REVIEW: Effect of Tau Protein on Mitochondrial Functions
Khoren K. Epremyan1,a*, Tatyana N. Goleva1, and Renata A. Zvyagilskaya1
1Bach Institute of Biochemistry, Federal Research Center of Biotechnology, Russian Academy of Sciences, 119071 Moscow, Russia* To whom correspondence should be addressed.
Received April 27, 2022; Revised July 4, 2022; Accepted July 6, 2022
Alzheimer’s disease is the most common age-related progressive neurodegenerative disorder of brain cortex and hippocampus leading to cognitive impairment. Accumulation of extracellular amyloid plaques and intraneuronal neurofibrillary tangles are believed to be the main hallmarks of the disease. Origin of Alzheimer’s disease is not totally clear, multiple initiator factors are likely to exist. Intracellular impacts of Alzheimer’s disease include mitochondrial dysfunction, oxidative stress, ER-stress, disruption of autophagy, severe metabolic challenges leading to massive neuronal apoptosis. Mitochondria are the key players in all these processes. This formed the basis for the so-called mitochondrial cascade hypothesis. This review provides current data on the molecular mechanisms of the development of Alzheimer’s disease associated with mitochondria. Special attention was paid to the interaction between Tau protein and mitochondria, as well as to the promising therapeutic approaches aimed at preventing development of neurodegeneration.
KEY WORDS: Alzheimer’s disease, Tau protein, bioenergetics, mitochondriaDOI: 10.1134/S0006297922080028
Abbreviations: AD, Alzheimer’s disease; Drp1, dynamin-related protein 1; ETC, electron transport chain; hTau, full-length human Tau protein; MitoQ, mitoquinone mesylate; OPA1, mitochondrial dynamin like GTPase; PINK1, PTEN-induced kinase 1; P-Tay, hyperphosphorylated Tau protein; ROS, reactive oxygen species; SkQ1, 10-(6′-plastoquinonyl)decyltriphenylphosphonium; TRAK2, trafficking kinesin-binding protein 2; UCHL-1, ubiquitin carboxy-terminal hydrolase L1.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disease leading to cognitive impairment (impaired cognitive function), one of the most common causes of dementia, morbidity, and mortality in aging population [1, 2]. The hereditary and sporadic forms of AD are thought to have the same markers – accumulation of misfolded and aggregated proteins, extracellular deposits of (Aβ)-amyloid senile plaques, and intracellular neurofibrillary tangles consisting mainly of aggregates of hyperphosphorylated Tau protein. It is believed that the increasing accumulation of Aβ aggregates triggers a cascade of cellular changes including hyperphosphorylation of the Tau protein and inflammation [1-3]. However, the amyloid cascade hypothesis, which postulates the key role of Aβ in AD development, cannot explain all the anomalies observed in AD [1, 2]. Formation of amyloid oligomers is necessary but not sufficient for the development of dementia, and additional factors are required, including involvement of peripheral immune cells, excessive production of pro-inflammatory mediators, chronic endoplasmic reticulum stress (ER stress), and especially chronic energy imbalance and mitochondrial damage [1, 3-7]. Postmitotic neurons are especially sensitive to mitochondrial dysfunction, disruption of their dynamics (fusion and fragmentation, intracellular localization), which causes dysfunction of synapses, the main site of adenosine triphosphate (ATP) utilization, and their subsequent loss. The loss of synapses correlates with the loss of cognitive functions and precedes the loss of neurons, first in the entorhinal cortex and hippocampus, and then in most of the cortex [1].
Recent studies show that there is an intrinsic relationship between Tau protein and mitochondria. Tau can influence mitochondrial function at three different levels: transport, morphology, and mitochondrial bioenergetics. For example, the truncated N-terminal Tau protein (20-22 kDa) is largely concentrated in the brain mitochondria in AD, and its amount in the nerve terminal fields correlates with pathological changes in the synapses [8]. Mitochondrial dysfunction was found in transgenic TauP301L mice carrying the Tau protein P301L mutation, which causes frontotemporal dementia [9]. Dysregulation of mitochondrial complex I during aging depends on Tau [10]. Other cellular mechanisms by which a hyperphosphorylated Tau induces loss of synapses by the retinal ganglion cells during glucolipotoxicity include microtubule destabilization and disruption of the microtubule-dependent synaptic transport of mRNA and mitochondria, disruption of energy generation by mitochondria in the synapses due to hyperphosphorylation of Tau in a GSK3β (glycogen synthase kinase 3-beta)-dependent manner [11].
Numerous mitochondrial anomalies have been identified using several cellular and animal models of tauopathy [4, 12]. These include increased production of reactive oxygen species (ROS), changes in mitochondrial dynamics, and impaired oxidative phosphorylation [13, 14].
Chronic stress resulted in atrophy of apical dendrites and loss of spines in the prefrontal cortex neurons, as well as significant memory impairments in the wild-type mice, while animals with the Tau-protein (Tau-KO) knockout had no such changes. Quantitative proteomic analysis of the synapse fraction of the prefrontal cortex combined with the analysis using transmission electron microscopy has suggested an important role of mitochondria in regulating the effects of stress. Particularly, the Tau-dependent changes in the levels of proteins involved in metabolite transport by mitochondria and oxidative phosphorylation have been detected in the animals under chronic stress, as well as in mitochondria localization in the prefrontal cortex synapses [15].
Aβ oligomers inhibited mitochondrial transport in axons in the primary neurons in the wild-type mice, while neurons with the reduced level of Tau protein had normal transport of mitochondria in axons [16]. These observations suggest that along with Aβ, Tau protein is also required to disrupt mitochondrial transport in axons, and a reduced level of Tau can protect against the Aβ-induced changes in the transport of mitochondria in axons [16]. Mitochondrial functional defects were detected in the 3xTg-AD mice (reduced respiration and activity of pyruvate dehydrogenase, elevated ROS production, and lipid peroxidation [17-19]), as well as impaired regulation of mitochondrial Krebs cycle proteins, metabolism of pyruvate, glycolysis, antioxidant proteins, transport of metabolites, oxidation of fatty acids, utilization of ketone bodies and oxidative phosphorylation system proteins, especially complexes I and IV of the mitochondrial electron transport chain (ETC) [10, 20]. In the study of the brains of AD patients and 3xTg-AD mice, it was found that the dynamin-related protein 1 (Drp1), responsible for mitochondrial fragmentation, is activated by hyperphosphorylated Tau protein (P-Tau) [21].
Recent studies have also shown that the Aβ- and P-Tau-induced inhibition of autophagy and mitophagy are important events in AD pathogenesis. The age-related increases in the levels of Aβ and P-Tau reduced the levels of several autophagy and mitophagy proteins [22]. Activating action of Aβ on the protein Drp1 [23, 24], P-Tau – on Drp1 [23-25], inhibiting action of Aβ – on PINK1/parkin [26, 27], and P-Tau – on PINK1/parkin [26] led to disruption of mitophagy and autophagy [22].
Thus, over the past decades, a lot of evidence has been accumulated indicating direct association of Tau protein with mitochondrial dysfunction in the development of neurodegeneration. However, for the most part, they revealed these pathological interactions at the cellular and subcellular level and established existence of morphological and bioenergetic changes. At the same time, interactions between Tau protein and mitochondria at the molecular level remained poorly understood.
In this review, we attempted to collect all the available data of recent years, allowing us to fill in the gaps in understanding molecular mechanisms of the Tau-induced mitochondrial dysfunction, as well as to trace general logic of pathological changes from the lowest level of interactions to a higher one, from molecular to cellular. In addition, the review presents the recently described promising therapeutic approaches aimed at attenuating or preventing the development of neurodegeneration in AD associated with mitochondria.
IMPACT OF TAU PROTEIN ON BIOENERGY PARAMETERS OF
MITOCHONDRIA
Exposure of the isolated mitochondria from human neuroblastoma SH-SY5Y cells to oligomeric Tau protein caused mitochondrial swelling, release of cytochrome c, and membrane potential collapse [28].
Localization of different forms of Tau (full-length sized human Tau protein (hTau), phosphorylated form, or N-terminal fragment cleaved by caspase) in mitochondria or their association with the outer mitochondrial membrane has been proven [14, 29], which implies the possibility of participation of “mitochondrial” Tau in cellular dysfunctions observed in tauopathies.
Mitochondrial membranes are obvious targets for toxic intracellular Tau species, not least in synapses, where mitochondria are particularly numerous [30]. The ability of aggregates of the full-length Tau-441 oligomers to penetrate lipid vesicles with a certain composition of membranes was established [31], mitochondrial membranes were found to be particularly vulnerable to protein aggregates [28]. Importantly, Tau proteins exhibited high affinity for the membranes enriched in cardiolipin (1,3-diphosphatidyl-sn-glycerol, CL), a characteristic phospholipid of mitochondrial membranes [32, 33], which plays fundamental role in formation of the respiratory chain supercomplexes to maintain optimal oxidative phosphorylation [34]; CL oxidation is a prerequisite for the release of cytochrome c from mitochondria [35], which predetermines onset of the irreversible stage of apoptosis.
Subcortical injection of the recombinant Tau-protein oligomers into the brain of mice caused mitochondrial dysfunction by reducing the activity of complex I and activating the mitochondrial-dependent apoptotic pathway leading to synaptic dysfunction [36].
