Modeling the Structure of Human tRNA-Guanine Transglycosylase in Complex with 7-Methylguanine and Revealing the Factors that Determine the Enzyme Interaction with Inhibitors
Sergey V. Pushkarev1, Valeriia A. Vinnik2, Irina V. Shapovalova1, Vytas K. Švedas1,3, and Dmitry K. Nilov3,a*
1Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119991 Moscow, Russia2Faculty of Fundamental Medicine, Lomonosov Moscow State University, 119991 Moscow, Russia
3Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received February 21, 2022; Revised March 22, 2022; Accepted March 23, 2022
tRNA-guanine transglycosylase, an enzyme catalyzing replacement of guanine with queuine in human tRNA and participating in the translation mechanism, is involved in the development of cancer. However, information on the small-molecule inhibitors that can suppress activity of this enzyme is very limited. Molecular dynamics simulations were used to determine the amino acid residues that provide efficient binding of inhibitors in the active site of tRNA-guanine transglycosylase. It was demonstrated using 7-methylguanine molecule as a probe that the ability of the inhibitor to adopt a charged state in the environment of hydrogen bond acceptors Asp105 and Asp159 plays a key role in complex formation. Formation of the hydrogen bonds and hydrophobic contacts with Gln202, Gly229, Phe109, and Met259 residues are also important. It has been predicted that introduction of the substituents would have a different effect on the ability to inhibit tRNA-guanine transglycosylase, as well as the DNA repair protein poly(ADP-ribose) polymerase 1, which can contribute to the development of more efficient and selective compounds.
KEY WORDS: tRNA-guanine transglycosylase, queuine, inhibitor, 7-methylguanine, molecular dynamics, dockingDOI: 10.1134/S0006297922050054
Abbreviations: 7-MG, 7-methylguanine; MD, molecular dynamics; PARP-1, poly(ADP-ribose) polymerase 1; TGT, tRNA-guanine transglycosylase.
INTRODUCTION
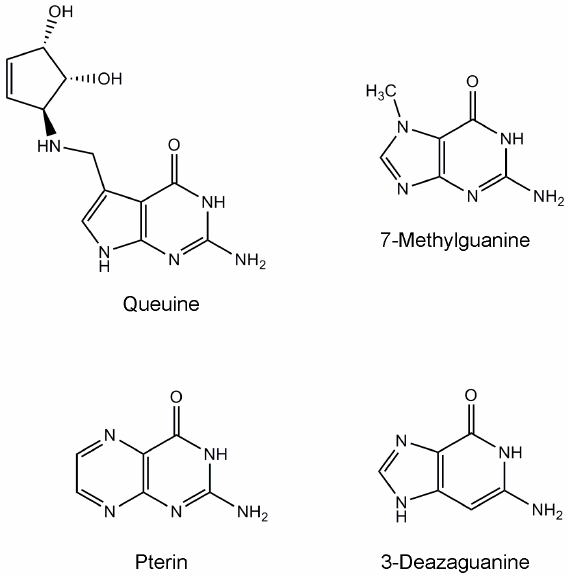
tRNA-guanine transglycosylase (TGT) catalyzes the replacement of guanine with queuine (a derivative of 7-deazaguanine, Fig. 1) in tRNAAsn, tRNAAsp, tRNAHis, and tRNATyr at position 34 (the first nucleotide of the anticodon) [1-4]. This modification presumably influences the efficiency of tRNA aminoacylation, codon–anticodon recognition, and translational speed and fidelity [5-7]. The eukaryotic TGT protein is a heterodimer consisting of the catalytic QTRT1 subunit and noncatalytic QTRT2 subunit [8]. Structure of the QTRT1 in complex with the queuine substrate was determined using X-ray crystallography [4]. According to the kinetic scheme proposed for the eukaryotic TGT enzyme, the queuine molecule binds first, tRNA binds second, and then guanine is cleaved with concomitant formation of an intermediate in which the tRNA is covalently bound to the active site residue Asp279. Further transformation of the intermediate leads to formation of the final reaction product, queuine-modified tRNA [9].
Fig. 1. Chemical structures of the substrate and known TGT inhibitors.
