Agonists in the Extended Conformation Stabilize the Active State of β-Adrenoceptors
Alexander V. Efimov1,a*, Olga V. Meshcheryakova2,b, and Alexey G. Ryazanov3,c
1Institute of Protein Research, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia2Institute of Biology of the Karelian Research Centre of the Russian Academy of Sciences, 185910 Petrozavodsk, Russia
3Department of Pharmacology, Rutgers Robert Wood Johnson Medical School, Piscataway, 08854 New Jersey, USA
* To whom correspondence should be addressed.
Received March 21, 2022; Revised June 21, 2022; Accepted June 21, 2022
In this study, we conducted a comparative analysis of the structure of agonists and antagonists of transmembrane (TM) β-adrenoceptors (β-ARs) and their interactions with the β-ARs and proposed the mechanism of receptor activation. A characteristic feature of agonist and antagonist molecules is the presence of a hydrophobic head (most often, one or two aromatic rings) and a tail with a positively charged amino group. All β-adrenergic agonists have two carbon atoms between the aromatic ring of the head and the nitrogen atom of the amino group. In antagonist molecules, this fragment can be either reduced or increased to four atoms due to the additional carbon and oxygen atoms. The agonist head, as a rule, has two H-bond donors or acceptors in the para- and meta-positions of the aromatic rings, while in the antagonist heads, these donors/acceptors are absent or located in other positions. Analysis of known three-dimensional structures of β-AR complexes with agonists showed that the agonist head forms two H-bonds with the TM5 helix, and the tail forms an ionic bond with the D3.32 residue of the TM3 helix and one or two H-bonds with the TM7 helix. The tail of the antagonist can form similar bonds, but the interaction between the head and the TM5 helix is much weaker. As a result of these interactions, the agonist molecule acquires an extended “strained string” conformation, in contrast to the antagonist molecule, which has a longer, bended, and flexible tail. The “strained string” of the agonist interacts with the TM6 helix (primarily with the W6.48 residue) and turns it, which leads to the opening of the G protein-binding site on the intracellular side of the receptor, while flexible and larger antagonist molecules do not have the same effect on the receptor.
KEY WORDS: β-adrenoceptor, GPCR, agonist, antagonist, activation mechanismDOI: 10.1134/S0006297922070057
INTRODUCTION
G protein-coupled receptors (GPCRs) constitute the largest family of cell membrane receptors that includes over 800 human proteins targeted by at least 30% of current medicines (see reviews [1, 2]). This might be the main reason why GPCRs have been extensively studied in several recent decades, resulting in the elucidation of many aspects of their biology, biochemistry, and pharmacology. Originally, the behavior of GPCRs was described in terms of a simple two-state model; however, there is growing body of evidence that GPCRs are not molecular switches, but rather molecular relays, i.e., dynamic proteins with multiple states between active and inactive conformations [4-8].
Recent crystallography data have provided the snapshots of both active and inactive functional states of GPCRs [9, 10]. GPCR structures solved so far share the same overall fold – a bundle of seven transmembrane (TM) α-helices with three extra- and three intracellular loops. The extracellular interface is responsible for the ligand binding, while the intracellular portion interacts with G proteins, β-arrestins, and other downstream effectors. Analysis of known GPCR structures indicates that receptor activation is associated with subtle changes in the extracellular portion of the protein and extensive rearrangement of the TM helices on the cytoplasmic side [11, 12]. Agonist binding at the GPCR extracellular interface results in the opening of the intracellular part for the G protein binding, which promotes G protein activation (GDP release) and initiates the signaling cascade.
The progress in membrane protein crystallography and related techniques in the past decade [7, 13] has allowed to elucidate many aspects of GPCR structure, activation, and physiology; however, some details of ligand recognition and receptor activation remain poorly understood. One of the main aims of this study was comparative analysis of the structures of β-adrenoceptor (β-AR) ligands. Based on the results of this analysis, we proposed that agonist molecules adopt an extended (“strained string”) conformation and stabilize the active state of β-AR, in contrast to antagonist molecules, which have longer and flexible tail and fail to produce the same effect on the receptors.
MATERIALS AND METHODS
As the main research approaches in this study, we used stereochemical analysis of the known three-dimensional structures of β-AR complexes with the corresponding ligands and comparative analysis of the chemical structure of agonists and antagonists and their conformations in the complexes. For this, we created a database of such complexes that included 64 structures determined by crystallography so far and a database of β-AR sequences from the Swiss-Prot UniProt [14]. The atomic coordinates of the complexes were taken from the Protein Data Bank (PDB, URL: https://www.rcsb.org) [15]. The three-dimensional structures of the receptors and their ligands were analyzed using RasMol [16] and PyMOL Molecular Graphics System Version 1.4.1 Schrödinger, LLC. β-AR subtypes designated using the nomenclature recommended by the NC-IUPHAR Subcommittee on Adrenergic Receptors. Amino acid residues in β-ARs were designated according to the Ballesteros and Weinstein nomenclature [17]. Multiple sequence alignment was performed with the Clustal Omega program (1.2.4) included in the UniProt resource [14]. Images of ligand molecules were taken from the Drug Informational Portal, ChEBI, and ChemSpider.
RESULTS AND DISCUSSION
Comparative analysis of chemical structures of β-AR ligands. In terms of receptor activation (intrinsic efficacy), GPCR ligands can be categorized into four groups: (i and ii) full and partial agonists that produce the maximal or sub-maximal functional response, respectively; (iii) inverse agonists that decrease the basal receptor activity (activity in the absence of ligand); and (iv) neutral antagonists, that compete with other ligands for the orthosteric binding site, but their interaction with the receptor does not result in the G protein binding.