In the transgenic mice overexpressing the mutant TauP301 functional analysis has confirmed mitochondrial dysfunction in the form of decreased mitochondrial respiration and ATP synthesis [9]. Proteomic analysis of the mitochondria additionally revealed decrease in the D subunit of ATP synthase in the TauP301L mice, which was also found in the postmortem brain of the individuals with a TauP301L mutation [9]. Activity of the mitochondrial cytochrome c-oxidase (complex IV) did not change in the transgenic mice with Tau pathology [9], but these changes were characteristic of the mice with AD overexpressing mutant forms of the amyloid protein precursor (APP) [37], as well as upon accumulation of Aβ oligomers and presence of Tau pathology [9, 36].
An intron mutation 10 + 16 in the MAPT gene encoding Tau caused frontotemporal dementia and parkinsonism associated with chromosome 17 (FTDP-17). The increased potential generated at the mitochondrial membrane in the FTDP-17 neurons led to the excessive production of ROS in mitochondria, which, in turn, caused oxidative stress and cell death. Excessive formation of mitochondrial ROS in these cells could be prevented by the mitochondria-targeted antioxidant mitoquinone (MitoQ) [38].
A pronounced mitochondrial dysfunction was observed in the 3xTg-AD mice, produced by interbreeding of the pR5 TauP301L transgenic mice in which neurofibrillary tangles were formed, and in the APP(sw)PS2(N141I) mice that produce amyloid plaques, which was manifested by the inhibition of I- and IV-ETC complexes, reduced ATP production, elevated ROS levels, compared to the mice with pathology of Aβ or Tau. Therefore, there was a synergy of pathological defects caused by Tau and Aβ with AD mutations [10, 39].
Tau protein mutations (TauP301L and TauV337M) causing frontotemporal dementia reduced ATP production due to ETC inhibition and significantly reduced interaction of the physiological full-length Tau protein with mitochondrial proteins [40].
The regulatory failure of Ca2+ transport also contributes to etiology of AD and is associated with mitochondrial dysfunction [41]. Overexpression of the pathological Tau form phosphorylated at Ser396/404 in the primary neurons of the rat cerebral cortex aggravated the Aβ-induced loss of the mitochondrial membrane potential due to impaired mitochondrial buffer capacity to maintain Ca2+ concentration. The Tau knockout (Tau–/–) protected primary mouse cortical neurons from the loss of mitochondrial membrane potential induced by low concentrations of Aβ42, but resulted in the significant increase in cytoplasmic Ca2+ concentration compared to the primary cortical neurons in the wild-type mice. In general, Tau exacerbated the Aβ-induced mitochondrial dysfunction independent on the Aβ-induced changes in the concentration of cytoplasmic Ca2+ [42]. Nematodes that express the wild-type full-size Tau protein had increased mitochondrial damage and defects in the cell movement compared to the control worms in the absence of accumulation of Tau protein aggregates in the larval stages; calcium chelation with EGTA restored mitochondrial activity and improved mobility of these larvae [43].
Hippocampus neurons from the 6-month-old transgenic tauopathy model mouse (THY-Tau22) expressing oligomeric Tau, contained elongated mitochondria and exhibited signs of cellular stress, but no apparent cytotoxicity compared to the control mice. Levels of several key mitochondrial proteins differed markedly between the THY-Tau22 hippocampus and control mice, including mitochondrial SIRT3, PTEN-induced kinase 1 (PINK1), adenine nucleotide translocase 1 (ANT-1), and division protein Drp1. Base excision repair of DNA (BER), primary system for correcting the effects of oxidative DNA damage, was upregulated in the 6-month-old transgenic mice. DNA polymerase β (Polβ), a key BER DNA polymerase, was detected in significant concentrations in the cytoplasm of hippocampal neurons in the 6-month-old transgenic mice and was localized with and within mitochondria. Polβ has also colocalized with the mitochondria in the AD human brain in the neurons containing oligomeric Tau. Most of these changes were specific for transgenic mice at the age of 6 months and were not detected in the 12 months old mice, when Tau pathology reached its maximum and Tau oligomeric forms were no longer detected [44].
The 3xTg-AD mice with 50% decrease in Polβ (3xTg-AD/Polβ+/–) and also containing a mutated version of the human Tau protein, exhibited pronounced AD phenotypes with impaired cellular bioenergetics and mitochondrial dysfunction. These studies show that Tau alterations, mitochondrial abnormalities, and BER deficiency may interact with each other, modulating the development of tauopathies [44].
Suppression of Tau expression by Tau–/– knockout in mice improved mitochondrial bioenergetic functions through enhancing ETC and ATP production, and, likely, through increasing of the levels of proteins involved in mitochondrial fusion processes, significantly reducing oxidative damage, and activating Nrf-2 and PGC-1α signaling, modulating mitochondrial antioxidant defense and biogenesis, and increasing ATP production in the hippocampus [45]. Recent work by the same authors has shown that deletion of the Tau protein gene prevented cognitive impairment in the Tau–/– mice and improved mitochondrial functions in the course of normal aging [46]. Since expression of the cyclophilin D and adenine nucleotide translocase genes, which are believed to form a nonspecific Ca2+/phosphate-dependent mitochondrial pore (mPTP), was decreased, it was suggested that mPTP could also be involved in these processes [46].
Thus, a common molecular mechanism could be elucidated from the above data showing that Tau protein disrupts bioenergetic parameters of the neurons in AD pathogenesis. Tau protein binding to cardiolipin provides the possibility of direct effect on mitochondrial proteins. This, in turn, results in the reduced ATP synthesis due to ETC complex I inhibition, increased production of ROS, impaired Ca2+ homeostasis, and loss of the membrane potential. While such bioenergetic problems are already sufficient for the development of pathology in neurons, which are most dependent on the energy homeostasis in cells, they also become the basis for pathological processes at higher levels, as discussed below.
INFLUENCE OF TAU PROTEIN ON TRANSPORT AND DISTRIBUTION OF
MITOCHONDRIA IN NEURONS
Mitochondria, as dynamic organelles, regulate cell viability and synapse morphology [47]. Dynamics of mitochondria is regulated by continuous fusion and division [48]. Fusion is mediated by mitofusins, Mfn1 and Mfn2, integral membrane proteins of the outer mitochondrial membrane [49], and mitochondrial dynamin-like GTPase (OPA1) associated with the inner mitochondrial membrane [50]. In mammalian cells, the dynamin-like protein Drp1 plays a key role in mitochondrial division [51]. Dynamics of mitochondria determines morphology, size, distribution, and functions of mitochondria [52].
Proteomics of subcellular organelles combined with bioenergetic estimation of transgenic mice expressing the hTau at the age preceding and coinciding with the onset of thauopathy revealed that Tau pathological forms are predominantly associated with the synaptic mitochondria, which coincides with the changes in bioenergetics that resemble aging synaptic mitochondrial phenotype in the wild-type mice. While the mitochondrial content has not changed, maximal respiration of mitochondria has been impaired in the hTau mice synaptosomes. It has also been determined that the Tau protein associated with mitochondria is bound to the outer mitochondrial membrane. This data show that accumulation of the non-mutant Tau in the synapse produces harmful effects on mitochondria, which probably contributes to the synaptic dysfunction observed in the context of tauopathies [53].
The hippocampus neurons expressing caspase-cleaved Tau truncated at Asp421 (T4C3) were shown to accumulate significantly on the mitochondrial population in the soma, and clear mitochondrial bioenergetic deficit was observed, including mitochondrial depolarization, oxidative stress, and significant decrease in ATP production. At the same time, expression of the mitochondrial kinesin-binding protein 2 (TRAK2, Trafficking kinesin-binding protein 2) required to bind mitochondria to kinesins and move to the cell periphery along microtubules, decreased in the immortalized and primary hippocampal neurons. Moreover, binding of TRAK2 to mitochondria increased compared to the neurons expressing full-length Tau, further inhibiting movement of the mitochondria. Thus, the truncated form of Tau could affect mitochondrial transport in neurons by suppressing TRAK2 expression, strengthening TRAK2 binding to mitochondria, and reducing production of ATP required to support movement of these organelles to synapses [54].
Tau protein inhibited the mitochondrial Rho GTPase 1 (RHOT1)-mediated mitochondrial anterograde movement. RHOT1 is another adapter that binds the outer mitochondrial membrane to TRAK2, described above forming a mitochondrial motor–adapter complex. Increased levels of RHOT1 prevented loss of synapses and reversed cognitive impairment in the mice with tauopathy by restoring mitochondrial populations in synapses [55].
Overexpression of hTau increased the level of proteins responsible for mitochondrial fusion, including OPA1, Mfn1, and Mfn2, and reduced Mfn2 ubiquitination, enhancing the fusion of mitochondria and their perinuclear accumulation in the HEK293 cells and primary neurons of the hippocampus of rats. The ShRNA-mediated reduction of mitofusins to ~45-52% of the control levels attenuated the hTau-enhanced mitochondrial fusion, while inhibition of OPA1 to ~50% of the control level had no positive effects. The hTau accumulation at later stages inhibited mitochondrial functions, which was manifested by the decrease in ATP level, ATP/ADP ratio, and complex I activity [13]. These observations suggest that intracellular accumulation of the full-length Tau protein at the early stages of pathology can impair the anterograde movement of mitochondria due to increased fusion and can cause neurodegeneration due to synaptic dysfunction associated with perinuclear accumulation of mitochondria.