Recent experiments have revealed an important role of TGT in the development of breast cancer [10, 11]. It was shown that TGT deficiency significantly reduced proliferation and migration of the cancer cells in vitro (MCF7 and MDA-MB-231 lines) and in vivo (nude BALB/c mice with the corresponding transplanted tumors). In particular, knockout/knockdown of the TGT gene in cancer cells reduced the number and volume of tumors in the xenograft model. It should also be mentioned that enhanced expression of TGT was found in lung cancer patients, and the expression level correlated with survival [12, 13]. Involvement of the human TGT in pathological processes indicates the need for the development and testing of selective inhibitors of this protein.
Inhibition of human TGT by low-molecular-weight compounds has been poorly investigated. The main source of information about potential inhibitors is publication devoted to testing structural analogues of the substrate against TGT from rabbit reticulocytes [14]. 7-Methylguanine (7-MG, natural nitrogenous base) was found to be one of the most effective inhibitors, competitive with respect to queuine (Fig. 1, Ki = 1 µM) [14, 15]. Some other studies have shown that 7-MG is also able to suppress the activity of bacterial TGT [16, 17]. Alignment of TGT sequences from different organisms, revealing conserved active site residues, is shown in Online Resource 1, Fig. S1. Interestingly, 7-MG suppresses activity of the human poly(ADP-ribose) polymerase 1 (PARP-1, Ki = 60 µM) [18-21] which plays an important role in pathologies of cardiovascular, nervous, and respiratory systems, as well as in cancer diseases [22-25]. A more selective effect on the target may be required for clinical use of TGT inhibitors, and therefore, structural optimization of 7-MG and its analogues is of great interest.
In the present work, interaction of the human TGT with the 7-MG inhibitor was modeled, and recommendations were given for further development of more effective and selective inhibitors.
MATERIALS AND METHODS
For molecular mechanical description of 7-MG in a cationic state, parameters of the Amber-compatible field ff99 [26] were used with exception of partial charges. Assignment of partial atomic charges was carried out by the RESP method (see in Online Resource 1, Fig. S2) [27, 28]. For this purpose, molecular electrostatic potential was preliminarily calculated at the HF/6-31G* level of theory using GAMESS [29]. Parameters of the uncharged 7-MG molecule were obtained in a similar way in our previous work [18]. Amino acid residues of the protein were described by the ff14SB force field with improved torsion parameters [30].
Molecular model of the human TGT was constructed based on the crystal structure of an enzyme–substrate complex containing a queuine molecule (PDB ID 6h45, chain A) [4]. Coordinates of the Arg68-His93 and Asn101-Asp105 regions were transferred from the 7nq4 structure [31]. Coordinates of the 7-MG inhibitor in the active site were obtained by superimposing the fused rings of 7-MG and queuine. Next, the structure of the enzyme-inhibitor complex was optimized and studied by molecular dynamics (MD) method using AmberTools 20 and Amber 20 [32-34]. Hydrogen atoms were added taking into account ionization properties of the amino acid residues; when protonating the histidine side chains, their molecular environment was considered. The structure was placed in a truncated octahedral TIP3P water box (minimum distance from the protein to the box edge was 12 Å); Cl– ions were added to neutralize the total charge.
At the first stage of energy minimization of the obtained system (2500 steepest descent steps + 2500 conjugate gradient steps), the protein and 7-MG coordinates were kept fixed by positional restraints of 2 kcal/(mol⋅Å2) on heavy atoms. The second stage of minimization (5000 steepest descent steps + 5000 conjugate gradient steps) was performed without any restraints. The system was then heated up from 0 to 300 K using positional restraints of 1 kcal/(mol⋅Å2) on the protein and 7-MG atoms (250 ps, constant volume) and equilibrated at 300 K (25 ns, constant pressure). Acquisition of the equilibrium conformation of the complex was confirmed by analyzing the root-mean-square deviation of the protein backbone atoms from the initial position (mobile loops and terminal residues of the protein were not taken into account). A 100 ns trajectory of the equilibrium MD simulation was then calculated and analyzed. More detailed simulation protocols are given in Online Resource 2, Table S1. The calculations were carried out on the supercomputer “Lomonosov-2” using graphics processing units (Nvidia Tesla K40M).
Docking of the 7-MG molecule into the TGT active site was carried out using the Lead Finder 1708 program in the “extra precision” mode [35, 36]; an energy grid map covered the binding site of the queuine molecule. The structures were visualized using the VMD 1.9 program [37].