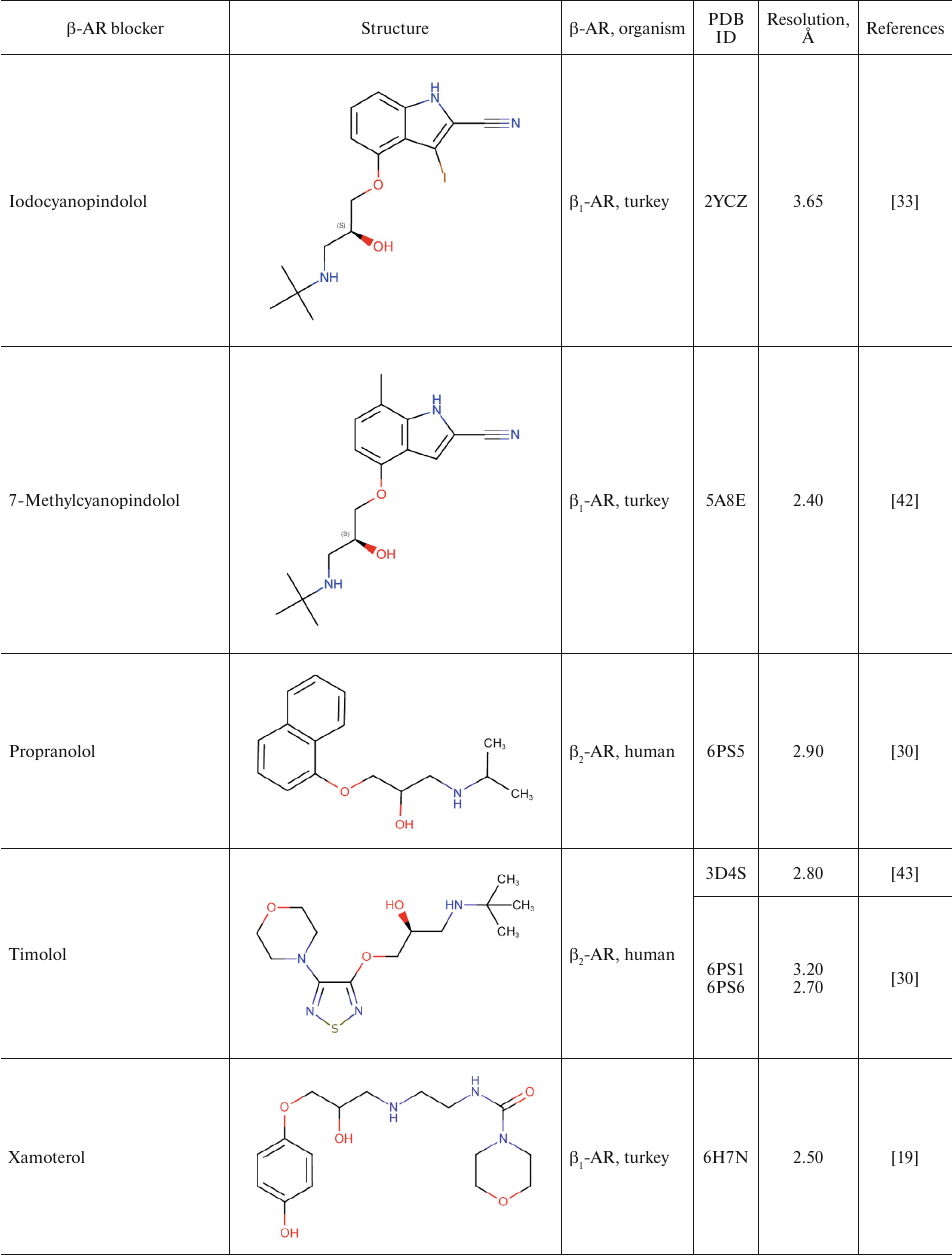
Table 1 shows chemical structures of β-AR agonists that have been co-crystallized with the receptor. A characteristic feature of these molecules is the presence of an aromatic head and a tail with a positively charged amine. The tail consists of ethanolamine core and various substitutes connected to the amine group. Hydroxyl groups in the para- and meta-positions of the catechol moieties and in the para-position in aromatic rings of non-catechol agonists can form hydrogen bonds with the receptor helices.
Aromatic heterocyclic heads of non-catechol agonists can have other donors and acceptors of hydrogen bonds that might be involved in the binding with the receptor. Hence, GPCR agonists have two centers of polar interactions with the receptor (donors/acceptors of H-bonds of the head and donors/acceptors and positively charged amine of the tail). One may ask if the distance between these centers has any influence on the specificity of receptor–ligand interactions. As seen in Table 1, the length of the tail fragment between the N-atom and the aromatic ring is the same in all agonists and comprises three covalent bonds. In other words, the N-atom is separated by 2 carbons from the