J. Med. Genet. A, 146A, 833-842, doi:
10.1002/ajmg.a.32236.
100.Li, L., Fan, D. B., Zhao, Y. T., Li, Y., Kong,
D. Q., et al. (2017) Two novel mutations identified in ADCC families
impair crystallin protein distribution and induce apoptosis in human
lens epithelial cells, Sci. Rep., 7, 17848, doi:
10.1038/s41598-017-18222-z.
101.Sun, W., Xiao, X., Li, S., Guo, X., and Zhang,
Q. (2011) Mutation analysis of 12 genes in Chinese families with
congenital cataracts, Mol. Vis., 17, 2197-2206.
102.Kong, X. D., Liu, N., Shi, H. R., Dong, J. M.,
Zhao, Z. H., et al. (2015) A novel 3-base pair deletion of the CRYAA
gene identified in a large Chinese pedigree featuring autosomal
dominant congenital perinuclear cataract, Genet. Mol. Res.,
14, 426-432, doi: 10.4238/2015.
103.Berry, V., Ionides, A., Pontikos, N., Georgiou,
M., Yu, J., et al. (2020) The genetic landscape of crystallins in
congenital cataract, Orphanet. J. Rare Dis., 15, 333,
doi: 10.1186/s13023-020-01613-3.
104.Chen, Q., Ma, J., Yan, M., Mothobi, M. E., Liu,
Y., et al. (2009) A novel mutation in CRYAB associated with autosomal
dominant congenital nuclear cataract in a Chinese family, Mol.
Vis., 15, 1359-1365.
105.Jiao, X., Khan, S. Y., Irum, B., Khan, A. O.,
Wang, Q., et al. (2015) Missense mutations in CRYAB are liable for
recessive congenital cataracts, PLoS One, 10, e0137973,
doi: 10.1371/journal.pone.0137973.
106.Liu, M., Ke, T., Wang, Z., Yang, Q., Chang, W.,
et al. (2006) Identification of a CRYAB mutation associated with
autosomal dominant posterior polar cataract in a Chinese family,
Invest. Ophthalmol. Vis. Sci., 47, 3461-3466, doi:
10.1167/iovs.05-1438.
107.Xia, X. Y., Wu, Q. Y., An, L. M., Li, W. W.,
Li, N., et al. (2014) A novel P20R mutation in the alpha-B crystallin
gene causes autosomal dominant congenital posterior polar cataracts in
a Chinese family, BMC Ophthalmol., 14, 108, doi:
10.1186/1471-2415-14-108.
108.Del Bigio, M. R., Chudley, A. E., Sarnat, H.
B., Campbell, C., Goobie, S., et al. (2011) Infantile muscular
dystrophy in Canadian aboriginals is an alphaB-crystallinopathy,
Ann. Neurol., 69, 866-871, doi: 10.1002/ana.22331.
109.Khan, A. O., Abu Safieh, L., and Alkuraya, F.
S. (2010) Later retinal degeneration following childhood surgical
aphakia in a family with recessive CRYAB mutation (p.R56W),
Ophthalmic Genet., 31, 30-36, doi:
10.3109/13816810903452047.
110.Fichna, J. P., Potulska-Chromik, A., Miszta,
P., Redowicz, M. J., Kaminska, A. M., et al. (2017) A novel dominant
D109A CRYAB mutation in a family with myofibrillar myopathy affects
alphaB-crystallin structure, BBA Clin., 7, 1-7, doi:
10.1016/j.bbacli.2016.11.004.
111.Brodehl, A., Gaertner-Rommel, A., Klauke, B.,
Grewe, S. A., Schirmer, I., et al. (2017) The novel alphaB-crystallin
(CRYAB) mutation p.D109G causes restrictive cardiomyopathy, Hum.
Mutat., 38, 947-952, doi: 10.1002/humu.23248.
112.Sacconi, S., Feasson, L., Antoine, J. C.,
Pecheux, C., Bernard, R., et al. (2012) A novel CRYAB mutation
resulting in multisystemic disease, Neuromuscul. Disord.,
22, 66-72, doi: 10.1016/j.nmd.2011.07.004.
113.Vicart, P., Caron, A., Guicheney, P., Li, Z.,
Prevost, M. C., et al. (1998) A missense mutation in the
alphaB-crystallin chaperone gene causes a desmin-related myopathy,
Nat. Genet., 20, 92-95, doi: 10.1038/1765.