RESULTS AND DISCUSSION
The human TGT model was constructed on the basis of the 6h45 crystal structure that describes interaction of the QTRT1 catalytic subunit with the queuine substrate. In general, this complex with a resolution of 2.4 Å was appropriate for preparing the starting model, however, it was decided to transfer some parts of the protein chain from the 7nq4 structure obtained by the same authors. The first region, Arg68-His93, is less ordered in the 6h45 structure compared to other TGT structures, which may be related to crystallization conditions. Through a network of hydrogen bonds, the Arg68-His93 residues affect conformation of another region, Asn101-Asp105, which was also transferred from the 7nq4 structure.
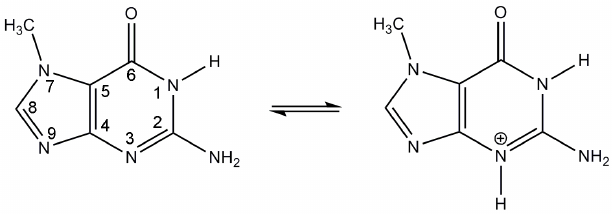
In modeling of the 7-MG binding to the TGT active site ionization properties of this inhibitor were taken into account. Previously, interaction of the structurally similar compounds with bacterial TGT was studied using isothermal calorimetry, site-directed mutagenesis, and X-ray crystallography. It was suggested that the presence of two negatively charged aspartate residues, Asp105 and Asp159 (human TGT numbering), promotes transition of the inhibitors to cationic form upon binding in the active site [38, 39]. The corresponding transition of 7-MG (protonation on the N3 atom) is shown in Fig. 2. The positively charged inhibitor is able to interact more efficiently with Asp105 and Asp159, so it was decided to model 7-MG in both cationic form and neutral form (as a control).
Fig. 2. Proposed transition of 7-MG to cationic form upon binding to the TGT active site.
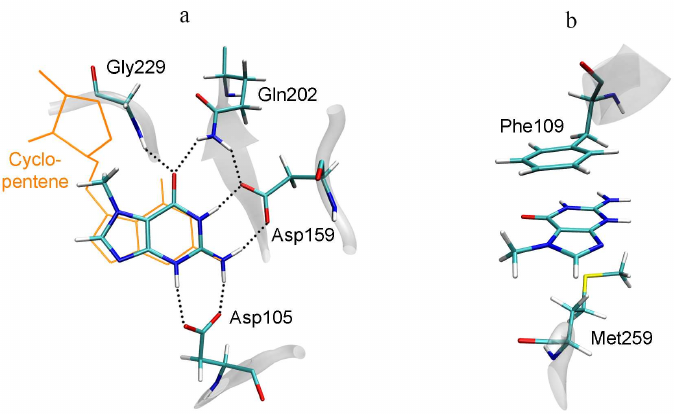
7-MG is a competitive inhibitor of TGT and a structural analogue of the queuine substrate, so it is expected to bind in a similar manner. The starting position of 7-MG in the active site of the TGT model corresponded to coordinates of the 7-deazaguanine fragment of queuine. The enzyme–inhibitor complex was then equilibrated and subjected to a 100 ns MD simulation to generate a trajectory, the analysis of which made it possible to characterize intermolecular interactions (Table, Fig. 3). First of all, formation of the hydrogen bonds of 7-MG with Asp159 and Asp105 should be mentioned, since interaction of the delocalized positive charge of the inhibitor with the negatively charged carboxyl groups could make a significant contribution to the binding energy. In that case one of the bonds with Asp105 is mediated by the protonated nitrogen atom N3. Additional hydrogen bonds are formed with the Gln202 and Gly229 residues. Purine rings of 7-MG form π-stacking with Phe109, as well as hydrophobic contact with the side chain of Met259. The methyl group of 7-MG is in contact with the benzene ring of Phe109.
Interactions of the cationic form of 7-MG in the active site of human
TGT revealed by a 100 ns MD simulation

Note. Mean distances are presented with standard deviations.
* Geometric center of 7-MG fused rings.
** Geometric center of the Phe109 benzene ring.
Fig. 3. Position of 7-MG in the active site of human TGT revealed by MD modeling. a) Interactions with polar residues; b) interactions with hydrophobic residues. The queuine molecule, transferred from the 6h45 structure following the Cα-atom superimposition, is shown in orange.