Similar effects of Tau protein have been observed in other models. Expression of the hTau protein (2N4R) or Tau protein 2N4RΔC20 cleaved by caspase-3 has been able to induce aggregation and accumulation of the hTau protein [56], causing perinuclear clustering of mitochondria [13, 57] and altering the level of expression of mitochondrial proteins responsible for fusion [13] in fibroblasts of the patients with sporadic atopic dermatitis, in the rat hippocampal neurons, as well as in several mammalian cell lines, which suggests close relationship between the intracellular localization of mitochondria and Tau. Tau overexpression activated mitochondrial proteins responsible for their fusion, leading to perinuclear accumulation of mitochondria accompanied by the decrease in the complex I activity and ATP levels [13]. Impaired transport of mitochondria in axons has been found in the sensory neurons of embryonic fish Danio rerio, a model organism in biomedical research, expressing different forms of Tau, including mutant TauP301L [58, 59]. Comparing the TauP301L transgenic mice without Tau protein and the double transgenic mice with knockout of the genes encoding Tau and Drp1 (TauP301L-Drp1+/–) showed that the double mutant mice had better cognitive abilities compared to the TauP301L mutants: they had increased dendritic spines, the number of mitochondria was reduced, while their length increased [60]. This may indicate that the increased mitochondrial fragmentation on exposure to Drp1-dependent Tau protein may be a major pathogenetic pathway in AD. Comparative analysis of the effect of hTau, prone to hyperphosphorylation, hyperphosphorylated Tau, and Aβ42 peptide, on the brain antioxidant defense system and on Marf (human Mfn2 homologue) and Drp1 in the transgenic Drosophila melanogaster revealed that, despite the deficiency of antioxidants caused by different types of Tau and Aβ42, Tau appears to have had a more toxic effect on the eye phenotype and the regulation of Marf and Drp1 [61]. Flies expressing the wild-type Tau protein exhibited a stronger expression of Marf, a fusion protein, while hyperphosphorylated Tau increased expression of Drp1, a division protein.
As for the protein OPA1 responsible for the fusion of the inner mitochondrial membranes, its direct relationship with the Tau protein has not yet been found [62].
From the above, it can be concluded that Tau protein plays a critical role in the modulation of mitochondrial dynamics during neurodegeneration. And while it was previously thought to be due to its natural ability to bind to microtubules, the recent studies suggest direct inhibition of the formation of the functional mitochondrial motor–adapter complex responsible for mitochondria movement to the periphery of neurons. In addition, in the early stages of pathology, the full-length Tau activates mitochondrial fusion proteins, leading to their perinuclear clustering and also preventing transport to the periphery. All this is exacerbated by the problems with bioenergetic functions of mitochondria, described in the first section, since the movement of loads along microtubules is an ATP-dependent process. All these pathological effects create the basis for synaptic dysfunction, as normal functioning of synapses requires energy produced by mitochondria and buffer capacity of calcium ions, which is impossible with the wrong quality, quantity, and localization of mitochondria in neurons.
TAU PROTEIN AND MITOPHAGY
Energy requirements for the neuronal excitability, synaptic activity, and plasticity are met almost exclusively by mitochondrial oxidative phosphorylation. But mitochondria, in addition to the well-known function of energy production, perform other key functions in the cell. Therefore, quantity and quality of mitochondria and their redox state must be carefully monitored by the cell. Along with the dynamics of mitochondria, their biogenesis, protein degradation systems, mitochondrial DNA repair enzymes, and mitophagy, the selective removal of damaged non-functioning mitochondria, are also responsible for controlling mitochondria quantity and quality.
New evidence suggests that mitophagy is impaired in AD. In animal and cellular AD models and in patients with sporadic late-onset AD, mitophagy impairment has been shown to contribute to synaptic dysfunction and cognitive deficits, causing Aβ and Tau accumulation and cellular energy deficiency; they, in turn, disrupt mitophagy [63]. Mitophagy deficiency and increased potential generated at the mitochondrial membrane have been reported in tauopathy models in vitro and in vivo [64].
The NH2-Tau fragment (20-22 kDa) located between the 26th and 230th amino acid residues of the longest human Tau isoform (also known as NH2hTau) has been found in the cellular and animal AD models, as well as in mitochondria of the synapses and cerebrospinal fluid from the patients with AD; it exhibits neurotoxicity in the primary neurons of hippocampus by disrupting mitochondrial metabolism both directly by inhibiting ANT-1-dependent ADP/ATP metabolism and indirectly by disrupting mitophagy. The hTau and the TauP301L mutant inhibited mitophagy in neuroblastoma cells by reducing the movement of parkin into mitochondria. In the nervous system of Caenorhabditis elegans, hTau expression reduced mitophagy, while TauP301L expression completely inhibited it. Tau specifically disrupted recruitment of parkin to the defective mitochondria, isolating it in the cytosol. This sequestration was mediated by the aberrant interactions of parkin with the Tau projection domain [65].
Parkin-dependent removal of the damaged mitochondria occurring in the postmitotic neurons expressing NH2hTau contributed to neuronal death, while cytosolic ubiquitin carboxy-terminal hydrolase L1 (UCHL-1) controlling ubiquitin homeostasis and thus synaptic physiological remodeling critically contributed to mitochondrial and synaptic deficiency in this AD model in vitro. Pharmacological or genetic suppression of “incorrect” mitophagy by inhibiting mitochondrial degradation via autophagosomes or by shRNA-mediated suppression of parkin or UCHL-1 gene expression provided partial but significant protection against the NH2hTau-induced neuronal death. Moreover, the endogenous NH2hTau is stably associated with parkin and UCHL-1 in mitochondria of human AD synapses. Taken altogether, this indicates causal relationship between the excessive removal of damaged mitochondria and the NH2hTau-induced neuronal death in vitro. Pathogenic shortening of Tau can contribute to degradation of synapses in AD due to aberrant recruitment of parkin and UCHL-1 into mitochondria, making them more prone to “harmful” autophagic clearance [66].
Conclusive in vitro and in vivo data have also shown that the free ubiquitin pool is a unifying node linking two major intracellular proteolytic systems: ubiquitin–proteasome system (UPS) and selective autophagy (mitophagy in particular) [67], and that strict control of ubiquitin levels is of vital importance for synapse homeostasis and neuronal survival [68]. The parkin-associated mitophagy pathway, which plays an important role in maintaining a competent mitochondrial network in postmitotic neurons, mainly in the ATP-consuming synapses [69-71], performs not only ubiquitination of several proteins of the mitochondrial outer membrane [72], but also direct recruitment and activation of UPS components in mitochondria [73]. Among the family of neuron-specific deubiquitinating enzymes, UCHL-1 is the most abundant (1-2% of soluble brain proteins) and multifunctional, acting as a hydrolase – removing and processing ubiquitin molecules from target proteins, and as a ubiquitin ligase – generating polyubiquitin chains, specific labels for subsequent utilization of the proteins. Moreover, UCHL-1 is also able to bind to ubiquitin, inhibiting its degradation in neurons [74], its primary role in synapses is to control structure and/or function of neurons, local maintenance of ubiquitin homeostasis both in vitro and in vivo [75-77]. It has been proven that the impaired UCHL-1 activity is associated with synaptic insufficiency in AD neurodegeneration [78, 79], and its immunoreactivity is markedly detected in the Tau-loaded tangle-bearing neurons located in the selectively affected areas [78]. Interestingly, E3 ubiquitin ligase and parkin have been identified as proteins interacting with UCHL-1 [80] suggesting that this enzyme may serve as a possible modifier of the parkin-dependent removal of damaged mitochondria in neurons.
Overexpression of the hTau induced mitophagy deficits in the HEK293 cells, primary neurons of the hippocampus, and in the brains of transgenic mice. At the same time, potential generated on the mitochondrial membrane increased, while the levels of PTEN-induced kinase 1 (PINK1) and parkin decreased in the mitochondrial fraction. A dose-dependent accumulation of Tau protein in the fraction of the outer mitochondrial membrane, along with its accumulation in the cytoplasm was observed. These data suggest that the intracellular hTau accumulation could cause mitophagy deficiency through direct insertion into the mitochondrial membrane and thus affect PINK1/parkin localization [64].
Hence, the Tau protein-mediated inhibition of the energy-producing function of mitochondria and the subsequent energy deficit described in the first two sections, on the one hand, creates a natural need for the cell to recycle (utilize) non-functional mitochondria. On the other hand, the hyperphosphorylated Tau directly activates mitochondrial fragmentation by inhibiting OPA1 and mitofusins – fusion proteins, and activating Drp1 – a division protein. At the same time, Tau protein blocks mitophagy by directly binding parkin and/or hindering its association with the outer mitochondrial membrane. This makes ubiquitination of membrane proteins and subsequent destruction of mitochondria impossible. Here we can suggest the path in which (under the influence of Tau protein at all stages) energy homeostasis of the cell is disturbed first followed by the development of mitochondria dysfunction and fragmentation, while their disposal through mitophagy is blocked.