114.Liu, Y., Zhang, X., Luo, L., Wu, M., Zeng, R.,
et al. (2006) A novel alphaB-crystallin mutation associated with
autosomal dominant congenital lamellar cataract, Invest. Ophthalmol.
Vis. Sci., 47, 1069-1075, doi: 10.1167/iovs.05-1004.
115.Selcen, D., and Engel, A. G. (2003)
Myofibrillar myopathy caused by novel dominant negative alpha
B-crystallin mutations, Ann. Neurol., 54, 804-810, doi:
10.1002/ana.10767.
116.Reilich, P., Schoser, B., Schramm, N., Krause,
S., Schessl, J., et al. (2010) The p. G154S mutation of the alpha-B
crystallin gene (CRYAB) causes late-onset distal myopathy,
Neuromuscul. Disord., 20, 255-259, doi:
10.1016/j.nmd.2010.01.012.
117.Inagaki, N., Hayashi, T., Arimura, T., Koga,
Y., Takahashi, M., et al. (2006) Alpha B-crystallin mutation in dilated
cardiomyopathy, Biochem. Biophys. Res. Commun., 342,
379-386, doi: 10.1016/j.bbrc.2006.01.154.
118.Berry, V., Francis, P., Reddy, M. A., Collyer,
D., Vithana, E., et al. (2001) Alpha-B crystallin gene (CRYAB) mutation
causes dominant congenital posterior polar cataract in humans, Am.
J. Hum. Genet., 69, 1141-1145, doi: 10.1086/324158.
119.Forrest, K. M., Al-Sarraj, S., Sewry, C., Buk,
S., Tan, S. V., et al. (2011) Infantile onset myofibrillar myopathy due
to recessive CRYAB mutations, Neuromuscul. Disord., 21,
37-40, doi: 10.1016/j.nmd.2010.11.003.
120.Marcos, A. T., Amoros, D., Munoz-Cabello, B.,
Galan, F., Rivas Infante, E., et al. (2020) A novel dominant mutation
in CRYAB gene leading to a severe phenotype with childhood onset,
Mol. Genet. Genomic Med., 8, e1290, doi:
10.1002/mgg3.1290.
121.Van der Smagt, J. J., Vink, A., Kirkels, J. H.,
Nelen, M., ter Heide, H., et al. (2014) Congenital posterior pole
cataract and adult onset dilating cardiomyopathy: Expanding the
phenotype of alphaB-crystallinopathies, Clin. Genet., 85,
381-385, doi: 10.1111/cge.12169.
122.Yu, Y., Xu, J., Qiao, Y., Li, J., and Yao, K.
(2021) A new heterozygous mutation in the stop codon of CRYAB (p.
X176Y) is liable for congenital posterior pole cataract in a Chinese
family, Ophthalmic Genet., 42, 139-143, doi:
10.1080/13816810.2020.1855665.
123.Muranova, L. K., Strelkov, S. V., and Gusev, N.
B. (2020) Effect of cataract-associated mutations in the N-terminal
domain of alphaB-crystallin (HspB5), Exp. Eye Res., 197,
108091, doi: 10.1016/j.exer.2020.108091.
124.Cobb, B. A., and Petrash, J. M. (2000)
Structural and functional changes in the alpha A-crystallin R116C
mutant in hereditary cataracts, Biochemistry, 39,
15791-15798, doi: 10.1021/bi001453j.
125.Kumar, L. V., Ramakrishna, T., and Rao, C. M.
(1999) Structural and functional consequences of the mutation of a
conserved arginine residue in alphaA and alphaB crystallins, J.
Biol. Chem., 274, 24137-24141, doi:
10.1074/jbc.274.34.24137.
126.Singh, D., Raman, B., Ramakrishna, T., and Rao,
Ch. M. (2006) The cataract-causing mutation G98R in human
alphaA-crystallin leads to folding defects and loss of chaperone
activity, Mol. Vis., 12, 1372-1379.
127.Bova, M. P., Yaron, O., Huang, Q., Ding, L.,
Haley, D. A., et al. (1999) Mutation R120G in alphaB-crystallin, which
is linked to a desmin-related myopathy, results in an irregular
structure and defective chaperone-like function, Proc. Natl. Acad.