substituted benzene or other aromatic ring. Moreover, in all agonists with six-membered aromatic rings, the O-atoms of hydroxyls located in the para- and meta-positions and the N-atoms of the tail are separated by seven and six covalent bonds, respectively.
Table 1. β-AR agonists that have been
co-crystallized with the receptor
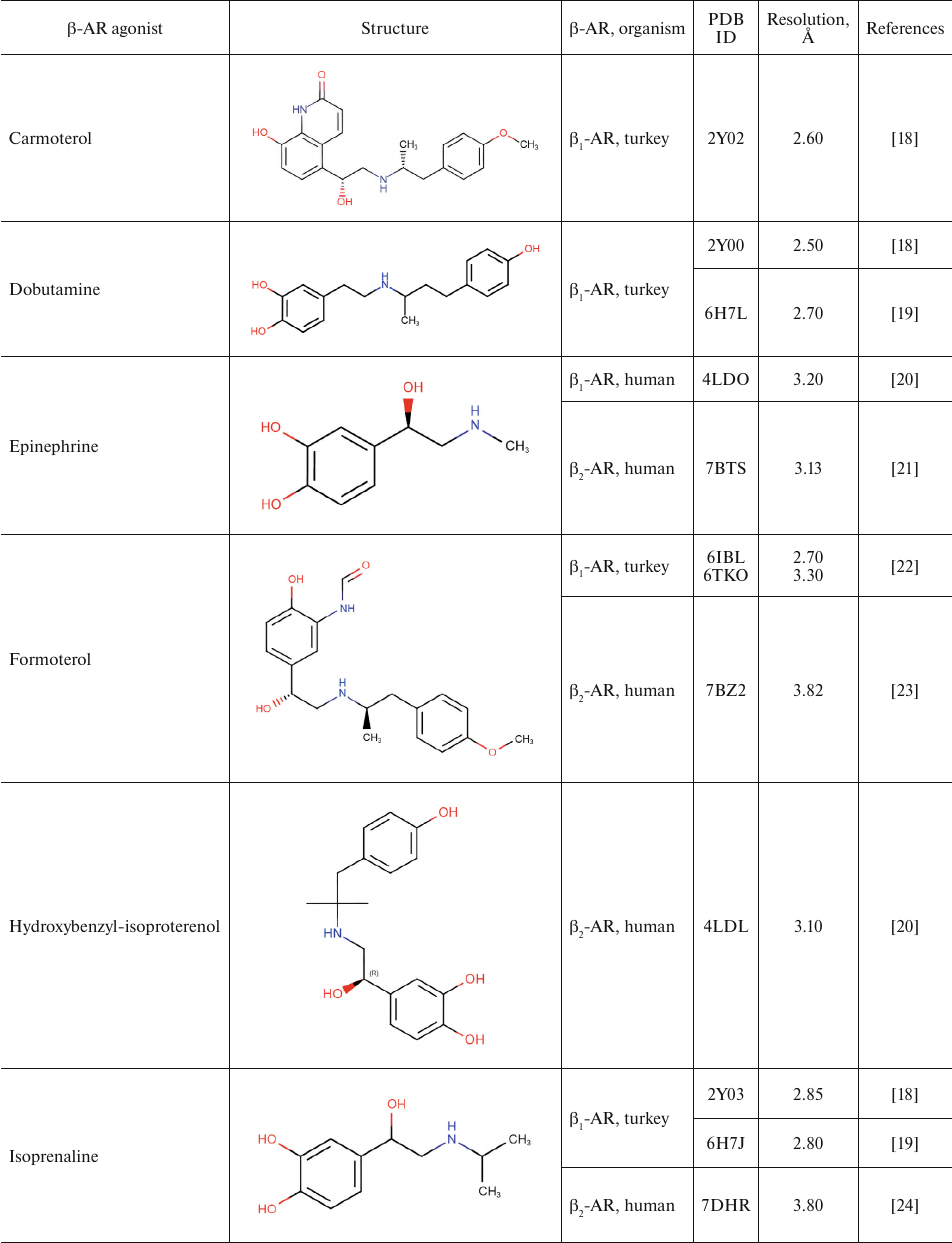
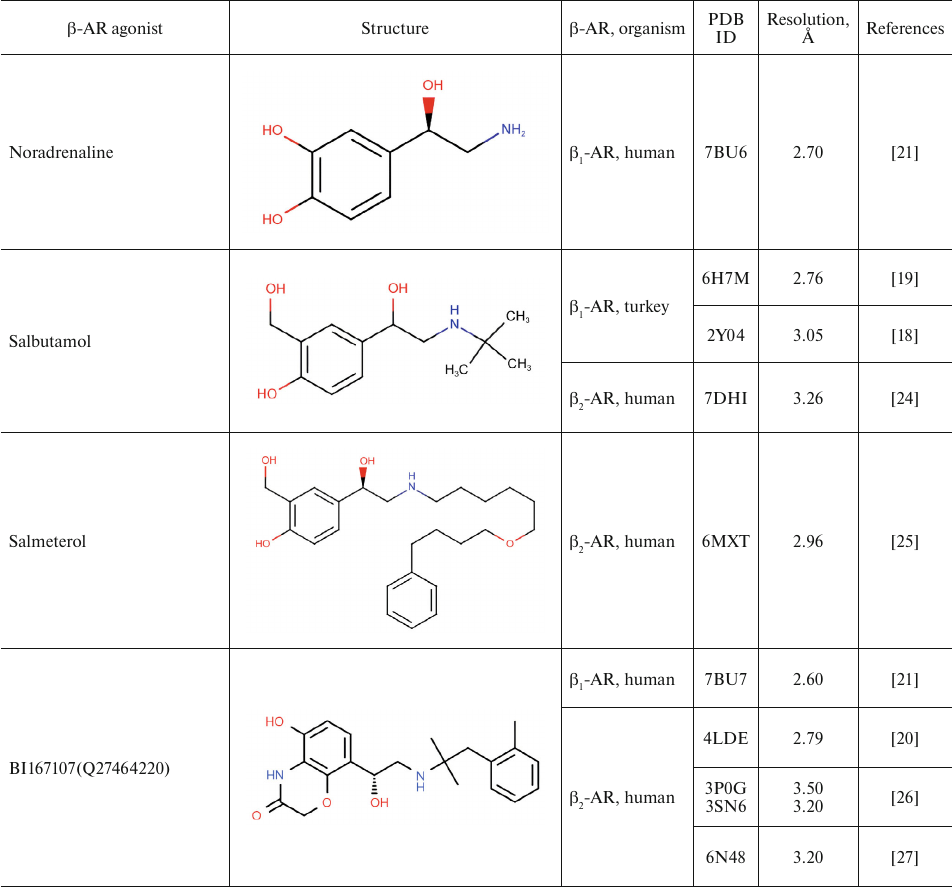
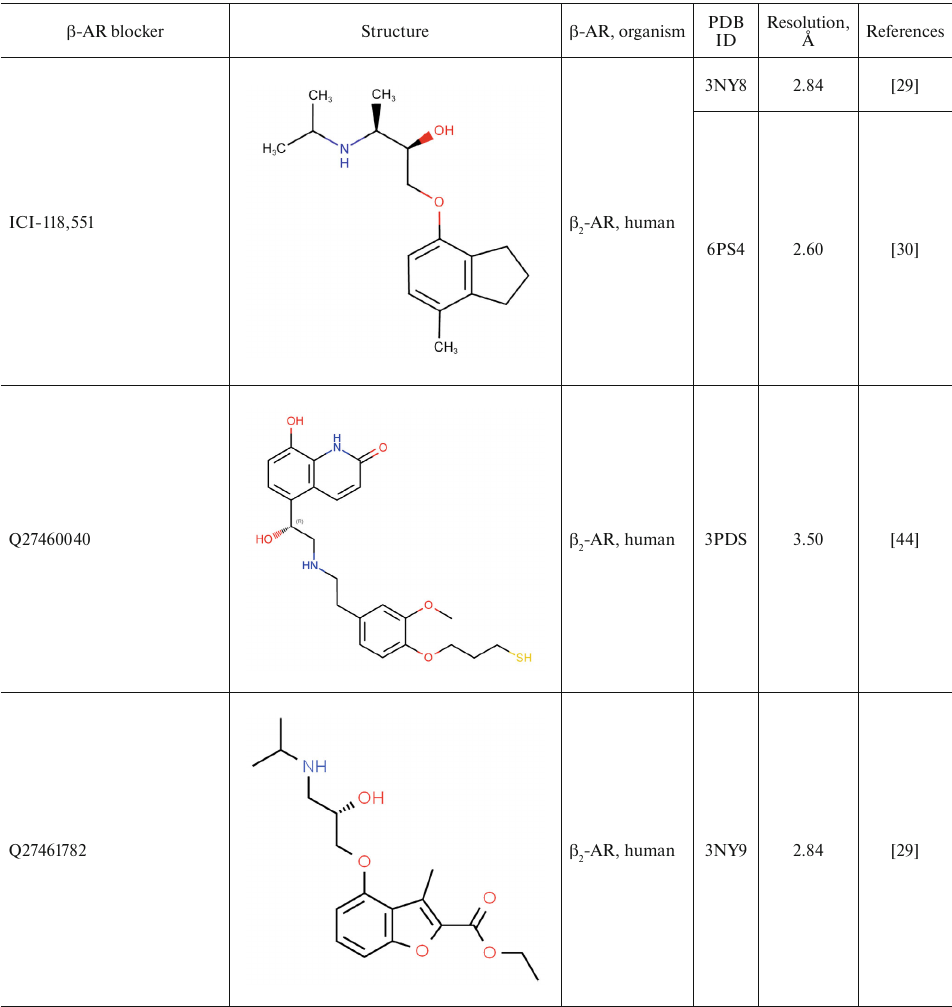
For comparison, Table 2 shows partial agonists, antagonists, and inverse agonists (referred to as β-blockers after Emtage et al. [28]. These compounds also have aromatic heads and tails with positively charged amine groups. However, compared to agonists, these ligands have either longer (Table 2) or shorter (e.g., doxepin and bretylium tosylate not shown here) tail fragments between the amine N-atom and the aromatic ring. In most cases, the N-atom of the amine group is separated from the aromatic ring by 4 atoms (often one of them is O). Moreover, antagonists and inverse agonists presented in Table 2 do not contain donors or acceptors of H-bonds in the para-position of the aromatic head or even lack them at all. Note that the NH-groups of cyanopindolol and carazolol are located in the meta-position relative to the tail. Another feature of β-blockers is that they typically have larger heads consisting of two and three rings as compared to the agonists.