Modeling of the TGT complex formation with the neutral form of 7-MG showed a significant decrease in the stability of interactions with the residues of the active site compared to its cationic form. Hydrogen bonds with the Asp159 and Gly229 residues were episodic, which affected the average distance between atoms, and bonds with Asp105 and Gln202 were actually lost (see Online Resource 2, Table S2). The side chain of Asp105 was found to be turned away from the inhibitor molecule (Online Resource 1, Fig. S3 and S4), which is apparently due to the absence of a proton on the N3 atom. It is noteworthy that the related compound 3-deazaguanine, in which the N3 atom is replaced by carbon (Fig. 1), has an inhibitory activity two orders of magnitude lower compared to 7-MG [14]. Thus, we can conclude that the effective binding of 7-MG molecule in the active site of TGT is possible due to its transition to a cationic form.
Since 7-MG also inhibits the DNA repair protein PARP-1, which is not similar in sequence to TGT, it is necessary to reveal structural factors that determine the possibility of its binding to two different targets, as well as possible ways of inhibitor modifications for a more selective action. Previously, it was experimentally shown that the important factor for effective binding of purine derivatives and analogues with TGT and PARP-1 is the presence of amino group at the position 2 and oxo group at the position 6 [14, 18]. In the case of TGT, the amino group forms the above-mentioned hydrogen bonds with Asp105 and Asp159 (Fig. 3), and in the case of PARP-1, it interacts with the key residue Gly863 (Fig. 4). The oxo group forms hydrogen bonds with the Gln202 and Gly229 residues of TGT and with the Gly863 and Ser904 residues of PARP-1. Special attention should be paid to high similarity of the π-stacking interaction: in the case of TGT, it is mediated by the Phe109 side chain, and in the case of PARP-1, by the Tyr907 side chain. Taken together, these interactions facilitate binding of the inhibitors to both TGT and PARP-1.
Fig. 4. Position of 7-MG in the active site of human PARP-1 revealed in previous work [19]. a) Interactions with polar residues; b) interactions with hydrophobic residues. NAD+ molecule, transferred from the 6bhv structure following the Cα-atom superimposition, is shown in orange.
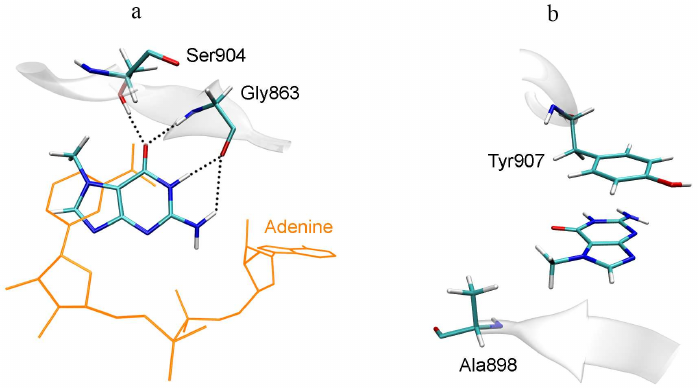
A possible way to optimize the structure of 7-MG and its analogues is to modify amino group at the position 2 or methyl group at the position 7. The first option is suitable for the PARP-1 inhibitors, since the introduced substituent is oriented towards the additional binding site of the NAD+ adenine group (Fig. 4a). In the case of TGT, the amino group forms hydrogen bonds with Asp105 and Asp159, therefore, any modification of this group will disrupt these interactions [40]. The second option (modification of the methyl group), on the contrary, is not suitable for PARP-1, since the corresponding binding pocket formed with participation of the Ala898 residue (Fig. 4b) is very limited in size. In the case of TGT, the 7-MG methyl group is oriented towards the binding site of cyclopentene moiety of the queuine substrate formed by the residues Val161-Ser164 and Gly229-Gly232. This site is able to interact with various groups of atoms, which is confirmed by the broad substrate specificity of human TGT, demonstrated using synthetic derivatives of 7-deazaguanine [41].