PROMISING THERAPIES FOR TAU-PATHOLOGIES ASSOCIATED WITH
MITOCHONDRIAL DYSFUNCTION AND OXIDATIVE STRESS
High level of integration of vital cell processes, variety of intracellular interactions and regulated signaling pathways, as well as proven involvement of mitochondria in pathological changes in AD from the earliest stages before the onset of clinical symptoms, make mitochondria one of the most promising therapeutic targets [81, 82]. Due to the fact that recently new mechanisms of mitochondrial dysfunction in AD related to their dynamics and distribution have been clarified, therapeutic approaches aimed at these new mechanisms have been reported more often [83]. For example, one recent work demonstrated mitophagy accelerators of high efficiency: urolithin A in combination with epigallocatechin gallate improved both bioenergetic and morphological parameters in the cellular model of AD (mTau-HT22) [84].
However, general focus of the works proposing new approaches has been shifted towards stimulation of mitochondrial bioenergetics. As described in the previous sections, although Tau directly affects the regulatory proteins of mitochondrial fusion/fission, mitophagy, and axonal transport, energy deficiency underlies all these higher-level pathological processes.
Currently, one of the most advanced approaches for improving bioenergy functions of mitochondria are mitochondria-targeted compounds consisting of a natural antioxidant molecule (ubiquinone or plastoquinone, respectively, components of the respiratory or photosynthetic electron transport chain) connected via a C10-aliphatic linker with the cation triphenylphosphonium for targeted delivery and accumulation in mitochondria. Mitochondrial lipophilic antioxidants have significant advantages over conventional antioxidants, as they are transported into cells and mitochondria in accordance with the membrane potential generated on the cytoplasmic and mitochondrial membranes, respectively. As a result, their concentration in mitochondria can increase by several orders of magnitude. This allows them to be used at low, non-toxic, micromolar or submicromolar concentrations. Moreover, lipophilic mitochondria-targeted antioxidants have high distribution coefficient in the membrane (104), resulting in a four-order of magnitude increase of its concentration in the lipid bilayer [85]. Finally, they can be restored (regenerated) by the components of the respiratory chain, which ensures their continuous functioning. Compounds most intensively studied and used in the treatment of various pathologies, especially neurodegenerative diseases, are MitoQ and SkQ1 [85-88].
In the 3xTg-AD mice expressing three human mutant genes, two of which cause early onset of AD (APPswe and PS1M146V) and one causing frontotemporal dementia (TauP301L) [89], administration of MitoQ during the period of initial manifestation of AD-like pathologies, preservation of cognitive functions was observed compared with the control mice, as well as a decrease in neuronal oxidative stress, loss of synapses, astrogliosis, microglial cell proliferation, Aβ accumulation, caspase activation, and Tau hyperphosphorylation. MitoQ treatment significantly increased lifespan of the 3xTg-AD mice [90]. MitoQ significantly reduced the amount of ROS and Tau aggregation in the HEK293T cells expressing the full-length Tau protein [91].
Administration of the mitochondria-targeted antioxidant SkQ1 to OXYS rats with increased oxidative stress mimicking key characteristics of the sporadic form of AD at the age of 12-18 months (during the period of active progression of AD) resulted in accumulation of this compound in various areas of the brain, mainly in neuronal mitochondria. By improving the structure and functional state of mitochondria, SkQ1 prevented neuron death and synapse damage, increased neurotrophic supply, and reduced levels of Aβ42 and Tau hyperphosphorylation in the hippocampus of OXYS rats resulting in improved memory and learning ability [92].
Another interesting trend is moderate inhibition of complex I of the mitochondrial respiratory chain. This leads to the increased AMP/ATP ratio causing activation of AMP-activated protein kinase, which, in turn, inhibited GSK3β and cyclin-dependent kinase 5 (CDK5), responsible for Tau hyperphosphorylation, while activating phosphatase PP2Ac, responsible for Tau dephosphorylation. In addition, moderate inhibition of the respiratory chain complex I can initiate an integrated stress response (ISR), which increases resistance of neurons to stress conditions, thereby exerting a neuroprotective effect [93]. The effect of a specific inhibitor of complex I, tricyclic pyrone compound CP2, was studied in the mouse models of AD. CP2 protected against synaptic dysfunction and cognitive impairment in the 3xTg-AD mice and lowered human P-Tau [94]. However, it is worth mentioning here that in the case of such a complicated pathology, even moderate inhibition could have unexpected effects, and ISR induced under certain conditions could direct the cell towards apoptosis.
A possible therapeutic potential has also been suggested for the mitochondria-targeted lithium, which possessed neuroprotective and neurotrophic properties that may be associated with the enhanced mitochondrial functions [95].
Intracerebroventricular microinjection of the diabetogenic drug streptozotocin (STZ) to Wistar rats leads to modeling of sporadic AD characterized by Tau pathology and concomitant cognitive decline, insulin resistance, neuroinflammation, oxidative stress, and mitochondrial dysfunction. Paeonol, an active phenolic component of some medicinal plants, improved cognitive functions in the rat model of AD induced by STZ, partially reduced the level of ROS and activity of tumor necrosis factor α (TNFα) and interleukin 6 (IL-6), enhanced overall antioxidant capacity, superoxide dismutase (SOD) and catalase activity, and increased mitochondrial membrane potential. Thus, paeonol may be of interest in the context of attenuating neuroinflammation, oxidative stress, and mitochondrial dysfunction [96].
Injection of stem cells from the human exfoliated deciduous teeth (SHED) was shown to improve cognitive abilities and reverse memory loss in the SAMP8 mouse model of AD. These effects were closely related to mitochondria. This treatment was accompanied by the increased membrane potential, ATP production, restoration of mitochondrial axonal transport, and inhibition of the formation of Aβ42 aggregates and hyperphosphorylated Tau [97].
Protection against mitochondrial dysfunction and effects of neurotoxicity by minocycline (broad-spectrum antibiotic with neuroprotective activity) that are based on modulation of the impaired protein kinase B (Akt) function, inhibiting Tau hyperphosphorylation under the action of GSK3β, may also be a promising therapeutic intervention [98].
Calorie restriction by 30-40% increased lifespan, improved condition of many organisms [99], slowed down many adverse effects of aging [100], such as increased ROS production and reduced rate of oxygen consumption [101, 102]. Reactive nitrogen species at concentrations 4-5 orders of magnitude lower than those capable of causing mitochondrial dysfunction or damage to neurons during caloric restriction increased mitochondrial biogenesis, thereby providing neuronal resistance to stress and neurodegenerative stimuli [103, 104]. Calorie restriction and intermittent fasting prevented cognitive depression in the triple transgenic 3xTg-AD mice [105].
Several other compounds have been described as neuroprotective and stimulating the energy-producing mitochondrial function. Fibroblast growth factor 21 (FGF21) was able to repair nerve damage in the diabetic and aging mice, weakening neurodegeneration by reducing neuroinflammation through regulation of the NF-κB pathway and the AMPKα/AKT pathway, protecting mitochondria in the neurons [106].
Boswellic acid improved antioxidant properties of mitochondria and increased activity of ETC complexes in the rat model of AD [107].
For the sake of completeness, reference should be made to other strategies used to combat AD. Nanoselenium chondroitin sulfate (CS@Se) with antioxidant properties reduced anxiety and improved spatial learning and memory in the mouse model mimicking AD, significantly reduced cell edema and pyknosis, protected mitochondria and prevented abnormal changes in the ultrastructure of synaptic hippocampus neurons in the mice with AD. It significantly increased the level of antioxidant enzymes, Na+/K+-ATPase and acetyltransferase (ChAT), reduced the levels of malondialdehyde (a marker of oxidative stress) and acetylcholinesterase (ChAE) in the AD mice, reduced excessive phosphorylation of Tau (Ser396/Ser404) by regulating GSK3β expression. CS@Se could activate the ERK1/2 signaling pathways regulated by the extracellular signal of kinases 1/2, and p38 MAPK, p38 mitogen-activated protein kinase, inhibiting nuclear translocation of NF-κB (kappa B transcription factor), thereby regulating expression of the pro-inflammatory cytokines [108].
The data have been accumulated on the protective role of TERT protein (telomerase reverse transcriptase) in the brain and postmitotic neurons: decreased toxicity of β-amyloid, pathological Tau protein, and α-synuclein, as well as activation of autophagy as an important process of degradation of toxic proteins of neurons have been noted [109].
Given the multifactorial nature of the Tau-associated pathologies, perhaps the most promising way to develop new therapeutic strategies is combined use of complementary technologies.
Thus, despite the fact that the new mechanisms and targets of Tau-mediated mitochondrial dysfunction have been clarified in recent years, most of the developed therapeutic approaches and compounds are aimed at maintaining and/or restoring the energy-producing function of mitochondria, inhibition of which underlies neurodegeneration, in general, and Tau pathologies, in particular.
CONCLUSIONS
In this review, we have sought not just to report recent discoveries of relationship between Tau protein and mitochondria during AD development, but to build general picture of pathological molecular processes. According to the recent data, Tau protein disrupts energy homeostasis due to its ability to bind to mitochondria and to cardiolipin, and directly inhibit ETC complex I, reduce membrane potential and ATP production, and increase ROS production (figure). Moreover, this is one of the earliest stages in AD pathogenesis, which forms the basis for the subsequent ones.