Sci. USA, 96, 6137-6142, doi: 10.1073/pnas.96.11.6137.
128.Perng, M. D., Wen, S. F., van IJssel, P.,
Prescott, A. R., and Quinlan, R. A. (2004) Desmin aggregate formation
by R120G alphaB-crystallin is caused by altered filament interactions
and is dependent upon network status in cells, Mol. Biol. Cell,
15, 2335-2346, doi: 10.1091/mbc.e03-12-0893.
129.Ghahramani, M., Yousefi, R., Krivandin, A.,
Muranov, K., Kurganov, B., et al. (2020) Structural and functional
characterization of D109H and R69C mutant versions of human
alphaB-crystallin: The biochemical pathomechanism underlying cataract
and myopathy development, Int. J. Biol. Macromol., 146,
1142-1160, doi: 10.1016/j.ijbiomac.2019.09.239.
130.Clark, A. R., Lubsen, N. H., and Slingsby, C.
(2012) sHSP in the eye lens: Crystallin mutations, cataract and
proteostasis, Int. J. Biochem. Cell Biol., 44, 1687-1697,
doi: 10.1016/j.biocel.2012.02.015.
131.Gerasimovich, E. S., Strelkov, S. V., and
Gusev, N. B. (2017) Some properties of three alphaB-crystallin mutants
carrying point substitutions in the C-terminal domain and associated
with congenital diseases, Biochimie, 142, 168-178, doi:
10.1016/j.biochi.2017.09.008.
132.Raju, I., and Abraham, E. C. (2011) Congenital
cataract causing mutants of alphaA-crystallin/sHSP form aggregates and
aggresomes degraded through ubiquitin–proteasome pathway, PLoS
One, 6, e28085, doi: 10.1371/journal.pone.0028085.
133.Ahsan, S. M., Bakthisaran, R., Tangirala, R.,
and Rao, C. M. (2021) Nucleosomal association and altered interactome
underlie the mechanism of cataract caused by the R54C mutation of
alphaA-crystallin, Biochim. Biophys. Acta Gen. Subj.,
1865, 129846, doi: 10.1016/j.bbagen.2021.129846.
134.Gong, B., Zhang, L. Y., Pang, C. P., Lam, D.
S., and Yam, G. H. (2009) Trimethylamine N-oxide alleviates the severe
aggregation and ER stress caused by G98R alphaA-crystallin, Mol.
Vis., 15, 2829-2840.
135.Raju, I., and Abraham, E. C. (2013) Mutants of
human alphaB-crystallin cause enhanced protein aggregation and
apoptosis in mammalian cells: Influence of co-expression of HspB1,
Biochem. Biophys. Res. Commun., 430, 107-112, doi:
10.1016/j.bbrc.2012.11.051.
136.Chavez Zobel, A. T., Loranger, A., Marceau, N.,
Theriault, J. R., Lambert, H., et al. (2003) Distinct chaperone
mechanisms can delay the formation of aggresomes by the
myopathy-causing R120G alphaB-crystallin mutant, Hum. Mol.
Genet., 12, 1609-1620, doi: 10.1093/hmg/ddg173.
137.Sanbe, A., Osinska, H., Saffitz, J. E., Glabe,
C. G., Kayed, R., et al. (2004) Desmin-related cardiomyopathy in
transgenic mice: a cardiac amyloidosis, Proc. Natl. Acad. Sci.
USA, 101, 10132-10136, doi: 10.1073/pnas.0401900101.
138.Hayes, V. H., Devlin, G., and Quinlan, R. A.
(2008) Truncation of alphaB-crystallin by the myopathy-causing Q151X
mutation significantly destabilizes the protein leading to aggregate
formation in transfected cells, J. Biol. Chem., 283,
10500-10512, doi: 10.1074/jbc.M706453200.
139.Mitzelfelt, K. A., Limphong, P., Choi, M. J.,
Kondrat, F. D., Lai, S., et al. (2016) The human 343delT HSPB5
chaperone associated with early-onset skeletal myopathy causes defects
in protein solubility, J. Biol. Chem., 291, 14939-14953,
doi: 10.1074/jbc.M116.730481.