The molecular model of human TGT enables searching for inhibitors by docking and virtual screening approaches. The most promising compounds can be selected from a large library by evaluating binding energies and analyzing interactions with the crucial amino acid residues [42-44]. As an illustrative example, we performed docking of the 7-MG molecule against the ensemble of TGT structures obtained by MD simulation. For this purpose, structures were extracted at 10 ns intervals over the 100 ns trajectory of the equilibrium TGT simulation, and water and inhibitor molecules were removed from them. Next, 7-MG was docked into the region covering the binding site of the queuine substrate, and thus the ensemble of enzyme–inhibitor complexes was obtained (Online Resource 1, Fig. S5). Analysis of the obtained models showed that the docking well reproduced interactions of 7-MG with the amino acid residues of TGT described above. The ensemble average value of the binding free energy ∆Gcalc was –7.4 kcal/mol. According to the formula ∆G = RT ln(Ki), this energy value corresponds to the inhibition constant of 4 µM, which is in good agreement with the experimental data (Ki = 1 µM) [14].
The conducted study made it possible to draw some conclusions that are important for designing inhibitors of human TGT, a promising target for the treatment of oncological diseases. (i) The inhibitor may adopt a charged state upon binding in the active site in the environment of Asp105 and Asp159, which leads to the formation of more stable hydrogen bonds with these residues. This feature is important for membrane transport, as inhibitor molecule becomes positively charged only upon binding to the target protein inside the cell. (ii) The possibility of hydrogen bonding and hydrophobic contacts with Gln202, Gly229, Phe109, and Met259 residues should also be taken into account in the inhibitor design. (iii) A more selective interaction of the inhibitor with TGT could be achieved by elongation of the structure towards the binding site of the substrate’s cyclopentene group formed by the Val161-Ser164 and Gly229-Gly232 residues.
Open access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Funding. This study was supported by the Russian Science Foundation (project no. 19-74-10072).
Acknowledgments. The research was carried out using the equipment of the shared research facilities of HPC computing resources at the Lomonosov Moscow State University.
Ethics declarations. The authors declare no conflicts of interest in financial or any other sphere. This article does not contain description of studies involving animals or human participants performed by any of the authors.
Electronic supplementary material. The online version contains supplementary material available at https://doi.org/10.1134/S0006297922050054.
REFERENCES
1.Nishimura, S. (1983) Structure, biosynthesis, and
function of queuosine in transfer RNA, Prog. Nucleic Acid Res. Mol.
Biol., 28, 49-73.
2.Morris, R. C., and Elliott, M. S. (2001) Queuosine
modification of tRNA: A case for convergent evolution, Mol. Genet.
Metab., 74, 147-159.
3.Eric Thomas, C., Chen, Y. C., and Garcia, G. A.
(2011) Differential heterocyclic substrate recognition by, and
pteridine inhibition of E. coli and human tRNA-guanine
transglycosylases, Biochem. Biophys. Res. Commun., 410,
34-39.
4.Johannsson, S., Neumann, P., and Ficner, R. (2018)
Crystal structure of the human tRNA guanine transglycosylase catalytic
subunit QTRT1, Biomolecules, 8, 81.
5.Fergus, C., Barnes, D., Alqasem, M. A., Kelly, V.
P. (2015) The queuine micronutrient: charting a course from microbe to
man, Nutrients, 7, 2897-2929.
6.Tuorto, F., Legrand, C., Cirzi, C., Federico, G.,
Liebers, R., et al. (2018) Queuosine-modified tRNAs confer nutritional
control of protein translation, EMBO J., 37, e99777.
7.Müller, M., Legrand, C., Tuorto, F., Kelly, V.
P., Atlasi, Y., et al. (2019) Queuine links translational control in
eukaryotes to a micronutrient from bacteria, Nucleic Acids Res.,
47, 3711-3727.
8.Chen, Y. C., Kelly, V. P., Stachura, S. V., and
Garcia, G. A. (2010) Characterization of the human tRNA-guanine
transglycosylase: Confirmation of the heterodimeric subunit structure,
RNA, 16, 958-968.
9.Alqasem, M. A., Fergus, C., Southern, J. M.,
Connon, S. J., and Kelly, V. P. (2020) The eukaryotic tRNA-guanine
transglycosylase enzyme inserts queuine into tRNA via a sequential
bi-bi mechanism, Chem. Commun. (Camb.), 56,
3915-3918.
10.Zhang, J., Lu, R., Zhang, Y., Matuszek, Ż.,
Zhang, W., Xia, Y., et al. (2020) tRNA queuosine modification enzyme
modulates the growth and microbiome recruitment to breast tumors,
Cancers (Basel), 12, 628.