Comprehensive Tau – mitochondria interactions leading to neurodegeneration in AD
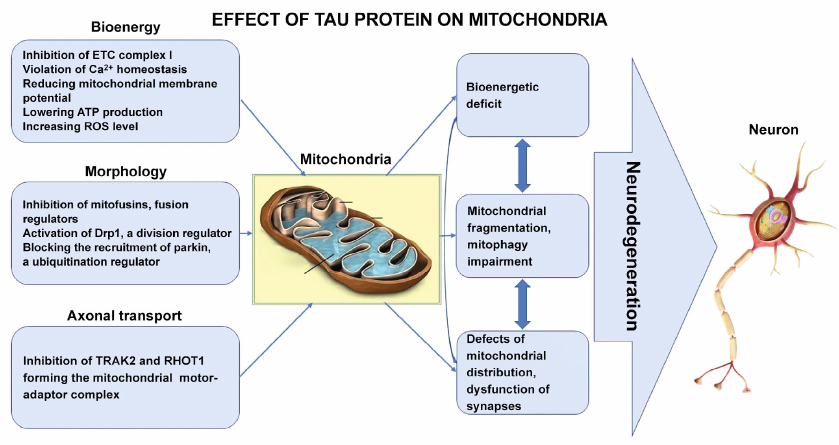
At the higher level, Tau protein, by directly affecting regulatory proteins, disrupts fusion/division balance and blocks mitophagy, which are critical processes for maintaining an adequate quantitative and functional state of mitochondria required to satisfy exceptional energy needs of the neurons (figure).
Finally, Tau protein promotes synaptic dysfunction, can directly inhibit mitochondria distribution in neurons, their anterograde movement towards synapses, where mitochondria provide energy, and towards Ca2+ depot for the release of neurotransmitters during synaptic transmission (figure).
We would like to pay special attention to the fact that all the higher-level pathological processes are initiated and aggravated due to disruption of energy homeostasis. In this regard, it becomes clear why most of the new approaches to AD therapy are focused on maintaining the energy-producing functions of mitochondria, organelles deeply integrated into the cell life processes. But this high level of integration makes mitochondria also the main target for the pathological action of Tau protein during AD development.
Contributions. K. K. Epremyan, R. A. Zvyagilskaya – concept and management; K. K. Epremyan – writing the text; R. A. Zvyagilskaya, T. N. Goleva – editing the text of the article.
Funding. This research was financially supported in part by the Russian Foundation for Basic Research (grant no. 19-34-90165).
Ethics declarations. The authors declare no conflicts of interest in financial or any other sphere. This article does not contain any studies with human participants or animals performed by any of the authors.
REFERENCES
1.Goleva, T., Rogov, A., and Zvyagilskaya, R. (2017)
Alzheimer’s disease: molecular hall marks and yeast models, J.
Alzheimer’s Dis. Parkinsonism, 7, 394-401, doi:
10.4172/2161-0460.1000394.
2.Soria Lopez, J. A., González, H. M., and
Léger, G. C. (2019) Alzheimer’s disease, Handb. Clin.
Neurol., 167, 231-255, doi:
10.1016/B978-0-12-804766-8.00013-3.
3.Eckert, A., Nisbet, R., Grimm, A., and Götz,
J. (2014) March separate, strike together – role of
phosphorylated TAU in mitochondrial dysfunction in Alzheimer’s
disease, Biochim. Biophys. Acta, 1842, 1258-1266, doi:
10.1016/j.bbadis.2013.08.013.
4.Manczak, M., and Reddy, P. H. (2012) Abnormal
interaction between the mitochondrial fission protein Drp1 and
hyperphosphorylated tau in Alzheimer’s disease neurons:
implications for mitochondrial dysfunction and neuronal damage, Hum.
Mol. Genet., 21, 2538-2547, doi: 10.1093/hmg/dds072.
5.Rai, S. N., Singh, C., Singh, A., Singh, M. P., and
Singh, B. K. (2020) Mitochondrial dysfunction: a potential therapeutic
target to treat Alzheimer’s disease, Mol. Neurobiol.,
57, 3075-3088, doi: 10.1007/s12035-020-01945-y.
6.John, A., and Reddy, P. H. (2021) Synaptic basis of
Alzheimer’s disease: focus on synaptic amyloid beta, P-tau and
mitochondria, Ageing Res. Rev., 65, 101208, doi:
10.1016/j.arr.2020.101208.
7.Briston, T., and Hicks, A. R. (2018) Mitochondrial
dysfunction and neurodegenerative proteinopathies: mechanisms and
prospects for therapeutic intervention, Biochem. Soc. Trans.,
46, 829-842, doi: 10.1042/BST20180025.
8.Amadoro, G., Corsetti, V., Stringaro, A., Colone,
M., D’Aguanno, S., et al. (2010) A NH2 tau fragment
targets neuronal mitochondria at AD synapses: possible implications for
neurodegeneration, J. Alzheimer’s Dis., 21,
445-470, doi: 10.3233/JAD-2010-100120.
9.David, D.C., Hauptmann, S., Scherping, I.,
Schuessel, K., Keil, U., et al. (2005) Proteomic and functional
analyses reveal a mitochondrial dysfunction in P301L tau transgenic
mice, J. Biol. Chem., 280, 23802-23814, doi:
10.1074/jbc.M500356200.
10.Rhein, V., Song, X., Wiesner, A., Ittner, L. M.,
Baysang, G., et al. (2009) Amyloid-b and tau synergistically impair the
oxidative phosphorylation system in triple transgenic Alzheimer’s
disease mice, Proc. Natl. Acad. Sci. USA, 106,
20057-20062, doi: 10.1073/pnas.0905529106.
11.Zhu, H., Zhang, W., Zhao, Y., Shu, X., Wang, W.,
et al. (2018) GSK3β-mediated tau hyperphosphorylation triggers
diabetic retinal neurodegeneration by disrupting synaptic and
mitochondrial functions, Mol. Neurodegener., 13, 62, doi:
10.1186/s13024-018-0295-z.
12.DuBoff, B., Feany, M., and Götz, J. (2013)
Why size matters – balancing mitochondrial dynamics in
Alzheimer’s disease, Trends Neurosci., 36, 325-335,
doi: 10.1016/j.tins.2013.03.002.
13.Li, X.C., Hu, Y., Wang, Z., Luo, Y., Zhang, Y.,
et al. (2016) Human wild-type full-length tau accumulation disrupts
mitochondrial dynamics and the functions via increasing mitofusins,
Sci. Rep., 6, 24756, doi: 10.1038/srep24756.
14.Pérez, M. J., Vergara-Pulgar, K., Jara,
C., Cabezas-Opazo, F., and Quintanilla, R. A. (2018) Caspase-cleaved
tau impairs mitochondrial dynamics in Alzheimer’s disease,
Mol. Neurobiol., 55, 1-15, doi:
10.1007/s12035-017-0385-x.
15.Lopes, S., Teplytska, L., Vaz-Silva, J., Dioli,
C., Trindade, R., et al. (2017) Tau deletion prevents stress-induced
dendritic atrophy in prefrontal cortex: role of synaptic mitochondria,
Cereb. Cortex, 27, 2580-2591, doi:
10.1093/cercor/bhw057.
16.Vossel, K. A., Zhang, K., Brodbeck, J., Daub, A.
C., Sharma, P., et al. (2010) Tau reduction prevents Abeta-induced
defects in axonal transport, Science, 330, 198, doi:
10.1126/science.1194653.
17.Yao, J., Irwin, R. W., Zhao, L., Nilsen, J.,
Hamilton, R. T., et al. (2009) Mitochondrial bioenergetic deficit
precedes Alzheimer’s pathology in female mouse model of
Alzheimer’s disease, Proc. Natl. Acad. Sci. USA,
106, 14670-14675, doi: 10.1073/pnas.0903563106.
18.Resende, R., Moreira, P. I., Proenca, T.,
Deshpande, A., Busciglio, J., et al. (2008) Brain oxidative stress in a
triple-transgenic mouse model of Alzheimer disease, Free Radic.
Biol. Med., 44, 2051-2057, doi:
10.1016/j.freeradbiomed.2008.03.012.
19.Sensi, S. L., Rapposelli, I. G., Frazzini, V.,
and Mascetra, N. (2008) Altered oxidant-mediated intraneuronal zinc
mobilization in a triple transgenic mouse model of Alzheimer’s
disease, Exp. Gerontol., 43, 488-492, doi:
10.1016/j.exger.2007.10.018.
20.Chou, J. L., Shenoy, D. V., Thomas, N.,
Choudhary, P. K., Laferla, F. M., et al. (2011) Early dysregulation of
the mitochondrial proteome in a mouse model of Alzheimer’s
disease, J. Proteom., 74, 466-479, doi:
10.1016/j.jprot.2010.12.012.
21.Kandimalla, R., Manczak, M., Fry, D., Suneetha,
Y., Sesaki, H., et al. (2016) Reduced dynamin-related protein 1
protects against phosphorylated Tau-induced mitochondrial dysfunction
and synaptic damage in Alzheimer’s disease, Hum. Mol.
Genet., 25, 4881-4897, doi: 10.1093/hmg/ddw312.
22.Reddy, P. H., and Oliver, D. M. (2019) Amyloid
beta and phosphorylated tau-induced defective autophagy and mitophagy
in Alzheimer’s disease, Cells, 8, 488, doi:
10.3390/cells8050488.