140.Zhang, H., Rajasekaran, N. S., Orosz, A., Xiao,
X., Rechsteiner, M., et al. (2010) Selective degradation of
aggregate-prone CryAB mutants by HSPB1 is mediated by
ubiquitin-proteasome pathways, J. Mol. Cell. Cardiol.,
49, 918-930, doi: 10.1016/j.yjmcc.2010.09.004.
141.Andley, U. P., Hamilton, P. D., and Ravi, N.
(2008) Mechanism of insolubilization by a single-point mutation in
alphaA-crystallin linked with hereditary human cataracts,
Biochemistry, 47, 9697-9706, doi: 10.1021/bi800594t.
142.Kore, R., Hedges, R. A., Oonthonpan, L.,
Santhoshkumar, P., Sharma, K. K., et al. (2012) Quaternary structural
parameters of the congenital cataract causing mutants of
alphaA-crystallin, Mol. Cell Biochem., 362, 93-102, doi:
10.1007/s11010-011-1131-8.
143.Khoshaman, K., Yousefi, R., Tamaddon, A. M.,
Abolmaali, S. S., Oryan, A., et al. (2017) The impact of different
mutations at Arg54 on structure, chaperone-like activity and
oligomerization state of human alphaA-crystallin: The pathomechanism
underlying congenital cataract-causing mutations R54L, R54P and R54C,
Biochim. Biophys. Acta Proteins Proteom., 1865, 604-618,
doi: 10.1016/j.bbapap.2017.02.003.
144.Murugesan, R., Santhoshkumar, P., and Sharma,
K. K. (2007) Cataract-causing alphaAG98R mutant shows
substrate-dependent chaperone activity, Mol. Vis., 13,
2301-2309.
145.Singh, D., Tangirala, R., Bakthisaran, R., and
Chintalagiri, M. R. (2009) Synergistic effects of metal ion and the
pre-senile cataract-causing G98R alphaA-crystallin: Self-aggregation
propensities and chaperone activity, Mol. Vis., 15,
2050-2060.
146.Wang, X., Osinska, H., Klevitsky, R., Gerdes,
A. M., Nieman, M., et al. (2001) Expression of R120G-alphaB-crystallin
causes aberrant desmin and alphaB-crystallin aggregation and
cardiomyopathy in mice, Circ. Res., 89, 84-91, doi:
10.1161/hh1301.092688.
147.Meehan, S., Berry, Y., Luisi, B., Dobson, C.
M., Carver, J. A., et al. (2004) Amyloid fibril formation by lens
crystallin proteins and its implications for cataract formation, J.
Biol. Chem., 279, 3413-3419, doi:
10.1074/jbc.M308203200.
148.Lee, C. F. (2009) Self-assembly of protein
amyloids: A competition between amorphous and ordered aggregation,
Phys. Rev. E Stat. Nonlin. Soft Matter Phys., 80, 031922,
doi: 10.1103/PhysRevE.80.031922.
149.Sandilands, A., Hutcheson, A. M., Long, H. A.,
Prescott, A. R., Vrensen, G., et al. (2002) Altered aggregation
properties of mutant gamma-crystallins cause inherited cataract,
EMBO J., 21, 6005-6014, doi: 10.1093/emboj/cdf609.
150.Meehan, S., Knowles, T. P., Baldwin, A. J.,
Smith, J. F., Squires, A. M., et al. (2007) Characterisation of amyloid
fibril formation by small heat-shock chaperone proteins human alphaA-,
alphaB- and R120G alphaB-crystallins, J. Mol. Biol., 372,
470-484, doi: 10.1016/j.jmb.2007.06.060.
151.Makley, L. N., McMenimen, K. A., DeVree, B. T.,
Goldman, J. W., McGlasson, B. N., et al. (2015) Pharmacological
chaperone for alpha-crystallin partially restores transparency in
cataract models, Science, 350, 674-677, doi:
10.1126/science.aac9145.
152.Zhao, L., Chen, X. J., Zhu, J., Xi, Y. B.,
Yang, X., et al. (2015) Lanosterol reverses protein aggregation in
cataracts, Nature, 523, 607-611, doi:
10.1038/nature14650.
153.Raju, M., Santhoshkumar, P., and Sharma, K.
(2016) Alpha-crystallin-derived peptides as therapeutic chaperones,
Biochim. Biophys. Acta, 1860, 246-251, doi:
10.1016/j.bbagen.2015.06.010.