Table 2. β-AR blockers (partial
agonists, antagonists, and inverse agonists) that have been
co-crystallized with the receptor
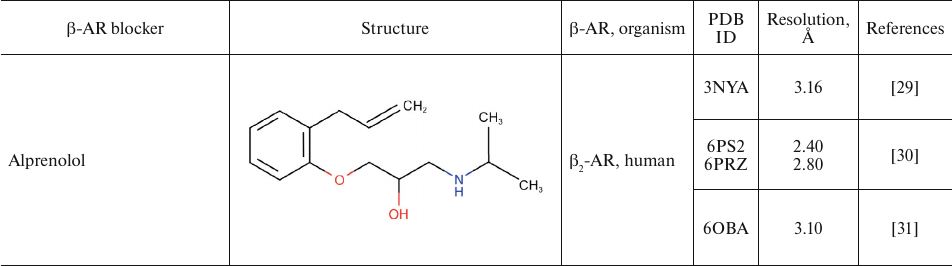
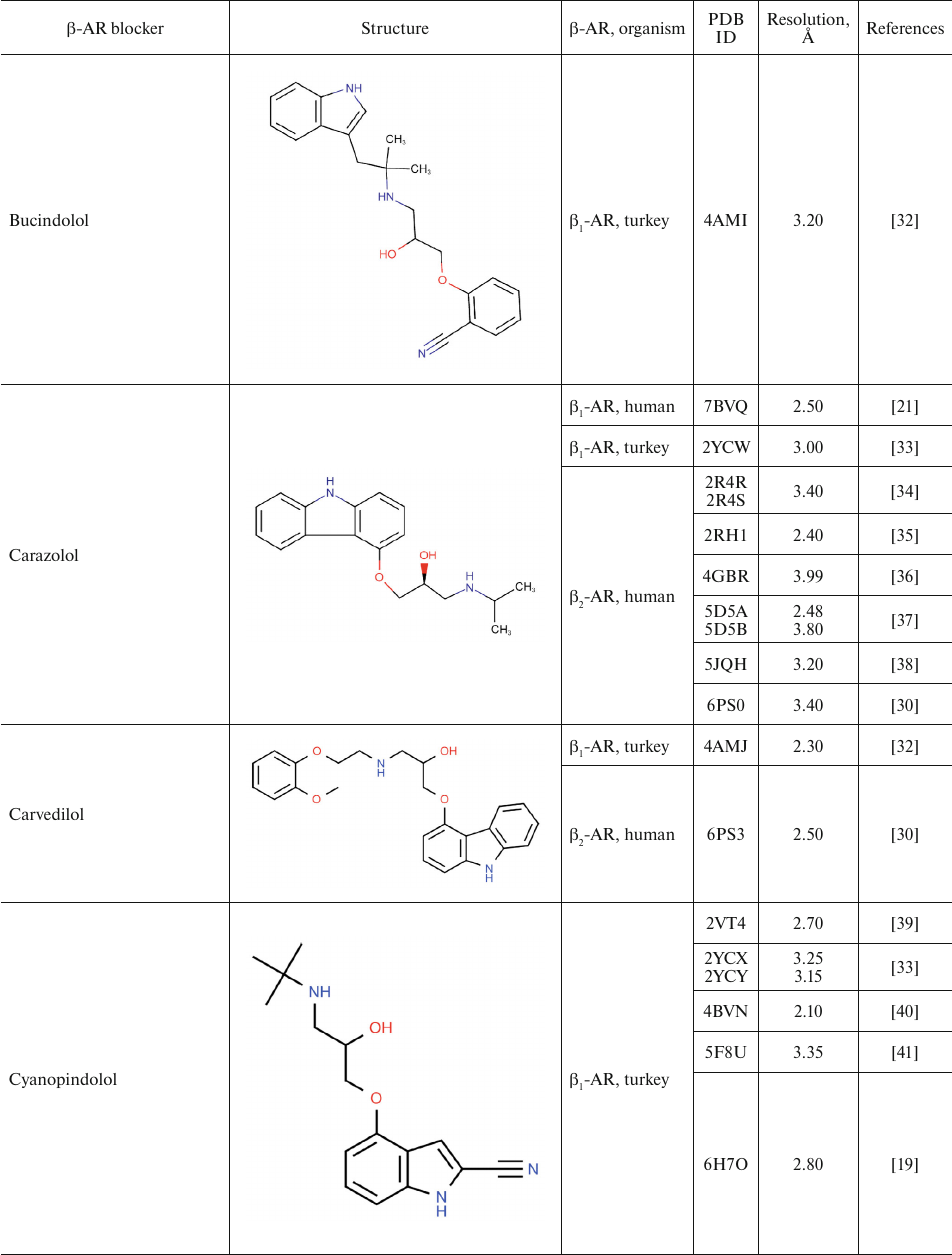
Two centers of polar interactions between β-ARs and their agonists. The orthosteric binding pocket of β-ARs is located within the TM region and is primarily composed of the extracellular fragments of the TM helices 3, 5, 6, and 7 (Fig. 1). Multiple biochemical and mutagenesis studies, as well as analysis of crystal structures of aminergic GPCRs, have allowed to locate critical amino acid residues in the TM helices [45]. It was demonstrated that the charged amine of the ligand interacts with Asp residue D3.32. GPCRs with D3.32 also have a Tyr residue at position Y7.43 and contain Asn at position N7.39, which suggests that their side chains are involved in the interactions with the positively charged moieties in the ligands. Therefore, these key amino acids form the center for the binding of amino groups and other polar groups of the ligand tail.