23.Medala, V.K., Gollapelli, B., Dewanjee, S.,
Ogunmokun, G., Kandimalla, R., et al. (2021) Mitochondrial dysfunction,
mitophagy, and role of dynamin-related protein 1 in Alzheimer’s
disease, J. Neurosci. Res., 99, 1120-1135, doi:
10.1002/jnr.24781.
24.Fuente-Muñoz, C. E., Rosas-Lemus, M.,
Moreno-Castilla, P., Bermúdez-Rattoni, F., Uribe-Carvajal, S.,
et al. (2020) Age-dependent decline in synaptic mitochondrial function
is exacerbated in vulnerable brain regions of female 3xTg-AD mice,
Int. J. Mol. Sci., 21, 8727, doi:
10.3390/ijms21228727.
25.Shefa, U., Jeong, N. Y., Song, I. O., Chung, H.
J., Kim, D., et al. (2019) Mitophagy links oxidative stress conditions
and neurodegenerative diseases, Neural Regen. Res., 14,
749-756, doi: 10.4103/1673-5374.249218.
26.Morton, H., Kshirsagar, S., Orlov, E., Bunquin,
L. E., Sawant, N., et al. (2021) Defective mitophagy and synaptic
degeneration in Alzheimer’s disease: focus on aging, mitochondria
and synapse, Free Radic. Biol. Med., 172, 652-667, doi:
10.1016/j.freeradbiomed.2021.07.013.
27.Reiss, A. B., Arain, H. A., Stecker, M. M.,
Siegart, N. M., and Kasselman, L. J. (2018) Amyloid toxicity in
Alzheimer’s disease, Rev. Neurosci., 29, 613-627,
doi: 10.1515/revneuro-2017-0063.
28.Camilleri, A., Ghio, S., Caruana, M., Weckbecker,
D., Schmidt, F., et al. (2020) Tau-induced mitochondrial membrane
perturbation is dependent upon cardiolipin, Biochim. Biophys. Acta
Biomembr., 1862, 183064, doi:
10.1016/j.bbamem.2019.183064.
29.Tang, Z., Ioja, E., Bereczki, E., Hultenby, K.,
Li, C., et al. (2015) mTor mediates tau localization and secretion:
implication for Alzheimer’s disease, Biochim. Biophys.
Acta, 1853, 1646-1657, doi:
10.1016/j.bbamcr.2015.03.003.
30.Amorim, J. A., Canas, P. M., Tome, A. R., Rolo,
A. P., Agostinho, P., et al. (2017) Mitochondria in excitatory and
inhibitory synapses have similar susceptibility to amyloid-beta
peptides modeling Alzheimer’s disease, J. Alzheimer’s
Dis., 60, 525-536, doi: 10.3233/JAD-170356.
31.Camilleri, A., Zarb, C., Caruana, M., Ostermeier,
U., Ghio, S., et al. (2013) Mitochondrial membrane perermeabilization
by amyloid aggregates and protection by polyphenols, Biochim.
Biophys. Acta, 1828, 2532-2543, doi:
10.1016/j.bbamem.2013.06.026.
32.Ardail, D., Privat, J. P., Egret-Charlier, M.,
Levrat, C., Lerme, F., et al. (1990) Mitochondrial contact sites. Lipid
composition and dynamics, J. Biol. Chem., 265,
18797-18802.
33.Paradies, G., Paradies, V., De Benedictis, V.,
Ruggiero, F. M., and Petrosillo, G. (2014) Functional role of
cardiolipin in mitochondrial bioenergetics, Biochim. Biophys.
Acta, 1837, 408-417, doi: 10.1016/j.bbabio.2013.10.006.
34.Suga, K., Hamasaki, A., Chinzaka, J., and
Umakoshi, H. (2016) Liposomes modified with cardiolipin can act as a
platform to regulate the potential flux of NADP+-dependent
isocitrate dehydrogenase, Metab. Eng. Commun., 3, 8-14,
doi: 10.1016/j.meteno.2015.11.002.
35.Schug, Z. T., and Gottlieb, E. (2009) Cardiolipin
acts as a mitochondrial signalling platform to launch apoptosis,
Biochim. Biophys. Acta, 1788, 2022-2031, doi:
10.1016/j.bbamem.2009.05.004.
36.Lasagna-Reeves, C. A., Castillo-Carranza, D. L.,
Sengupta, U., Clos, A. L., Jackson, G. R., et al. (2011) Tau oligomers
impair memory and induce synaptic and mitochondrial dysfunction in
wild-type mice, Mol. Neurodegener., 6, 39, doi:
10.1186/1750-1326-6-39.
37.Du, H., Guo, L., Yan, S., Sosunov, A. A.,
McKhann, G. M., and Yan, S. S. (2010) Early deficits in synaptic
mitochondria in an Alzheimer’s disease mouse model, Proc.
Natl. Acad. Sci. USA, 107, 18670-18675, doi:
10.1073/pnas.1006586107.
38.Esteras, N., Rohrer, J. D., Hardy, J., Wray, S.,
and Abramov, A. Y. (2017) Mitochondrial hyperpolarization in
iPSC-derived neurons from patients of FTDP-17 with 10+16 MAPT mutation
leads to oxidative stress and neurodegeneration, Redox Biol.,
12, 410-422, doi: 10.1016/j.redox.2017.03.008.
39.Eckert, A., Schulz, K.L., Rhein, V., and Gotz, J.
(2010) Convergence of amyloid-beta and tau pathologies on mitochondria
in vivo, Mol. Neurobiol., 41, 107-114, doi:
10.1007/s12035-010-8109-5.
40.Tracy, T. E., Madero-Pérez, J., Swaney, D.
L., Chang, T. S., Moritz, M., et al. (2022) Tau interactome maps
synaptic and mitochondrial processes associated with neurodegeneration,
Cell, 185, 712-728, doi: 10.1016/j.cell.2021.12.041.
41.Sanz-Blasco, S., Valero, R. A., Rodriguez-Crespo,
I., Villalobos, C., and Nunez, L. (2008) Mitochondrial Ca2+
overload underlies Abeta oligomers neurotoxicity providing an
unexpected mechanism of neuroprotection by NSAIDs, PLoS One,
3, e2718, doi: 10.1371/journal.pone.0002718.
42.Pallo, S. P., and Johnson, G. V. W. (2015) Tau
facilitates Aβ-induced loss of mitochondrial membrane potential
independent of cytosolic calcium fluxes in mouse cortical neurons,
Neurosci. Lett., 597, 32-37, doi:
10.1016/j.neulet.2015.04.021.
43.Palikaras, K., Achanta, K., Choi, S., Akbari, M.,
and Bohr, V. A. (2021) Alteration of mitochondrial homeostasis is an
early event in a C. elegans model of human tauopathy, Aging
(Albany NY), 13, 23876-23894, doi:
10.18632/aging.203683.
44.Zheng, J., Akbari, M., Schirmer, C., Reynaert, M.
L., Loyens, A., et al. (2020) Hippocampal tau oligomerization early in
tau pathology coincides with a transient alteration of mitochondrial
homeostasis and DNA repair in a mouse model of tauopathy, Acta
Neuropathol. Commun., 8, 25, doi:
10.1186/s40478-020-00896-8.
45.Jara, C., Aránguiz, A., Cerpa, W.,
Tapia-Rojas, C., and Quintanilla, R. A. (2018) Genetic ablation of tau
improves mitochondrial function and cognitive abilities in the
hippocampus, Redox Biol., 18, 279-294, doi:
10.1016/j.redox.2018.07.010.
46.Jara, C., Cerpa, W., Tapia-Rojas, C., and
Quintanilla, R. A. (2021) Tau deletion prevents cognitive impairment
and mitochondrial dysfunction age associated by a mechanism dependent
on cyclophilin-D, Front. Neurosci., 14, 586710, doi:
10.3389/fnins.2020.586710.
47.Jagasia, R., Grote, P., Westermann, B., and
Conradt, B. (2005) DRP-1-mediated mitochondrial fragmentation during
EGL-1-induced cell death in C. elegans, Nature,
433, 754-760, doi: 10.1038/nature03316.
48.Malka, F., Guillery, O., Cifuentes-Diaz, C.,
Guillou, E., Belenguer, P., et al. (2006) Separate fusion of outer and
inner mitochondrial membranes, EMBO Rep., 6, 853-859,
doi: 10.1038/sj.embor.7400488.
49.Chen, H., Detmer, S. A., Ewald, A. J., Griffin,
E. E., Fraser, S. E., et al. (2003) Mitofusins Mfn1 and Mfn2
coordinately regulate mitochondrial fusion and are essential for
embryonic development, J. Cell Biol., 160, 189-200, doi:
10.1083/jcb.200211046.
50.Ishihara, N., Fujita, Y., Oka, T., and Mihara, K.
(2006) Regulation of mitochondrial morphology through proteolytic
cleavage of OPA1, EMBO J., 25, 2966-2977, doi:
10.1038/sj.emboj.7601184.
51.Otara, H., Wang, C., Cleland, M. M., Setoguchi,
K., Yokota, S., et al. (2010) Mff is an essential factor for
mitochondrial recruitment of Drp1 during mitochondrial fission in
mammalian cells, J. Cell Biol., 191, 1141-1158, doi:
10.1083/jcb.201007152.
52.Chan, D. C. (2006) Mitochondria: dynamic
organelles in disease, aging, and development, Cell, 125,
1241-1252, doi: 10.1016/j.cell.2006.06.010.