11.Kumari, K., Groza, P., and Aguilo, F. (2021)
Regulatory roles of RNA modifications in breast cancer, NAR
Cancer, 3, zcab036.
12.Ma, Q., and He, J. (2020) Enhanced expression of
queuine tRNA-ribosyltransferase 1 (QTRT1) predicts poor prognosis in
lung adenocarcinoma, Ann. Transl. Med., 8, 1658.
13.Bian, M., Huang, S., Yu, D., and Zhou, Z. (2021)
tRNA metabolism and lung cancer: beyond translation, Front. Mol.
Biosci., 8, 659388.
14.Farkas, W. R., Jacobson, K. B., and Katze, J. R.
(1984) Substrate and inhibitor specificity of tRNA-guanine
ribosyltransferase, Biochim. Biophys. Acta, 781,
64-75.
15.Muralidhar, G., Utz, E. D., Elliott, M. S.,
Katze, J. R., and Trewyn, R. W. (1988) Identifying inhibitors of
queuine modification of tRNA in cultured cells, Anal. Biochem.,
171, 346-351.
16.Hoops, G. C., Townsend, L. B., and Garcia, G. A.
(1995) tRNA-guanine transglycosylase from Escherichia coli:
structure-activity studies investigating the role of the aminomethyl
substituent of the heterocyclic substrate PreQ1, Biochemistry,
34, 15381-15387.
17.Goodenough-Lashua, D. M., and Garcia, G. A.
(2003) tRNA-guanine transglycosylase from E. coli: A ping-pong
kinetic mechanism is consistent with nucleophilic catalysis, Bioorg.
Chem., 31, 331-344.
18.Nilov, D. K., Tararov, V. I., Kulikov, A. V.,
Zakharenko, A. L., Gushchina, I. V., et al. (2016) Inhibition of
poly(ADP-ribose) polymerase by nucleic acid metabolite 7-methylguanine,
Acta Naturae, 8, 108-115.
19.Nilov, D., Maluchenko, N., Kurgina, T.,
Pushkarev, S., Lys, A., et al. (2020) Molecular mechanisms of PARP-1
inhibitor 7-methylguanine, Int. J. Mol. Sci., 21,
2159.
20.Nilov, D. K., Pushkarev, S. V., Gushchina, I. V.,
Manasaryan, G. A., Kirsanov, K. I., et al. (2020) Modeling of the
enzyme-substrate complexes of human poly(ADP-ribose) polymerase 1,
Biochemistry (Moscow), 85, 99-107.
21.Manasaryan, G., Suplatov, D., Pushkarev, S.,
Drobot, V., Kuimov, A., et al. (2021) Bioinformatic analysis of the
nicotinamide binding site in poly(ADP-ribose) polymerase family
proteins, Cancers (Basel), 13, 1201.
22.Alemasova, E. E., and Lavrik, O. I. (2019)
Poly(ADP-ribosyl)ation by PARP1: reaction mechanism and regulatory
proteins, Nucleic Acids Res., 47, 3811-3827.
23.Lord, C. J., Tutt, A. N., and Ashworth, A. (2015)
Synthetic lethality and cancer therapy: Lessons learned from the
development of PARP inhibitors, Annu. Rev. Med., 66,
455-470.
24.Henning, R. J., Bourgeois, M., and Harbison, R.
D. (2018) Poly(ADP-ribose) polymerase (PARP) and PARP inhibitors:
Mechanisms of action and role in cardiovascular disorders,
Cardiovasc. Toxicol., 18, 493-506.
25.Berger, N. A., Besson, V. C., Boulares, A. H.,
Bürkle, A., Chiarugi, A., et al. (2018) Opportunities for the
repurposing of PARP inhibitors for the therapy of non-oncological
diseases, Br. J. Pharmacol., 175, 192-222.
26.Wang, J., Cieplak, P., and Kollman, P. A. (2000)
How well does a restrained electrostatic potential (RESP) model perform
in calculating conformational energies of organic and biological
molecules? J. Comput. Chem., 21, 1049-1074.
27.Bayly, C. I., Cieplak, P., Cornell, W. D., and
Kollman, P. A. (1993) A well-behaved electrostatic potential based
method using charge restraints for deriving atomic charges: The RESP
model, J. Phys. Chem., 97, 10269-10280.