In the second center of polar interactions, all catecholamine receptors have Ser residues at positions S5.42 and S5.46; most of them also have Ser at position S5.43, so that these residues can form H-bonds with the donors and/or acceptors of the aromatic head. Recent data [10] indicate that Thr at position T3.37 also can interact with polar groups of the ligand head.
Fig. 1. Typical structure of the agonist complex with TM helices 3, 5 6, and 7 forming the binding site in β-ARs. The key amino acid residues interacting with the ligand are shown as circles.
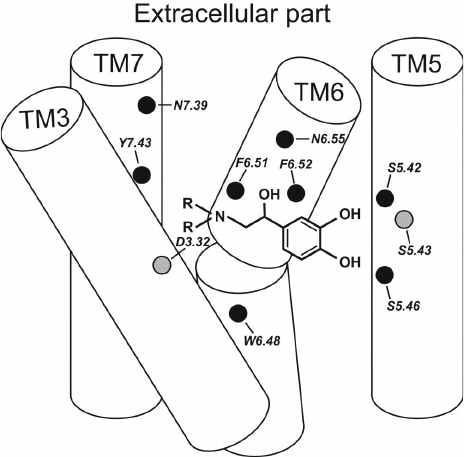
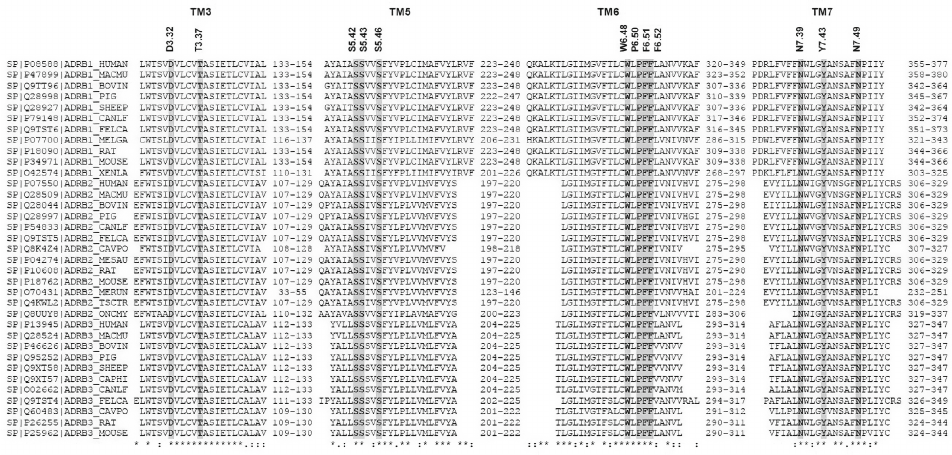
Figure 2 shows the alignment of amino acid sequences of TM helices 3, 5, 6, and 7 in β-AR subtypes β1, β2, and β3 from different animal species. All key amino acid residues mentioned above are highly conserved in all β-ARs.
Fig. 2. Multiple sequence alignment of TM3, TM5, TM6, and TM7 domains of β-ARs for different animal species. Domain information and protein sequences were taken from Swiss-Prot UniProt Knowledgebase (35 proteins) [14]. Key amino acids in the sequences are shown with a gray background.
Comparison of conformations of agonists, antagonists, and inverse agonists bound to β-ARs. Figure 3 shows crystal structures of the agonist isoprenaline (panel a) and neutral antagonist cyanopindolol (panel b) bound in the main binding pocket of β1-Ars [18, 33]. The figure shows the side chains of the amino acid residues forming the two centers of polar interactions as well as ligand conformations. As seen in Fig. 3b, the O-C-C-C group of atoms in cyanopindolol tail acquires a gauche-conformation (i.e., forms a kink in the tail), while the C-C-C-N group of atoms in the isoprenaline tail has a trans-conformation (Fig. 3a).
Fig. 3. The main binding pocket of the β1-AR with (a) agonist (isoprenaline; PDB ID, 2Y03) and (b) antagonist (cyanopindolol; PDB ID, 2YCY). Images are generated using PyMOL Molecular Graphics System.
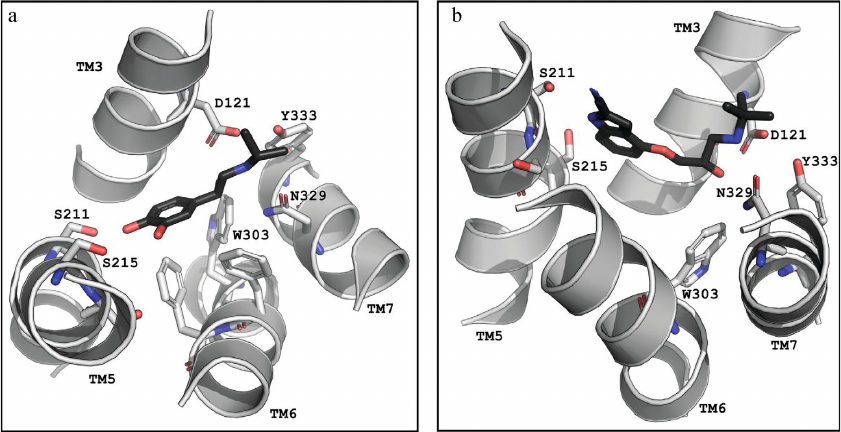
Figure 4 shows the superimposed structures from Fig. 3. It is clearly seen that the O-C-C-C group of atoms forms a kink in the cyanopindolol tail, and the isoprenaline tail has an extended conformation. Analysis shows that the same picture is observed in other complexes of agonists and antagonists with the receptors (Tables 1 and 2).