53.Trease, A. J., George, J. W., Roland, N. J.,
Lichter, E. Z., Emanuel, K., et al. (2022) Hyperphosphorylated human
tau accumulates at the synapse, localizing on synaptic mitochondrial
outer membranes and disrupting respiration in a mouse model of
tauopathy, Front. Mol. Neurosci., 15, 852368, doi:
10.3389/fnmol.2022.852368.
54.Quintanilla, R. A., Tapia-Monsalves, C., Vergara,
E. H., Pérez, M. J., and Aranguiz, A. (2020) Truncated tau
induces mitochondrial transport failure through the impairment of TRAK2
protein and bioenergetics decline in neuronal cells, Front. Cell
Neurosci., 14, 175, doi: 10.3389/fncel.2020.00175.
55.Jeong, Y. Y., Jia, N., Ganesan, D., and Cai, Q.
(2022) Broad activation of the PRKN pathway triggers synaptic failure
by disrupting synaptic mitochondrial supply in early tauopathy,
Autophagy, 18, 1472-1474, doi:
10.1080/15548627.2022.2039987.
56.Jarero-Basulto, J. J., Luna-Munoz, J., Mena, R.,
Kristofikova, Z., Ripova, D., et al. (2013) Proteolytic cleavage of
polymeric tau protein by caspase-3: implications for Alzheimer’s
disease, J. Neuropathol. Exp. Neurol., 72, 1145-1161,
doi: 10.1097/NEN.0000000000000013.
57.Kopeikina, K. J., Carlson, G. A., Pitstick, R.,
Ludvigson, A. E., Peters, A., et al. (2011) Tau accumulation causes
mitochondrial distribution deficits in neurons in a mouse model of
tauopathy and in human Alzheimer’s disease brain, Am. J.
Pathol., 179, 2071-2082, doi:
10.1016/j.ajpath.2011.07.004.
58.Plucinska, G., Paquet, D., Hruscha, A., Godinho,
L., Haass, C., et al. (2012) In vivo imaging of
disease-related mitochondrial dynamics in a vertebrate model system,
J. Neurosci., 32, 16203-16212, doi:
10.1523/JNEUROSCI.1327-12.2012.
59.Shahpasand, K., Uemura, I., Saito, T., Asano, T.,
Hata, K., et al. (2012) Regulation of mitochondrial transport and
inter-microtubule spacing by tau phosphorylation at the sites
hyperphosphorylated in Alzheimer’s disease, J. Neurosci.,
32, 2430-2441, doi: 10.1523/JNEUROSCI.5927-11.2012.
60.Kandimalla, R., Manczak, M., Pradeepkiran, J. A.,
Morton, H., and Reddy, P. H. (2022) A partial reduction of Drp1
improves cognitive behavior and enhances mitophagy, autophagy and
dendritic spines in a transgenic Tau mouse model of Alzheimer disease,
Hum. Mol. Genet., 31, 1788-1805, doi:
10.1093/hmg/ddab360.
61.Abtahi, S. L., Masoudi, R., and Haddadi, M.
(2020) The distinctive role of tau and amyloid beta in mitochondrial
dysfunction through alteration in Mfn2 and Drp1 mRNA levels: a
comparative study in Drosophila melanogaster,
Gene, 754, 144854, doi: 10.1016/j.gene.2020.144854.
62.Alavi, M. V. (2021) Tau phosphorylation and OPA1
proteolysis are unrelated events: implications for Alzheimer’s
disease, Biochim. Biophys. Acta Mol. Cell. Res., 1868,
119116, doi: 10.1016/j.bbamcr.2021.119116.
63.Kerr, J. S., Adriaanse, B. A., Greig, N. H.,
Mattson, M. P., Cader, M. Z., et al. (2017) Mitophagy and
Alzheimer’s disease: cellular and molecular mechanisms, Trends
Neurosci., 40, 151-166, doi: 10.1016/j.tins.2017.01.002.
64.Hu, Y., Li, X. C., Wang, Z. H., Luo, Y., Zhang,
X., et al. (2016) Tau accumulation impairs mitophagy via increasing
mitochondrial membrane potential and reducing mitochondrial Parkin,
Oncotarget, 7, 17356-17368, doi:
10.18632/oncotarget.7861.
65.Cummins, N., Tweedie, A., Zuryn, S.,
Bertran-Gonzalez, J., and Götz, J. (2019) Disease-associated tau
impairs mitophagy by inhibiting Parkin translocation to mitochondria,
EMBO J., 38, e99360, doi: 10.15252/embj.201899360.
66.Corsetti, V., Florenzano, F., Atlante, A., Bobba,
A., Ciotti, M. T., et al. (2015) NH2-truncated human tau
induces deregulated mitophagy in neurons by aberrant recruitment of
Parkin and UCHL-1: implications in Alzheimer’s disease, Hum.
Mol. Genet., 24, 3058-3081, doi: 10.1093/hmg/ddv059.
67.Escobar-Henriques, M., and Langer, T. (2014)
Dynamic survey of mitochondria by ubiquitin, EMBO Rep.,
15, 231-243, doi: 10.1002/embr.201338225.
68.Hegde, A. N., and DiAntonio, A. (2002) Ubiquitin
and the synapse, Nat. Rev. Neurosci., 3, 854-861, doi:
10.1038/nrn961.
69.Zhu, X., Perry, G., Smith, M. A., and Wang, X.
(2013) Abnormal mitochondrial dynamics in the pathogenesis of
Alzheimer’s disease, J. Alzheimer’s Dis., 33,
253-262, doi: 10.3233/JAD-2012-129005.
70.Amadoro, G., Corsetti, V., Florenzano, F.,
Atlante, A., Bobba, A., et al. (2014) Morphological and bioenergetic
demands underlying the mitophagy in post-mitotic neurons: the
pink-parkin pathway, Front. Aging Neurosci., 6, 18, doi:
10.3389/fnagi.2014.00018.
71.Reddy, P. H., Tripathi, R., Troung, Q., Tirumala,
K., Reddy, T. P., et al. (2012) Abnormal mitochondrial dynamics and
synaptic degeneration as early events in Alzheimer’s disease:
implications to mitochondria-targeted antioxidant therapeutics,
Biochim. Biophys. Acta, 1822, 639-649, doi:
10.1016/j.bbadis.2011.10.011.
72.Koyano, F., Okatsu, K., Ishigaki, S., Fujioka,
Y., Kimura, M., et al. (2013) The principal PINK1 and Parkin cellular
events triggered in response to dissipation of mitochondrial membrane
potential occur in primary neurons, Genes Cells., 18,
672-681, doi: 10.1111/gtc.12066.
73.Bingol, B., Tea, J. S., Phu, L., Reichelt, M.,
Bakalarski, C. E., et al. (2014) The mitochondrial deubiquitinase USP30
opposes parkin-mediated mitophagy, Nature, 510, 370-375,
doi: 10.1038/nature13418.
74.Osaka, H., Wang, Y.L., Takada, K., Takizawa, S.,
Setsuie, R., et al. (2003) Ubiquitin carboxy-terminal hydrolase L1
binds to and stabilizes monoubiquitin in neuron, Hum. Mol.
Genet., 12, 1945-1958, doi: 10.1093/hmg/ddg211.
75.Liu, Y., Fallon, L., Lashuel, H. A., Liu, Z., and
Lansbury, P. T., Jr. (2002) The UCH-L1 gene encodes two opposing
enzymatic activities that affect alpha-synuclein degradation and
Parkinson’s disease susceptibility, Cell, 111,
209-218, doi: 10.1016/s0092-8674(02)01012-7.
76.Cartier, A. E., Djakovic, S. N., Salehi, A.,
Wilson, S. M., Masliah, E., et al. (2009) Regulation of synaptic
structure by ubiquitin C-terminal hydrolase L1, J.
Neurosci., 29, 7857-7868, doi:
10.1523/JNEUROSCI.1817-09.2009.
77.Chen, F., Sugiura, Y., Myers, K. G., Liu, Y., and
Lin, W. (2010) Ubiquitin carboxyl-terminal hydrolase L1 is required for
maintaining the structure and function of the neuromuscular junction,
Proc. Natl. Acad. Sci. USA, 107, 1636-1641, doi:
10.1073/pnas.0911516107.
78.Guglielmotto, M., Monteleone, D., Boido, M.,
Piras, A., Giliberto, L., et al. (2012) Aβ1-42-mediated
down-regulation of Uch-L1 is dependent on NF-κB activation and
impaired BACE1 lysosomal degradation, Aging Cell, 11,
834-844, doi: 10.1111/j.1474-9726.2012.00854.x.
79.Poon, W. W., Carlos, A. J., Aguilar, B. L.,
Berchtold, N. C., Kawano, C. K., et al. (2013) β-Amyloid (Aβ)
oligomers impair brain-derived neurotrophic factor retrograde
trafficking by down-regulating ubiquitin C-terminal hydrolase,
UCH-L1, J. Biol. Chem., 288, 16937-16948, doi:
10.1074/jbc.M113.463711.
80.Zanon, A., Rakovic, A., Blankenburg, H.,
Doncheva, N. T., Schwienbacher, C., et al. (2013) Profiling of Parkin-
binding partners using tandem affinity purification, PLoS One,
8, e78648, doi: 10.1371/journal.pone.0078648.