28.Dupradeau, F. Y., Pigache, A., Zaffran, T.,
Savineau, C., Lelong, R., et al. (2010) The R.E.D. tools: Advances in
RESP and ESP charge derivation and force field library building,
Phys. Chem. Chem. Phys., 12, 7821-7839.
29.Barca, G. M. J., Bertoni, C., Carrington, L.,
Datta, D., De Silva, N., et al. (2020) Recent developments in the
general atomic and molecular electronic structure system, J. Chem.
Phys., 152, 154102.
30.Maier, J. A., Martinez, C., Kasavajhala, K.,
Wickstrom, L., Hauser, K. E., et al. (2015) ff14SB: Improving the
accuracy of protein side chain and backbone parameters from ff99SB,
J. Chem. Theory Comput., 11, 3696-3713.
31.Sievers, K., Welp, L., Urlaub, H., and Ficner, R.
(2021) Structural and functional insights into human tRNA guanine
transgylcosylase, RNA Biol., 18, 382-396.
32.Case, D. A., Belfon, K., Ben-Shalom, I. Y.,
Brozell, S. R., Cerutti, D. S., et al. (2020) AMBER 2020.
University of California, San Francisco.
33.Salomon-Ferrer, R., Götz, A. W., Poole, D.,
Le Grand, S., and Walker, R. C. (2013) Routine microsecond molecular
dynamics simulations with AMBER on GPUs. 2. Explicit solvent Particle
Mesh Ewald, J. Chem. Theory Comput., 9, 3878-3888.
34.Roe, D. R., and Cheatham, T. E. 3rd. (2013) PTRAJ
and CPPTRAJ: Software for processing and analysis of molecular dynamics
trajectory data, J. Chem. Theory Comput., 9,
3084-3095.
35.Stroganov, O. V., Novikov, F. N., Stroylov, V.
S., Kulkov, V., and Chilov, G. G. (2008) Lead finder: an approach to
improve accuracy of protein-ligand docking, binding energy estimation,
and virtual screening, J. Chem. Inf. Model., 48,
2371-2385.
36.Novikov, F. N., Stroylov, V. S., Zeifman, A. A.,
Stroganov, O. V., Kulkov, V., et al. (2012) Lead Finder docking and
virtual screening evaluation with Astex and DUD test sets, J.
Comput. Aided Mol. Des., 26, 725-735.
37.Humphrey, W., Dalke, A., and Schulten, K. (1996)
VMD: Visual molecular dynamics, J. Mol. Graph., 14,
33-38.
38.Neeb, M., Czodrowski, P., Heine, A., Barandun, L.
J., Hohn, C., et al. (2014) Chasing protons: How isothermal titration
calorimetry, mutagenesis, and pKa calculations trace the locus of
charge in ligand binding to a tRNA-binding enzyme, J. Med.
Chem., 57, 5554-5565.
39.Hohn, C., Härtsch, A., Ehrmann, F. R.,
Pfaffeneder, T., Trapp, N., et al. (2016) An immucillin-based
transition-state-analogous inhibitor of tRNA-guanine transglycosylase
(TGT), Chemistry, 22, 6750-6754.
40.Barandun, L. J., Immekus, F., Kohler, P. C.,
Tonazzi, S., Wagner, B., et al. (2012) From lin-benzoguanines to
lin-benzohypoxanthines as ligands for Zymomonas mobilis
tRNA-guanine transglycosylase: Replacement of protein-ligand hydrogen
bonding by importing water clusters, Chemistry, 18,
9246-9257.
41.Fergus, C., Al-Qasem, M., Cotter, M., McDonnell,
C.M., Sorrentino, E., et al. (2021) The human tRNA-guanine
transglycosylase displays promiscuous nucleobase preference but strict
tRNA specificity, Nucleic Acids Res., 49, 4877-4890.
42.Li, Q., and Shah, S. (2017) Structure-based
virtual screening, Methods Mol. Biol., 1558, 111-124.
43.Batool, M., Ahmad, B., and Choi, S. (2019) A
structure-based drug discovery paradigm, Int. J. Mol. Sci.,
20, 2783.
44.Gushchina, I. V., Polenova, A. M., Suplatov, D.
A., Švedas, V. K., and Nilov, D. K. (2020) vsFilt: A tool to
improve virtual screening by structural filtration of docking poses,
J. Chem. Inf. Model., 60, 3692-3696.