Fig. 4. Comparison of structures of the agonist (isoprenaline, orange) and antagonist (cyanopindolol, dark cyan) complexes with β1-AR (PDB ID, 2Y03, yellow, and 2YCY, blue). a) View from the receptor extracellular side; important residues are labeled according to Ballesteros–Weinstein notation [17]; b) overview of bound ligand conformation. The images are generated using PyMOL Molecular Graphics System.
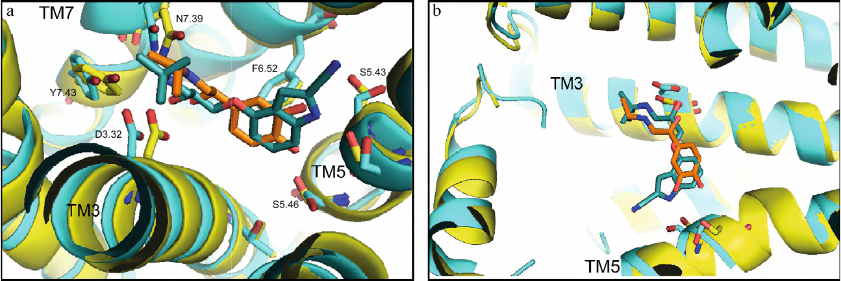
The amine groups of both ligands are located in the amine-binding center (D3.32, N7.39, Y7.43), while catechol hydroxyls of isoprenaline and NH- and cyanogroups of cyanopindolol are situated in the other center of polar interactions (S5.42, S5.43, S5.46). However, the overall geometries of the agonist and antagonist molecules are different. In isoprenaline, the tail has an extended (trans-) conformation, and the molecule resembles a “strained string” that stabilizes the active state of the receptor. The tail of cyanopindolol is bent between the amine N-atom and the aromatic ring, resulting in the zigzag-like conformation (Figs. 3 and 4). Under other conditions (for example, in a native membrane, and not in a crystal), the tail of the antagonist can be transformed into an extended conformation and back, i.e., TM helices in the receptor complexes with antagonists can have a greater dynamic mobility than in the complexes with agonists (see Nygaard et al. (2013) [6]).
We have examined other agonists bound to the corresponding aminergic GPCRs and, indeed, the tail fragment between the amine N-atom and the aromatic ring in these complexes also has an extended conformation (PDB ID: 2Y00, 2Y02, 2Y04, 3P0G, 3PDS, 3SN6, and other structures presented in Table 1). On the other hand, antagonist molecules are bent in the tail fragments between the amine N-atom and the aromatic ring (PDB ID: 2VT4, 2YCW, 2RH1, 3D4S, 3NY8, 3NY9, 3NYA, 3PBL, 3RZE, and other structures presented in Table 2). These data suggest that the extended conformation of the agonist tail is of particular importance in the active state stabilization in β-ARs and other aminergic GPCRs. We can also speculate that the distance between the two centers of polar interactions with the agonists should be equal (or close) to the distance between the corresponding centers in the activated GPCR.
The role of the agonist “strained string” conformation in the β-AR activation. Simple geometry considerations outlined above suggest that the agonist molecule can act as a “strained string” that stabilizes the arrangement of TM helices 3, 5, 6, and 7 corresponding to the receptor active state (Fig. 1). It appears that the interaction with the agonist bring the TM5 helix closer to the TM3 and TM7 helices. In particular, this “strained string” interacts with Trp at position W6.48 and Phe residues at positions F6.51 and F6.52. These interactions are likely to be responsible for the rearrangement of TM6, i.e., for its rotation and/or vertical see-saw movement around a pivot in the middle of the membrane, which results in the opening of the intracellular portion for the G protein binding [46-48]. The interactions with W6.48 result in a subtle rotation of TM6 in the extracellular portion, which is amplified towards the cytoplasmic side by the characteristic kink in the helix introduced by Pro residue P6.50 [11]. Unlike an agonist, an antagonist or an inverse agonist has a longer tail fragments with a loose conformation, a kink in the middle of the tail, and weak polar interactions of the head with the TM5 helix. Therefore, they cannot act as a “strained string”, but instead occupy the active site of the receptor due to the polar and hydrophobic interactions.
Based on these findings, we propose several predictions on how minor differences in the ligand structure can influence its functional characteristics:
1. Elongation of the agonist tail between the amine N-atom and the aromatic ring could result in the agonist conversion into antagonist.
2. Shortening of the tail fragment in the antagonist or inverse agonist with polar moieties in the corresponding positions of their heads may result in their transformation into agonists or partial agonists.
3. Removal of catechol hydroxyls or the corresponding polar substitutes in other aromatic heads could reduce the agonist activity. Modifications of these polar substitutes with aromatic or aliphatic groups substitutes, especially bulky ones, are likely to have a similar effect.
4. Modification of amine groups of ligands (both agonists and antagonists) with bulky substitutes reduce or even prevent ligand binding to GPCRs.
5. The trans-conformation (extended conformation) of the agonist tail can be transformed into a gauche- or cis-conformation by chemical modification, which can result in the reduction of agonist activity or even transform it into an antagonist.
Funding. This work was supported by the Russian Foundation for Basic Research (project no. 20-04-00453).
Ethics declarations. The authors declare no conflicts of interest. This article does not contain descriptions of studies involving humans or animals as study subjects.
Open access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third-party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
REFERENCES
1.Drews, J. (2000) Drug discovery: a historical
perspective, Science, 287, 1960-1964, doi:
10.1126/science.287.5460.1960.
2.Eiger, D. S., Pham, U., Gardner, J., Hicks, C., and
Rajagopal, S. (2022) GPCR systems pharmacology: a different perspective
on the development of biased therapeutics, Am. J. Physiol. Cell.
Physiol., 322, 887-895, doi: 10.1152/ajpcell.00449.2021.
3.Costanzi, S., Siegel, J., Tikhonova, I. G., and
Jacobson, K. A. (2009) Rhodopsin and the others: a historical
perspective on structural studies of G protein-coupled receptors,
Curr. Pharm. Des., 15, 3994-4002, doi:
10.2174/138161209789824795.