81.Shevtsova, E. F., Maltsev, A. V., Vinogradova, D.
V., Shevtsov, P. N., and Bachurin, S. O. (2021) Mitochondria as a
promising target for developing novel agents for treating
Alzheimer’s disease, Med. Res. Rev., 41, 803-827,
doi: 10.1002/med.21715.
82.Johri, A. (2021) Disentangling mitochondria in
Alzheimer’s disease, Int. J. Mol. Sci., 22, 11520,
doi: 10.3390/ijms222111520.
83.Mary, A., Eysert, F., Checler, F., and Chami, M.
(2022) Mitophagy in Alzheimer’s disease: molecular defects and
therapeutic approaches, Mol. Psychiatry, doi:
10.1038/s41380-022-01631-6.
84.Kshirsagar, S., Sawant, N., Morton, H., Reddy, A.
P., and Reddy, P. H. (2021) Mitophagy enhancers against phosphorylated
Tau-induced mitochondrial and synaptic toxicities in Alzheimer’s
disease, Pharmacol. Res., 174, 105973, doi:
10.1016/j.phrs.2021.105973.
85.Feniouk, B. A., and Skulachev, V. P. (2017)
Cellular and molecular mechanisms of action of mitochondria-targeted
antioxidants, Curr. Aging Sci., 10, 41-48, doi:
10.2174/1874609809666160921113706.
86.Plotnikov, E. Y., Silachev, D. N., Jankauskas, S.
S., Rokitskaya, T. I., Chupyrkina, A. A., et al. (2012) Mild uncoupling
of respiration and phosphorylation as a mechanism providing nephro- and
neuroprotective effects of penetrating cations of the SkQ family,
Biochemistry (Moscow), 77, 1029-1037, doi:
10.1134/s0006297912090106.
87.Lukashev, A. N., Skulachev, M. V., Ostapenko, V.,
Savchenko, A. Y., Pavshintsev, V. V., et al. (2014) Advances in
development of rechargeable mitochondrial antioxidants, Prog. Mol.
Biol. Transl. Sci., 127, 251-265, doi:
10.1016/b978-0-12-394625-6.00010-6.
88.Isaev, N. K., Stelmashook, E. V., Genrikhs, E.
E., Korshunova, G. A., Sumbatyan, N. V., et al. (2016) Neuroprotective
properties of mitochondria-targeted antioxidants of the SkQ-type,
Rev. Neurosci., 27, 849-855, doi:
10.1515/revneuro-2016-0036.
89.Oddo, S., Caccamo, A., Shepherd, J. D., Murphy,
M. P., Golde, T. E., et al. (2003) Triple-transgenic model of
Alzheimer’s disease with plaques and tangles: intracellular
Aβ and synaptic dysfunction, Neuron, 39, 409-421,
doi: 10.1016/s0896-6273(03)00434-3.
90.Young, M. L., and Franklin, J. L. (2019) The
mitochondria-targeted antioxidant MitoQ inhibits memory loss,
neuropathology, and extends lifespan in aged 3xTg-AD mice, Mol. Cell
Neurosci., 101, 103409, doi: 10.1016/j.mcn.2019.103409.
91.Samluk, L., Ostapczuk, P., and Dziembowska, M.
(2022) Long-term mitochondrial stress induces early steps of Tau
aggregation by increasing reactive oxygen species levels and affecting
cellular proteostasis, Mol. Biol. Cell, 33, ar67, doi:
10.1091/mbc.E21-11-0553.
92.Stefanova, N. A., Muraleva, N. A., Maksimova, K.
Y., Rudnitskaya, E. A., Kiseleva, E., et al. (2016) An antioxidant
specifically targeting mitochondria delays progression of
Alzheimer’s disease-like pathology, Aging (Albany NY),
8, 2713-2733, doi: 10.18632/aging.101054.
93.Trushina, E., Trushin, S., and Hasan, F. (2022)
Mitochondrial complex I as a therapeutic target for Alzheimer’s
disease, Acta Pharm. Sin. B, 12, 483-495, doi:
10.1016/j.apsb.2021.11.003.
94.Stojakovic, A., Chang, S. Y., Nesbitt, J.,
Pichurin, N. P., Ostroot, M. A., et al. (2021) Partial inhibition of
mitochondrial complex I reduces tau pathology and improves energy
homeostasis and synaptic function in 3xTg-AD mice, J.
Alzheimer’s Dis., 79, 335-353, doi:
10.3233/JAD-201015.
95.Singulani, M. P., De Paula, V. J. R., and
Forlenza, O. V. (2021) Mitochondrial dysfunction in Alzheimer’s
disease: therapeutic implications of lithium, Neurosci. Lett.,
760, 136078, doi: 10.1016/j.neulet.2021.136078.
96.Tayanloo-Beik, A., Kiasalari, Z., and Roghani, M.
(2022) Paeonol ameliorates cognitive deficits in streptozotocin murine
model of sporadic Alzheimer’s disease via attenuation of
oxidative stress, inflammation, and mitochondrial dysfunction, J.
Mol. Neurosci., 72, 336-348, doi:
10.1007/s12031-021-01936-1.
97.Guo, W., Zeng, Z., Xing, C., Zhang, J., Bi, W.,
et al. (2022) Stem cells from human exfoliated deciduous teeth affect
mitochondria and reverse cognitive decline in a senescence-accelerated
mouse prone 8 model, Cytotherapy, 24, 59-71, doi:
10.1016/j.jcyt.2021.07.018.
98.Salehi, P., Shahmirzadi, Z. Y., Mirrezaei, F. S.,
Boushehri, F. S., Mayahi, F., et al. (2019) A hypothetic role of
minocycline as a neuroprotective agent against methylphenidate-induced
neuronal mitochondrial dysfunction and tau protein
hyper-phosphorylation: possible role of PI3/Akt/GSK3β signaling
pathway, Med. Hypotheses, 128, 6-10, doi:
10.1016/j.mehy.2019.04.017.
99.Colman, R. J., Anderson, R. M., Johnson, S. C.,
Kastman, E. K., Kosmatka, K. J., et al. (2009) Caloric restriction
delays disease onset and mortality in rhesus monkeys, Science,
325, 201-204, doi: 10.1126/science.1173635.
100.Sohal, R. S., and Weindruch, R. (1996)
Oxidative stress, caloric restriction, and aging, Science,
273, 59-63, doi: 10.1126/science.273.5271.59.
101.Sanz, A., Caro, P., Ibanez, J., Gomez, J.,
Gredilla, R., et al. (2005) Dietary restriction at old age lowers
mitochondrial oxygen radical production and leak at complex I and
oxidative DNA damage in rat brain, J. Bioenerg. Biomembr.,
37, 83-90, doi: 10.1007/s10863-005-4131-0.
102.Singh, R., Lakhanpal, D., Kumar, S., Sharma,
S., Kataria, H., et al. (2012) Late-onset intermittent fasting dietary
restriction as a potential intervention to retard age-associated brain
function impairments in male rats, Age (Dordr), 34,
917-933, doi: 10.1007/s11357-011-9289-2.
103.Cerqueira, F. M., Cunha, F. M., Laurindo, F.
R., and Kowaltowski, A. J. (2012) Calorie restriction increases
cerebral mitochondrial respiratory capacity in a
NO•-mediated mechanism: impact on neuronal survival,
Free Radic. Biol. Med., 52, 1236-1241, doi:
10.1016/j.freeradbiomed.2012.01.011.
104.Lambert, A. J., Wang, B., Yardley, J., Edwards,
J., and Merry, B. J. (2004) The effect of aging and caloric restriction
on mitochondrial protein density and oxygen consumption, Exp.
Gerontol., 39, 289-295, doi:
10.1016/j.exger.2003.12.009.
105.Halagappa, V. K. M., Guo, Z., Pearson, M.,
Matsuoka, Y., Cutler, R. G., et al. (2007) Intermittent fasting and
caloric restriction ameliorate age-related behavioral deficits in the
triple-transgenic mouse model of Alzheimer’s disease,
Neurobiol. Dis., 26, 212-220, doi:
10.1016/j.nbd.2006.12.019.
106.Kang, K., Xu, P., Wang, M., Chunyu, J., Sun,
X., et al. (2020) FGF21 attenuates neurodegeneration through modulating
neuroinflammation and oxidant-stress, Biomed. Pharmacother.,
129, 110439, doi: 10.1016/j.biopha.2020.110439.
107.Mohamed, T. M., Youssef, M. A. M., Bakry, A.
A., and El-Keiy, M. M. (2021) Alzheimer's disease improved through the
activity of mitochondrial chain complexes and their gene expression in
rats by boswellic acid, Metab. Brain Dis., 36, 255-264,
doi: 10.1007/s11011-020-00639-7.
108.Ji, D., Wu, X., Li, D., Liu, P., Zhang, S., et
al. (2020) Protective effects of chondroitin sulphate nano-selenium on
a mouse model of Alzheimer’s disease, Int. J. Biol.
Macromol., 154, 233-245, doi:
10.1016/j.ijbiomac.2020.03.079.
109.Saretzki, G., and Wan, T. (2021) Telomerase in
brain: the new kid on the block and its role in neurodegenerative
diseases, Biomedicines, 9, 490, doi:
10.3390/biomedicines9050490.