4.Kahsai, A. W., Xiao, K., Rajagopal, S., Ahn, S.,
Shukla, A. K., et al. (2011) Multiple ligand-specific conformations of
the β2-adrenergic receptor, Nat. Chem. Biol., 7,
692-700, doi: 10.1038/nchembio.634.
5.Katritch, V., Cherezov, V., and Stevens, R. C.
(2012) Diversity and modularity of G protein-coupled receptor
structures, Trends Pharmacol. Sci., 33, 17-27, doi:
10.1016/j.tips.2011.09.003.
6.Nygaard, R., Zou, Y., Dror, R. O., Mildorf, T. J.,
Arlow, D. H., et al. (2013) The dynamic process of
β2-adrenergic receptor activation, Cell,
152, 532-542, doi: 10.1016/j.cell.2013.01.008.
7.Weis, W. I., and Kobilka, B. K. (2018) The
molecular basis of G protein-coupled receptor activation, Annu. Rev.
Biochem., 87, 897-919, doi:
10.1146/annurev-biochem-060614-033910.
8.Frei, J. N., Broadhurst, R. W., Bostock, M. J.,
Solt, A., Jones, A. J. Y., et al. (2020) Conformational plasticity of
ligand-bound and ternary GPCR complexes studied by 19F NMR
of the β1-adrenergic receptor, Nat. Commun.,
11, 669, doi: 10.1038/s41467-020-14526-3.
9.Lebon, G., Warne, T., and Tate, C. G. (2012)
Agonist-bound structures of G protein-coupled receptors, Curr. Opin.
Struct. Biol., 22, 482-490, doi:
10.1016/j.sbi.2012.03.007.
10.Wang, C., Jiang, Y., Ma, J., Wu, H., Wacker, D.,
et al. (2013) Structural basis for molecular recognition at serotonin
receptors, Science, 340, 610-614, doi:
10.1126/science.1232807.
11.Standfuss, J., Edwards, P. C., D’Antona,
A., Fransen, M., Xie, G., et al. (2011) The structural basis of
agonist-induced activation in constitutively active rhodopsin,
Nature, 471, 656-660, doi: 10.1038/nature09795.
12.Venkatakrishnan, A. J., Deupi, X., Lebon, G.,
Tate, C. G., Schertler, G. F., et al. (2013) Molecular signatures of
G-protein-coupled receptors, Nature, 494, 185-194, doi:
10.1038/nature11896.
13.Granier, S., and Kobilka, B. (2012) A new era of
GPCR structural and chemical biology, Nat. Chem. Biol.,
8, 670-673, doi: 10.1038/nchembio.1025.
14.The UniProt Consortium (2021) UniProt: the
universal protein knowledgebase in 2021, Nucleic Acids Res.,
49, D480-D489, doi: 10.1093/nar/gkaa1100.
15.Berman, H. M., Westbrook, J., Feng, Z.,
Gilliland, G., Bhat, T. N., et al. (2000) The protein data bank,
Nucleic Acids Res., 28, 235-242, doi:
10.1093/nar/28.1.235.
16.Sayle, R. A., and Milner-White, E. J. (1995)
RASMOL: biomolecular graphics for all, Trends Biochem. Sci.,
20, 374, doi: 10.1016/s0968-0004(00)89080-5.
17.Ballesteros, J. A., and Weinstein, H. (1995)
Integrated methods for the construction of three-dimensional models and
computational probing of structure-function relations in G
protein-coupled receptors, in Methods in Neurosciences (Sealfon,
S. C., ed.) Academic Press, USA, pp. 366-428, doi:
10.1016/S1043-9471(05)80049-7.
18.Warne, T., Moukhametzianov, R., Baker, J. G.,
Nehmé, R., Edwards, P. C., et al. (2011) The structural basis
for agonist and partial agonist action on a
β1-adrenergic receptor, Nature, 469,
241-244, doi: 10.1038/nature09746.
19.Warne, T., Edwards, P. C., Doré, A. S.,
Leslie, A. G. W., and Tate, C. G. (2019) Molecular basis for
high-affinity agonist binding in GPCRs, Science, 364,
775-778, doi: 10.1126/science.aau5595.
20.Ring, A. M., Manglik, A., Kruse, A. C., Enos, M.
D., Weis, W. I., et al. (2013) Adrenaline-activated structure of
β2-adrenoceptor stabilized by an engineered nanobody,
Nature, 502, 575-579, doi: 10.1038/nature12572.
21.Xu, X., Kaindl, J., Clark, M. J., Hübner,
H., Hirata, K., et al. (2021) Binding pathway determines norepinephrine
selectivity for the human β1AR over
β2AR, Cell Res., 31, 569-579, doi:
10.1038/s41422-020-00424-2.
22.Lee, Y., Warne, T., Nehmé, R., Pandey, S.,
Dwivedi-Agnihotri, H., et al. (2020) Molecular basis of β-arrestin
coupling to formoterol-bound β1-adrenoceptor,
Nature, 583, 862-866, doi: 10.1038/s41586-020-2419-1.
23.Zhang, Y., Yang, F., Ling, S., Lv, P., Zhou, Y.,
et al. (2020) Single-particle cryo-EM structural studies of the
β2AR-Gs complex bound with a full agonist formoterol,
Cell Discov., 6, 4, doi: 10.1038/s41421-020-0176-9.
24.Yang, F., Ling, S. L., Zhou, Y. X., Zhang, Y. N.,
Lv, P., et al. (2021) Different conformational responses of the
beta2-adrenergic receptor-Gs complex upon binding of the partial
agonist salbutamol or the full agonist isoprenaline, Natl. Sci.
Rev., 8, nwaa284, doi: 10.1093/nsr/nwaa284.
25.Masureel, M., Zou, Y., Picard, L. P., Van der
Westhuizen, E., Mahoney, J. P., et al. (2018) Structural insights into
binding specificity, efficacy and bias of a β2AR
partial agonist, Nat. Chem. Biol., 14, 1059-1066, doi:
10.1038/s41589-018-0145-x.
26.Rasmussen, S. G., DeVree, B. T., Zou, Y., Kruse,
A. C., Chung, K. Y., et al. (2011) Crystal structure of the
β2 adrenergic receptor-Gs protein complex,
Nature, 477, 549-555, doi: 10.1038/nature10361.
27.Liu, X., Masoudi, A., Kahsai, A. W., Huang, L.
Y., Pani, B., et al. (2019) Mechanism of β2AR
regulation by an intracellular positive allosteric modulator,
Science, 364, 1283-1287, doi:
10.1126/science.aaw8981.
28.Emtage, A. L., Mistry, S. N., Fischer, P. M.,
Kellam, B., and Laughton, C. A. (2017) GPCRs through the keyhole: the
role of protein flexibility in ligand binding to β-adrenoceptors,
J. Biomol. Struct. Dyn., 35, 2604-2619, doi:
10.1080/07391102.2016.1226197.
29.Wacker, D., Fenalti, G., Brown, M. A., Katritch,
V., Abagyan, R., et al. (2010) Conserved binding mode of human
β2 adrenergic receptor inverse agonists and antagonist
revealed by X-ray crystallography, J. Am. Chem. Soc.,
132, 11443-11445, doi: 10.1021/ja105108q.
30.Ishchenko, A., Stauch, B., Han, G. W., Batyuk,
A., Shiriaeva, A., et al. (2019) Toward G protein-coupled receptor
structure-based drug design using X-ray lasers, IUCrJ, 24
(Pt 6), 1106-1119, doi: 10.1107/S2052252519013137.
31.Liu, X., Kaindl, J., Korczynska, M.,
Stößel, A., Dengler, D., Stanek, M., et al. (2020) An
allosteric modulator binds to a conformational hub in the
β2 adrenergic receptor, Nat. Chem. Biol.,
16, 749-755, doi: 10.1038/s41589-020-0549-2.
32.Warne, T., Edwards, P. C., Leslie, A. G., and
Tate, C. G. (2012) Crystal structures of a stabilized
β1-adrenoceptor bound to the biased agonists bucindolol
and carvedilol, Structure, 20, 841-849, doi:
10.1016/j.str.2012.03.014.
33.Moukhametzianov, R., Warne, T., Edwards, P. C.,
Serrano-Vega, M. J., Leslie, A. G., et al. (2011) Two distinct
conformations of helix 6 observed in antagonist-bound structures of a
β1-adrenergic receptor, Proc. Natl. Acad. Sci.
USA, 108, 8228-8232, doi: 10.1073/pnas.1100185108.
34.Rasmussen, S. G., Choi, H. J., Rosenbaum, D. M.,
Kobilka, T. S., Thian, F. S., et al. (2007) Crystal structure of the
human β2 adrenergic G-protein-coupled receptor,
Nature, 450, 383-387, doi: 10.1038/nature06325.
35.Cherezov, V., Rosenbaum, D. M., Hanson, M. A.,
Rasmussen, S. G., Thian, F. S., et al. (2007) High-resolution crystal
structure of an engineered human β2-adrenergic G
protein-coupled receptor, Science, 23, 1258-1265, doi:
10.1126/science.1150577.
36.Zou, Y., Weis, W. I., and Kobilka, B. K. (2012)
N-terminal T4 lysozyme fusion facilitates crystallization of a G
protein coupled receptor, PLoS One, 7, e46039, doi:
10.1371/journal.pone.0046039.
37.Huang, C. Y., Olieric, V., Ma, P., Howe, N.,
Vogeley, L., et al. (2016) In meso in situ serial X-ray
crystallography of soluble and membrane proteins at cryogenic
temperatures, Acta Crystallogr. D Struct. Biol., 72 (Pt
1), 93-112, doi: 10.1107/S2059798315021683.
38.Staus, D. P., Strachan, R. T., Manglik, A., Pani,
B., Kahsai, A. W., et al. (2016) Allosteric nanobodies reveal the
dynamic range and diverse mechanisms of G-protein-coupled receptor
activation, Nature, 535, 448-452, doi:
10.1038/nature18636.
39.Warne, T., Serrano-Vega, M. J., Baker, J. G.,
Moukhametzianov, R., Edwards, P. C., et al. (2008) Structure of a
β1-adrenergic G-protein-coupled receptor,
Nature, 454, 486-491, doi: 10.1038/nature07101.
40.Miller-Gallacher, J. L., Nehmé, R., Warne,
T., Edwards, P. C., Schertler, G. F., et al. (2014) The 2.1 Å
resolution structure of cyanopindolol-bound
β1-adrenoceptor identifies an intramembrane
Na+ ion that stabilises the ligand-free receptor, PLoS
One, 9, e92727, doi: 10.1371/journal.pone.0092727.
41.Leslie, A. G., Warne, T., and Tate, C. G. (2015)
Ligand occupancy in crystal structure of β1-adrenergic
G protein-coupled receptor, Nat. Struct. Mol. Biol., 22,
941-942, doi: 10.1038/nsmb.3130.
42.Sato, T., Baker, J., Warne, T., Brown, G. A.,
Leslie, A. G., et al. (2015) Pharmacological analysis and structure
determination of 7-methylcyanopindolol-bound
β1-adrenergic receptor, Mol. Pharmacol.,
88, 1024-1034, doi: 10.1124/mol.115.101030.
43.Hanson, M. A., Cherezov, V., Griffith, M. T.,
Roth, C. B., Jaakola, V. P., et al. (2008) A specific cholesterol
binding site is established by the 2.8 Å structure of the human
β2-adrenergic receptor, Structure, 16,
897-905, doi: 10.1016/j.str.2008.05.001.
44.Rosenbaum, D. M., Zhang, C., Lyons, J. A., Holl,
R., Aragao, D., et al. (2011) Structure and function of an irreversible
agonist-β2 adrenoceptor complex, Nature,
469, 236-240, doi: 10.1038/nature09665.
45.Conner, A. C., Barwell, J., Poyner, D. R., and
Wheatley, M. (2011) The use of site-directed mutagenesis to study
GPCRs, Methods Mol. Biol., 746, 85-98, doi:
10.1007/978-1-61779-126-0_5.
46.Rasmussen, S. G., Choi, H. J., Fung, J. J.,
Pardon, E., Casarosa, P., et al. (2011) Structure of a
nanobody-stabilized active state of the β2
adrenoceptor, Nature, 469, 175-180, doi:
10.1038/nature09648.
47.Chung, K. Y. (2013) Structural aspects of GPCR-G
protein coupling, Toxicol. Res., 29, 149-155, doi:
10.5487/TR.2013.29.3.149.
48.Wu, Y., Zeng, L., and Zhao, S. (2021) Ligands of
adrenergic receptors: a structural point of view, Biomolecules,
11, 936, doi: 10.3390/biom11070936.